

Abstract
The ability to sense and adapt to changes in pO2 is crucial for basic metabolism in most organisms, leading to elaborate pathways for sensing hypoxia (low pO2). This review focuses on the mechanisms utilized by mammals and bacteria to sense hypoxia. While responses to acute hypoxia in mammalian tissues lead to altered vascular tension, the molecular mechanism of signal transduction is not well understood. In contrast, chronic hypoxia evokes cellular responses that lead to transcriptional changes mediated by the hypoxia inducible factor (HIF), which is directly controlled by post-translational hydroxylation of HIF by the non-heme Fe(II)/αKG-dependent enzymes FIH and PHD2. Research on PHD2 and FIH is focused on developing inhibitors and understanding the links between HIF binding and the O2 reaction in these enzymes. Sulfur speciation is a putative mechanism for acute O2-sensing, with special focus on the role of H2S. This sulfur-centered model is discussed, as are some of the directions for further refinement of this model. In contrast to mammals, bacterial O2-sensing relies on protein cofactors that either bind O2 or oxidatively decompose. The sensing modality for bacterial O2-sensors is either via altered DNA binding affinity of the sensory protein, or else due to the actions of a two-component signaling cascade. Emerging data suggests that proteins containing a hemerythrin-domain, such as FBXL5, may serve to connect iron sensing to O2-sensing in both bacteria and humans. As specific molecular machinery becomes identified, these hypoxia sensing pathways present therapeutic targets for diseases including ischemia, cancer, or bacterial infection.
Keywords: HIF, hypoxia, oxygen sensing, cysteine, FNR, FixL
1. Introduction
Oxygen is the terminal electron acceptor used during aerobic respiration, making it essential for the survival of nearly all plants, animals, and single-celled organisms. During respiration, the reduction of O2 into H2O is coupled to the production of cellular energy in the form of ATP. Too little oxygen results in inefficient respiration and a switchover to anaerobic metabolism or cell death if the proper adaptive response is not elicited. Conversely, too much oxygen can lead to oxidative stress and the production of reactive oxygen species (ROS) that can damage the cellular machinery and cause cellular death. As a result of the necessity to regulate O2 availability, organisms have evolved complex and elegant systems for sensing and maintaining O2 homeostasis. O2 sensing pathways typically utilize metalloproteins to either reversibly bind O2 or to catalyze irreversible O2-dependent reactions, leading to transcriptional and metabolic changes in response to varied pO2. With few exceptions, disparate O2-sensing pathways are utilized by mammals and bacteria, leading us to treat O2-sensing in these organisms separately in this review.
The atmosphere is composed of ~ 21 % oxygen, leading to an appreciable concentration of dissolved O2 in water under ambient conditions (288 μM at 20 °C)[1]. However, due to the limits of diffusion and metabolic demands, intracellular O2 levels are often less than 10% of this ambient value in mammalian tissues. There can be further variation according to the tissue, cell type, and developmental stage of the mammal[2]. It's estimated that O2 concentrations are ~5% of the ambient level in typical tissues from adults [3], which leads to a number of finely tuned responses to variations over a narrow range of pO2. Like mammals, bacteria must also adapt to changes in [O2] (or, equivalently, pO2) in order to maintain proper cellular function. As many bacteria are facultative anaerobes and their entire metabolism can change in response to the availability of terminal electron acceptors, in many ways bacterial responses are more extreme than those of mammals. Furthermore, pathogenic bacteria must distinguish changes in pO2 from changes in ROS, as they experience wide variations in pO2 as well as large pulses of ROS due to host immune response.
Biological literature typically refers to pO2 sensing from the perspective of a norm in which [O2] is sufficient – making detection a matter of sensing O2 deficiency, or hypoxia. Importantly, hypoxia has a significant effect on the pathogenesis of many human diseases, including chronic heart and lung disease, myocardial ischemia, pulmonary hypertension, and cancer, all of which are major contributors to the mortality rate worldwide [3-7]. Similarly, due to the important role of O2 in bacterial metabolism, bacterial hypoxia sensing plays a significant role in bacterial proliferation. Having an understanding of the different strategies and cellular mechanisms to sense and regulate pO2 can help improve therapeutic development targeting these pathways, either for treating diseases related to poor oxygenation or for combating pathogens.
2.1 Acute Hypoxia Sensing by Mammalian Tissue
Mammals follow distinct sensing strategies for acute hypoxia (time scale of minutes) and chronic hypoxia (time scale of hours to days). Acute hypoxia sensing starts at the tissue level with specialized cells responding to changes in pO2 through the effects of potassium and calcium ion channels on the membrane potential of the cells [2, 8, 9]. The two dominant physiological responses to acute hypoxia are hypoxic systemic vasodilation and hypoxic pulmonary vasoconstriction, which change vascular tension to modify blood flow. Vasodilation is a response that increases the perfusion of blood to the affected tissue while vasoconstriction is a regulatory mechanism that balances perfusion to ventilation. Although these acute responses are crucial for tissues experiencing hypoxia, and are thought to be mediated within the cell by ion channel signaling pathways, the specific molecular mechanisms linking changes in pO2 to ion channel activity are not well understood.
2.1.1 Neuroepithelial bodies (NEBs) and type 1 glomus cells
The first step in acute hypoxia response is facilitated by specific chemosensory cells termed neuroepithelial bodies (NEBs), and type 1 glomus cells of the carotid body. NEBs are the first responders to pO2 as they line the mucosa of mammalian airways. NADPH oxidase (NOX) has been experimentally demonstrated to mediate the hypoxic response in NEBs via ROS generation [10]. Activation of NOX by protein kinase C (PKC) leads to superoxide (O2-) production which may be the signal to close K+ channels. Counter intuitively, hypoxia results in increased production of O2- by NOX [11]. Whether the chemical signal is O2- or H2O2 is ambiguous as O2- can dismutate to H2O2 and direct addition of H2O2 boosts the whole cell K+ channel current in NEBs (Fig 1.) [12]. The shift in membrane potential due to K+ channel closure causes an influx of Ca2+, triggering neurotransmitter release (serotonin and neuropeptides) from NEBs (Fig. 2) [13]. Although activation of NOX by PKC led to hypoxia sensitive K+ channel closure in the immortalized model NEB H146 cell line [14] NOX inhibitors failed to fully suppress this response, suggesting that multiple mechanisms may be utilized by these cells to sense O2 [15].
Figure 1. Proposed mechanism for ion channel O2 sensing in NEBs.
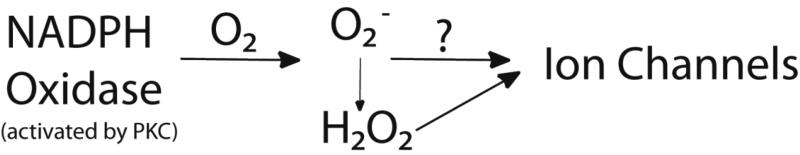
NADPH oxidase mediates the hypoxic response in NEBs via O2- or H2O2.
Figure 2. Acute hypoxia sensing in mammalian tissues.

Acute hypoxia causes inhibition of K+ channels in type 1 glomus cells and NEBs. The depolarization increases intracellular [Ca2+], leading to neurotransmitter release and improved ventilation. In systemic SMCs, acute hypoxia causes KATP channels to open, inhibiting calcium influx and causing vasodilation. Inhibition of K+ channels in pulmonary artery SMCs causes depolarization and calcium influx that results in vasoconstriction.
Type 1 glomus cells are specialized chemoreceptor cells belonging to the carotid body, and are similar to NEBs. In type 1 glomus cells, hypoxia results in closure of K+ channels, which depolarizes the cellular membrane. The resultant membrane depolarization triggers an influx of Ca2+ and the release of neurotransmitters (dopamine) to the carotid-sinus nerve that relays the information to the central nervous system and stimulates ventilation (Fig. 2) [9, 16]. In contrast to the role NOX plays in NEBs, NOX suppresses K+ channel closure in type 1 glomus cells [11, 17], indicating that the mechanisms for O2-sensing is subtly tweaked from tissue to tissue. As the same ion channel types can be either potentiated or inhibited depending on cell types, this would allow individual tissues to fine tune the hypoxic response.
2.1.2 Smooth muscle cells (SMCs)
SMCs work in concert with NEBs and type 1 glomus cells to facilitate tissue-specific responses to acute hypoxia. In pulmonary artery SMCs, acute hypoxia closes the K+ channels which leads to an increase in intracellular [Ca2+], triggering neurotransmitter release and vasoconstriction [18]. In contrast, the hypoxic response in many other tissues is vasodilation due to the function of systemic SMCs. In systemic SMCs, hypoxia causes ATP sensitive K+ (KATP) channels to open and hyperpolarize the membrane, inhibiting Ca2+ influx and causing muscle cell relaxation [18, 19]. These findings and the observation that inhibiting oxidative phosphorylation drastically influences O2 sensing in NEBs and SMCs led to the model that the energy demands of the cell ultimately govern the response to hypoxia, however the scientific literature contains conflicting data regarding this model. For example, myocite relaxation can occur at pO2 levels that don't compromise energy metabolism, suggesting that decreased energy metabolism is not a requirement for hypoxia sensing by ion channels [8]. For a more in depth discussion of this evidence the reader is directed to more focused reviews on that subject [2, 8].
Adaptation to hypoxia may involve vasodilation or vasoconstriction, depending on the affected tissue; however, both result from changes in membrane channels that cause myocite contraction or relaxation. In this manner, adaptation to acute hypoxia relies on a carefully orchestrated response that involves neuroepithelial cells, type 1 glomus cells, and smooth muscle cells. The cellular machinery that play a role in the acute hypoxic response are just now emerging and understanding of the molecular details awaits further research.
2.1.3 Heme oxygenase (HO)
One challenge in this area of study is distinguishing O2-sensing processes from those that merely consume O2. An excellent example of this is HO, an O2-consuming enzyme. One isoform of HO (HO-2) has been proposed to sense hypoxia, based on data from genetic knockouts in mice, but the literature is conflicting. Heme oxygenase oxidatively cleaves heme into CO and biliverdin, consuming O2 and NADPH [20], however this does not necessarily mean that HO senses O2.
There are three isoforms of HO identified of which the first two have been well characterized and reported to have a high affinity for O2 (P50(O2) values: HO-1 = 0.036 μM , HO-2 = 0.013 μM) [2, 21]. HO-1 is an inducible heme oxygenase that is expressed in response to stresses such as hypoxia [22], whereas HO-2 is constitutively expressed and is proposed to modulate Ca2+ activated K+ channels in the membranes of glomus cells[23]. While the extremely low P50(O2) of HO-1 and -2 indicates that these enzymes will be saturated with respect to [O2] over the physiological range of O2, and incapable of a response proportionate to changes in pO2, HO-2 has intriguing connections to cellular hypoxia responses.
HO-2 was proposed to respond to hypoxia due to the HO-2 produced CO opening K+ channels [23]. This model requires that hypoxia leads to decreased [CO], thereby closing the K+ channels and leading to Ca2+ influx and neurotransmitter release [24]. While the very low P50(O2) value for HO-2 appears to eliminate a direct link between CO production and physiologically relevant changes in [O2], a number of observations suggest a connection between HO-2 and responses to hypoxia. At the organismal level, HO-2 deficient mice were reported to be hypoxaemic (decreased pO2 in the blood) and exhibited signs of vasoconstriction activity [25]. Cell-based assays showed that both HO-2 activity and CO could open K+ channels, implicating HO-2 in the O2 sensing pathway for pulmonary artery SMCs [25, 26]. In contrast, other cell-based assays produced conflicting data on the role of HO-2 in glomus cells [25, 27], which may be due to the use of cells from different species in these studies. These findings underscore the need to resolve the mechanisms linking hypoxia to ion channel function, in order to have a more comprehensive understanding of O2 sensing in mammalian tissue.
An important aspect of acute hypoxia sensing that remains elusive is a molecular view connecting decreased pO2 with K+ channel activity. The proposals described above are further muddied by patch-clamp experiments indicating that K+ ion channels may sense O2 directly as there is an absence of detectable modifications to the cytosolic environment, such as changes in pH, ATP levels or [Ca2+] [8]. Although NOX regulates ion channels [10-12], the specific molecular players connecting NOX to K+ channels remain to be clarified. Whether CO, O2-, or the recently proposed H2S (see below), the chemistry underneath acute hypoxia sensing promises to be a fertile field for investigation.
2.2 H2S as an O2 Sensor
A recent proposal for hypoxia sensing in higher organisms is that hydrogen sulfide (H2S), or some other sulfur species, is the direct sensor for acute hypoxia in many tissues of higher organisms [28, 29]. While controversial, there are compelling correlations between O2 and H2S biochemistry, suggesting a connection between these gases. H2S elicits responses similar to those caused by hypoxia in many tissues [28], and the molecular players are more fully identified than for the CO and O2- models discussed above. The key features of this hypothesis are: the O2-sensitive speciation of sulfur into reduced and oxidized pools to signal changes in pO2; and the transduction of this signal by an unknown mechanism into cellular responses to hypoxia.
At a very basic level, the speciation of sulfur into reduced (H2S) and oxidized (SOx) pools depends on the availability of O2, leading to a correlation between hypoxia and elevated [H2S] within cells [30]. While a simplified view suggests that this is due to the balance between the cytosolic metabolism of S-containing compounds to produce H2S and the mitochondrial oxidation of H2S to SSO32- and SO42- (Fig. 3), the story is somewhat more complex. In particular, the distribution of various enzymes involved in sulfur metabolism may be more varied than previously thought. As oxidation to form SSO32- and SO42- are slowed under conditions of low pO2, the reduced sulfur pool increases under hypoxic conditions. But other factors, such as H2S consumption by ROS [31-33] and H2S production promoted by elevated glutathione levels [34] indicate that H2S levels do not respond solely to changes in pO2. This interplay between various redox pools, pO2 and [H2S], combined with the challenges in measuring different sulfur species [31, 35, 36], makes it difficult to establish a clear causal link between hypoxia and elevated levels of reduced sulfur species.
Figure 3. H2S production and oxidation in the cytosol and mitochondria.

Key compounds are cysteine (Cys), homocysteine (hCys), methionine (Met), and glutathione persulfide (GSSH).
A simplified view of the production of H2S centers on the transsulfuration pathway and on cysteine catabolism [31, 32, 37, 38]. In the transsulfuration pathway, H2S is liberated from cysteine, homocysteine, and cystathionine by the PLP-dependent enzymes cystathione β-synthase (CBS) and cystathione γ-lyase (CSE), which are typically cytosolic enzymes [39]. However, data suggests that CBS and CSE translocate to mitochondria under cellular stress, which may account for cysteine metabolism within the mitochondria [40, 41]. H2S is also produced from Cys by the sequential action of the enzymes cysteine aminotransferase (CAT), which uses αKG as a co-substrate, and mercaptopyruvate sulfotransferase (MST); while CAT and MST are predominantly cytosolic, MST is also found within the mitochondrion [42]. Whereas oxidation of mitochondrial H2S to SO42- leads to excretion, the fate of cytosolic H2S is less clear (Fig. 3). Connections between the metabolism of glutathione (GSH), cysteine, and H2S (as reviewed by Gojon [31]), imply that cytosolic H2S equivalents are fed into the mitochondria for oxidation. Indeed, it was recently proposed that H2S oxidation rather than H2S production is the crucial step in hypoxia sensing [43].
Mitochondrial enzymes oxidize H2S within the mitochondrial matrix, connecting H2S catabolism to respiration through the quinone pool in the inner membrane [37]. The membrane enzyme sulfide quino-oxidoreductase (SQR) oxidizes S2- to S0 using the quinone pool, transferring the S0 equivalent to glutathione to form a persulfide (GSSH) and sending electrons into the respiratory redox chain via catechols. The S0 equivalent is then oxidized to the S4+ oxidation state (sulfite) by the enzyme persulfide dioxygenase (ETHE1) [37, 38, 44] with subsequent conversion either by sulfite oxidase to sulfate, or by rhodanese, which combines a S0 equivalent (RSSH) with sulfite to form thiosulfate (SSO32-). The non-enzymatic reduction of thiosulfate by GSH has been shown to rapidly produce H2S under hypoxic conditions using purified reagents or mammalian tissues, suggesting that this may be a reaction that will rapidly increase the H2S level under acute hypoxia [34].
Cells can both produce and consume H2S in proportion to varied pO2, with GSH, thiosulfate, and sulfite as key players in H2S metabolism. Notably, the rate of H2S consumption decreases when [O2] drops below ~ 20 μM in bovine tissues [30], suggesting that acute hypoxia leads to elevated H2S. Thiosulfate has been proposed to serve as a pool for the rapid reductive release of H2S by reducing agents such as glutathione (GSH), which could lead to the production of H2S under hypoxic conditions [34]. The mitochondrial enzyme persulfide dioxygenase (ETHE1) catalyzes a key O2-consuming step in the catabolism of H2S, producing sulfite [44]. Precisely how the concentrations of these molecular species vary with pO2 is crucial for the H2S-centered model for acute hypoxia sensing. Intriguingly, ETHE1 is a non-heme Fe(II)-dependent oxygenase [44] which binds to the Fe(II) cofactor using a His2Asp facial triad; this is similar to the cofactor structure for the well-established hypoxia sensors factor inhibiting HIF (FIH) and prolyl hydroxylase (PHD), which control the transcriptional activity of HIF, the hypoxia inducible factor (see below). If H2S is a transient hypoxia sensor, it would be a remarkable development if yet another non-heme Fe(II) dioxygenase were to sense O2 in cells. This, however, remains an open question at present.
Exogenously administered H2S (or N-acetyl cysteine) mimics the hypoxic response in a variety of tissues, such as vasoconstriction or vasodilation depending on the organism [30]. However, the responses to elevated [H2S] and low [O2] may be complementary rather than identical, with much cross-talk between the two stressors [32]. While it has been asserted that this indicates that H2S and hypoxia are mediated by the same effector pathways within cells [28], the molecular pathways linking H2S to tissue responses have not yet been identified for testing. An intriguing study from 2011 found that, while HIF-1 was required for the transcriptional effects of chronic H2S in C. elegans, the identity of genes regulated by chronic H2S administration were distinct from those regulated by hypoxia [45]. It is possible that better discrimination of responses to acute hypoxia from those due to chronic hypoxia would help to clarify the specific effector pathways induced by H2S.
Although the proposal that H2S levels report on acute hypoxia is consistent with a number of correlations, the molecular basis for signal transduction is unclear. Two key questions moving forward are: How is sulfur speciation transduced as a signal? How does the mitochondrial sulfur pool connect with the cytosolic sulfur pool? It has been suggested that post-translational protein modification via sulfhydration is the molecular basis for signaling increased [H2S] [46]. In sulfhydration, a protein-bound persulfide (R-SSH) is formed by one of several possible mechanisms [31, 47]. There is an oxidant-promoted reaction (RSH + H2S → RSSH + 2 H+ + 2 e-), or sulfane transfer from an endogenous persulfide (GSSH + RSH → GSH + RSSH). Notably, this signaling mode suggests that sulfane sulfur (S0) may be the sulfur signaling agent rather than H2S [47]. We were unable to find any tests of sulfhydration connected to hypoxia sensing at the time of this review.
The connections between cytosolic and mitochondrial sulfur speciation are not completely understood. Under some conditions the persulfide form of the cytosolic enzyme MST may be imported into the mitochondria [42], thereby importing S0 equivalents in limited concentrations. However, in order to excrete the sulfide formed within the cytosol, there must be bulk mitochondrial import followed by oxidation, or else excretion into the circulatory system. Alternatively, O2-sensing may be solely a function of sulfur speciation within the mitochondrion [43]. Despite these gaps, the proposal that H2S senses acute hypoxia is much richer in molecular detail than the proposals connecting K+ ion channels to hypoxia via CO and O2- signaling.
2.3 Hypoxia Inducible Factor (HIF) and the HIF hydroxylases (FIH and PHD)
The response to acute hypoxia is largely a matter of vascular tension to modify blood delivery to tissues, however chronic hypoxia leads to responses within cells at the transcriptional level. HIF is the most important monitor of pO2 as it controls the expression of hundreds of genes over a wide range of physiological O2 tensions [48]. HIF also links acute and chronic responses to hypoxia by controlling a number of adaptations to pO2 including erythropoiesis, angiogenesis, glucose metabolism, cell proliferation, and glycolysis [3, 49]. Several isoforms of HIF are known, with HIF-1 being the dominant player in the human hypoxia response.
After the discovery of HIF-1 in the nuclear extracts of hypoxic cells in 1992 [50], it was soon realized that HIF-1 was a heterodimeric protein consisting of α and β subunits [51], and that HIF-1α levels and transcriptional activity increased with decreasing cellular O2 concentrations [52]. Three isoforms of the HIFα transcription factor have been identified, HIF-1α -3α, which are evolutionarily conserved in all metazoans [53] but absent from bacteria, yeast, and plants. While the abundance of HIF-α increase under hypoxia [49, 51], HIF-1β is a constitutively expressed protein that is also known as the aryl hydrocarbon receptor nuclear translocator (ARNT). Although both HIF-1α and HIF-2α are capable of initiating transcriptional activity, HIF-1α is believed to be the key regulator of the hypoxic response as the other two isoforms have varied expression levels depending on cell types [3]. HIF-3α, which is incapable of initiating hypoxia induced transcriptional activity, is not closely related to HIF-1α or HIF-2α and appears to serve no direct role in hypoxic sensing [48, 54].
The transcriptional activity of HIF-1α decreases with increasing pO2 due to the competition between nuclear translocation and the post-translational hydroxylation of HIF-1α. HIF-1α accumulates during hypoxia, permitting it enough time to translocate into the nucleus. Once there, it can dimerize with HIF-1β to bind the transcriptional co-activator protein p300 at the promoter regions of genes that allow the cell to survive hypoxia. HIF-1α undergoes O2-dependent post-translational modifications that either marks it for proteasomal degradation or prevents its interaction with p300, halting HIF-1 mediated gene expression (Fig 4). These post-translational modifications to HIF-1α take the form of hydroxylations to specific Pro and Asn residues, the rates of which are proportional to [O2]. Consequently, the transcriptional activity of HIF-1α decreases when the hydroxylation rate exceeds the rate of nuclear localization.
Figure 4. Regulation of HIF-1α by FIH and PHD2.

Posttranslational regulation of HIF-1α by FIH and PHD2 control HIF-1α transcriptional activity and stability under normoxic conditions. During hypoxia, HIF-1α forms a transcription complex with HIF-1β and p300, and initiates target gene expression.
The regulatory enzymes that control HIF-1 activity via posttranslational hydroxylation of asparagine and proline residues are the proteins FIH and PHD. Only one isoform of FIH is currently known, but there are 3 isoforms of PHD (PHD1-3) identified to date, of which PHD2 is recognized as the main regulator of HIF-1α [55]. Both PHD2 (KM(O2) = 250 μM) [56] and FIH (KM(O2) = 90 μM) [49] are O2 sensors as they have an absolute requirement for O2 in order for chemistry to occur, and their activity is proportional to variations in pO2 over the physiological range. The dual regulation of HIF-1α likely ensures strict control of the hypoxic response over a wide range of pO2 levels.
FIH and PHD2 belong to a large superfamily of non-heme Fe(II)/αKG-dependent oxygenases. FIH and PHD2 are proposed to follow the consensus ordered sequential mechanism for this superfamily, in which the co-substrate αKG binds first, then substrate (a domain of HIF-1α) binds second, followed by O2 (Fig. 5). Substrate binding stimulates O2 reactivity, which is typically attributed to the creation of an open coordination site due to aquo release. Oxidative decarboxylation generates a high-valent ferryl intermediate that abstracts hydrogen from an un-activated carbon, followed by rebound chemistry to hydroxylate the substrate (Fig 5). It is worth noting that the ferryl intermediate has been observed for neither FIH nor PHD2, suggesting that the chemistry of these two enzymes may differ in subtle ways from that of the broader αKG oxygenase superfamily. For more comprehensive reviews on the chemistry and structural conservation of αKG oxygenases see reviews [57] and [58].
Figure 5.

Proposed consensus mechanism of the αKG-dependent hydroxylases.
PHD2 and FIH hydroxylate specific residues in the O2-dependent degradation domain (ODD) and the C-terminal transactivation domain (CTAD) of HIF-1α, which leads to decreased transcriptional activity for HIF. Hydroxylation of either of two proline residues (Pro402, Pro564) in the ODD by PHD2 is a prerequisite of HIF-1α recognition by the von Hippel-Lindau protein (pVHL) (Fig. 4) [59], with hydroxylation at Pro564 being ten-fold faster than at Pro402 [60-62]. pVHL is the recognition component of an E3 ubiquitin-protein ligase which targets HIF-1α for rapid proteasomal degradation after successive rounds of ubiquitinylation [63]. Hydroxylation of Asn803 in the CTAD by FIH blocks p300 from binding, stopping expression of HIF-1α target genes (Fig. 4) [64]. O2 mediated regulation of HIF-1α varies slightly between different cells, depending on the O2 requirements in specific cell types [65].
HIF controls the expression of several genes that help mediate the adaptive response to fluctuating pO2, such as NOX [66], HO-1 [22], GLUT-1 [7] and PHD2 [55], placing it at the center of O2–sensing and homeostasis. Although the transcriptional activity of HIF is linked to both acute and chronic hypoxia sensing, the true sensors are FIH and PHD2, which use molecular oxygen to hydroxylate HIFα. Interestingly, HIF controls the expression of PHD2, which is induced under hypoxia. As the KM(O2) for PHD2 is high in relation to physiological pO2 within cells [49, 56], PHD2 has low activity during hypoxia, but its expression would ensure that the proper balance to HIF-1α levels are restored upon reoxygenation of the cell. Whilst the critical players (FIH and PHD2) in HIF mediated pO2 sensing have been identified, key questions that remain to be answered include: How is the chemistry of O2 activation stimulated by enzyme/HIF-1α binding? Are there physiological effectors of PHD2/FIH activity, including other gases such as H2S, CO, and NO? How can we selectively inhibit or increase PHD2/FIH activity? These would facilitate therapeutic targeting of these enzymes, as well as fundamental insight into the chemistry of this superfamily of non-heme Fe(II) αKG-dependent hydroxylases.
The O2-activation by FIH and PHD2 is significantly stimulated by substrate binding [67], as seen for other aKG hydroxylases. This is crucial to their role as O2-sensors because O2-activation in the absence of HIF leads to enzyme inactivation [68]. The best supported model for substrate-stimulated O2-activation in these enzymes focuses on the coordination geometry of the Fe(II) cofactor. The Fe(II) switches from predominantly 6-coordinate to 5-coordinate upon substrate binding due to aquo release, as shown by electronic spectroscopy and computational results for several enzymes from the aKG-dependent hydroxylase family [67, 69, 70]. By creating a coordination site on Fe(II), O2 binding is then forced to follow substrate binding. Although the chemical mechanism of O-O bond cleavage is unclear, back bonding from αKG to the metal likely plays a role in stimulating O2 activation at Fe(II) [69, 70].
Despite the coordination changes observed under non-turnover conditions, certain features of the above model need to be refined. For example, the kinetics of ligand exchange from the Fe(II) cofactor is not known – unless aquo exchange from the Fe(II) is very slow, O2 would be able to access the metal prior to substrate binding. Further, the redox potential of the Fe(II) has been proposed to shift upon substrate binding, thereby facilitating its reaction with O2 [71]. These two points suggest that substrate binding may, in fact, simply increase the equilibrium binding affinities for O2 relative to that of H2O. Another broader question about this model that remains under study is how substrate binding alters contacts within the active site to stimulate O2 activation. Answering these questions will elucidate much about the specific chemistry involved in O-O cleavage as related to hypoxia sensing.
Recent work on the HIF hydroxylases has provided some insight into the molecular mechanism of pO2 activation by the HIF hydroxylases. The mechanical linkage between HIF-1α binding and O2-activation remain unclear in FIH, however this is central to O2 sensing. Although FIH will inactivate in vitro through an auto-oxidation reaction in the absence of HIF-1α [68, 72], HIF-1α binding stimulates the O2 reactivity of FIH many-fold. MCD and CD data of FIH in solution revealed that the Fe(II) cofactor geometry shifts from 6-coordinate to a mixture of 5/6-coordinate upon HIF-1α binding [67], in agreement with the X-ray crystal structure of HIF-1α bound FIH [73] and suggesting that aquo release may limit the O2 reactivity [74]. As the consensus mechanism for this family of enzymes requires that the Fe(II) be 5-coordinate prior to binding O2 [58], the MCD data showing only partial formation of the 5-coordinate Fe(II) suggests that O2 binding or activation may be sluggish in FIH. Further support for this notion comes from a suite of mechanistic data which points to rate-limiting O2 activation for FIH under accessible pO2 [75], which would lead to a direct correlation between enzyme activity and pO2. Notably, modeling and mutation studies demonstrate that the second coordination sphere residues are pivotal for directing O2 reactivity in FIH [67, 76]. The picture which emerges is one in which HIF-1α binding induces structural changes near the Fe(II), including aquo release, which stimulates O2-activation.
PHD2 activity also appears to be limited by the rate of O2-activation, consistent with its role as an O2-sensor. Pre-steady state kinetics indicated that PHD2 reacted with O2 very slowly when compared to other αKG oxygenases, and related freeze-quench experiments failed to isolate the putative ferryl intermediate [77], which was consistent with O2-activation as the rate-limiting step in PHD2. Steady-state kinetics further supports rate-limiting O2-activation in PHD2 [78]. In order for the HIF hydroxylases to be good O2 sensors, O2 binding or a step subsequent to O2 binding should be rate limiting as this would lead to proportionate changes in activity with pO2. The KM(O2) being higher than the physiologically relevant pO2 is also consistent with the notion of the HIF hydroxylases being O2 sensors as this would allow the rate of their chemistry be proportionate to pO2 near physiological levels.
A number of endogenously produced molecules, such as NO, CO, and H2S, are intriguing as putative effectors of the HIF hydroxylases as they may bind in place of O2 and are known to regulate pathways related to hypoxia responses. Nitric oxide (NO) regulates vascular tone, and has been shown to inhibit PHD2 [79] and FIH [80], hinting that this and other transient species could be physiological effectors of the HIF hydroxylases. Although CO binding to the HIF hydroxylases has yet to be demonstrated, endogenous CO has been shown to exhibit anti-apoptotic, anti-inflammatory, and protective responses against hyperoxia-induced lung injury [27]. Unpublished results show that H2S inhibits FIH (IC50 ~ 150 μM) (Taabazuing and Knapp, unpublished) – although this level of H2S is likely to be supraphysiological, it nevertheless further implicates H2S in the overall hypoxia response.
Methods to inhibit the HIF hydroxylases would be highly desired, as they would permit therapeutic targeting of the hypoxia sensing machinery. Although a number of synthetic compounds have been reported to inhibit these enzymes [81-83], challenges include improving both selectivity [81] and the relatively low affinity of the inhibitors (IC50 > 1μM). Inhibitors for FIH and PHD2 are typically structural mimics of αKG, able to bind to the Fe(II) cofactor via one or two ligation points, opening up the likelihood of cross-reactivity with other αKG oxygenases. Inclusion of a chiral center near the Fe(II) binding group improved inhibitor specificity in one study [84], suggesting that further improvements in inhibitors may be eminently achievable with further synthesis and screening efforts.
3. Bacterial O2 Sensing
Bacterial responses to changes in pO2 are crucial for balancing central metabolism as well as inducing cellular machinery needed for specialized metabolic pathways. These pathways include nitrogen fixation, hydrogen production and sulfate reduction, which utilize enzymes with metal cofactors that are highly sensitive to O2. Bacteria rely largely on sensory proteins containing [Fe-S] clusters or hemes to regulate gene expression in response to changes in pO2. In contrast to the oxygenase chemistry of FIH and PHD in higher organisms, which lead to altered protein/protein binding affinities, bacterial sensors typically report on the presence of O2 by changing the sensor/DNA binding affinity.
Bacterial sensor proteins bind O2 to the iron cofactor leading to altered downstream effects. Reversible binding of O2 to the heme protein sensors induces structural changes that affect binding to DNA or other proteins, allowing for reversible and dynamic gene regulation based on the O2-binding equilibrium. Heme-based sensors have been classified by their heme-binding-domains into four families [85]. Well studied heme-sensors include FixL [86], EcDos [87], AxPDEA1 [88], HemAT [89], and the CO-binding sensor CooA [90]. In contrast, O2 binding to [Fe-S] clusters leads to cluster degradation and diminished DNA binding affinity of the sensory proteins, making O2 sensing responsive to multiple steps, such as sensor protein production, cluster degradation, and metallocluster formation. [Fe-S] proteins such as FNR [91], NreB [92], ArnR [93], and WhiB [94] all sense O2; other reactive species such as CO, NO, and reactive oxygen species are sensed in a similar way.
Two modes of bacterial O2 sensing are direct transcriptional control and two-component signaling cascades. The well studied O2-sensor FNR exemplifies direct transcriptional control, as the [Fe4S4]2+ cofactor is degraded by O2, leading to attenuated DNA binding affinity [91]. The classic example of a two component signal transduction pathway for O2 sensing is the FixL/FixJ system, in which the O2 sensing protein (FixL) relays its response to varying pO2 through a histidine kinase (HK) domain to the response regulator protein (FixJ). The sensing proteins in these latter systems are typically heme-based sensing proteins, with exceptions to this including the [Fe-S] two component systems NreBC [92, 95, 96] and AirSR [97].
3.1 Sensing of O2 by transcriptional regulators
Direct O2 sensing at the transcriptional level occurs when the sensor serves the dual roles of O2 binding and DNA binding. FNR is one of the most extensively studied O2-sensing DNA binding proteins. E. coli FNR is a 53 kDa homodimeric protein [98] under anaerobic conditions, with each monomer containing a [Fe4S4]2+ cluster coordinated by four cysteine residues [91, 98, 99]. Dimeric FNR binds to DNA to promote the expression of genes for proteins involved in anaerobic metabolism and to repress those involved in aerobic metabolism [100].
The cofactor of FNR is highly sensitive to O2 in vitro, with ~95% activity at 0.3 μM O2 but only ~50% activity at 6 μM O2 [101], which mirrors in vivo measurements [102]. Upon exposure to O2, the [Fe4S4]2+ cluster of FNR rapidly degrades to [Fe2S2]2+ (Fig. 6) [100, 103, 104]. The rapid [Fe4S4]2+ to [Fe2S2]2+ cluster conversion leads to dissociation of homodimerric FNR into its subunits, ultimately weakening DNA-binding [100, 105]. There are two proposed mechanisms for oxidative decomposition of the [Fe4S4]2+ clusters: metal oxidation [106-109] and sulfur-based oxidation [99, 110, 111]. Regardless of the mechanism for cluster conversion, when exposed to superoxide, [Fe2S2]2+ FNR converts to apo FNR (Fig. 6) [104]. In vivo, reactivation of apo-FNR to [Fe4S4]2+ FNR is possible under anaerobic conditions [112], most likely utilizing the Isc pathway proteins [113].
Figure 6. O2 sensing by FNR.

A) FNR protein domain arrangement. B) FNR [Fe4S4]2+ cluster degradation. In the presence of O2, the FNR [Fe4S4]2+ cofactor is rapidly oxidized to [Fe2S2]2+, leading to dissociation of dimeric FNR. Reconstitution of the [Fe4S4]2+ cluster in FNR occurs through the Isc pathway in vivo, but can also be reconstituted in vitro by exogenous Fe2+ and DTT [111].
3.2 Two component signal transduction
Two component signal transduction is a fundamental signaling pathway primarily found in prokaryotes, with limited examples in eukaryotic systems [114-116]. The two components referred to in these systems are: 1) a sensory protein; and 2) a response-regulator protein. The sensing protein is a multidomain protein with a sensing domain and a histidine kinase (HK) domain. Ligand binding to the sensing domain induces an ATP dependent autophosphorylation on the HK domain. Subsequent phosphoryl group transfer from the HK domain to the response-regulator protein transduces the signal. As phosphorylation of the response-regulator protein can stimulate or repress gene expression of downstream targets depending on the specific system, two-component signal transduction can lead to a wide variety of sensory responses.
Representative bacterial O2-sensing two-component signal transduction systems include Aer2 [117], DosS-R, DosT-R [118], and FixL/FixJ [119]. The FixL/FixJ two component signal transduction sensor pathway exemplifies many of the key features needed for O2 sensing. FixL/FixJ was first identified in Sinorhizobium meliloti (SmFixL) [120] but has been characterized in other bacterial systems including Bradyrhizobium japonicum (BjFixL) and Rhizobium etli (ReFixL), with a homologous FixL in the algae Chlamydomonas reinhardtii [121]. These two-component regulatory systems are found widely, even in Mycobacterium tuberculosis, a pathogen responsible for causing tuberculosis [122].
FixL proteins contain a HK domain and at least one PAS (Per-ARNT-Sim) domain, which contains the heme that binds O2 (Fig. 7) [119]. FixL has a relatively low O2 affinity (KD ~ 100 μM) [123-125], allowing for physiological changes in pO2 to alter the transcriptional activity of FixJ. Under hypoxic conditions, the unligated five coordinate high spin Fe(II) of FixL induces gene expression [126-130] as this state of FixL autophosphorylates a conserved His residue within its HK domain. Phosphoryl transfer to a conserved Asp residue in the response regulator FixJ [86] induces a conformational change that leads to dimerization, thereby increasing affinity for the fixK promoter and inducing gene expression [131, 132]. Upon binding of O2, the Fe(II) converts to low spin six coordinate geometry, flattening the heme ring and disrupting hydrogen bonding interactions to the heme propionate groups. Most notable is the distal arginine residue breaking a salt bridge with a heme propionate group to help stabilize O2, initiating the conformational change of FixL's FG loop needed to inhibit phosphorylation [133-135].
Figure 7.

A) Protein crystal structure of the heme binding PAS domain of FixL from B. japonicum in the unliganded Fe3+ oxidation state (PDB 1LSW) [134], with the conformationally mobile FG loop labelled in black. B) The heme active site of BjFixL. Hydrogen bond interactions to the heme propionate groups have a significant role in shifting the FG loop conformation for oxy-FixL, slowing autophosphorylation [124, 133-135, 150-152]. C) Protein constructs of BjFixL, ReFixL and SmFixL. The empty N-terminal PAS domain likely tunes O2 affinity and influences signal transduction [153-156].
4. Hemerythrin-domain Proteins
The previously discussed O2-sensing strategies used by bacteria and higher organisms are notable for their distinct cofactor utilization. Heme and iron sulfur cluster based O2 sensing are the two most prevalent mechanisms utilized by bacteria, whereas non-heme Fe(II), ion channels, and (possibly) H2S are central to hypoxia sensing in mammalian cells. Recent research shows that hemerythrin-like diiron proteins are emerging as O2 sensors in both bacteria and humans. While hemerythrin (Hr) is a classic O2 binding protein from invertebrates, proteins containing an Hr-like domain have been found in bacteria and mammalian cells [136-138] where they are proposed to sense O2 and/or Fe levels. Two notable examples are bacterial hemerythrin DcrH-Hr found in Desulfovibrio vulgaris, and FBXL5 found in human cells.
DcrH-Hr is thought to function as an O2-sensor, using the [Fe(II) Fe(II)] cofactor found within its Hr-like domain. Invertebrate Hrs reversibly bind O2 to the reduced form of the cofactor to form oxy-Hr: [Fe(II) Fe(II)] + O2 ⇔ [Fe(III) Fe(II) O2-] [139]. As invertebrate oxy-Hr is relatively stable, O2 can be delivered before oxy-Hr undergoes the much slower auto-oxidation process which forms met-Hr: [Fe(II) Fe(III) O2-] → [Fe(III) Fe(III)] + H2O2. In contrast, oxy DcrH-Hr rapidly auto-oxidizes to form an extremely stable met-DcrH-Hr, suggesting a role in O2-sensing rather than reversible O2 delivery [140-142]. Structural features identified in DcrH-Hr that are proposed to promote rapid auto-oxidation and lead to its function as a sensor include a large solvent channel that would facilitate O2 and water access [142].
The signal transduction mechanism for O2-sensing for proteins containing a bacterial Hr domain likely relies on an oxidation-state dependent conformational change in the Hr domain. Recently, O2-sensing was demonstrated for the protein known as Bhr-DGC, a bacterial hemerythrin from Vibrio cholerae [143]. The rate for producing cyclic di-GMP, a secondary messenger in bacteria, by Bhr-DGC decreased by 10-fold when the protein was oxidized to the [Fe(III) Fe(III)] oxidation state, indicating that oxidation by O2 to form met Bhr-DGC led to a detectable change in an important signaling molecule [143].
F-box and leucine-rich repeat protein 5 (FBXL5) contains a HR-like domain that imparts O2-dependent stabilization to the protein. Although it is not clear as to whether or not FBXL5 serves to sense physiological changes in [O2], this protein does connect Fe homeostasis to O2 levels. FBXL5 is an adaptor protein that binds to IRP2 and an SCF E3 ubiquitin ligase, imparting O2 and Fe sensitivity to the IRP2 protein in human cells. This leads to the proteasomal degradation of IRP2 in the presence of Fe and O2 [136, 144] as a cellular signal that [Fe] levels are sufficiently high.
The N-terminal HR domain of FBXL5 reversibly binds a [Fe(II) Fe(II)] cofactor which closely resembles the structure of the canonical Hr [145, 146]. As the reduced form of FBXL5 is in equilibrium with unfolded FBXL5, its degraded by the cell, allowing IRP2 to accumulate. Upon exposure to O2, the cofactor oxidizes to the [Fe(III) Fe(III)] oxidation state which locks in the structure of FBXL5, thereby stabilizing this protein. Oxidized FBXL5 binds IRP2 and an SCF E3 ubiquitin ligase, leading to IRP2 ubiquitinylation and degradation. Consequently, FBXL5 responds to both [Fe] and [O2], with IRP2 degradation as the signal for sufficient levels of these essential compounds. Although it is not clear whether FBXL5 responds to physiological-relevant changes in [O2], it is clear that this protein connects Fe and O2 homeostasis.
It appears there are at least two distinct sensing mechanisms among Hr-like domains. Bacterial Hr-like proteins bind O2 and undergo rapid auto-oxidation [141, 143, 147], forming a stable [Fe(III) Fe(III)] cofactor. In contrast, FBXL5 does not form an observable O2-adduct, suggesting that cofactor oxidation may involve an alternate chemical mechanism. Comparison of the crystal structures of FBXL5 (PDB 3V5Z, 3V5Y, 3V5X) [148] and DcrH-Hr (PBD 2AWC) [142] show the binding pocket of FBXL5 is not large enough for O2 binding, suggesting outer sphere electron transfer between O2 and the [Fe(II) Fe(II)] center [148]. Unlike DcrH-Hr, which has been demonstrated to directly bind O2, there is strong experimental evidence that FBXL5 does not bind O2 at the dinuclear center [145, 148], including NMR studies ruling out a significant conformational changes upon exposure of FBXL5 to O2 [149].
4. Conclusion
Acute and chronic changes in pO2 lead to distinct adaptive responses in mammals, ranging in scale from vascular changes through transcriptional changes. While several biochemical pathways for pO2 sensing are known with varied levels of detail, a number of outstanding research questions remain which focus on the chemical interactions that underlay sensing. Tissue level responses to changed pO2 appear to be mediated by ion-channels, however the molecular mechanisms for transducing pO2 into ion channel activity remain to be defined. Similarly, the proteins that control the transcriptional activity of HIF through post-translational hydroxylation (FIH and PHD enzymes) are well defined, leading to questions that largely center on the chemical mechanisms of O-O bond cleavage and the identification of enzyme regulators/inhibitors. Emerging data suggests that the speciation of sulfur compounds report on hypoxia, with two large questions being the connections between cytosolic and mitochondrial sulfur pools, and the mechanisms for transducing the signal. Continuing research in these areas holds promise in treatments for diseases including ischemia and cancer, where vasculature development and anaerobic metabolism play significant roles in cell growth.
Bacterial hypoxia sensing is mediated by direct transcriptional control over gene expression, and is much better understood than hypoxia sensing in mammals. Two component signaling systems allow for signal transduction based on pO2, classically mediated by O2 binding directly to heme domains. An alternative approach relies on O2-mediated degradation of Fe-S clusters to change protein oligomerization, thereby changing transcriptional activity. As O2 sensing in bacteria is directed toward allowing the organism to adapt metabolically to altered pO2, these pathways could aid in targeting pathogenic bacteria.
Highlights.
Hypoxia sensing leads to important metabolic and transcriptional changes.
Hypoxia sensing occurs by O2 binding, chemical modifications, and two-component signaling.
A variety of metabolites are implicated in hypoxia sensing.
The HIF pathway is the dominant pathway for long-term hypoxia sensing in human cells.
FIH and PHD are the central players in human hypoxia sensing.
Acknowledgements
We thank the NIH for funding (R01-GM077413). JAH and CYT were partially funded by CBI fellowships (T32-GM008515).
Abbreviations
- ARNT
aryl hydrocarbon receptor nuclear translocator
- bHLH
basic helix-loop-helix
- CAT
cysteine aminotransferase
- CBS
cystathione β-synthase (CBS)
- CSE
cystathione γ-lyase
- CTAD
C-terminal transactivation domain
- ETHE1
persulfide dioxygenase
- FBXL5
F-box and Leucine-rich Repeat Protein 5
- FIH
factor inhibiting HIF;
- FNR
fumarate and nitrate reduction regulator
- GSSH
glutathione persulfide
- GST
glutathione S-transferase
- hCys
homocysteine
- HIF
hypoxia inducible factor;
- HK
histidine kinase
- HO
heme oxygenase
- Hr
hemerythrin
- Isc
iron-sulfur cluster
- LC-ESI MS
liquid chromatography-electrospray ionization mass spectrometry
- MST
mercaptopyruvate sulfurtransferase
- NEB
neuroepithelial bodies
- NOX
NADPH oxidase
- ODD
oxygen degradation domain
- PAS
Per-Arnt-Sim
- PHD2
HIF prolyl hydroxylase domain 2
- PKC
protein kinase C
- pVHL
von-Hippel Lindau protein
- ROS
reactive oxygen species
- SMC
smooth muscle cells
- αKG
α-ketoglutarate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lide DR. FL Press; 79, CRC Press Boca Raton: 1998-1999. [Google Scholar]
- 2.Ward JPT. Biochim. Biophys. Acta. 2008;1777:1–14. doi: 10.1016/j.bbabio.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semenza GL. Trends Pharmacol. Sci. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hewitson KS, McNeill LA, Schofield CJ. Curr. Pharm. Des. 2004;10:821–833. doi: 10.2174/1381612043452884. [DOI] [PubMed] [Google Scholar]
- 6.Gerczuk PZ, Kloner RA. J. Am. Coll. Cardiol. 2012;59:969–978. doi: 10.1016/j.jacc.2011.07.054. [DOI] [PubMed] [Google Scholar]
- 7.Semenza GL. Nat. Rev. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Barneo J, Pardal R, Ortega-Saenz P. Annu. Rev. Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- 9.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL, Engl N. J. Med. 2005;353:2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu XW, Wang D, Nurse CA, Dinauer MC, Cutz E. Proc. Natl. Acad. Sci. U.S.A. 2000;97:4374–4379. doi: 10.1073/pnas.97.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, Dinger B, Sanders K, Hoidal J, Obeso A, Stensaas L, Fidone S, Gonzalez C. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;289:L916–L924. doi: 10.1152/ajplung.00015.2005. [DOI] [PubMed] [Google Scholar]
- 12.Kemp JP, Peers C. Adv. Exp. Med. Biol. 2009;648:39–48. doi: 10.1007/978-90-481-2259-2_4. [DOI] [PubMed] [Google Scholar]
- 13.Cutz E, Pan J, Yeger H, Domnik NJ, Fisher JT. Semin. Cell Dev. Biol. 2013;24:40–50. doi: 10.1016/j.semcdb.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 14.O'Kelly I, Peers C, Kemp PJ. Lahiri S, Prabhakar NR, Forster RE, editors. Oxygen Sensing: Molecule to Man. Advances in Experimental Medicine, Biology. 2000;475:611–622. [PubMed] [Google Scholar]
- 15.O'Kelly I, Peers C, Kemp PJ. Biochem. Biophys. Res. Commun. 2001;283:1131–1134. doi: 10.1006/bbrc.2001.4919. [DOI] [PubMed] [Google Scholar]
- 16.Lopez-Barneo J. Curr. Opin. Neurobiol. 2003;13:493–499. doi: 10.1016/s0959-4388(03)00093-x. [DOI] [PubMed] [Google Scholar]
- 17.Hayashida Y, Gonzalez C, Kondo H, He L, Dinger B, Gonzalez C, Obeso A, Fidone S. Adv. Exp. Med. Biol. Vol. 580. Springer; US: 2006. Arterial Chemorecept. pp. 155–160. [DOI] [PubMed] [Google Scholar]
- 18.Brij SO, Peacock AJ. Thorax. 1998;53:1075–1079. doi: 10.1136/thx.53.12.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dart C, Standen NB, Physiol J. 1995;483:29–39. doi: 10.1113/jphysiol.1995.sp020565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maines MD. Annu. Rev. Pharmacol. Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 21.Migita CT, Matera KM, Ikeda-Saito M, Olson JS, Fujii H, Yoshimura T, Zhou H, Yoshida T. J. Biol. Chem. 1998;273:945–949. doi: 10.1074/jbc.273.2.945. [DOI] [PubMed] [Google Scholar]
- 22.Jarmi T, Agarwal A. Curr. Hypertens. Rep. 2009;11:56–62. doi: 10.1007/s11906-009-0011-z. [DOI] [PubMed] [Google Scholar]
- 23.Williams SEJ, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 24.Kemp PJ. Biochem. Biophys. Res. Commun. 2005;338:648–652. doi: 10.1016/j.bbrc.2005.08.110. [DOI] [PubMed] [Google Scholar]
- 25.Adachi T, Ishikawa K, Hida W, Matsumoto H, Masuda T, Date F, Ogawa K, Takeda K, Furuyama K, Zhang YZ, Kitamuro T, Ogawa H, Maruyama Y, Shibahara S. Biochem. Biophys. Res. Commun. 2004;320:514–522. doi: 10.1016/j.bbrc.2004.05.195. [DOI] [PubMed] [Google Scholar]
- 26.Zhang F, Kaide JI, Yang LM, Jiang HL, Quan S, Kemp R, Gong WY, Balazy M, Abraham NG, Nasjletti A. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H137–H144. doi: 10.1152/ajpheart.00678.2002. [DOI] [PubMed] [Google Scholar]
- 27.Ryter SW, Alam J, Choi AMK. Physiol. Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 28.Olson KR. Clin Chem Lab Med. 2013;51:623–632. doi: 10.1515/cclm-2012-0551. [DOI] [PubMed] [Google Scholar]
- 29.Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, Madden JA. J Exp Biol. 2006;209:4011–4023. doi: 10.1242/jeb.02480. [DOI] [PubMed] [Google Scholar]
- 30.Olson KR, Whitfield NL, Bearden SE, Leger JS, Nilson E, Gao Y, Madden JA. Am J Physiol-Reg I. 2010;298:R51–R60. doi: 10.1152/ajpregu.00576.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Predmore BL, Lefer DJ, Gojon G. Antioxid Redox Sign. 2012;17:119–140. doi: 10.1089/ars.2012.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein A, Bailey SM. Redox Biol. 2013;1:32–39. doi: 10.1016/j.redox.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, Szoleczky P, Chang TJ, Zhou ZM, Wu LY, Wang R, Papapetropoulos A, Szabo C. P Natl Acad Sci USA. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson KR, DeLeon ER, Gao Y, Hurley K, Saduskas V, Batz C, Stoy G. Faseb J. 2013;27 doi: 10.1152/ajpregu.00421.2012. [DOI] [PubMed] [Google Scholar]
- 35.Ubuka T. J Chromatogr B. 2002;781:227–249. doi: 10.1016/s1570-0232(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 36.Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Am J Physiol-Reg I. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- 37.Hildebrandt TM, Grieshaber MK. Febs J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 38.Kabil O, Banerjee R. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agrawal N, Banerjee R. Plos One. 2008;3 doi: 10.1371/journal.pone.0004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Proc. Natl. Acad. Sci. U.S.A. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Proc. Natl. Acad. Sci. U.S.A. 2013;110:12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagahara N, Ito T, Kitamura H, Nishino T. Histochem. Cell Biol. 1998;110:243–250. doi: 10.1007/s004180050286. [DOI] [PubMed] [Google Scholar]
- 43.Olson KR. Resp. Physiol. Neurobiol. 2013;186:173–179. doi: 10.1016/j.resp.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 44.Kabil O, Banerjee R. J Biol Chem. 2012;287:44561–44567. doi: 10.1074/jbc.M112.407411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller DL, Budde MW, Roth MB. Plos One 6. 2011 doi: 10.1371/journal.pone.0025476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu WT, Gazi SK, Barrow RK, Yang GD, Wang R, Snyder SH. Sci Signal. 2009;2 doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toohey JI. Anal Biochem. 2011;413:1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 48.Loenarz C, Chowdhury R, Schofield CJ, Flashman E. Pure Appl. Chem. 2008;80:1837–1847. [Google Scholar]
- 49.Schofield CJ, Ratcliffe PJ. Nat. Rev. Mol. Cell Bio. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 50.Semenza GL, Wang GL. Mol. Cell. Bio. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc. Natl. Acad. Sci. U.S.A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. J. Biol. Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- 53.Loenarz C, Coleman ML, Boleininger A, Schierwater B, Holland PH, Ratcliffe PJ, Schofield CJ. Embo Reports. 2011;12:63–70. doi: 10.1038/embor.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Makino Y, Renhai C, Svensson K, Bertisson G, Asman M, Tanaka H, Yihai C, Berkenstam A, Poellinger L. Nature. 2001;414:550. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 55.Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smirnova NA, Hushpulian DM, Speer RE, Gaisina IN, Ratan RR, Gazaryan IG. Biochem-Mosc. 2012;77:1108–1119. doi: 10.1134/S0006297912100033. [DOI] [PubMed] [Google Scholar]
- 57.Hangasky JA, Taabazuing CY, Valliere MA, Knapp MJ. Metallomics. 2013;5:287–301. doi: 10.1039/c3mt20153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hausinger RP. Crit. Rev. Biochem. Mol. Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 59.Ivan M, Kondo K, Yang HF, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 60.Pektas S, Knapp MJ. J. Inorg. Biochem. 2013;126:55–60. doi: 10.1016/j.jinorgbio.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flashman E, Bagg EAL, Chowdhury R, Mecinovic J, Loenarz C, McDonough MA, Hewitson KS, Schofield CJ. J. Biol. Chem. 2008;283:3808–3815. doi: 10.1074/jbc.M707411200. [DOI] [PubMed] [Google Scholar]
- 62.Pappalardi MB, McNulty DE, Martin JD, Fisher KE, Jiang Y, Burns MC, Zhao H, Thau H, Sweitzer S, Schwartz B, Annan RS, Copeland RA, Tummino PJ, Luo L. Biochem. J. 2011;436:363–369. doi: 10.1042/BJ20101201. [DOI] [PubMed] [Google Scholar]
- 63.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 65.Semenza GL. J. Appl. Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 66.Yuan GX, Khan SA, Luo WB, Nanduri J, Semenza GL, Prabhakar NR. J. Cell. Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Light KM, Hangasky JA, Knapp MJ, Solomon EI. J. Am. Chem. Soc. 2013;135:9665–9674. doi: 10.1021/ja312571m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y-H, Comeaux LM, Herbst RW, Saban E, Kennedy DC, Maroney MJ, Knapp MJ. J. Inorg. Biochem. 2008;102:2120–2129. doi: 10.1016/j.jinorgbio.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solomon EI, Decker A, Lehnert N. PNAS. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem. Rev. 1999;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 71.Costas M, Mehn MP, Jensen MP, Que L. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 72.Saban E, Flagg SC, Knapp MJ. Journal of Inorganic Biochemistry. 2011;105:630–636. doi: 10.1016/j.jinorgbio.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. J. Biol. Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 74.Neidig ML, Solomon EI. Chem. Comm. 2005:5843–5863. doi: 10.1039/b510233m. [DOI] [PubMed] [Google Scholar]
- 75.Hangasky JA, Saban E, Knapp MJ. Biochemistry. 2013;52:1594–1602. doi: 10.1021/bi3015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saban E, Chen Y-H, Hangasky JA, Taabazuing CY, Holmes BE, Knapp MJ. Biochemistry. 2011;50:4733–4740. doi: 10.1021/bi102042t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Jr., Krebs C, Schofield CJ. FEBS J. 2010;277:4089–4099. doi: 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Flagg SC, Giri N, Pektas S, Maroney MJ, Knapp MJ. Biochemistry. 2012;51:6654–6666. doi: 10.1021/bi300229y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sandau KB, Zhou J, Kietzmann T, Brune B. J. Biol. Chem. 2001;276:39805–39811. doi: 10.1074/jbc.M107689200. [DOI] [PubMed] [Google Scholar]
- 80.Park Y-K, Ahn D-R, Oh M, Lee T, Yang EG, Son M, Park H. Mol. Pharmacol. 2008;74:236–245. doi: 10.1124/mol.108.045278. [DOI] [PubMed] [Google Scholar]
- 81.McDonough MA, McNeill LA, Tilliet M, Papamicael CA, Chen QY, Banerji B, Hewitson KS, Schofield CJ. J. Am. Chem. Soc. 2005;127:7680–7681. doi: 10.1021/ja050841b. [DOI] [PubMed] [Google Scholar]
- 82.Banerji B, Garcia AC, McNeill LA, McDonough MA, Buck MRG, Hewitson KS, Oldham NJ, Schofield CJ. Chem. Comm. 2005:5438–5440. doi: 10.1039/b510707e. [DOI] [PubMed] [Google Scholar]
- 83.Flagg SC, Martin CB, Taabazuing CY, Holmes BE, Knapp MJ. J. Inorg. Biochem. 2012;113:25–30. doi: 10.1016/j.jinorgbio.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rose NR, McDonough MA, King ONF, Kawamura A, Schofield CJ. Chem. Soc. Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 85.Gilles-Gonzalez MA, Gonzalez G. J. Inorg. Biochem. 2005;99:1–22. doi: 10.1016/j.jinorgbio.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 86.Gilles-Gonzalez MA, Gonzalez G. J. Biol. Chem. 1993;268:16293–16297. [PubMed] [Google Scholar]
- 87.Delgado-Nixon VM, Gonzalez G, Gilles-Gonzalez M-A. Biochemistry. 2000;39:2685–2691. doi: 10.1021/bi991911s. [DOI] [PubMed] [Google Scholar]
- 88.Chang AL, Tuckerman JR, Gonzalez G, Mayer R, Weinhouse H, Volman G, Amikam D, Benziman M, Gilles-Gonzalez M-A. Biochemistry. 2001;40:3420–3426. doi: 10.1021/bi0100236. [DOI] [PubMed] [Google Scholar]
- 89.Hou S, Larsen RW, Boudko D, Riley CW, Karatan E, Zimmer M, Ordal GW, Alam M. Nature. 2000;403:540–544. doi: 10.1038/35000570. [DOI] [PubMed] [Google Scholar]
- 90.Shelver D, Kerby RL, He Y, Roberts GP. Proceedings of the National Academy of Sciences. 1997;94:11216–11220. doi: 10.1073/pnas.94.21.11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kiley PJ, Beinert H. FEMS Microbiol. Rev. 1998;22:341–352. doi: 10.1111/j.1574-6976.1998.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 92.Muellner M, Hammel O, Mienert B, Schlag S, Bill E, Unden G. Biochemistry. 2008;47:13921–13932. doi: 10.1021/bi8014086. [DOI] [PubMed] [Google Scholar]
- 93.Nishimura T, Teramoto H, Vertès AA, Inui M, Yukawa H. J. Bacteriol. 2008;190:3264–3273. doi: 10.1128/JB.01801-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bhat SA, Singh N, Trivedi A, Kansal P, Gupta P, Kumar A. Free Radical Biol. Med. 2012;53:1625–1641. doi: 10.1016/j.freeradbiomed.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 95.Fedtke I, Kamps A, Krismer B, Gotz F, Bacteriol J. 2002;184:6624–6634. doi: 10.1128/JB.184.23.6624-6634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamps A, Achebach S, Fedtke I, Unden G, Gotz F. Mol. Microbiol. 2004;52:713–723. doi: 10.1111/j.1365-2958.2004.04024.x. [DOI] [PubMed] [Google Scholar]
- 97.Sun F, Ji Q, Jones MB, Deng X, Liang H, Frank B, Telser J, Peterson SN, Bae T, He C. J. Am. Chem. Soc. 2012;134:305–314. doi: 10.1021/ja2071835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Green J, Sharrocks AD, Green B, Geisow M, Guest JR. Mol. Microbiol. 1993;8:61–68. doi: 10.1111/j.1365-2958.1993.tb01203.x. [DOI] [PubMed] [Google Scholar]
- 99.Khoroshilova N, Popescu C, Munck E, Beinert H, Kiley PJ. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6087–6092. doi: 10.1073/pnas.94.12.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ. J. Biol. Chem. 1996;271:2762–2768. doi: 10.1074/jbc.271.5.2762. [DOI] [PubMed] [Google Scholar]
- 101.Jervis AJ, Crack JC, White G, Artymiuk PJ, Cheesman MR, Thomson AJ, Le Brun NE, Green J. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4659–4664. doi: 10.1073/pnas.0804943106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Becker S, Holighaus G, Gabrielczyk T, Unden G, Bacteriol J. 1996;178:4515–4521. doi: 10.1128/jb.178.15.4515-4521.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jordan PA, Thomson AJ, Ralph ET, Guest JR, Green J. FEBS Lett. 1997;416:349–352. doi: 10.1016/s0014-5793(97)01219-2. [DOI] [PubMed] [Google Scholar]
- 104.Sutton VR, Stubna A, Patschkowski T, Münck E, Beinert H, Kiley PJ. Biochemistry. 2003;43:791–798. doi: 10.1021/bi0357053. [DOI] [PubMed] [Google Scholar]
- 105.Lazazzera BA, Bates DM, Kiley PJ. Genes Dev. 1993;7:1993–2005. doi: 10.1101/gad.7.10.1993. [DOI] [PubMed] [Google Scholar]
- 106.Crack J, Green J, Thomson AJ. J. Biol. Chem. 2004;279:9278–9286. doi: 10.1074/jbc.M309878200. [DOI] [PubMed] [Google Scholar]
- 107.Crack JC, Gaskell AA, Green J, Cheesmant MR, Le Brun NE, Thomson AJ. J. Am. Chem. Soc. 2008;130:1749–1758. doi: 10.1021/ja077455+. [DOI] [PubMed] [Google Scholar]
- 108.Crack JC, Green J, Cheesman MR, Le Brun NE, Thomson AJ. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2092–2097. doi: 10.1073/pnas.0609514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Crack JC, Green J, Le Brun NE, Thomson AJ. J. Biol. Chem. 2006;281:18909–18913. doi: 10.1074/jbc.C600042200. [DOI] [PubMed] [Google Scholar]
- 110.Sutton VR, Mettert EL, Beinert H, Kiley PJ. J. Bacteriol. 2004;186:8018–8025. doi: 10.1128/JB.186.23.8018-8025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang B, Crack JC, Subramanian S, Green J, Thomson AJ, Le Brun NE, Johnson MK. Proc. Natl. Acad. Sci. U.S.A. 2012;109:15734–15739. doi: 10.1073/pnas.1208787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mettert EL, Kiley PJ. J. Mol. Biol. 2005;354:220–232. doi: 10.1016/j.jmb.2005.09.066. [DOI] [PubMed] [Google Scholar]
- 113.Mettert EL, Outten FW, Wanta B, Kiley PJ. J. Mol. Biol. 2008;384:798–811. doi: 10.1016/j.jmb.2008.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gao R, Stock AM. Annu. Rev. Microbiol. 2009;63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stock AM, Robinson VL, Goudreau PN. Annu. Rev. Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 116.West AH, Stock AM. Trends Biochem. Sci. 2001;26:369–376. doi: 10.1016/s0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 117.Airola MV, Huh D, Sukomon N, Widom J, Sircar R, Borbat PP, Freed JH, Watts KJ, Crane BR. J. Mol. Biol. 2013;425:886–901. doi: 10.1016/j.jmb.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Green J, Crack JC, Thomson AJ, LeBrun NE. Curr. Opin. Microbiol. 2009;12:145–151. doi: 10.1016/j.mib.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 119.Monson EK, Weinstein M, Ditta GS, Helinski DR. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4280–4284. doi: 10.1073/pnas.89.10.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dephilip P, Batut J, Boistard P. J. Bacteriol. 1990;172:4255–4262. doi: 10.1128/jb.172.8.4255-4262.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murthy UMN, Wecker MSA, Posewitz MC, Gilles-Gonzalez M-A, Ghirardi ML. FEBS Lett. 2012;586:4282–4288. doi: 10.1016/j.febslet.2012.06.052. [DOI] [PubMed] [Google Scholar]
- 122.Cho HY, Cho HJ, Kim YM, Oh JI, Kang BS. J. Biol. Chem. 2009;284:13057–13067. doi: 10.1074/jbc.M808905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gilles-Gonzalez MA, Gonzalez G, Perutz MF, Kiger L, Marden MC, Poyart C. Biochemistry. 1994;33:8067–8073. doi: 10.1021/bi00192a011. [DOI] [PubMed] [Google Scholar]
- 124.Dunham CM, Dioum EM, Tuckerman JR, Gonzalez G, Scott WG, Gilles-Gonzalez MA. Biochemistry. 2003;42:7701–7708. doi: 10.1021/bi0343370. [DOI] [PubMed] [Google Scholar]
- 125.Tanaka A, Nakamura H, Shiro Y, Fujii H. Biochemistry. 2006;45:2515–2523. doi: 10.1021/bi051989a. [DOI] [PubMed] [Google Scholar]
- 126.Anthamatten D, Hennecke H. Mol. Gen. Genet. 1991;225:38–48. doi: 10.1007/BF00282640. [DOI] [PubMed] [Google Scholar]
- 127.David M, Daveran ML, Batut J, Dedieu A, Domergue O, GHAI J, Hertig C, Boistard P, Kahn D. Cell. 1988;54:671–683. doi: 10.1016/s0092-8674(88)80012-6. [DOI] [PubMed] [Google Scholar]
- 128.Fischer HM. Microbiol. Rev. 1994;58:352–386. doi: 10.1128/mr.58.3.352-386.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Virts EL, Stanfield SW, Helinski DR, Ditta GS. Proc. Natl. Acad. Sci. U.S.A. 1988;85:3062–3065. doi: 10.1073/pnas.85.9.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gillesgonzalez MA, Gonzalez G, Perutz MF. Biochemistry. 1995;34:232–236. doi: 10.1021/bi00001a027. [DOI] [PubMed] [Google Scholar]
- 131.Galinier A, Garnerone AM, Reyrat JM, Kahn D, Batut J, Boistard P. J. Biol. Chem. 1994;269:23784–23789. [PubMed] [Google Scholar]
- 132.Gouet P, Fabry B, Guillet V, Birck C, Mourey L, Kahn D, Samama JP. Struct. Fold Des. 1999;7:1517–1526. doi: 10.1016/s0969-2126(00)88342-2. [DOI] [PubMed] [Google Scholar]
- 133.Gong WM, Hao B, Chan MK. Biochemistry. 2000;39:3955–3962. doi: 10.1021/bi992346w. [DOI] [PubMed] [Google Scholar]
- 134.Hao B, Isaza C, Arndt J, Soltis M, Chan MK. Biochemistry. 2002;41:12952–12958. doi: 10.1021/bi020144l. [DOI] [PubMed] [Google Scholar]
- 135.Hiruma Y, Kikuchi A, Tanaka A, Shiro Y, Mizutani Y. Biochemistry. 2007;46:6086–6096. doi: 10.1021/bi062083n. [DOI] [PubMed] [Google Scholar]
- 136.Salahudeen AA, Thompson JW, Ruiz JC, Ma H-W, Kinch LN, Li Q, Grishin NV, Bruick RK. Science. 2009;326:722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bailly X, Vanin S, Chabasse C, Mizuguchi K, Vinogradov SN. Bmc Evol. Biol. 8. 2008 doi: 10.1186/1471-2148-8-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.French CE, Bell JML, Ward FB. Fems Microbiol. Lett. 2008;279:131–145. doi: 10.1111/j.1574-6968.2007.01011.x. [DOI] [PubMed] [Google Scholar]
- 139.Kurtz DM, Jr, McCleverty JA, Meyer TJ. Comprehensive Coordination Chemistry II. Oxford; Pergamon: 2003. pp. 229–260. [Google Scholar]
- 140.Onoda A, Okamoto Y, Sugimoto H, Shiro Y, Hayashi T. Inorg. Chem. 2011;50:4892–4899. doi: 10.1021/ic2001267. [DOI] [PubMed] [Google Scholar]
- 141.Xiong JJ, Kurtz DM, Ai JY, Sanders-Loehr J. Biochemistry. 2000;39:5117–5125. doi: 10.1021/bi992796o. [DOI] [PubMed] [Google Scholar]
- 142.Isaza CE, Silaghi-Dumitrescu R, Iyer RB, Kurtz DM, Jr., Chan MK. Biochemistry. 2006;45:9023–9031. doi: 10.1021/bi0607812. [DOI] [PubMed] [Google Scholar]
- 143.Schaller RA, Ali SK, Klose KE, Kurtz DM., Jr. Biochemistry. 2012;51:8563–8570. doi: 10.1021/bi3011797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA. Science. 2009;326:718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shu C, Sung MW, Stewart MD, Igumenova TI, Tan X, Li P. ChemBioChem. 2012;13:788–791. doi: 10.1002/cbic.201200043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thompson JW, Salahudeen AA, Chollangi S, Ruiz JC, Brautigam CA, Makris TM, Lipscomb JD, Tomchick DR, Bruick RK. J. Biol. Chem. 2012;287:7357–7365. doi: 10.1074/jbc.M111.308684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kao W-C, Wang VCC, Huang Y-C, Yu SSF, Chang T-C, Chan SI. J. Inorg. Biochem. 2008;102:1607–1614. doi: 10.1016/j.jinorgbio.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 148.Thompson JW, Salahudeen AA, Chollangi S, Ruiz JC, Brautigam CA, Makris TM, Lipscomb JD, Tomchick DR, Bruick RK. J. Biol. Chem. 2012;287:7357–7365. doi: 10.1074/jbc.M111.308684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chollangi S, Thompson JW, Ruiz JC, Gardner KH, Bruick RK. J. Biol. Chem. 2012;287:23710–23717. doi: 10.1074/jbc.M112.360404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gong WM, Hao B, Mansy SS, Gonzalez G, Gilles-Gonzalez MA, Chan MK. Proc. Natl. Acad. Sci. U.S.A. 1998;95:15177–15182. doi: 10.1073/pnas.95.26.15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Balland V, Bouzhir-Sima L, Kiger L, Marden MC, Vos MH, Liebl U, Mattioli TA. J. Biol. Chem. 2005;280:15279–15288. doi: 10.1074/jbc.M413928200. [DOI] [PubMed] [Google Scholar]
- 152.Reynolds MF, Ackley L, Blizman A, Lutz Z, Manoff D, Miles M, Pace M, Patterson J, Pozzessere N, Saia K, Sato R, Smith D, Tarves P, Weaver M, Sieg K, Lukat-Rodgers GS, Rodgers KR. Arch. Biochem. Biophys. 2009;485:150–159. doi: 10.1016/j.abb.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 153.Key J, Moffat K. Biochemistry. 2005;44:4627–4635. doi: 10.1021/bi047942r. [DOI] [PubMed] [Google Scholar]
- 154.Ayers RA, Moffat K. Biochemistry. 2008;47:12078–12086. doi: 10.1021/bi801254c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Moeglich A, Ayers RA, Moffat K. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sousa EHS, Tuckerman JR, Gondim ACS, Gonzalez G, Gilles-Gonzalez M-A. Biochemistry. 2013;52:456–465. doi: 10.1021/bi300991r. [DOI] [PubMed] [Google Scholar]