

Abstract
Alzheimer’s disease and other related neurodegenerative diseases are highly debilitating disorders that affect millions of people worldwide. Efforts towards developing effective treatments for these disorders have shown limited efficacy at best, with no true cure to this day being present. Recent work, both clinical and experimental, indicates that many neurodegenerative disorders often display a coexisting metabolic dysfunction which may exacerbate neurological symptoms. It stands to reason therefore that metabolic pathways may themselves contain promising therapeutic targets for major neurodegenerative diseases. In this review, we provide an overview of some of the most recent evidence for metabolic dysregulation in Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease, and discuss several potential mechanisms that may underlie the potential relationships between metabolic dysfunction and etiology of nervous system degeneration. We also highlight some prominent signaling pathways involved in the link between peripheral metabolism and the central nervous system that are potential targets for future therapies, and we will review some of the clinical progress in this field. It is likely that in the near future, therapeutics with combinatorial neuroprotective and ‘eumetabolic’ activities may possess superior efficacies compared to less pluripotent remedies.
Keywords: Neurodegenerative diseases, metabolic dysfunction, bodyweight, diabetes, glucose homeostasis, insulin, leptin, ghrelin, adiponectin, glucagon-like peptide 1, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease
INTRODUCTION
Aging is a highly complex, evolutionarily conserved process that is subject to modification by a myriad of genetic and environmental factors. Aging can be defined as the progressive loss of an organism’s optimal function, which continues until its eventual failure and death. Aging and many aging-associated disorders involve perturbed energy balance. Metabolism, including glucose regulation and appetite balance, is controlled by both central regulatory inputs (primarily via the hypothalamus) and peripheral signals such as insulin, ghrelin, cholecystokinin, and adipokines (e.g. leptin, adiponectin, resistin). Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD) are debilitating aging-related neurodegenerative disorders, with severe cognitive and/or motor symptoms that progressively worsen over time, causing reduced quality of life, increased medical costs and eventual death. Neurodegenerative diseases mainly affect the middle-aged and elderly and represent a significant burden on patients and healthcare systems [1, 2]. Although AD, HD, and PD are neurodegenerative disorders, current therapies designed to treat these disorders that target the central nervous system often demonstrate limited efficacy. Increasing evidence suggests a link between the incidence and progression of some neurodegenerative disorders and metabolic dysfunction. Some recent data has demonstrated that therapies targeted at restoring metabolic homeostasis may improve cognitive and motor function as well as increase lifespan in neurodegenerative diseases [3, 4]. Uncontrolled, progressive weight loss and abnormal glucose tolerance are common metabolic dysfunctions observed in AD, HD, and PD, which appear to negatively impact overall prognosis through an, as of yet, poorly defined series of mechanisms [5, 6]. Whether alterations in systemic metabolism are etiologically linked to AD, HD, and PD, or are a consequence of the disease processes itself is still presently unclear. However, it is noteworthy that the disease loci of AD, HD, and PD often involve the hypothalamus, a key regulatory brain region for global energy homeostasis [7]. This review will focus on the multiple metabolic risk factors and disruptions associated with AD, HD, and PD, the mechanisms of abnormal energy balance in these disorders, and potential available therapies that could target metabolic pathways (‘eumetabolic’ agents) to treat neurodegeneration (see Fig. 1 for overview).
Fig. 1.
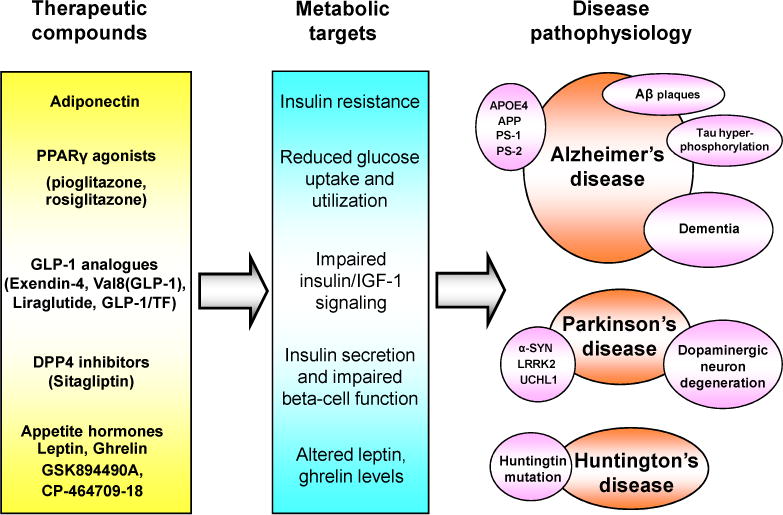
An overview of some of the relationships between metabolic dysfunction, neurodegenerative disorders, and potential therapies. Metabolic dysfunction including impaired glucose metabolism, insulin resistance and abnormal appetite regulation are now known to be comorbid with AD and some other chronic neurodegenerative disorders, such as HD and PD. Treatments that target metabolic malfunctions may be effective for modifying neurodegenerative-disease pathology and symptoms. IGF-1 (insulin-like growth factor-1); PPARs (peroxisome proliferator-activated receptors); DPP-4 (dipeptidyl peptidase-4); APP (amyloid precursor protein); PS 1 (presenilin 1); PS 2 (presenilin 2); APOE (apolipoprotein E); α-SYN (α-synuclein); LRRK2 (leucine-rich repeat kinase 2); UCHL1 (ubiquitin carboxy-terminal hydrolase L1) [202, 203].
ALZHEIMER’S DISEASE (AD)
AD is the sixth leading cause of death in the United States and is the most common form of dementia and cognitive impairment [8–10]. Symptoms of AD include progressive cognitive decline and memory loss, as well as an inability to perform routine daily activities. AD progression has been demonstrated to involve the intricate alterations of complex protein networks that still need further elucidation [11]. It is estimated that < 1% of AD cases are caused by rare genetic variations found in a small number of families, involving chromosome 21 on the gene for amyloid precursor protein (APP), chromosome 14 on the gene for the presenilin-1 (PS-1), and chromosome 1 on the gene for presenilin-2 (PS-2) [12]. In these inherited forms of AD, often referred to as ‘early-onset’ AD, symptoms tend to develop before the age of 65. The vast majority of AD cases however are sporadic and therefore lack a simple genetic cause, although both early-onset and sporadic AD share similar behavioral symptoms and pathophysiological mechanisms. Progressive memory loss and reduced cognitive function are associated with two primary neurodegenerative lesions, accumulations of beta-amyloid (Aβ) known as plaques and neurofibrillary tangles (NFTs) composed of the microtubule protein tau [13–15]. The generation and accumulation of Aβ is considered pivotal for the development of AD [14], because Aβ accumulation can set forth a cascade of events, including hyper-phosphorylation of tau, which in turn results in the formation of neurofibrillary tangles. Although most cases of AD are sporadic, alterations in the expression of the lipid-trafficking molecule, apolipoprotein E4 (ApoE4), has also been demonstrated to be a genetic risk factor for AD [16, 17]. Interestingly, ApoE4 alterations have also been suggested to be a risk factor for diabetes and hyperinsulinemia [18]. Despite a wealth of information concerning AD pathophysiology, the initial events that trigger Aβ plaque formation are unclear and there are currently no effective treatments that can stop or reverse the AD-related neurodegeneration. The currently available treatments for AD are only modestly effective, and some recent clinical trials in AD patients have failed to demonstrate benefit for current medications, or have even indicated harm. Failure to treat AD effectively may be due to the fact that the mechanisms that cause the disease are not fully understood, or that existing diagnostic criteria exclude factors of etiologic importance. It is now clear that disruptions in metabolism are present in AD and a greater understanding of these disrupted metabolic pathways may offer new insights into more efficacious treatments for AD.
AD AND BODY WEIGHT
Numerous studies have shown that excess body weight during middle-age is linked to an increased risk of developing AD. Clinically, obesity (Body Mass Index – BMI greater than 30) at 40–45 years of age is associated with a 3-fold increase in the risk of developing AD, while being overweight (BMI between 25 and 30) is associated with a 2-fold increase in AD risk, compared to individuals with a normal BMI [19]. The Baltimore Longitudinal Study of Aging, a landmark study of aging and age-related disorders, demonstrated that among women, being obese in midlife was associated with a hazard ratio of 6.57 for AD and among men, weight gain resulting in a BMI greater than the 90th percentile, between ages 30 and 50, was associated with an increased AD risk [20]. Kivipelto et al. also found similar results in a cohort of 1449 individuals followed for approximately 21 years, and midlife obesity was a significant risk factor for developing AD [21].
High BMI and obesity are not the only weight changes associated with an increased risk of developing AD. In a cohort of 299 men and women aged 50–79 years old, in which 60 individuals developed AD after 20 years of follow-up, weight loss of 5 kg or more reliably predicted AD development [22]. In contrast, Hughes et al. demonstrated that a higher baseline BMI and a slower rate of BMI decline were protective against dementia [23]. It is unclear exactly why excess body weight in midlife represents a risk factor for developing AD later in life, whereas in old age it appears to be protective. It must be noted however, that most studies use non-specific measures of body composition, such as total body weight and BMI (calculated as weight in kilograms divided by height in meters squared), rather than specific measures of body fat and muscle mass composition. Normal aging is associated with increases in body fat and decreases in lean muscle mass, therefore non-specific adiposity measures, such as BMI, may have limited accuracy when describing the relationship between body weight and the risk of developing AD. Yet, the studies summarized previously still strongly suggest a link between obesity, global energy regulation, and AD pathogenesis, which needs to be further elucidated in order to fully understand AD pathology.
ALTERATIONS IN BRAIN GLUCOSE METABOLISM IN AD
It is possible that the association between increased risk of developing AD and excess body weight in midlife reflects a diet high in simple sugars and fats and a sedentary lifestyle. A recent study showed that adherence to a ‘Mediterranean diet’ and intense physical exercise can be protective against AD [24]. Similarly, reducing caloric intake increases health-span, reduces damage in the brain due to aging, and provides greater maintenance of various brain functions, potentially through ‘hormetic’ mechanisms [25–28]. Experimental results indeed can corroborate clinical findings, as it has been shown that rats fed a high-fat/glucose diet, to induce insulin resistance, were found to exhibit impaired spatial learning ability, reduced hippocampal dendritic spine density, and reduced long-term potentiation in the CA1 region [29]. Glucotoxicity, or disrupted insulin signaling, are two of the potential mechanisms thought to mediate changes in hippocampal function observed from a high-fat diet, implying that diet-induced insulin resistance/ hyperinsulinemia may be one of the links between obesity and AD. Epidemiological studies have indicated an association between type 2 diabetes mellitus (DM) and an increased risk of developing AD. The Rotterdam study, the first of its kind to probe for a connection between type 2 DM and AD, revealed an approximate two-fold increase in risk of developing AD in patients with diabetes, compared to patients without the condition [30]. Furthermore, in the same study, DM requiring insulin treatment was associated with a four-fold increase in incidence of AD. The presence of type 2 DM and the ApoE4 allele together has also been shown to increase the risk of developing AD, to more than five-fold, compared to individuals without those two conditions [18]. Additionally, Luchsinger et al. demonstrated that hyperinsulinemia is associated with a doubled risk of developing AD [31]. Moreover, a thorough review of a registry of AD patients revealed that 80% had either type 2 DM or impaired fasting glucose measurements [32].
Abnormal glucose homeostasis is linked to cognitive dysfunction in such that patients with either type 1 or type 2 DM display significant memory impairment and attention deficits on cognitive testing compared to control subjects [33]. Hyperglycemia increases the number of mental subtraction errors in individuals with diabetes [34] and poor glycemic control (as evidenced by high hemoglobin A1C levels), has been associated with low scores on neuropsychological testing [35]. There are a number of mechanisms through which dysglycemia can lead to cognitive dysfunction. Hyperglycemia can lead to the activation of the polyol pathway, formation of advanced glycation end products, activation of protein kinase C, increased glucose shunting in the hexosamine pathway, and it is also possible that the increase in reactive oxygen species (ROS) associated with these mechanisms are then, in-part, responsible for altered brain function [36–38]. In animal models, global alterations in functional neurotransmission have also been linked to hyperglycemia, including abnormal N-methyl-D-aspartate (NMDA), acetylcholine, serotonin, dopamine and norepinephrine neurotransmission [39–42]. Whether these abnormalities lead to irreversible neuronal damage is presently unclear. There is also evidence that hyperglycemia may directly contribute to the pathophysiology of AD. Administration of high amounts of glucose can induce tau cleavage and apoptosis, and db/db mice, which are commonly used to model DM, exhibit an increase in tau phosphorylation compared to controls [43]. Although the exact mechanism is not entirely known, numerous reports therefore support the notion that metabolic dysfunction may worsen neurodegeneration in AD patients.
METABOLIC HORMONES AND THEIR THERAPEUTIC POTENTIAL IN AD
Insulin and AD
Recent work has begun to outline the presence of insulin signaling dysfunction in AD. Insulin receptors are abundant in many brain regions, including the hippocampus, although the physiological role of insulin in the brain is not fully understood [44, 45]. Insulin is now believed to influence regional glucose metabolism, as evidenced by rodent studies and one human study utilizing PET imaging [46–49]. Many studies have also shown that insulin plays a role in regulating learning and memory processes [50–52]. Clinically, there is a higher density of insulin receptors in the brain of AD patients compared to control subjects, possibly reflecting upregulation of the receptor in an attempt to compensate for the decreased functionality of insulin [53]. Furthermore, hyperinsulinemia (a marker of insulin resistance in the metabolic disease spectrum) can decrease the availability of insulin degrading enzyme (IDE), which is essential for the degradation and clearance of Aβ in the brain [54]. Additionally, mouse models of type 1 and type 2 DM are associated with increased tau phosphorylation, which is likely secondary to glucotoxicity [43].
It is therefore likely that therapies that manipulate insulin signaling may be beneficial for treating the neurological symptoms of AD. Peroxisome proliferator-activated receptors (PPARs), which belong to the steroid, thyroid and retinoid receptor superfamily, are ligand-inducible transcription factors. The PPARs subfamily is comprised of three isoforms: PPARα, PPARβ/δ and PPARγ. Two PPARγ agonists, pioglitazone and rosiglitazone, are currently widely prescribed for the treatment of type 2 DM. PPARγ agonists improve both lipid and glucose metabolism, mainly by increasing peripheral insulin sensitivity, which ameliorates the metabolic dysfunction brought on by the diabetic pathophysiology [55]. There is increasing evidence demonstrating the efficacy of PPARγ agonists for the treatment of AD. PPARγ activation suppresses the expression of inflammatory genes, which, clinically, has been shown to ameliorate neurodegeneration [56]. Experimentally, treatment with PPARγ agonists has been associated with both reduced Aβ plaque load and improved behavioral outcomes in an animal model of AD [57]. Clinical studies have corroborated this finding; i.e. treatment with a PPARγ agonist reduces disease-related pathology, improves learning and memory, and enhances attention in AD patients [57]. The cyclooxygenase inhibitor Ibuprofen (iso-butyl-propanoic-phenolic acid), which can activate PPARγ, has been demonstrated to significantly reduce amyloid pathology and reduce microglial-mediated inflammation in a mouse model of AD, potentially via PPARγ signaling [58, 59]. In addition, PPARγ agonists have been shown to reduce Aβ plaque burden and Aβ42 (a specifically toxic form of Aβ) levels in the brain by approximately 20–25%, restore insulin responsiveness and lower glucocorticoid levels in mouse models of AD [60, 61]. These results suggest that PPARγ agonists may be useful for the treatment of AD, a hypothesis greatly strengthened by both experimental and clinical studies demonstrating that rosiglitazone can attenuate learning and memory deficits in AD [3, 61, 62]. It is worth noting that PPARγ also transcriptionally induces IDE expression, which could explain the effectiveness of PPARγ agonists in treating both type 2 DM and AD [63]. PPARδ, which is expressed at higher levels in the brain than PPARγ, also plays a role in regulating lipid and glucose metabolism. In a recent study, treatment with the PPARδ agonist, GW742, reduced amyloid burden, an effect thought to be mediated by alterations in amyloid clearance [64]. Current data suggest that insulin and its downstream signaling cascades play an important role in AD pathogenesis. It is unclear whether treatments that target insulin signaling demonstrate efficacy due to direct or indirect mechanisms, because insulin directly affects the brain and a systemic metabolic dysfunction is often co-morbid with AD. Nevertheless, these studies provide convincing evidence that the insulin signaling pathway may be a novel therapeutic target for the treatment of AD.
Leptin and AD
Leptin, which is primarily synthesized in adipose tissue, plays a pivotal role in the control of food intake, body weight, fat storage, immune system function, reproductive function, insulin sensitivity, and neuronal protection [65–67]. Obesity is associated with leptin resistance/hyperleptinemia in addition to insulin resistance. Within the central nervous system, leptin crosses the blood-brain-barrier to bind to specific receptors in the hypothalamus to mediate food intake, body weight and energy expenditure [68]. A small number of studies have begun to outline a potential connection between abnormal leptin levels and AD. AD patients with significant weight loss display lower plasma leptin levels than weight-stable AD patients, and disruption of homeostasis between leptin and cortisol is also observed in some AD patients [69, 70]. In the Framingham study, a landmark longitudinal study, lower plasma leptin levels were associated with a higher risk of incident AD, corresponding to an absolute risk over a 12-year follow-up of 25% for persons in the lowest quartile versus 6% in the highest [71]. Leptin may be directly involved in the development of AD symptoms by exerting effects on the brain, as high expression of leptin receptors are found in the hippocampus, suggesting that leptin plays a role in controlling learning and memory. Indeed, leptin has been shown to be crucial for the maintenance of normal hippocampal synaptic plasticity [72, 73]. Impairments in long-term potentiation in the CA1 region of the hippocampus and poor spatial memory compared to controls, are noted in leptin receptor-deficient rodent models, implying an involvement of leptin and/or its receptor in normal memory function [74]. If leptin is indeed required for normal hippocampal function, it stands to reason that it may represent a reasonable therapeutic target for AD, a disease in which the hippocampus is particularly vulnerable. These data indicate that leptin – besides its role in energy regulation – may directly regulate the behavioral and pathological progression of AD. Leptin is structurally and functionally similar to a class of signaling molecules known as proinflammatory cytokines, and it plays a regulatory role in innate and adaptive immunity [75]. AD pathogenesis in the brain includes overexpression of cytokines and upregulation of the innate immune system. Multiple studies have shown that manipulating leptin levels can ameliorate AD pathology by affecting Aβ plaques. Leptin treatment can promote Aβ clearance by reducing β-secretase activity and increasing ApoE-dependent Aβ uptake [76], and it also improves memory performance in AD animal models. Leptin can also reduce tau phosphorylation through inactivation of GSK-3β [77]. AMP-activated protein kinase (AMPK) is emerging as a central modulator of the major pathological hallmarks of AD in the brain, and leptin deficiency in AD can contribute to down-regulation of the AMPK system, causing increases in Aβ and phosphorylated tau [78, 79]. As leptin appears to play a significant functional role in the progression of AD, a growing body of evidence supports the use of leptin-modifying drugs to treat AD. Leptin administration improves memory in SAMP-8 mice, an accelerated senescence rodent model that develops amyloid plaques [80]. Chronic leptin treatment has also been shown to significantly reduce the levels of Aβ and phosphorylated tau without an untoward inflammatory reaction in two different transgenic models of AD [76, 81]. These biochemical and pathological changes were also correlated with behavioral improvements, suggesting that leptin not only reduces AD pathology but also ameliorates cognitive symptoms [81]. Direct administration of leptin into the brain has been observed to facilitate hippocampal long-term potentiation and improve memory performance in mice [82]. Thus, abnormal leptin signaling – either as a consequence of leptin resistance or secondary to reduced leptin levels – may be one of the primary mechanisms underlying hippocampal dysfunction in AD. The studies summarized here suggest that leptin could also serve as a therapy that increases learning and memory capacity in AD patients and reduces Aβ plaques in the brain, while helping maintain adequate metabolic control.
Ghrelin and AD
Ghrelin is a hormone produced by the stomach which assists in the promotion of sensations of hunger [83]. While its interactions with the neurons of lateral, paraventricular, and arcuate nuclei of the hypothalamus to regulate energy balance and growth hormone release have been well documented [84], recent evidence suggests that ghrelin also binds to and activates the ghrelin receptors expressed on the pyramidal neurons of layer V in the sensorimotor area and in the cingulate gyrus of the cerebral cortex [85]. Furthermore, studies have demonstrated that ghrelin, in addition to its role in promoting energy intake, may also affect cognition as well. Carlini et al. demonstrated that intracerebroventricular (i.c.v.) injections of ghrelin increased memory retention in rats [86]. This group also showed that ghrelin potentiates a dose-dependent increase in memory retention, with the maximal effect occurring in the hippocampus [87]. It was recently shown that circulating ghrelin binds to neurons of the hippocampal formation, promoting dendritic spine formation and generation of long-term potentiation (LTP) [84]. Targeted disruption of ghrelin signaling resulted in a decreased number of spine synapses in the stratum radiatum and impaired performance in behavioral memory testing, both of which were rapidly restored by ghrelin administration [84]. These data strongly support a role for ghrelin signaling in maintaining normal memory function and provide a motivation for the study of ghrelin regulation in AD. Clinical studies have indicated a role for ghrelin in various metabolic disorders as well as AD [88, 89]. Ghrelin levels in the brain are altered in some Alzheimer’s patients, suggesting that changes to the ghrelin signaling system may indeed contribute to AD pathophysiology [90]. Recently, two non-peptide ghrelin receptor agonists (GSK894490A and CP-464709-18) were shown to significantly improve performance in the novel object recognition and modified water maze tests in male Lister hooded rats, indicating the potential of ghrelin for treating cognitive dysfunction [91]. These studies suggest that ghrelin plays a vital role in not only regulating metabolic control, but also in regulating cognitive function and memory capacity, and that abnormal ghrelin signaling could be caused by, and/or worsen AD and its symptomology [92, 93].
Adiponectin and AD
Adiponectin, the most abundant adipocytokine secreted by adipose tissue, is a hormone that plays a role in regulating insulin sensitivity and energy expenditure [94]. Targeted deletion of the adiponectin gene can lead to insulin resistance [95], and continuous systemic infusion of adiponectin can enhance insulin sensitivity in type 2 diabetic mice [96]. Adiponectin abnormalities have been implicated in various metabolic disorders [97–99], all of which could also act as risk factors for the development of AD [100]. Recent studies have shown that the adiponectin receptors AdipoR1 and AdipoR2 are expressed throughout the central nervous system (CNS) [101]. However, there is still some debate about whether adiponectin crosses the blood-brain-barrier or not [102, 103]. Few studies to date have focused on a potential correlation between adiponectin and AD. Recently, one clinical study demonstrated that some AD patients have elevated levels of adiponectin in both plasma and cerebrospinal fluid (CSF) [104], suggesting that it may play a role in mediating AD progression, possibly through its effects on peripheral or brain metabolism. It is also noteworthy that elevated interleukin-6 has been detected in the brains of some AD patients [105, 106], and that treatment with adiponectin can reduce the secretion of the centrally active interleukin-6 from brain endothelial cells [103]. Due to its relatively recent discovery, there are currently no known clinical trials of adiponectin modifying drugs related to AD therapy. However, it is likely that adiponectin system-targeted compounds could offer potential as therapeutic targets for the metabolic component of AD due to its well documented role in diabetes and other metabolic disorders [99, 107].
Glucagon-Like Peptide 1 and AD
Glucagon-like peptide 1 (GLP-1) enhances pancreatic islet beta-cell proliferation and glucose-dependent insulin secretion, lowers blood glucose and food intake, and is an effective incretin-based therapy for type 2 DM [108]. GLP-1 and its cognate receptor are both expressed throughout the brain and stimulation of this system appears to be neuroprotective and can reduce neuronal degeneration in various animal models [4, 109, 110]. A body of literature suggests the potential therapeutic relevance of GLP-1 to CNS disorders such as AD [111]. GLP-1 and exendin-4, a natural and stable long-acting analogue of GLP-1, possess neurotrophic properties and protect neurons against Aβ and oxidative insults [112]. GLP-1 can reduce amyloid-beta peptide levels in vivo and decreases levels of amyloid precursor protein in cultured neuronal cells, implying that GLP-1 could be effective at reducing plaque load in AD [113]. In addition, GLP-1 receptor agonists protect neurons against Aβ and glutamate-induced apoptosis in cells and attenuate cholinergic neuron atrophy in the basal forebrain of the rat following an excitotoxic lesion [114]. A recent study using GLP-1 receptor knock-out mice demonstrated that GLP-1 receptor signaling can play an important role in the control of synaptic plasticity [115]. The novel GLP-1 analogue (Val8(GLP-1)) has also been shown to enhance synaptic plasticity and to reverse the impairment of long-term potentiation (LTP) induced by Aβ fragments, further strengthening the hypothesis that GLP-1 may be useful for treating AD [116]. Other stable GLP-1 analogues such as Liraglutide, Asp7GLP-1, N-glyc-GLP-1, and Pro9GLP-1 were recently found to have facillitatory effects on LTP by the same group [116]. Additionally, the novel long-acting GLP-1 analogue GLP-1/Tf (GLP-1 fused to transferrin) potentially also shows promise for the treatment of metabolic dysfunction in AD [117]. Attenuation of the activity levels of dipeptidyl peptidase 4 (DPP-4, the enzyme that cleaves and inactivates GLP-1), can stabilize the plasma levels of the bioactive GLP-1. A recent study has demonstrated that sitagliptin, a DPP-4 inhibitor, could significantly delay some forms of AD pathology, including amyloid deposition, when administrated early in the disease course in a mouse model of AD [118].
Brain-Derived Neurotrophic Factor and AD
Brain-derived neurotrophic factor (BDNF) a 13-kDa protein, is a member of the family of neutrophins [119]. Besides its well-established roles in neuronal development, neuronal maintenance and survival as well as promoting synaptic plasticity, BDNF also plays a considerable role in regulating global metabolic function. BDNF deficiency in rodents leads to hyperphagia, obesity, hyperinsulinemia, and hyperleptinemia [120, 121]. There is also evidence that BDNF deficiency in humans can lead to metabolic dysfunction [122]. BDNF signaling has been shown to be impaired in AD, which could be relevant considering the neurological and metabolic abnormalities noted in AD. Studies have demonstrated low brain BDNF mRNA expression in patients with AD, including the hippocampus [123], neocortex and in the nucleus basalis of Meynert [124–126]. A recent study on the role of BDNF in AD investigated the sorting protein-related receptor with A-type repeats (SORLA). SORLA regulates APP intracellular trafficking and processing into Aβ, and when overexpressed, can reduce amyloid plaque formation [127, 128]. BDNF was found to be a major inducer of SORLA gene transcription through the extracellular regulated kinase (ERK) pathway [129]. Circulating plasma BDNF levels have been shown to associate with multiple cardiovascular markers of age-related pathophysiology and also to significantly decrease with age [130], and it is therefore possible that a gradual decrease in BDNF levels may contribute to the increase in the risk of developing AD with advancing age.
METABOLIC DYSFUNCTION IN HUNTINGTON’S DISEASE
Huntington’s disease is an autosomal dominant genetic disorder that affects approximately 5–7 individuals per 100,000 [131]. The disease is characterized by progressive chorea, dystonia, cognitive dysfunction and psychiatric and behavioral symptoms [132]. Individuals with HD usually become symptomatic between the fourth and fifth decades of life and invariably die from complications of the disease within one to two decades after initial diagnosis [133]. The neuropathologic hallmarks of HD include severe cell loss and atrophy in the caudate and putamen [134]. The underlying genetic defect in HD is the presence of expanded repeats of the trinucleotide CAG in exon 1 of the HD gene, which encodes for the protein huntingtin [135]. Huntingtin (htt) is expressed ubiquitously in mammalian cells but its specific functions are not fully understood. Htt is believed to influence a number of cellular functions, including BDNF expression, vesicle trafficking, axonal transport and transcriptional regulation [136–139]. The genetic defect that leads to the development of HD causes the formation of a mutant form of huntingtin, which contains polyglutamine expansions that make the protein more susceptible to a pathological proteolysis process that generates cytotoxic amino-terminal htt fragments and also promotes subsequent aggregation of the remaining htt protein [140]. Aggregates of mutant huntingtin appear to be neurotoxic, as they interfere with the normal function of several nuclear and cytoplasmic proteins that regulate transcription, apoptosis, mitochondrial function and axonal transport [141–144]. Postmortem brains of HD patients demonstrate reduced BDNF levels in the caudate and putamen, whereas wild-type htt stimulates BDNF gene transcription through promoter II activation [145–147]. In the presence of mutant htt, a transcriptional repressor inhibits BDNF gene transcription [146, 148]. In addition, as mentioned above, mutant htt may also interfere with BDNF vesicle transport [149]. Mutant htt aggregates are also found outside of the central nervous system, and animal studies suggest that these aggregates can cause cell toxicity in peripheral organs as well. For example, in the R6/2 and N171-82Q mouse models of HD, mutant htt aggregates are present in pancreatic islet cells [150, 4], and these HD mice display decreased pancreatic beta-cell mass and exhibit impaired glycemic control.
HD and Body Weight
Weight loss is a well-recognized manifestation of HD [151–153]. Patients with clinical HD and presymptomatic gene carriers typically have a lower BMI than control subjects [154]. Although poor dietary intake from severe dysphagia can occur in HD, weight loss is common in patients who have an intact appetite and high caloric intake [155, 156]. Total energy expenditure is higher in HD patients compared to controls, which in part reflects their present hyperkinetic state (chorea and dystonia) [157–159]. While hyperkinesis appears to be an important contributing factor, it does not entirely account for all weight loss observed in HD. A cross-sectional study of individuals with early-stage HD showed that BMI was significantly lower in HD patients than control subjects and that neither disease duration, dystonia, nor chorea scores were significantly associated with BMI [153]. If indeed hyperkinesis is not a sine qua non for weight loss in HD, it has been speculated that HD patients experience increased energy expenditure, secondary to a hypermetabolic state. A recent study demonstrated that clinically unaffected individuals with a CAG repeat length greater or equal to 37 required an increased caloric intake to maintain their BMI compared to individuals with less than 37 CAG repeats [160], suggesting a deficit in energy homeostasis not accounted for by excessive motor activity. In addition, among a group of 517 patients with early stage HD followed for 3 years, a higher CAG repeat number was associated with a lower initial mean BMI and faster rate of BMI decline [5]. Increasing numbers of CAG repeats correspond to increased disease severity, indicating more substantial aggregation of mutant htt in central and peripheral organs involved in energy regulation. Post-mortem studies in HD patients have demonstrated neuronal loss in regions of the hypothalamus involved in energy balance, including both the paraventricular and ventromedial nuclei. Although the hypothalamus plays an important role in energy regulation, attributing a high rate of energy expenditure to the presence of aggregates in the hypothalamus is too simplistic. Direct deleterious effects of mutant htt on peripheral tissues are likely to contribute to the dysregulation of the energy balance system. Abnormalities in key metabolic tissues such as adipose tissue [161, 162], pancreas [163, 164], and skeletal muscle [165, 166] have all been described in mouse models of HD. Furthermore, Mochel et al. demonstrated that HD patients with weight loss have lower levels of the branched chain amino acids, valine, leucine, and isoleucine, suggesting a critical need for Krebs cycle energy substrates [154], and reflecting mitochondrial dysfunction. Additionally, Popovic et al. found high plasma ghrelin and low plasma leptin in HD patients (~ 6 years of disease duration) compared with healthy controls, indicating a state of negative energy balance [167]. Low leptin levels have also been noted in a mouse model of HD [4], suggesting that metabolic hormones are an important aspect of the energy dysregulation observed in HD.
HD and Altered Glucose Homeostasis
Studies have found an association between HD and an increased risk for the development of type 2 DM [168]. In one study, seven out of 14 patients with documented HD demonstrated an abnormal response in standard glucose tolerance tests [169]. Additionally, some HD patients with normal glucose tolerance can still display an increase in insulin resistance [170]. Glycosuria and glucose intolerance develop in mouse models of HD beginning as early as 9 weeks and as mentioned above, these mice also express mutant htt aggregates in the pancreas, implying that mutant htt pathology can also mediate metabolic dysfunction [171]. Therefore it has been suggested that an effective treatment for HD should target the symptoms that affect the entire body, including the loss of motor coordination and dysglycemia [172]. In addition to directly impairing insulin secretion due to the formation of aggregates in the pancreas, mutant htt may also affect insulin resistance via pathophysiological functionality of the hypothalamus [4]. Blockade of hypothalamic insulin receptors by phosphoinositide 3-kinase inhibitors leads to hepatic insulin resistance and increased hepatic glucose production, implying that the hypothalamus is necessary for normal insulin signaling [173]. Martin et al. demonstrated that treatment with insulin failed to correct hyperglycemia, although treatment with exendin-4, a drug commonly used for the treatment of type 2 DM, improved the glycemic profile and reduced mutant huntingtin aggregates in both the brain and pancreas, while also prolonging lifespan in a mouse model of HD [4]. These findings suggest that targeting the peripheral metabolic dysfunction observed in HD could be beneficial for treating some aspects of this disorder, and further support the notion that new treatments that target the entire body – rather than just the CNS – are likely to be more effective for treating HD.
METABOLIC DYSFUNCTION IN PARKINSON’S DISEASE
Parkinson’s disease (PD) is a neurodegenerative movement disorder, only slightly less prevalent than AD, that usually affects individuals in their sixth decade of life [174]. Classical PD symptoms include bradykinesia, resting tremor, rigidity, and gait instability. Pathologically, PD is characterized by a profound neuronal loss in the pars-compacta of the substantia nigra. In addition, in PD there is often an accumulation of ubiquinated protein deposits in the cytoplasm of neurons (Lewy bodies) and neurites (Lewy neurites), usually containing aggregates of the protein α-synuclein [175–177]. Low levels of BDNF have been observed in the substantia nigra pars compacta of PD patients compared to controls and experimentally, two pathogenic mutations linked to α-synuclein have been associated with a loss of BDNF synthesis in rodents [124, 178]. Furthermore, mice with a conditional deletion of BDNF in the midbrain and hindbrain have been shown to exhibit reduced numbers of dopaminergic neurons in the substantia nigra [179], indicating that BDNF is necessary for normal development of those neurons.
PD and Body Weight
Weight loss is a well-recognized manifestation of PD. In general, individuals with PD possess a lower BMI than age-matched controls [180, 181]. PD patients lose weight in the years preceding their diagnosis and continue to lose weight thereafter [182]. While impaired hand-mouth coordination, dysphagia, and hyposmia have been listed as contributing factors, there is some debate over whether decreased energy intake is a significant contributor to the weight loss observed in PD [182, 183]. Levi et al. and Markus et al. observed an increased resting energy expenditure in PD patients, compared to healthy controls, using indirect calorimetry [184, 185]. However Toth et al. reported a lower daily energy expenditure in PD patients using double-labeled water technique [186]. A recent study using double-labeled water to measure daily energy expenditure in PD patients displaying weight loss or a stable-weight found no significant energetic difference between the two groups [183]. Unlike AD, obesity is not considered to be a risk factor for PD development [187, 188]. However, at least one study reported that in a cohort of over 10,000 men, subjects who lost at least 0.5 units of BMI in the decade following college entry had a significantly increased risk of developing PD compared with individuals with stable weight. It is unclear however if the weight loss during young adulthood seen in the study represented an early manifestation of PD. Leptin tends to be low in PD patients who experience weight loss, which may reflect a decrease in body fat. Evidente et al. found a trend towards low plasma leptin levels in PD patients with unintended weight loss [189]. These results were corroborated by another study that demonstrated that PD patients who lost body weight possessed lower serum leptin levels than weight-stable PD patients [190]. In addition to lower plasma leptin levels, lower plasma ghrelin levels were also noted in PD patients who experienced weight loss [191]. While low plasma leptin is expected to accompany loss of body fat, the finding of low plasma ghrelin is less intuitive and warrants further investigation. A recent study showed that peripheral ghrelin is neuroprotective for nigrostriatal dopamine function through activation of UCP2-dependent alterations in mitochondrial respiration [192]. Additionally, ghrelin or ghrelin receptor knockout mice are more susceptible to dopaminergic neuron loss in the substantia nigra and striatum after MPTP treatment, suggesting that abnormal ghrelin signaling may represent a predisposing factor for nigrostriatal dopamine dysfunction.
PD and Altered Glucose Homeostasis
Recent studies have demonstrated the presence of dysglycemia and abnormal glucose tolerance in patients with PD [193, 194]. However, there is some debate regarding a direct association between PD and type 2 DM. One large epidemiologic study, which utilized the UK-based General Practice Research Database (GPRD) which contains computerized medical records of over 5 million people, found that the prevalence of diabetes was similar in patients with and without PD. Furthermore, the study concluded that the risk of developing diabetes was lower in PD patients than in subjects without PD [195]. A second major epidemiological study, the Physician’s Health Study, which includes a cohort of U.S. male physicians, indicated that although individuals with diabetes had an increased risk of developing PD, the highest risk was found in individuals with short-duration, older-onset diabetes without complications. Thus, it is presently unclear whether diabetes increases the likelihood of developing PD and further studies are therefore needed to investigate a potential link. There is also some evidence for abnormal insulin signaling in PD. Although insulin receptors are normally abundant in the substantia nigra, some PD patients display a loss of insulin receptors in this brain region [196, 197]. In rat models with streptozotocin-induced diabetes, low levels of insulin have been associated with decreased amounts of dopamine transporter mRNA and tyrosine hydroxylase mRNA in the substantia nigra [198]. Moreover, increased insulin receptor substrate-2 phosphorylation, a sign of insulin resistance, has been observed in the dopamine-depleted striatum in PD [199].
CONCLUSIONS
AD, PD and HD all represent diseases that progress in an age-dependent, but accelerated, manner. All of these disorders display accumulation of products of oxidative reactions which lead to wide-spread lipid and protein damage [200]. These neurodegenerative disorders mimic advanced aging on multiple levels, including cognitive and motor decline as well as molecular signaling defects such as impaired insulin signaling. It is striking that all three disorders display a metabolic dysfunction phenotype in conjunction with a neurodegenerative pathology. While damage to the hypothalamus via abnormal protein aggregates, such as mutant htt or Aβ plaques, may be part of the cause of energy dysregulation, hypothalamic disease on its own does not explain the whole process entirely. A more complete and acceptable hypothesis that explains the metabolic changes combined with neuronal damage in these diseases includes a description of a disrupted connection between peripheral organs that manage energy regulation and the CNS through alterations in leptin, ghrelin, GLP-1 and insulin signaling. Although these metabolic hormones are traditionally thought to be involved primarily in metabolic disorders, it is now clear, as discussed above, that alterations in these hormones can have severe implications for the etiology of several prominent neurodegenerative disorders. Furthermore, it is also clear that therapeutic targets that manipulate these metabolic factors are showing promise for the treatment of neurodegenerative disorders (see Fig. 1 for overview). Current research is starting to suggest that successful treatments for neurodegenerative disorders should ideally target the whole body, rather than focusing on the CNS alone. It is likely that multiple endocrinological factors play a combinatorial role in facilitating AD, HD, and PD pathophysiology; hence, studies that attempt to understand the overall effects of the combination of these hormones, rather than just the individual, are necessary [201]. It is important to note that many animal studies are confounded by a series of factors, including housing conditions and diet, that can make these subjects more prone to developing metabolic dysfunctions and cause challenges in ascertaining whether the metabolic phenotype is associated with the disease progression or with the animal’s lifestyle [28]. In order to better understand disease processes and better serve patients, we must attempt to design studies that look into combinatorial effects of metabolic hormones and neuronal signaling molecules in accurate animal models, allowing us to characterize and discover the potential that metabolic factors hold, in treating both metabolic disorders and various neurodegenerative diseases.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
ABBREVIATIONS
- Aβ
Beta-amyloid
- AD
Alzheimer’s disease
- AMPK
AMP-activated protein kinase
- ApoE4
Apolipoprotein E4
- APP
Amyloid precursor protein
- BDNF
Brain-derived neurotrophic factor
- BMI
Body Mass Index
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- DM
Diabetes mellitus
- DPP4
Dipeptidyl peptidase-4
- ERK
Extracellular regulated kinase
- GLP-1
Glucagon-like peptide 1
- GPRD
General Practice Research Database
- GSK-3β
Glycogen synthase kinase-3β
- HD
Huntington’s disease
- htt
Huntingtin
- IDE
Insulin degrading enzyme
- IGF-1
Insulin-like growth factor-1
- LRRK2
Leucine-rich repeat kinase 2
- LTP
Long-term potentiation
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NFTs
Neurofibrillary tangles
- NMDA
N-methyl-D-aspartate
- PD
Parkinson’s disease
- PPARs
Peroxisome proliferator-activated receptors
- PS-1
Presenilin-1
- PS-2
Presenilin-2
- ROS
Reactive oxygen species
- SORLA
Sorting protein-related receptor with A-type repeats
- UCHL1
Ubiquitin carboxy-terminal hydrolase L1
- UCP2
Uncoupling protein 2
- α-SYN
α-synuclein
Footnotes
CONFLICT OF INTEREST
None
The authors have no conflicts of scientific interest with respect to the manuscript.
References
- 1.Alzheimer’s Association. 2010 Alzheimer’s disease facts and figures. Alzheimer’s and Dementia. 2010;6:158–94. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Oster E, Dorsey ER, Bausch J, Shinaman A, Kayson E, Oakes D, et al. Fear of health insurance loss among individuals at risk for Huntington disease. American Journal of Medical Genetics Part A. 2008;146A:2070–7. doi: 10.1002/ajmg.a.32422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–8. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- 4.Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, et al. Exendin-4 Improves Glycemic Control, Ameliorates Brain and Pancreatic Pathologies, and Extends Survival in a Mouse Model of Huntington’s Disease. Diabetes. 2009;58:318–28. doi: 10.2337/db08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aziz N, van der Marck M, Pijl H, Olde Rikkert M, Bloem B, Roos R. Weight loss in neurodegenerative disorders. J Neurol. 2008;255:1872–80. doi: 10.1007/s00415-009-0062-8. [DOI] [PubMed] [Google Scholar]
- 6.Papapetropoulos S, Ellul J, Argyriou AA, Talelli P, Chroni E, Papapetropoulos T. The effect of vascular disease on late onset Parkinson’s disease. Eur J Neurol. 2004;11:231–5. doi: 10.1046/j.1468-1331.2003.00748.x. [DOI] [PubMed] [Google Scholar]
- 7.Standaert DG, Lee VM, Greenberg BD, Lowery DE, Trojanowski JQ. Molecular features of hypothalamic plaques in Alzheimer’s disease. Am J Pathol. 1991;139:681–91. [PMC free article] [PubMed] [Google Scholar]
- 8.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer Disease in the US Population: Prevalence Estimates Using the 2000 Census. Arch Neurol. 2003;60:1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 9.Ashford J. APOE genotype effects on alzheimer’s disease onset and epidemiology. J Mol Neurosci. 2004;23:157–65. doi: 10.1385/JMN:23:3:157. [DOI] [PubMed] [Google Scholar]
- 10.Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–94. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Martin B, Brenneman R, Becker KG, Gucek M, Cole RN, Maudsley S. iTRAQ Analysis of Complex Proteome Alterations in 3xTgAD Alzheimer’s Mice: Understanding the Interface between Physiology and Disease. PLoS ONE. 2008;3:e2750. doi: 10.1371/journal.pone.0002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selkoe DJ. Alzheimer’s Disease–Genotypes, Phenotype, and Treatments. Science. 1997;275:630–1. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–8. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 14.Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 15.Maudsley S, Mattson MP. Protein twists and turns in Alzheimer disease. Nat Med. 2006;12:392–3. doi: 10.1038/nm0406-392. [DOI] [PubMed] [Google Scholar]
- 16.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 17.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peila R, Rodriguez BL, Launer LJ. Type 2 Diabetes, APOE Gene, and the Risk for Dementia and Related Pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–62. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 19.Whitmer RA, Gunderson EP, Quesenberry CP, Jr, Zhou J, Yaffe K. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr Alzheimer Res. 2007;4:103–9. doi: 10.2174/156720507780362047. [DOI] [PubMed] [Google Scholar]
- 20.Zonderman AB. Predicting Alzheimer’s disease in the Baltimore longitudinal study of aging. J Geriatr Psychiatry Neurol. 2005;18:192–5. doi: 10.1177/0891988705281863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kareholt I, Winblad B, et al. Obesity and Vascular Risk Factors at Midlife and the Risk of Dementia and Alzheimer Disease. Arch Neurol. 2005;62:1556–60. doi: 10.1001/archneur.62.10.1556. [DOI] [PubMed] [Google Scholar]
- 22.Barrett-Connor E, Edelstein SL, Corey-Bloom J, Wiederholt WC. Weight loss precedes dementia in community-dwelling older adults. J Am Geriatr Soc. 1996;44:1147–52. doi: 10.1111/j.1532-5415.1996.tb01362.x. [DOI] [PubMed] [Google Scholar]
- 23.Hughes TF, Borenstein AR, Schofield E, Wu Y, Larson EB. Association between late-life body mass index and dementia: The Kame Project. Neurology. 2009;72:1741–6. doi: 10.1212/WNL.0b013e3181a60a58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scarmeas N, Luchsinger JA, Schupf N, Brickman AM, Cosentino S, Tang MX, et al. Physical Activity, Diet, and Risk of Alzheimer Disease. JAMA. 2009;302:627–37. doi: 10.1001/jama.2009.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin B, Mattson MP, Maudsley S. Caloric restriction and intermittent fasting: Two potential diets for successful brain aging. Ageing Res Rev. 2006;5:332–53. doi: 10.1016/j.arr.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin B, Golden E, Egan JM, Mattson MP, Maudsley S. Reduced energy intake: the secret to a long and healthy life? IBS J Sci. 2007;2:35–9. [PMC free article] [PubMed] [Google Scholar]
- 27.Martin B, Golden E, Carlson OD, Egan JM, Mattson MP, Maudsley S. Caloric restriction: Impact upon pituitary function and reproduction. Ageing Res Rev. 2008;7:209–24. doi: 10.1016/j.arr.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin B, Ji S, Maudsley S, Mattson MP. “Control” laboratory rodents are metabolically morbid: Why it matters. Proc Natl Acad Sci. 2010;107:6127–33. doi: 10.1073/pnas.0912955107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–8. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–42. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 31.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–92. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 32.Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes. 2004;53:474–81. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 33.Kodl CT, Seaquist ER. Cognitive Dysfunction and Diabetes Mellitus. Endocr Rev. 2008;29:494–511. doi: 10.1210/er.2007-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox DJ, Kovatchev BP, Gonder-Frederick LA, Summers KH, McCall A, Grimm KJ, et al. Relationships Between Hyperglycemia and Cognitive Performance Among Adults With Type 1 and Type 2 Diabetes. Diabetes Care. 2005;28:71–7. doi: 10.2337/diacare.28.1.71. [DOI] [PubMed] [Google Scholar]
- 35.Cukierman-Yaffe T, Gerstein HC, Williamson JD, Lazar RM, Lovato L, Miller ME, et al. Relationship Between Baseline Glycemic Control and Cognitive Function in Individuals With Type 2 Diabetes and Other Cardiovascular Risk Factors. Diabetes Care. 2009;32:221–6. doi: 10.2337/dc08-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biessels GJ, van der Heide LP, Kamal A, Bleys RLAW, Gispen WH. Ageing and diabetes: implications for brain function. Eur J Pharm. 2002;441:1–14. doi: 10.1016/s0014-2999(02)01486-3. [DOI] [PubMed] [Google Scholar]
- 37.Brownlee M. The Pathobiology of Diabetic Complications. Diabetes. 2005;54:1615–25. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 38.Klein JP, Waxman SG. The brain in diabetes: molecular changes in neurons and their implications for end-organ damage. Lancet Neurol. 2003;2:548–54. doi: 10.1016/s1474-4422(03)00503-9. [DOI] [PubMed] [Google Scholar]
- 39.Ramakrishnan R, Sheeladevi R, Suthanthirarajan N. PKC-alpha. mediated alterations of indoleamine contents in diabetic rat brain. Brain Res Bull. 2004;64:189–94. doi: 10.1016/j.brainresbull.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Kamal A, Biessels GJ, Urban IJA, Gispen WH. Hippocampal synaptic plasticity in streptozotocin-diabetic rats: impairment of long-term potentiation and facilitation of long-term depression. Neuroscience. 1999;90:737–45. doi: 10.1016/s0306-4522(98)00485-0. [DOI] [PubMed] [Google Scholar]
- 41.Welsh B, Wecker L. Effects of streptozotocin-induced diabetes on acetylcholine metabolism in rat brain. Neurochem Res. 1991;16:453–60. doi: 10.1007/BF00965566. [DOI] [PubMed] [Google Scholar]
- 42.Biessels GJ, Kappelle AC, Bravenboer B, Erkelens DW, Gispen WH. Cerebral function in diabetes mellitus. Diabetologia. 1994;37:643–50. doi: 10.1007/BF00417687. [DOI] [PubMed] [Google Scholar]
- 43.Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased Tau Phosphorylation and Cleavage in Mouse Models of Type 1 and Type 2 Diabetes. Endocrinology. 2009;150:5294–301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marks JL, Porte DJ, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology. 1990;127:3234–6. doi: 10.1210/endo-127-6-3234. [DOI] [PubMed] [Google Scholar]
- 45.Unger J, McNeill TH, Moxley RT, 3rd, White M, Moss A, Livingston JN. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience. 1989;31:143–57. doi: 10.1016/0306-4522(89)90036-5. [DOI] [PubMed] [Google Scholar]
- 46.Doyle P, Cusin I, Rohner-Jeanrenaud F, Jeanrenaud B. Four-day hyperinsulinemia in euglycemic conditions alters local cerebral glucose utilization in specific brain nuclei of freely moving rats. Brain Res. 1995;684:47–55. doi: 10.1016/0006-8993(95)00402-c. [DOI] [PubMed] [Google Scholar]
- 47.Lucignani G, Namba H, Nehlig A, Porrino LJ, Kennedy C, Sokoloff L. Effects of insulin on local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab. 1987;7:309–14. doi: 10.1038/jcbfm.1987.68. [DOI] [PubMed] [Google Scholar]
- 48.Marfaing P, Penicaud L, Broer Y, Mraovitch S, Calando Y, Picon L. Effects of hyperinsulinemia on local cerebral insulin binding and glucose utilization in normoglycemic awake rats. Neurosci Lett. 1990;115:279–85. doi: 10.1016/0304-3940(90)90469-p. [DOI] [PubMed] [Google Scholar]
- 49.Bingham EM, Hopkins D, Smith D, Pernet A, Hallett W, Reed L, et al. The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes. 2002;51:3384–90. doi: 10.2337/diabetes.51.12.3384. [DOI] [PubMed] [Google Scholar]
- 50.Park CR, Seeley RJ, Craft S, Woods SC. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Phy Beh. 2000;68:509–14. doi: 10.1016/s0031-9384(99)00220-6. [DOI] [PubMed] [Google Scholar]
- 51.Craft S, Asthana S, Newcomer JW, Wilkinson CW, Matos IT, Baker LD, et al. Enhancement of Memory in Alzheimer Disease With Insulin and Somatostatin, but Not Glucose. Arch Gen Psychiatry. 1999;56:1135–40. doi: 10.1001/archpsyc.56.12.1135. [DOI] [PubMed] [Google Scholar]
- 52.Fehm HL, Perras B, Smolnik R, Kern W, Born J. Manipulating neuropeptidergic pathways in humans: a novel approach to neuropharmacology? Eur J Pharmacol. 2000;405:43–54. doi: 10.1016/s0014-2999(00)00540-9. [DOI] [PubMed] [Google Scholar]
- 53.Frölich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Trans. 1998;105:423–38. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 54.Craft S, Stennis Watson G. Insulin and neurodegenerative disease: shared and specific mechanisms. The Lancet Neurology. 2004;3:169–78. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 55.Waugh J, Keating GM, Plosker GL, Easthope S, Robinson DM. Pioglitazone: a review of its use in type 2 diabetes mellitus. Drugs. 2006;66:85–109. doi: 10.2165/00003495-200666010-00005. [DOI] [PubMed] [Google Scholar]
- 56.Daynes RA, Jones DC. Emerging roles of PPARS in inflammation and immunity. Nat Rev Immunol. 2002;2:748–59. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 57.Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5:481–9. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci. 2000;20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–10. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 60.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–53. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 61.Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–73. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 62.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–54. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 63.Du J, Zhang L, Liu S, Zhang C, Huang X, Li J, et al. PPARgamma transcriptionally regulates the expression of insulin-degrading enzyme in primary neurons. Biochem Biophys Res Commun. 2009;383:485–90. doi: 10.1016/j.bbrc.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 64.Kalinin S, Richardson JC, Feinstein DL. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2009;6:431–7. doi: 10.2174/156720509789207949. [DOI] [PubMed] [Google Scholar]
- 65.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 66.Harvey J. Leptin: a diverse regulator of neuronal function. J Neurochem. 2007;100:307–13. doi: 10.1111/j.1471-4159.2006.04205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–6. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 68.Jéquier E. Leptin signaling, adiposity, and energy balance. Ann N Y Acad Sci. 2002;967:379–88. doi: 10.1111/j.1749-6632.2002.tb04293.x. [DOI] [PubMed] [Google Scholar]
- 69.Power DA, Noel J, Collins R, O’Neill D. Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dement Geriatr Cogn Disord. 2001;12:167–70. doi: 10.1159/000051252. [DOI] [PubMed] [Google Scholar]
- 70.Olsson T, Nasman B, Rasmuson S, Ahren B. Dual relation between leptin and cortisol in humans is disturbed in Alzheimer’s disease. Biol Psychiatry. 1998;44:374–6. [PubMed] [Google Scholar]
- 71.Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA. 2009;302:2565–72. doi: 10.1001/jama.2009.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doherty GH, Oldreive C, Harvey J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience. 2008;154:1297–307. doi: 10.1016/j.neuroscience.2008.04.052. [DOI] [PubMed] [Google Scholar]
- 73.Shanley LJ, Irving AJ, Harvey J. Leptin Enhances NMDA Receptor Function and Modulates Hippocampal Synaptic Plasticity. J Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113:607–15. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- 75.Lam QL, Lu L. Role of leptin in immunity. Cell Mol Immunol. 2007;4:1–13. [PubMed] [Google Scholar]
- 76.Fewlass DC, Noboa K, Pi-Sunyer FX, Johnston JM, Yan SD, Tezapsidis N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004;18:1870–8. doi: 10.1096/fj.04-2572com. [DOI] [PubMed] [Google Scholar]
- 77.Greco SJ, Sarkar S, Casadesus G, Zhu X, Smith MA, Ashford JW, et al. Leptin inhibits glycogen synthase kinase-3[beta] to prevent tau phosphorylation in neuronal cells. Neuroscience Letters. 2009;455:191–4. doi: 10.1016/j.neulet.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Greco SJ, Sarkar S, Johnston JM, Zhu X, Su B, Casadesus G, et al. Leptin reduces Alzheimer’s disease-related tau phosphorylation in neuronal cells. Biochem Biophys Res Commun. 2008;376:536–41. doi: 10.1016/j.bbrc.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farr SA, Banks WA, Morley JE. Effects of leptin on memory processing. Peptides. 2006;27:1420–5. doi: 10.1016/j.peptides.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 81.Greco SJ, Bryan KJ, Sarkar S, Zhu X, Smith MA, Ashford JW, et al. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1155–67. doi: 10.3233/JAD-2010-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harvey J, Solovyova N, Irving A. Leptin and its role in hippocampal synaptic plasticity. Prog Lipid Res. 2006;45:369–78. doi: 10.1016/j.plipres.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Vriese C, Delporte C. Influence of ghrelin on food intake and energy homeostasis. Curr Opin Clin Nutr Metab Care. 2007;10:615–9. doi: 10.1097/MCO.0b013e32829fb37c. [DOI] [PubMed] [Google Scholar]
- 84.Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–8. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 85.Hou Z, Miao Y, Gao L, Pan H, Zhu S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regul Pept. 2006;134:126–31. doi: 10.1016/j.regpep.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 86.Carlini VP, Monzon ME, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, et al. Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun. 2002;299:739–43. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- 87.Carlini VP, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR. Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun. 2004;313:635–41. doi: 10.1016/j.bbrc.2003.11.150. [DOI] [PubMed] [Google Scholar]
- 88.Pedrosa C, Oliveira B, Albuquerque I, Simoes-Pereira C, Vaz-de-Almeida M, Correia F. Obesity and metabolic syndrome in 7–9 years-old Portuguese schoolchildren. Diabetology & Metabolic Syndrome. 2010;2:40. doi: 10.1186/1758-5996-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tesauro M, Schinzari F, Caramanti M, Lauro R, Cardillo C. Cardiovascular and metabolic effects of ghrelin. Curr Diabetes Rev. 2010;6:228–35. doi: 10.2174/157339910791658871. [DOI] [PubMed] [Google Scholar]
- 90.Gahete MD, Rubio A, Córdoba-Chacón J, Gracia-Navarro F, Kineman RD, Avila J, et al. Expression of the Ghrelin and Neurotensin Systems is Altered in the Temporal Lobe of Alzheimer’s Disease Patients. Journal of Alzheimer’s Disease. 2010;22:819–28. doi: 10.3233/JAD-2010-100873. [DOI] [PubMed] [Google Scholar]
- 91.Atcha Z, Chen WS, Ong AB, Wong FK, Neo A, Browne ER, et al. Cognitive enhancing effects of ghrelin receptor agonists. Psychopharmacology (Berl) 2009;206:415–27. doi: 10.1007/s00213-009-1620-6. [DOI] [PubMed] [Google Scholar]
- 92.Giordano V, Peluso G, Iannuccelli M, Benatti P, Nicolai R, Calvani M. Systemic and brain metabolic dysfunction as a new paradigm for approaching Alzheimer’s dementia. Neurochem Res. 2007;32:555–67. doi: 10.1007/s11064-006-9125-8. [DOI] [PubMed] [Google Scholar]
- 93.Cong WN, Golden E, Pantaleo N, White CM, Maudsley S, Martin B. Ghrelin receptor signaling: a promising therapeutic target for metabolic syndrome and cognitive dysfunction. CNS Neurol Disord Drug Targets. 2010;9:557–63. doi: 10.2174/187152710793361513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dridi S, Taouis M. Adiponectin and energy homeostasis: consensus and controversy. J Nutr Biochem. 2009;20:831–9. doi: 10.1016/j.jnutbio.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 95.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–6. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 96.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–6. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 97.Ukkola O, Santaniemi M. Adiponectin: a link between excess adiposity and associated comorbidities? Journal of Molecular Medicine. 2002;80:696–702. doi: 10.1007/s00109-002-0378-7. [DOI] [PubMed] [Google Scholar]
- 98.Diez JJ, Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol. 2003;148:293–300. doi: 10.1530/eje.0.1480293. [DOI] [PubMed] [Google Scholar]
- 99.Renaldi O, Pramono B, Sinorita H, Purnomo LB, Asdie RH, Asdie AH. Hypoadiponectinemia: a risk factor for metabolic syndrome. Acta Med Indones. 2009;41:20–4. [PubMed] [Google Scholar]
- 100.Kroner Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern Med Rev. 2009;14:373–9. [PubMed] [Google Scholar]
- 101.Alim I, Fry WM, Walsh MH, Ferguson AV. Actions of adiponectin on the excitability of subfornical organ neurons are altered by food deprivation. Brain Res. 2010;1330:72–82. doi: 10.1016/j.brainres.2010.02.076. [DOI] [PubMed] [Google Scholar]
- 102.Kos K, Harte AL, da Silva NF, Tonchev A, Chaldakov G, James S, et al. Adiponectin and resistin in human cerebrospinal fluid and expression of adiponectin receptors in the human hypothalamus. J Clin Endocrinol Metab. 2007;92:1129–36. doi: 10.1210/jc.2006-1841. [DOI] [PubMed] [Google Scholar]
- 103.Spranger J, Verma S, Gohring I, Bobbert T, Seifert J, Sindler AL, et al. Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells. Diabetes. 2006;55:141–7. [PubMed] [Google Scholar]
- 104.Une K, Takei YA, Tomita N, Asamura T, Ohrui T, Furukawa K, et al. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur J Neurol. 2010;2010:18. doi: 10.1111/j.1468-1331.2010.03194.x. [DOI] [PubMed] [Google Scholar]
- 105.Hüll M, Strauss S, Berger M, Volk B, Bauer J. The participation of interleukin-6, a stress-inducible cytokine, in the pathogenesis of Alzheimer’s disease. Behav Brain Res. 1996;78:37–41. doi: 10.1016/0166-4328(95)00213-8. [DOI] [PubMed] [Google Scholar]
- 106.Rösler N, Wichart I, Jellinger KA. Clinical significance of neurobiochemical profiles in the lumbar cerebrospinal fluid of Alzheimer’s disease patients. J Neural Transm. 2001;108:231–46. doi: 10.1007/s007020170091. [DOI] [PubMed] [Google Scholar]
- 107.Nedvídková J, Smitka K, Kopský V, Hainer V. Adiponectin, an adipocyte-derived protein. Physiol Res. 2005;54:133–40. [PubMed] [Google Scholar]
- 108.Gallwitz B. Glucagon-like peptide-1-based therapies for the treatment of type 2 diabetes mellitus. Treat Endocrinol. 2005;4:361–70. doi: 10.2165/00024677-200504060-00005. [DOI] [PubMed] [Google Scholar]
- 109.Biswas SC, Buteau J, Greene LA. Glucagon-like peptide-1 (GLP-1) diminishes neuronal degeneration and death caused by NGF deprivation by suppressing Bim induction. Neurochem Res. 2008;33:1845–51. doi: 10.1007/s11064-008-9646-4. [DOI] [PubMed] [Google Scholar]
- 110.Chapter MC, White CM, DeRidder A, Chadwick W, Martin B, Maudsley S. Chemical modification of class II G protein-coupled receptor ligands: frontiers in the development of peptide analogs as neuroendocrine pharmacological therapies. Pharmacol Ther. 2010;125:39–54. doi: 10.1016/j.pharmthera.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1205–19. doi: 10.3233/JAD-2010-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Perry T, Greig NH. The glucagon-like peptides: a new genre in therapeutic targets for intervention in Alzheimer’s disease. J Alzheimers Dis. 2002;4:487–96. doi: 10.3233/jad-2002-4605. [DOI] [PubMed] [Google Scholar]
- 113.Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72:603–12. doi: 10.1002/jnr.10611. [DOI] [PubMed] [Google Scholar]
- 114.Perry T, Greig NH. Enhancing central nervous system endogenous GLP-1 receptor pathways for intervention in Alzheimer’s disease. Curr Alzheimer Res. 2005;2:377–85. doi: 10.2174/1567205054367892. [DOI] [PubMed] [Google Scholar]
- 115.Abbas T, Faivre E, Holscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: Interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res. 2009;205:265–71. doi: 10.1016/j.bbr.2009.06.035. [DOI] [PubMed] [Google Scholar]
- 116.McClean PL, Gault VA, Harriott P, Holscher C. Glucagon-like peptide-1 analogues enhance synaptic plasticity in the brain: a link between diabetes and Alzheimer’s disease. Eur J Pharmacol. 2010;630:158–62. doi: 10.1016/j.ejphar.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 117.Kim B-J, Zhou J, Martin B, Carlson OD, Maudsley S, Greig NH, et al. Transferrin Fusion Technology: A Novel Approach to Prolonging Biological Half-Life of Insulinotropic Peptides. Journal of Pharmacology and Experimental Therapeutics. 2010;334:682–92. doi: 10.1124/jpet.110.166470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.D’Amico M, Di Filippo C, Marfella R, Abbatecola AM, Ferraraccio F, Rossi F, et al. Long-term inhibition of dipeptidyl peptidase-4 in Alzheimer’s prone mice. Exp Gerontol. 2010;45:202–7. doi: 10.1016/j.exger.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 119.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15239–44. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–57. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 122.Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–9. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 123.Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 124.Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 125.Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–20. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 126.Murer MG, Boissiere F, Yan Q, Hunot S, Villares J, Faucheux B, et al. An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience. 1999;88:1015–32. doi: 10.1016/s0306-4522(98)00219-x. [DOI] [PubMed] [Google Scholar]
- 127.Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–6. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, et al. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rohe M, Synowitz M, Glass R, Paul SM, Nykjaer A, Willnow TE. Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J Neurosci. 2009;29:15472–8. doi: 10.1523/JNEUROSCI.3960-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Golden E, Emiliano A, Maudsley S, Windham BG, Carlson OD, Egan JM, et al. Circulating brain-derived neurotrophic factor and indices of metabolic and cardiovascular health: data from the Baltimore Longitudinal Study of Aging. PLoS One. 2010;5:e10099. doi: 10.1371/journal.pone.0010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958–63. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Walker FO. Huntington’s disease. Lancet. 2007;369:218–28. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 133.Wojaczynska-Stanek K, Adamek D, Marszal E, Hoffman-Zacharska D. Huntington disease in a 9-year-old boy: clinical course and neuropathologic examination. J Child Neurol. 2006;21:1068–73. doi: 10.1177/7010.2006.00244. [DOI] [PubMed] [Google Scholar]
- 134.Macdonald V, Halliday GM, Trent RJ, McCusker EA. Significant loss of pyramidal neurons in the angular gyrus of patients with Huntington’s disease. Neuropathol Appl Neurobiol. 1997;23:492–5. doi: 10.1111/j.1365-2990.1997.tb01326.x. [DOI] [PubMed] [Google Scholar]
- 135.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 136.Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J Neurosci. 2007;27:6972–83. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–81. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 138.Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, et al. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–9. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- 139.Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, et al. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- 140.Wellington CL, Leavitt BR, Hayden MR. Huntington disease: new insights on the role of huntingtin cleavage. J Neural Transm Suppl. 2000:1–17. doi: 10.1007/978-3-7091-6284-2_1. [DOI] [PubMed] [Google Scholar]
- 141.Cha JH. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. 2000;23:387–92. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- 142.Hickey MA, Chesselet MF. Apoptosis in Huntington’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:255–65. doi: 10.1016/S0278-5846(03)00021-6. [DOI] [PubMed] [Google Scholar]
- 143.Panov AV, Burke JR, Strittmatter WJ, Greenamyre JT. In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: relevance to Huntington’s disease. Arch Biochem Biophys. 2003;410:1–6. doi: 10.1016/s0003-9861(02)00585-4. [DOI] [PubMed] [Google Scholar]
- 144.Charrin BC, Saudou F, Humbert S. Axonal transport failure in neurodegenerative disorders: the case of Huntington’s disease. Pathol Biol (Paris) 2005;53:189–92. doi: 10.1016/j.patbio.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 145.Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–61. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- 146.Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 147.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–8. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 148.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 149.Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–38. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 150.Björkqvist M, Fex M, Renström E, Wierup N, Petersén A, Gil J, et al. The R6/2 transgenic mouse model of Huntington’s disease develops diabetes due to deficient beta-cell mass and exocytosis. Hum Mol Genet. 2005;14:565–74. doi: 10.1093/hmg/ddi053. [DOI] [PubMed] [Google Scholar]
- 151.Sanberg PR, Fibiger HC, Mark RF. Body weight and dietary factors in Huntington’s disease patients compared with matched controls. Med J Aust. 1981;1:407–9. doi: 10.5694/j.1326-5377.1981.tb135681.x. [DOI] [PubMed] [Google Scholar]
- 152.Farrer LA, Meaney FJ. An anthropometric assessment of Huntington’s disease patients and families. Am J Phys Anthropol. 1985;67:185–94. doi: 10.1002/ajpa.1330670304. [DOI] [PubMed] [Google Scholar]
- 153.Djousse L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH. Weight loss in early stage of Huntington’s disease. Neurology. 2002;59:1325–30. doi: 10.1212/01.wnl.0000031791.10922.cf. [DOI] [PubMed] [Google Scholar]
- 154.Mochel F, Charles P, Seguin F, Barritault J, Coussieu C, Perin L, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One. 2007;2:e647. doi: 10.1371/journal.pone.0000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morales LM, Estevez J, Suarez H, Villalobos R, Chacin de Bonilla L, Bonilla E. Nutritional evaluation of Huntington disease patients. Am J Clin Nutr. 1989;50:145–50. doi: 10.1093/ajcn/50.1.145. [DOI] [PubMed] [Google Scholar]
- 156.Trejo A, Tarrats RM, Alonso ME, Boll MC, Ochoa A, Velasquez L. Assessment of the nutrition status of patients with Huntington’s disease. Nutrition. 2004;20:192–6. doi: 10.1016/j.nut.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 157.Pratley RE, Salbe AD, Ravussin E, Caviness JN. Higher sedentary energy expenditure in patients with Huntington’s disease. Ann Neurol. 2000;47:64–70. [PubMed] [Google Scholar]
- 158.Stoy N, McKay E. Weight loss in Huntington’s disease. Ann Neurol. 2000;48:130–1. [PubMed] [Google Scholar]
- 159.Gaba AM, Zhang K, Marder K, Moskowitz CB, Werner P, Boozer CN. Energy balance in early-stage Huntington disease. Am J Clin Nutr. 2005;81:1335–41. doi: 10.1093/ajcn/81.6.1335. [DOI] [PubMed] [Google Scholar]
- 160.Marder K, Zhao H, Eberly S, Tanner CM, Oakes D, Shoulson I. Dietary intake in adults at risk for Huntington disease: analysis of PHAROS research participants. Neurology. 2009;73:385–92. doi: 10.1212/WNL.0b013e3181b04aa2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fain JN, Del Mar NA, Meade CA, Reiner A, Goldowitz D. Abnormalities in the functioning of adipocytes from R6/2 mice that are transgenic for the Huntington’s disease mutation. Hum Mol Genet. 2001;10:145–52. doi: 10.1093/hmg/10.2.145. [DOI] [PubMed] [Google Scholar]
- 162.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–62. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 163.Andreassen OA, Dedeoglu A, Stanojevic V, Hughes DB, Browne SE, Leech CA, et al. Huntington’s disease of the endocrine pancreas: insulin deficiency and diabetes mellitus due to impaired insulin gene expression. Neurobiol Dis. 2002;11:410–24. doi: 10.1006/nbdi.2002.0562. [DOI] [PubMed] [Google Scholar]
- 164.Hurlbert MS, Zhou W, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington’s disease gene develop diabetes. Diabetes. 1999;48:649–51. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- 165.Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–26. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- 166.Strand AD, Aragaki AK, Shaw D, Bird T, Holton J, Turner C, et al. Gene expression in Huntington’s disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14:1863–76. doi: 10.1093/hmg/ddi192. [DOI] [PubMed] [Google Scholar]
- 167.Popovic V, Svetel M, Djurovic M, Petrovic S, Doknic M, Pekic S, et al. Circulating and cerebrospinal fluid ghrelin and leptin: potential role in altered body weight in Huntington’s disease. Eur J Endocrinol. 2004;151:451–5. doi: 10.1530/eje.0.1510451. [DOI] [PubMed] [Google Scholar]
- 168.Podolsky S, Leopold NA. Abnormal glucose tolerance and arginine tolerance tests in Huntington’s disease. Gerontology. 1977;23:55–63. doi: 10.1159/000212174. [DOI] [PubMed] [Google Scholar]
- 169.Podolsky S, Leopold NA, Sax DS. Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet. 1972;1:1356–8. doi: 10.1016/s0140-6736(72)91092-6. [DOI] [PubMed] [Google Scholar]
- 170.Lalic NM, Maric J, Svetel M, Jotic A, Stefanova E, Lalic K, et al. Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol. 2008;65:476–80. doi: 10.1001/archneur.65.4.476. [DOI] [PubMed] [Google Scholar]
- 171.Hunt MJ, Morton AJ. Atypical diabetes associated with inclusion formation in the R6/2 mouse model of Huntington’s disease is not improved by treatment with hypoglycaemic agents. Exp Brain Res. 2005;166:220–9. doi: 10.1007/s00221-005-2357-z. [DOI] [PubMed] [Google Scholar]
- 172.Martin B, Golden E, Keselman A, Stone M, Mattson MP, Egan JM, et al. Therapeutic perspectives for the treatment of Huntington’s disease: treating the whole body. Histol Histopathol. 2008;23:237–50. doi: 10.14670/hh-23.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–82. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 174.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–66. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 175.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183–91. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 176.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 177.Mezey E, Dehejia AM, Harta G, Suchy SF, Nussbaum RL, Brownstein MJ, et al. Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Molecular Psychiatry. 1998;3:493–9. doi: 10.1038/sj.mp.4000446. [DOI] [PubMed] [Google Scholar]
- 178.Kohno R, Sawada H, Kawamoto Y, Uemura K, Shibasaki H, Shimohama S. BDNF is induced by wild-type alpha-synuclein but not by the two mutants, A30P or A53T, in glioma cell line. Biochem Biophys Res Commun. 2004;318:113–8. doi: 10.1016/j.bbrc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 179.Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–9. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Abbott RA, Cox M, Markus H, Tomkins A. Diet, body size and micronutrient status in Parkinson’s disease. Eur J Clin Nutr. 1992;46:879–84. [PubMed] [Google Scholar]
- 181.Beyer PL, Palarino MY, Michalek D, Busenbark K, Koller WC. Weight change and body composition in patients with Parkinson’s disease. J Am Diet Assoc. 1995;95:979–83. doi: 10.1016/S0002-8223(95)00269-3. [DOI] [PubMed] [Google Scholar]
- 182.Chen H, Zhang SM, Hernan MA, Willett WC, Ascherio A. Weight loss in Parkinson’s disease. Ann Neurol. 2003;53:676–9. doi: 10.1002/ana.10577. [DOI] [PubMed] [Google Scholar]
- 183.Delikanaki-Skaribas E, Trail M, Wong WW, Lai EC. Daily energy expenditure, physical activity, and weight loss in Parkinson’s disease patients. Mov Disord. 2009;24:667–71. doi: 10.1002/mds.22372. [DOI] [PubMed] [Google Scholar]
- 184.Levi S, Cox M, Lugon M, Hodkinson M, Tomkins A. Increased energy expenditure in Parkinson’s disease. BMJ. 1990;301:1256–7. doi: 10.1136/bmj.301.6763.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Markus HS, Cox M, Tomkins AM. Raised resting energy expenditure in Parkinson’s disease and its relationship to muscle rigidity. Clin Sci (Lond) 1992;83:199–204. doi: 10.1042/cs0830199. [DOI] [PubMed] [Google Scholar]
- 186.Toth MJ, Fishman PS, Poehlman ET. Free-living daily energy expenditure in patients with Parkinson’s disease. Neurology. 1997;48:88–91. doi: 10.1212/wnl.48.1.88. [DOI] [PubMed] [Google Scholar]
- 187.Chen H, Zhang SM, Schwarzschild MA, Hernan MA, Willett WC, Ascherio A. Obesity and the risk of Parkinson’s disease. Am J Epidemiol. 2004;159:547–55. doi: 10.1093/aje/kwh059. [DOI] [PubMed] [Google Scholar]
- 188.Logroscino G, Sesso HD, Paffenbarger RS, Jr, Lee IM. Body mass index and risk of Parkinson’s disease: a prospective cohort study. Am J Epidemiol. 2007;166:1186–90. doi: 10.1093/aje/kwm211. [DOI] [PubMed] [Google Scholar]
- 189.Evidente VG, Caviness JN, Adler CH, Gwinn-Hardy KA, Pratley RE. Serum leptin concentrations and satiety in Parkinson’s disease patients with and without weight loss. Mov Disord. 2001;16:924–7. doi: 10.1002/mds.1165. [DOI] [PubMed] [Google Scholar]
- 190.Lorefalt B, Toss G, Granerus AK. Weight loss, body fat mass, and leptin in Parkinson’s disease. Mov Disord. 2009;24:885–90. doi: 10.1002/mds.22466. [DOI] [PubMed] [Google Scholar]
- 191.Fiszer U, Michalowska M, Baranowska B, Wolinska-Witort E, Jeske W, Jethon M, et al. Leptin and ghrelin concentrations and weight loss in Parkinson’s disease. Acta Neurol Scand. 2010;121:230–6. doi: 10.1111/j.1600-0404.2009.01185.x. [DOI] [PubMed] [Google Scholar]
- 192.Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29:14057–65. doi: 10.1523/JNEUROSCI.3890-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci. 1993;69:125–30. doi: 10.3109/00207459309003322. [DOI] [PubMed] [Google Scholar]
- 194.Lipman IJ, Boykin ME, Flora RE. Glucose intolerance in Parkinson’s disease. J Chronic Dis. 1974;27:573–9. doi: 10.1016/0021-9681(74)90031-9. [DOI] [PubMed] [Google Scholar]
- 195.Becker C, Brobert GP, Johansson S, Jick SS, Meier CR. Diabetes in patients with idiopathic Parkinson’s disease. Diabetes Care. 2008;31:1808–12. doi: 10.2337/dc08-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Moroo I, Yamada T, Makino H, Tooyama I, McGeer PL, McGeer EG, et al. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson’s disease. Acta Neuropathol. 1994;87:343–8. doi: 10.1007/BF00313602. [DOI] [PubMed] [Google Scholar]
- 197.Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003;964:107–15. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- 198.Figlewicz DP, Brot MD, McCall AL, Szot P. Diabetes causes differential changes in CNS noradrenergic and dopaminergic neurons in the rat: a molecular study. Brain Res. 1996;736:54–60. doi: 10.1016/0006-8993(96)00727-5. [DOI] [PubMed] [Google Scholar]
- 199.Morris JK, Zhang H, Gupte AA, Bomhoff GL, Stanford JA, Geiger PC. Measures of striatal insulin resistance in a 6-hydroxydopamine model of Parkinson’s disease. Brain Res. 2008;1240:185–95. doi: 10.1016/j.brainres.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Baquer NZ, Taha A, Kumar P, McLean P, Cowsik SM, Kale RK, et al. A metabolic and functional overview of brain aging linked to neurological disorders. Biogerontology. 2009;10:377–413. doi: 10.1007/s10522-009-9226-2. [DOI] [PubMed] [Google Scholar]
- 201.Martin B, Brenneman R, Golden E, Walent T, Becker KG, Prabhu VV, et al. Growth factor signals in neural cells: coherent patterns of interaction control multiple levels of molecular and phenotypic responses. J Biol Chem. 2009;284:2493–511. doi: 10.1074/jbc.M804545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.St George-Hyslop PH. Molecular genetics of Alzheimer’s disease. Biol Psychiatry. 2000;47:183–99. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- 203.Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord. 2007;22(Suppl 17):S343–50. doi: 10.1002/mds.21676. [DOI] [PubMed] [Google Scholar]