

Abstract
Atherosclerotic cardiovascular diseases, chronic inflammatory diseases of multifactorial etiology, are the leading cause of death worldwide. In the last decade, more infectious agents, labeled as “infectious burden”, rather than any single pathogen, have been showed to contribute to the development of atherosclerosis through different mechanisms. Some microorganisms, such as Chlamydia pneumoniae (C. pneumoniae), human cytomegalovirus, etc. may act directly on the arterial wall contributing to endothelial dysfunction, foam cell formation, smooth muscle cell proliferation, platelet aggregation as well as cytokine, reactive oxygen specie, growth factor, and cellular adhesion molecule production. Others, such as Helicobacter pylori (H. pylori), influenza virus, etc. may induce a systemic inflammation which in turn may damage the vascular wall (e.g., by cytokines and proteases). Moreover, another indirect mechanism by which some infectious agents (such as H. pylori, C. pneumoniae, periodontal pathogens, etc.) may play a role in the pathogenesis of atherosclerosis is molecular mimicry. Given the complexity of the mechanisms by which each microorganism may contribute to atherosclerosis, defining the interplay of more infectious agents is far more difficult because the pro-atherogenic effect of each pathogen might be amplified. Clearly, continued research and a greater awareness will be helpful to improve our knowledge on the complex interaction between the infectious burden and atherosclerosis.
Keywords: Infectious burden, Atherosclerosis, Bacteria, Virus, Pathogenetic mechanisms
Core tip: Several studies support the hypothesis that the infectious burden (IB) may be more involved in the pathogenesis of atherosclerosis than any single pathogen. However, because of the complexity of the interplay of more infectious agents in the host and the limitations of the methods available for the assessment of IB, the role of IB in the pathogenesis of atherosclerosis may have been underestimated.
INTRODUCTION
Atherosclerosis, a chronic inflammatory disease of multifactorial etiology, may be considered as a multistage process, starting from the endothelial injury to the fibrous cap and thrombus formation in the advanced plaque. Key process in the development of atherosclerosis is low density lipoprotein (LDL) oxidation and accumulation in vascular cells, promoting foam cell formation as well as increased secretion of mediators of inflammation, such as interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF)-α[1]. The inflammatory state, in turn, can induce oxidative stress by enhancing the production of reactive oxygen species (ROS) in the vascular wall[1], contributing to the progression and destabilization of atherosclerotic plaque and consequently to cardiovascular diseases (CVDs). It is well known that CVDs are the leading cause of death worldwide, accounting for approximately 17.3 million deaths per year[2].
Current opinion is that increased incidence of CVDs is probably the result of a high prevalence of both traditional risk factors such as hypertension, dyslipidemia, etc. and nontraditional risk factors including inflammation, oxidative stress, and infectious agents[3]. In the last decade, infectious agents have acquired a growing importance, since they are able to induce inflammation and/or oxidative stress[4].
More recently, several studies have provided evidence that more infectious agents, for example, Chlamydia pneumonia (C. pneumoniae), Helicobacter pylori (H. pylori), human cytomegalovirus (HCMV), Herpes simplex virus (HSV), labeled as “infectious burden” (IB), rather than any single pathogen, may be involved in the development of atherosclerosis and the subsequent cardiovascular events.
EVIDENCE LINKING INFECTIOUS BURDEN WITH ATHEROSCLEROSIS
Zhu et al[5] were the first to show the association between increasing risk of coronary artery disease (CAD) and increasing number of infectious agents including C. pneumoniae, H. pylori, HCMV, HSV-1 and 2, and hepatitis A virus (HAV). Indeed, the prevalence of CAD was 48%, 69% and 85% in individuals with seropositivity to ≤ 2 pathogens, to 3 or 4 pathogens and to 5 pathogens respectively. Since then, several serological studies found a prospective relation between increasing number of infectious agents (HSV-1 and 2, HCMV, Epstein Barr virus, EBV, Haemophilus influenzae, C. pneumoniae, Mycoplasma pneumoniae, and H. pylori) and CVD outcomes[6-8]. At the same time, serological assessments demonstrated the association between increasing number of infectious agents (C. pneumoniae, H. pylori, M. pneumoniae, H. influenzae, HCMV, EBV, HSV-1 and 2) and progression of atherosclerosis[9,10]. Again, cross-sectional and case-control studies confirmed the relationship between the seropositivity to C. pneumoniae, H. pylori, HAV, HCMV, HSV-1 and 2, and atherosclerosis[11,12].
The evidence for a direct contribution of IB in the pathogenesis of atherosclerosis is based on the simultaneous detection of two pathogens in the atherosclerotic plaque (C. pneumoniae and H. pylori or M. pneumoniae and C. pneumoniae or C. pneumoniae and HCMV)[13-15]. Better yet is the evidence for a synergistic effect of C. pneumoniae and H. pylori, M. pneumoniae and C. pneumoniae, HCMV and C. pneumoniae in initiating or aggravating atherosclerosis in several animal models[16-18]. Similarly, there are some data showing the synergistic effect of the co-infection with C. pneumoniae and HCMV on the expression of atherogenic factors including IL-6, IL-8 and basic fibroblast growth factor in vascular smooth muscle cells (VSMCs) involved in advanced plaque formation[19]. Also seropositivity for both C. pneumoniae and HCMV infections was found to be associated with premature myocardial infarction even after adjustment for coronary risk factors and socioeconomic status[20].
Interestingly, the significant association between the increasing number of infectious agents together with elevated IL-6, C-reactive protein (CRP), and fibrinogen levels, and CAD prevalence, supports the hypothesis that inflammation may be one pathway by which more infectious agents and CVDs are linked[5,8,12].
The involvement of IB in the pathogenesis of atherosclerosis is expected since numerous infectious agents have been shown to play a role in the development and progression of atherosclerosis[4].
C. pneumoniae, an obligate intracellular bacterium, is responsible for respiratory infections such as sinusitis, pharyngitis and pneumonia. Exposure to C. pneumoniae is extremely common and epidemiological studies indicate that anti-C. pneumoniae antibody prevalence is 50% by the age of 20 and increases with increasing age[21]. C. pneumoniae is characterized by the ability to systematically disseminate from the lungs through peripheral blood mononuclear cells and to localize in several extrapulmonary tissues[22-25]. In recent years, it has been demonstrated that C. pneumoniae, in response to several stress conditions (iron or essential amino acid starvation, interferon (IFN)-γ or antibiotic treatment), can generate a persistent form during its developmental cycle[26-29]. Chlamydial persistent form may endure for a long time inside host cells since it is more suited to evade the host immune response and is more difficult to eradicate with antibiotics, leading to a chronic inflammatory state[26].
Cumulative evidence on the involvement of C. pneumoniae and atherosclerosis has been provided by seroepidemiological studies[30-32], C. pneumoniae DNA detection in the atherosclerotic plaque[31-33], the isolation of viable bacteria from the atheroma[4,32] and in vivo studies, demonstrating that C. pneumoniae infection may accelerate the progression of atherosclerotic lesion in animal models[4,31,34]. Lastly, in vitro studies have evidenced that C. pneumoniae is able to multiply within vascular cells, such as macrophages, endothelial cells, SMCs and platelets, and to induce chronic inflammation through the elicitation of inflammatory cytokines (e.g., IL-6, IL-1β and TNF-α)[31,32,35]. Furthermore, once inside the vascular tissue, C. pneumoniae has been shown to induce the production of ROS leading to oxidative stress, which contributes to LDL oxidation and accumulation within vascular cells and to foam cell formation[36].
Periodontal pathogens, such as Porphyromonas gingivalis (P. gingivalis), Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans), Tannerella forsythia (T. forsythia), Prevotella intermedia, Fusobacterium nucleatum (F. nucleatum), Treponema denticola, Campylobacter rectus, Streptococcus sanguis, and Streptococcus mutans, are responsible of a complex group of chronic oral inflammatory diseases like periodontitis or gingivitis. Over the last years, different lines of evidence have supported the role of periodontal bacteria in cardiovascular diseases. First of all, it has been demonstrated that oral bacteria can disseminate in the blood stream causing bacteriemia[37] and localize in vascular wall. Indeed, DNA, RNA and antigens of a variety of oral bacterial species (e.g., P. gingivalis, A. actinomycetemcomitans, T. forsythia and F. nucleatum) have been detected in atherosclerotic plaques[4]. More importantly, evidence of live P. gingivalis and A. actinomycetemcomitans in the atheroma[38], supports the direct involvement of these pathogens in the pathogenesis of atherosclerosis. Moreover, in vivo studies have shown the ability of P. gingivalis to accelerate atherosclerosis in murine models[4,39] and to induce aortic and coronary lesions in both normocholesterolemic and hypercholesterolemic pigs[40]. In vitro studies have demonstrated that periodontal pathogens are able to infect endothelial cells, SMCs and macrophages, eliciting the production of proinflammatory cytokines and chemokines (e.g., IL-6 and monocyte chemoattractant protein (MCP)-1) and the formation of foam cells, hence contributing to atherosclerosis[41,42].
H. pylori, a common cause of chronic gastritis as well as a risk factor for gastric cancer, is widespread in the general population. In the last decade, it has been considered as a possible risk factor for atherosclerosis, since H. pylori DNA has been found in the atherosclerotic plaque[4,14]. Several seroepidemiological studies have confirmed a relationship between H. pylori and atherosclerosis although others have failed to demonstrate such an association[43-46]. Controversial are data showing the ability of H. pylori to accelerate the atherosclerotic lesion development in mouse models[4]. However, H. pylori may also contribute to the systemic inflammation underlying atherosclerosis through the elicitation of acute-phase reactants (e.g., CRP) and inflammatory cytokines (e.g., IL-6)[47].
Other bacteria, such as M. pneumoniae, have been proposed as possible pathogens in atherosclerosis with controversial results. Several seroepidemiological studies have found the association between CVDs and M. pneumoniae[48,49]. Furthermore, an in vivo study has demonstrated that M. pneumoniae infection aggravated atherosclerosis in hypercholesterolemic mice[18]. However, pathological studies have not supported the association between this microorganism and atherosclerosis, since M. pneumoniae DNA has been detected in atherosclerotic tissues as well as in healthy vessels[4].
Lifelong persistent infection with HCMV has been also associated with atherosclerosis. HCMV was first detected in human atheromatous tissue by Benditt et al[50] in 1983. Experimental data have shown the ability of HCMV to infect the human vascular wall, resulting in altered function of the endothelium[51]. Furthermore, both antigen and nucleic acid sequence of HCMV have been detected in SMCs from carotid artery plaques[52-54].
In addition, HCMV DNA has been more often detected in arterial samples from patients with atherosclerosis than in control subjects[55]. Similarly, higher prevalence as well as higher titer of HCMV antibody have been observed in patients undergoing vascular surgery for atherosclerosis than in control subjects[56]. In addition, a meta-analysis study has reported a significant increased coronary heart disease risk for patients infected with HCMV[57].
Recently it has been suggested that HSV-2, but not HSV-1, was associated with premature CVD[58]. Consistent with a potential relationship between HSV-2 and CVD, Raza-Ahmad et al[59] previously examined coronary artery specimens of patients undergoing coronary artery bypass grafting and found 45% of them positive for HSV-2 and only 1% positive for HSV-1. Likewise, a large cross sectional study linked HSV-2 to hypertension, but it did not find any association with HSV-1. The reasons of the association with HSV-2 and not with HSV-1 are unclear.
There is also evidence supporting the role of influenza as a trigger for cardiovascular events[60]. However, data are debated. Some authors think that influenza (A and B) seropositivity is not a predictor of risk for CAD. Others propose that influenza virus might play a role in atherogenesis or atherothrombosis and that influenza vaccination might reduce the risk of recurrent myocardial infarction[60,61]. Recently, a correlation between influenza B virus infection and acute myocardial infarction has been reported[62].
Although there have been positive associations of antibody titers or viral antigens of the hepatitis viruses with CVD[63-65], many recent studies have reported no association. Zhu et al[63] has suggested a causal role for HAV infection in atherogenesis, on the basis of a significantly higher prevalence of CAD among subjects living in the Washington, DC, area who had serum IgG antibodies to HAV. The same research group has reported a high relative hazard for myocardial infarction or death among individuals positive for IgG antibodies to HAV. However, some authors believe that epidemiological evidence argues against a significant role for HAV infection in atherogenesis, since in countries where HAV infection is far less frequent, such as northern European countries and Australia, the incidence of cardiovascular diseases is remarkably higher than that detected in countries showing an high HAV infection prevalence[66].
Several studies have also investigated the association of atherosclerosis with hepatitis C virus (HCV) infection, with conflicting results. Some studies have reported that the presence of antibody against HCV was associated with an increased risk of carotid artery plaque in the general population[65]. In addition, positive-strand HCV RNA has been detected in carotid plaque tissues from anti-HCV antibody-positive patients but it was not detected in anti-HCV antibody-negative patients[67,68]. Furthermore, multivariate logistic regression analysis has showed that HCV core protein positivity was an independent predictor of carotid plaque, supporting the possible link between persistent HCV infection and carotid atherosclerosis in subjects without severe liver dysfunction[69]. Patients with chronic HCV infection are known to develop not only hepatitis, but also various metabolic disorders[70,71]. Indeed, HCV affects both glucose and lipid metabolism. Recent population-based studies have demonstrated hypolipidemia in subjects with chronic HCV infection[72,73]. Although altered lipid metabolism is linked to atherosclerosis, the effect of HCV on atherosclerosis remains controversial[73-75]. A systematic review published by Roed et al[76] has suggested an increased risk of CAD in HCV infected individuals. Recently, a study has revealed that chronic HCV infection was associated with increased insulin resistance and with mild atherosclerosis, thus underlining the complexity of this association[77].
A growing body of literature reports that human immunodeficiency virus (HIV) infected patients suffer from an elevated risk for both subclinical atherosclerotic disease and CVD events than uninfected individuals[78-81]. However, the results of a meta-analysis as well as a number of independent studies have questioned this association[82,83]. Furthermore, antiretroviral therapy (ART) has been shown to have independent effects on lesion development in several experimental studies, and some compounds, such as protease inhibitors, are associated with lipodystrophy, central adiposity, hyperlipidaemia, and endothelial dysfunction, all recognized risk factors for CVD[84-86]. However, the risk of CVD associated with HIV infection is not fully accounted for by the effects of antiretrovirals in these studies. Indeed, other papers have suggested that direct HIV infection of endothelial cells could contribute to atherosclerosis by causing endothelial dysfunction[87]. Furthermore, Hsue et al[88] have shown that increased atherosclerosis can occur in the absence of ART in HIV-infected patients. Recently, Desvarieux et al89] have further emphasized the role of HIV in atherosclerosis, reporting the preponderant association of HIV infection (rather than ART) with increased atherosclerosis in never smokers, thus also determining the validity of the relationship independent of this important confounder.
POSSIBLE MECHANISMS UNDERLYING INFECTIOUS BURDEN RELATED TO ATHEROSCLEROSIS
A substantial body of evidence supports the hypothesis that more infectious agents rather than a single pathogen may contribute to atherosclerosis through different mechanisms (Figure 1). Some microorganisms, such as A. actinomycetemcomitans, may act directly on the arterial wall contributing to endothelial dysfunction, foam cell formation, SMC proliferation, platelet aggregation and cytokine production[4,41]. Otherwise, microorganisms, such as H. pylori, may induce a systemic inflammation which in turn may damage the vascular wall (e.g., by cytokines and proteases). Indeed, many observational studies have reported the association of the seropositivity to H. pylori with a sensitive marker of systemic inflammation and even predictor of acute cardiovascular events such as CRP[90,91].
Figure 1.
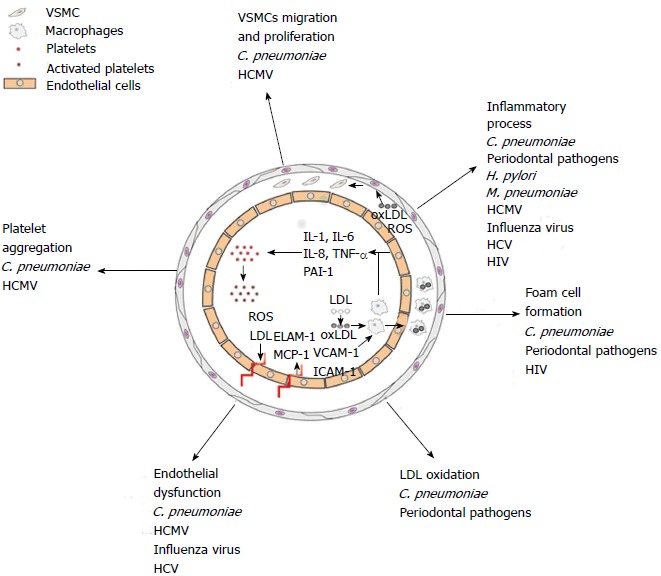
Schematic representation of transversal artery section. Possible etiopathogenetic mechanisms of the infectious agents in atherosclerotic plaque development. C. pneumoniae: Chlamydia pneumoniae; HCMV: Human cytomegalovirus; H. pylori: Helicobacter pylori; M. pneumoniae: Mycoplasma pneumoniae; HCV: Hepatitis C virus; HIV: Human immunodeficiency virus; SMC; Smooth muscle cell; ROS; Reactive oxygen species; LDL; Low-density lipoprotein; Ox-LDL: Oxidized low-density lipoprotein; IL: Interleukin; TNF-α: Tumor necrosis factor; PAI: Plasminogen activator inhibitor-1; ELAM-1: Endothelial-leukocyte adhesion molecule-1; VCAM-1: Vascular cell adhesion molecule-1; ICAM-1: Intercellular adhesion molecule-1; MCP-1: Monocyte chemoattractant protein-1.
Furthermore, there are also infectious agents, such as C. pneumoniae and P. gingivalis, that may contribute to atherosclerosis by both direct and indirect mechanisms. As a direct effect, these microorganisms have been shown to infect macrophages, SMCs and endothelial cells inducing the production of ROS, cytokines (IL-6, IL-1β and TNF-α, etc.), growth factors (basic fibroblast growth factor, bFGF, tumor growth factor (TGF)-β, etc.) and cellular adhesion molecules (vascular cell adhesion molecule-1, VCAM-1, intercellular adhesion molecule-1, ICAM-1, endothelial-leukocyte adhesion molecule-1, ELAM-1, etc.), all responsible for the typical pathological changes of the atherosclerotic plaque[36,41,42]. On the other hand, C. pneumoniae and P. gingivalis can contribute to atherosclerosis indirectly by inducing systemic inflammation[92,93]. Indeed, circulating cytokines (IL-6) and acute phase proteins (serum amyloid A), produced in response to systemic infection of animal models with C. pneumoniae or P. gingivalis, have been associated with the progression and destabilization of atherosclerotic lesions[94,95]. Also, in human, increases in circulating CRP levels and P. gingivalis or C. pneumoniae antibody levels have been associated with an increased risk of CAD[4,96].
Another indirect mechanism by which infectious agents play a role in the pathogenesis of atherosclerosis is molecular mimicry. There is evidence that the humoral immune response against the heat shock proteins (HSPs) found in C. pneumoniae, H. pylori and P. gingivalis, may cross-react with human HSPs in vascular cells, initiating an autoimmune process, responsible for vascular endothelial injury[97-99]. In fact, antibody levels against HSPs have been associated with early and advanced atherosclerosis[100]. In addition, in vivo studies have also confirmed that the T-cell immune response against HSP, derived from H. pylori, C. pneumoniae and P. gingivalis, could promote atherogenesis[101-103].
As far as concern viral agents, data supporting a direct effect of these agents on the pathogenesis of atherosclerosis are usually weak; infections with viruses are more likely to have an indirect effect on the initiation and progression of atherosclerosis.
Relative to HCMV, it has been observed that SMCs isolated from atherosclerotic coronary lesions, harbor HCMV DNA sequences and express immediate early proteins, such as IE84, one of the immediate early proteins of the virus that binds and inhibits p53[104]. Inhibition of p53 by the virus is held responsible for the enhanced proliferation of SMCs and impaired apoptosis, either of which may contribute to restenosis[104]. Furthermore, persistent infection of HCMV in endothelial cells leads to dysfunction of these cells and activates proinflammatory signaling pathways, which promote enhanced proliferation and migration of monocytes and SMCs into intima of the vascular wall as well as lipid accumulation and expansion of the atherosclerotic lesion[105,106].
The precise mechanism by which influenza virus infection contributes to atherosclerosis is unclear, however inflammation and coagulopathy seem to be key factors. Specifically, the potential mechanisms may include: (1) antigenic cross-reactivity; (2) an increase in pro-inflammatory and prothrombotic cytokines, such as IL-2, IL-6, IL-10 and IL-18; (3) pronounced expression of inflammatory cytokines by infected monocytes and reduced clotting time; (4) increased trafficking of macrophages into the arterial wall; and (5) induction of procoagulant activity in infected endothelial cells, reduced clotting time, and increased expression of tissue factor[60,62]. Repeated influenza virus infection may injure vascular endothelial cells and initiate the inflammatory response that is required to accelerate and enhance the development of atherosclerosis.
It has been suggested that influenza virus may trigger the destabilization of already present vulnerable plaques. Naghavi et al[107] have showed that inoculation of influenza virus A in atherosclerotic apolipoprotein E-deficient mice led to a marked increase in inflammation and thrombosis in plaques but not in normal area. Influenza virus infection may cause the production of IL-2, IL-6, IL-10, IL-18, IFN-γ and TNF-α, which induces endothelial cells to release endothelin (ET)-1, sICAM-1 and sVCAM-1. These inflammatory cytokines may trigger the destabilization of existing vulnerable plaques and lead to an acute myocardial infarction without being involved in the development or progression of atherosclerosis[108].
As stated before, the role of HCV in atherosclerosis is widely debated. A role of chronic inflammation in atherogenesis has been suggested[109] because chronic HCV infection has been associated with vasculitis and mixed cryoglobulinemia, which may cause vascular injury as well as cerebrovascular damage[110]. Concentrations of sICAM-1 have been reported to be higher in HCV patients than in control subjects[111], and Cacoub et al[112] have reported a possible association of anti-endothelial cell autoantibodies, commonly observed in HCV patients but not in other viral diseases, with vasculitis. However, a recent paper has demonstrated a favorable effect of HCV on atherosclerosis[77] probably due to the alteration in lipid parameters of the subjects with chronic HCV infection caused by the progression of liver disease and partly by a metabolic process associated with HCV replication.
Several papers have reported that atherosclerosis is consistently higher among the HIV positive patients, with or without treatment. Recently Shrestha et al[113] have postulated three key sequential biological processes that lead to accelerate progression of atherosclerosis: (1) inflammation leads to the recruitment of monocytes; (2) monocytes migrate to the endothelium and differentiate to macrophages and foam cells; and (3) apoptosis of foam cells leads to plaque development through calcium-dependent endoplasmic reticulum stress. The HIV itself, or together with treatment, affects this progression by increasing inflammation, promoting the transformation of monocytes, and increasing apoptosis through ER stress and an imbalance of calcium.
Given the complexity of the mechanisms by which each microorganism may play a role in the pathogenesis of atherosclerosis, defining the interplay of more infectious agents is far more difficult because the pro-atherogenic effect of each pathogen might be amplified.
ASSESSMENT OF INFECTIOUS BURDEN RELATED TO ATHEROSCLEROSIS
The main unanswered question is the definition of IB. Several infectious agents, such as C. pneumoniae, H. influenzae, H. pylori, M. pneumoniae, HCMV, HSV, EBV and HAV, etc. have been proposed as constituting the IB related to atherosclerosis, but, to date, there is no consensus both on the number and on which microorganisms should be considered.
The majority of the infectious agents involved in the IB are widespread, as evidenced from the high prevalence of antibodies in the general population; more than half of the world population is seropositive, for example, to C. pneumoniae, H. pylori, HCMV and HSV. Again, HSV, HCMV, C. pneumoniae and H. pylori infections could be acquired early in life, and persist over time. The situation is further complicated by the fact that the infectious agents involved in IB are responsible for persistent infection (e.g., HIV and C. pneumoniae), repeated infection (e.g., influenza virus), latent infection followed by life-long reactivation (e.g., HSV and HCMV) or chronic infection (e.g., HBV, HCV and, H. pylori).
Nowadays, the assessment of the IB related to atherosclerosis is based mainly on serological methods. The main limitations of serology are to define whether the antibody response reflects a past or chronic infection and to identify the differences in seropositivity between patients and general population, especially if seropositivity is common. In addition, serological diagnostic methods are not appropriate for the detection of novel or rare pathogens. Lastly, most serological assays are designed for diagnostic testing in clinical settings, and not for the assessment of the burden of infections acquired through life. Notably, most of the infectious agents involved in the IB, such as C. pneumoniae, H. pylori, HSV, and HCMV, can cause asymptomatic infections that are not routinely investigated. As a result, these undiagnosed infections, if left untreated, can contribute to the development of severe complications, including CVDs.
Other technical obstacles in the assessment of the IB related to atherosclerosis include difficulties in obtaining atherosclerotic plaques and in isolating and culturing certain infectious agents. Indeed, atherosclerotic plaques are obtained too late during the course of the disease to be of clinical use.
Another intriguing issue is the interaction of more infectious agents with host factors, such as age, gender, ethnicity, and other concomitant infections or clinical conditions that may impair the host immune system, thus potentially modifying the establishment, progression and outcome of the infection. Moreover, genome-wide association studies have now convincingly shown that the susceptibility to an infection as well as the diverse outcomes (for example the resolution of infection, the clinical deterioration to severe disease, or the progression from acute infection to persistent infection) can be, at least, partly explained by genetic variation[114]. In this regard, a recent study has showed that IL-6 gene polymorphisms appear to influence the susceptibility to the atherogenic effect of more infectious agents including C. pneumoniae, CMV, H. pylori and HSV-1[115].
CONCLUSION
Based on the extent of the issues previously described, the role of the IB in the pathogenesis of atherosclerosis may have been substantially underestimated, so that the true impact of IB is likely to be much greater than it is currently recognized.
Different approaches could be taken to address the problem; one possibility may be to conceive a well-designed protocol that includes the number and the type of infectious agents, the antibody response (IgG and/or IgA) as well as the monitoring of antibody titer, atherosclerotic biological markers and cytokines. The latter are particularly critical in chronic viral infections, such as HIV infection, in which two monocytes surface markers (CD11b and chemokine (C-X-C motif) receptor (CXCR)-1) have been proposed as predictors of CVD[116].
Clearly, continued research and a better awareness of this problem will be helpful to improve our knowledge on the complex interaction between IB and atherosclerosis.
Footnotes
P- Reviewers: He JY, Stover CM S- Editor: Song XX L- Editor: A E- Editor: Wu HL
Supported by Grants to R. Sessa from Center for Social Disease Research, “Sapienza” University, Rome
References
- 1.Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med. 2010;14:70–78. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Global Atlas on Cardiovascular Disease Prevention and Control. Mendis S, Puska P, Norrving, B (Editors) Available from: http://whqlibdoc.who.int/publications/2011/9789241564373_eng.pdf.
- 3.Balagopal PB, de Ferranti SD, Cook S, Daniels SR, Gidding SS, Hayman LL, McCrindle BW, Mietus-Snyder ML, Steinberger J. Nontraditional risk factors and biomarkers for cardiovascular disease: mechanistic, research, and clinical considerations for youth: a scientific statement from the American Heart Association. Circulation. 2011;123:2749–2769. doi: 10.1161/CIR.0b013e31821c7c64. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld ME, Campbell LA. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost. 2011;106:858–867. doi: 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- 5.Zhu J, Quyyumi AA, Norman JE, Csako G, Waclawiw MA, Shearer GM, Epstein SE. Effects of total pathogen burden on coronary artery disease risk and C-reactive protein levels. Am J Cardiol. 2000;85:140–146. doi: 10.1016/s0002-9149(99)00653-0. [DOI] [PubMed] [Google Scholar]
- 6.Rupprecht HJ, Blankenberg S, Bickel C, Rippin G, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of viral and bacterial infectious burden on long-term prognosis in patients with coronary artery disease. Circulation. 2001;104:25–31. doi: 10.1161/hc2601.091703. [DOI] [PubMed] [Google Scholar]
- 7.Smieja M, Gnarpe J, Lonn E, Gnarpe H, Olsson G, Yi Q, Dzavik V, McQueen M, Yusuf S. Multiple infections and subsequent cardiovascular events in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation. 2003;107:251–257. doi: 10.1161/01.cir.0000044940.65226.1f. [DOI] [PubMed] [Google Scholar]
- 8.Mundkur LA, Rao VS, Hebbagudi S, Shanker J, Shivanandan H, Nagaraj RK, Kakkar VV. Pathogen burden, cytomegalovirus infection and inflammatory markers in the risk of premature coronary artery disease in individuals of Indian origin. Exp Clin Cardiol. 2012;17:63–68. [PMC free article] [PubMed] [Google Scholar]
- 9.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002;33:2581–2586. doi: 10.1161/01.str.0000034789.82859.a4. [DOI] [PubMed] [Google Scholar]
- 10.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Rippin G, Victor A, Hafner G, Schlumberger W, Meyer J. Impact of infectious burden on extent and long-term prognosis of atherosclerosis. Circulation. 2002;105:15–21. doi: 10.1161/hc0102.101362. [DOI] [PubMed] [Google Scholar]
- 11.Elkind MS, Ramakrishnan P, Moon YP, Boden-Albala B, Liu KM, Spitalnik SL, Rundek T, Sacco RL, Paik MC. Infectious burden and risk of stroke: the northern Manhattan study. Arch Neurol. 2010;67:33–38. doi: 10.1001/archneurol.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nazmi A, Diez-Roux AV, Jenny NS, Tsai MY, Szklo M, Aiello AE. The influence of persistent pathogens on circulating levels of inflammatory markers: a cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis. BMC Public Health. 2010;10:706. doi: 10.1186/1471-2458-10-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higuchi Mde L, Reis MM, Sambiase NV, Palomino SA, Castelli JB, Gutierrez PS, Aiello VD, Ramires JA. Coinfection with Mycoplasma pneumoniae and Chlamydia pneumoniae in ruptured plaques associated with acute myocardial infarction. Arq Bras Cardiol. 2003;81:12–22, 1-11. doi: 10.1590/s0066-782x2003000900001. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan M, Yavuz SS, Cinar B, Koksal V, Kut MS, Yapici F, Gercekoglu H, Demirtas MM. Detection of Chlamydia pneumoniae and Helicobacter pylori in atherosclerotic plaques of carotid artery by polymerase chain reaction. Int J Infect Dis. 2006;10:116–123. doi: 10.1016/j.ijid.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Virok D, Kis Z, Kari L, Barzo P, Sipka R, Burian K, Nelson DE, Jackel M, Kerenyi T, Bodosi M, et al. Chlamydophila pneumoniae and human cytomegalovirus in atherosclerotic carotid plaques--combined presence and possible interactions. Acta Microbiol Immunol Hung. 2006;53:35–50. doi: 10.1556/AMicr.53.2006.1.3. [DOI] [PubMed] [Google Scholar]
- 16.Burnett MS, Gaydos CA, Madico GE, Glad SM, Paigen B, Quinn TC, Epstein SE. Atherosclerosis in apoE knockout mice infected with multiple pathogens. J Infect Dis. 2001;183:226–231. doi: 10.1086/317938. [DOI] [PubMed] [Google Scholar]
- 17.Liuba P, Pesonen E, Paakkari I, Batra S, Andersen L, Forslid A, Ylä-Herttuala S, Persson K, Wadström T, Wang X, et al. Co-infection with Chlamydia pneumoniae and Helicobacter pylori results in vascular endothelial dysfunction and enhanced VCAM-1 expression in apoE-knockout mice. J Vasc Res. 2003;40:115–122. doi: 10.1159/000070708. [DOI] [PubMed] [Google Scholar]
- 18.Damy SB, Higuchi ML, Timenetsky J, Reis MM, Palomino SP, Ikegami RN, Santos FP, Osaka JT, Figueiredo LP. Mycoplasma pneumoniae and/or Chlamydophila pneumoniae inoculation causing different aggravations in cholesterol-induced atherosclerosis in apoE KO male mice. BMC Microbiol. 2009;9:194. doi: 10.1186/1471-2180-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prochnau D, Straube E, Figulla HR, Rödel J. Supra-additive expression of interleukin-6, interleukin-8 and basic fibroblast growth factor in vascular smooth muscle cells following coinfection with Chlamydia pneumoniae and cytomegalovirus as a novel link between infection and atherosclerosis. Can J Infect Dis Med Microbiol. 2012;23:e26–e30. doi: 10.1155/2012/987476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gattone M, Iacoviello L, Colombo M, Castelnuovo AD, Soffiantino F, Gramoni A, Picco D, Benedetta M, Giannuzzi P. Chlamydia pneumoniae and cytomegalovirus seropositivity, inflammatory markers, and the risk of myocardial infarction at a young age. Am Heart J. 2001;142:633–640. doi: 10.1067/mhj.2001.118118. [DOI] [PubMed] [Google Scholar]
- 21.Grayston JT. Background and current knowledge of Chlamydia pneumoniae and atherosclerosis. J Infect Dis. 2000;181 Suppl 3:S402–S410. doi: 10.1086/315596. [DOI] [PubMed] [Google Scholar]
- 22.Sessa R, Di Pietro M, Schiavoni G, Santino I, Cipriani P, Romano S, Penco M, del Piano M. Prevalence of Chlamydia pneumoniae in peripheral blood mononuclear cells in Italian patients with acute ischaemic heart disease. Atherosclerosis. 2001;159:521–525. doi: 10.1016/s0021-9150(01)00537-8. [DOI] [PubMed] [Google Scholar]
- 23.Wang SS, Tondella ML, Bajpai A, Mathew AG, Mehranpour P, Li W, Kacharava AG, Fields BS, Austin H, Zafari AM. Circulating Chlamydia pneumoniae DNA and advanced coronary artery disease. Int J Cardiol. 2007;118:215–219. doi: 10.1016/j.ijcard.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Di Pietro M, Schiavoni G, Sessa V, Pallotta F, Costanzo G, Sessa R. Chlamydia pneumoniae and osteoporosis-associated bone loss: a new risk factor? Osteoporos Int. 2013;24:1677–1682. doi: 10.1007/s00198-012-2217-1. [DOI] [PubMed] [Google Scholar]
- 25.Di Pietro M, Filardo S, Cazzavillan S, Segala C, Bevilacqua P, Bonoldi E, D’Amore ES, Rassu M, Sessa R. Could past Chlamydial vascular infection promote the dissemination of Chlamydia pneumoniae to the brain? J Biol Regul Homeost Agents. 2013;27:155–164. [PubMed] [Google Scholar]
- 26.Schoborg RV. Chlamydia persistence -- a tool to dissect chlamydia--host interactions. Microbes Infect. 2011;13:649–662. doi: 10.1016/j.micinf.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Pietro M, Tramonti A, De Santis F, De Biase D, Schiavoni G, Filardo S, Zagaglia C, Sessa R. Analysis of gene expression in penicillin G induced persistence of Chlamydia pneumoniae. J Biol Regul Homeost Agents. 2012;26:277–284. [PubMed] [Google Scholar]
- 28.Di Pietro M, De Santis F, De Biase D, Sessa R. The elusive but pathogenic peptidoglycanof Chlamydiae. Eur J Inflamm. 2013;11:257–260. [Google Scholar]
- 29.Di Pietro M, Filardo S, De Santis F, Sessa R. New insights into Chlamydiae persistence: an energy metabolism strategy? Int J Immunopathol Pharmacol. 2013;26:525–528. doi: 10.1177/039463201302600227. [DOI] [PubMed] [Google Scholar]
- 30.Sessa R, Di Pietro M, Santino I, del Piano M, Varveri A, Dagianti A, Penco M. Chlamydia pneumoniae infection and atherosclerotic coronary disease. Am Heart J. 1999;137:1116–1119. doi: 10.1016/s0002-8703(99)70371-6. [DOI] [PubMed] [Google Scholar]
- 31.Sessa R, Nicoletti M, Di Pietro M, Schiavoni G, Santino I, Zagaglia C, Del Piano M, Cipriani P. Chlamydia pneumoniae and atherosclerosis: current state and future prospectives. Int J Immunopathol Pharmacol. 2009;22:9–14. doi: 10.1177/039463200902200102. [DOI] [PubMed] [Google Scholar]
- 32.Joshi R, Khandelwal B, Joshi D, Gupta OP. Chlamydophila pneumoniae infection and cardiovascular disease. N Am J Med Sci. 2013;5:169–181. doi: 10.4103/1947-2714.109178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sessa R, Di Pietro M, Schiavoni G, Santino I, Benedetti-Valentini F, Perna R, Romano S, del Piano M. Chlamydia pneumoniae DNA in patients with symptomatic carotid atherosclerotic disease. J Vasc Surg. 2003;37:1027–1031. doi: 10.1067/mva.2003.200. [DOI] [PubMed] [Google Scholar]
- 34.Chen S, Shimada K, Zhang W, Huang G, Crother TR, Arditi M. IL-17A is proatherogenic in high-fat diet-induced and Chlamydia pneumoniae infection-accelerated atherosclerosis in mice. J Immunol. 2010;185:5619–5627. doi: 10.4049/jimmunol.1001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Pietro M, Schiavoni G, Del Piano M, Shaik Y, Boscolo P, Caraffa A, Teté S, Conti F, Sessa R. Chlamydia pneumoniae and atherosclerosis: the role of mast cells. J Biol Regul Homeost Agents. 2009;23:65–69. [PubMed] [Google Scholar]
- 36.Di Pietro M, Filardo S, De Santis F, Sessa R. Chlamydia pneumoniae infection in atherosclerotic lesion development through oxidative stress: a brief overview. Int J Mol Sci. 2013;14:15105–15120. doi: 10.3390/ijms140715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castillo DM, Sánchez-Beltrán MC, Castellanos JE, Sanz I, Mayorga-Fayad I, Sanz M, Lafaurie GI. Detection of specific periodontal microorganisms from bacteraemia samples after periodontal therapy using molecular-based diagnostics. J Clin Periodontol. 2011;38:418–427. doi: 10.1111/j.1600-051X.2011.01717.x. [DOI] [PubMed] [Google Scholar]
- 38.Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. 2005;25:e17–e18. doi: 10.1161/01.ATV.0000155018.67835.1a. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi C, Viereck J, Hua N, Phinikaridou A, Madrigal AG, Gibson FC, Hamilton JA, Genco CA. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis. 2011;215:52–59. doi: 10.1016/j.atherosclerosis.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brodala N, Merricks EP, Bellinger DA, Damrongsri D, Offenbacher S, Beck J, Madianos P, Sotres D, Chang YL, Koch G, et al. Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2005;25:1446–1451. doi: 10.1161/01.ATV.0000167525.69400.9c. [DOI] [PubMed] [Google Scholar]
- 41.Rodrigues PH, Reyes L, Chadda AS, Bélanger M, Wallet SM, Akin D, Dunn W, Progulske-Fox A. Porphyromonas gingivalis strain specific interactions with human coronary artery endothelial cells: a comparative study. PLoS One. 2012;7:e52606. doi: 10.1371/journal.pone.0052606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaik-Dasthagirisaheb YB, Huang N, Baer MT, Gibson FC. Role of MyD88-dependent and MyD88-independent signaling in Porphyromonas gingivalis-elicited macrophage foam cell formation. Mol Oral Microbiol. 2013;28:28–39. doi: 10.1111/omi.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christodoulou DK, Milionis HJ, Pappa P, Katsanos KH, Sigounas D, Florentin M, Elisaf M, Tsianos EV. Association of Helicobacter pylori infection with cardiovascular disease--is it just a myth? Eur J Intern Med. 2011;22:191–194. doi: 10.1016/j.ejim.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Park MJ, Choi SH, Kim D, Kang SJ, Chung SJ, Choi SY, Yoon DH, Lim SH, Kim YS, Yim JY, et al. Association between Helicobacter pylori Seropositivity and the Coronary Artery Calcium Score in a Screening Population. Gut Liver. 2011;5:321–327. doi: 10.5009/gnl.2011.5.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schöttker B, Adamu MA, Weck MN, Müller H, Brenner H. Helicobacter pylori infection, chronic atrophic gastritis and major cardiovascular events: a population-based cohort study. Atherosclerosis. 2012;220:569–574. doi: 10.1016/j.atherosclerosis.2011.11.029. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Segers S, Blaser MJ. Association between Helicobacter pylori and mortality in the NHANES III study. Gut. 2013;62:1262–1269. doi: 10.1136/gutjnl-2012-303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogha M, Nikvarz M, Pourmoghaddas Z, Shirneshan K, Dadkhah D, Pourmoghaddas M. Is helicobacter pylori infection a risk factor for coronary heart disease? ARYA Atheroscler. 2012;8:5–8. [PMC free article] [PubMed] [Google Scholar]
- 48.Reunanen A, Roivainen M, Kleemola M. Increased titer of antibodies to Mycoplasma pneumoniae may be associated with coronary heart disease. Atherosclerosis. 2005;180:209–210. doi: 10.1016/j.atherosclerosis.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 49.Daxböck F, Assadian A, Watkins-Riedel T, Assadian O. Persistently elevated IgA antibodies to Mycoplasma pneumoniae in patients with internal carotid artery stenosis. GMS Krankenhhyg Interdiszip. 2011;6:Doc04. doi: 10.3205/dgkh000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benditt EP, Barrett T, McDougall JK. Viruses in the etiology of atherosclerosis. Proc Natl Acad Sci USA. 1983;80:6386–6389. doi: 10.1073/pnas.80.20.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Dam-Mieras MC, Bruggeman CA, Muller AD, Debie WH, Zwaal RF. Induction of endothelial cell procoagulant activity by cytomegalovirus infection. Thromb Res. 1987;47:69–75. doi: 10.1016/0049-3848(87)90241-6. [DOI] [PubMed] [Google Scholar]
- 52.Melnick JL, Petrie BL, Dreesman GR, Burek J, McCollum CH, DeBakey ME. Cytomegalovirus antigen within human arterial smooth muscle cells. Lancet. 1983;2:644–647. doi: 10.1016/s0140-6736(83)92529-1. [DOI] [PubMed] [Google Scholar]
- 53.Yi L, Wang DX, Feng ZJ. Detection of human cytomegalovirus in atherosclerotic carotid arteries in humans. J Formos Med Assoc. 2008;107:774–781. doi: 10.1016/S0929-6646(08)60190-4. [DOI] [PubMed] [Google Scholar]
- 54.Xenaki E, Hassoulas J, Apostolakis S, Sourvinos G, Spandidos DA. Detection of cytomegalovirus in atherosclerotic plaques and nonatherosclerotic arteries. Angiology. 2009;60:504–508. doi: 10.1177/0003319708322390. [DOI] [PubMed] [Google Scholar]
- 55.Hendrix MG, Salimans MM, van Boven CP, Bruggeman CA. High prevalence of latently present cytomegalovirus in arterial walls of patients suffering from grade III atherosclerosis. Am J Pathol. 1990;136:23–28. [PMC free article] [PubMed] [Google Scholar]
- 56.Adam E, Melnick JL, Probtsfield JL, Petrie BL, Burek J, Bailey KR, McCollum CH, DeBakey ME. High levels of cytomegalovirus antibody in patients requiring vascular surgery for atherosclerosis. Lancet. 1987;2:291–293. doi: 10.1016/s0140-6736(87)90888-9. [DOI] [PubMed] [Google Scholar]
- 57.Ji YN, An L, Zhan P, Chen XH. Cytomegalovirus infection and coronary heart disease risk: a meta-analysis. Mol Biol Rep. 2012;39:6537–6546. doi: 10.1007/s11033-012-1482-6. [DOI] [PubMed] [Google Scholar]
- 58.Mendy A, Vieira ER, Gasana J. Seropositivity to herpes simplex virus type 2, but not type 1 is associated with premature cardiovascular diseases: a population-based cross-sectional study. Atherosclerosis. 2013;231:18–21. doi: 10.1016/j.atherosclerosis.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 59.Raza-Ahmad A, Klassen GA, Murphy DA, Sullivan JA, Kinley CE, Landymore RW, Wood JR. Evidence of type 2 herpes simplex infection in human coronary arteries at the time of coronary artery bypass surgery. Can J Cardiol. 1995;11:1025–1029. [PubMed] [Google Scholar]
- 60.Madjid M, Naghavi M, Litovsky S, Casscells SW. Influenza and cardiovascular disease: a new opportunity for prevention and the need for further studies. Circulation. 2003;108:2730–2736. doi: 10.1161/01.CIR.0000102380.47012.92. [DOI] [PubMed] [Google Scholar]
- 61.Lavallée P, Perchaud V, Gautier-Bertrand M, Grabli D, Amarenco P. Association between influenza vaccination and reduced risk of brain infarction. Stroke. 2002;33:513–518. doi: 10.1161/hs0202.102328. [DOI] [PubMed] [Google Scholar]
- 62.Guan X, Yang W, Sun X, Wang L, Ma B, Li H, Zhou J. Association of influenza virus infection and inflammatory cytokines with acute myocardial infarction. Inflamm Res. 2012;61:591–598. doi: 10.1007/s00011-012-0449-3. [DOI] [PubMed] [Google Scholar]
- 63.Zhu J, Quyyumi AA, Norman JE, Costello R, Csako G, Epstein SE. The possible role of hepatitis A virus in the pathogenesis of atherosclerosis. J Infect Dis. 2000;182:1583–1587. doi: 10.1086/317613. [DOI] [PubMed] [Google Scholar]
- 64.Alyan O, Kacmaz F, Ozdemir O, Deveci B, Astan R, Celebi AS, Ilkay E. Hepatitis C infection is associated with increased coronary artery atherosclerosis defined by modified Reardon severity score system. Circ J. 2008;72:1960–1965. doi: 10.1253/circj.cj-08-0459. [DOI] [PubMed] [Google Scholar]
- 65.Ishizaka N, Ishizaka Y, Takahashi E, Toda Ei E, Hashimoto H, Ohno M, Nagai R, Yamakado M. Increased prevalence of carotid atherosclerosis in hepatitis B virus carriers. Circulation. 2002;105:1028–1030. doi: 10.1161/hc0902.105718. [DOI] [PubMed] [Google Scholar]
- 66.Cainelli F, Concia E, Vento S. Hepatitis A virus infection and atherosclerosis. J Infect Dis. 2001;184:390–391. doi: 10.1086/322026. [DOI] [PubMed] [Google Scholar]
- 67.Boddi M, Abbate R, Chellini B, Giusti B, Giannini C, Pratesi G, Rossi L, Pratesi C, Gensini GF, Paperetti L, et al. Hepatitis C virus RNA localization in human carotid plaques. J Clin Virol. 2010;47:72–75. doi: 10.1016/j.jcv.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 68.Boddi M, Abbate R, Chellini B, Giusti B, Solazzo V, Soft F, Pratesi G, Pratesi C, Gensini G, Zignego AL. HCV infection facilitates asymptomatic carotid atherosclerosis: preliminary report of HCV RNA localization in human carotid plaques. Dig Liver Dis. 2007;39 Suppl 1:S55–S60. doi: 10.1016/s1590-8658(07)80012-0. [DOI] [PubMed] [Google Scholar]
- 69.Ishizaka Y, Ishizaka N, Takahashi E, Unuma T, Tooda E, Hashimoto H, Nagai R, Yamakado M. Association between hepatitis C virus core protein and carotid atherosclerosis. Circ J. 2003;67:26–30. doi: 10.1253/circj.67.26. [DOI] [PubMed] [Google Scholar]
- 70.Weinman SA, Belalcazar LM. Hepatitis C: a metabolic liver disease. Gastroenterology. 2004;126:917–919. doi: 10.1053/j.gastro.2003.01.001. [DOI] [PubMed] [Google Scholar]
- 71.Kawaguchi T, Izumi N, Charlton MR, Sata M. Branched-chain amino acids as pharmacological nutrients in chronic liver disease. Hepatology. 2011;54:1063–1070. doi: 10.1002/hep.24412. [DOI] [PubMed] [Google Scholar]
- 72.Miyazaki T, Honda A, Ikegami T, Saitoh Y, Hirayama T, Hara T, Doy M, Matsuzaki Y. Hepatitis C virus infection causes hypolipidemia regardless of hepatic damage or nutritional state: An epidemiological survey of a large Japanese cohort. Hepatol Res. 2011;41:530–541. doi: 10.1111/j.1872-034X.2011.00803.x. [DOI] [PubMed] [Google Scholar]
- 73.Mostafa A, Mohamed MK, Saeed M, Hasan A, Fontanet A, Godsland I, Coady E, Esmat G, El-Hoseiny M, Abdul-Hamid M, et al. Hepatitis C infection and clearance: impact on atherosclerosis and cardiometabolic risk factors. Gut. 2010;59:1135–1140. doi: 10.1136/gut.2009.202317. [DOI] [PubMed] [Google Scholar]
- 74.Ishizaka N, Ishizaka Y, Takahashi E, Tooda Ei, Hashimoto H, Nagai R, Yamakado M. Association between hepatitis C virus seropositivity, carotid-artery plaque, and intima-media thickening. Lancet. 2002;359:133–135. doi: 10.1016/s0140-6736(02)07339-7. [DOI] [PubMed] [Google Scholar]
- 75.Yang KC, Chen MF, Su TC, Jeng JS, Hwang BS, Lin LY, Liau CS, Lee YT. Hepatitis B virus seropositivity is not associated with increased risk of carotid atherosclerosis in Taiwanese. Atherosclerosis. 2007;195:392–397. doi: 10.1016/j.atherosclerosis.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 76.Roed T, Lebech AM, Kjaer A, Weis N. Hepatitis C virus infection and risk of coronary artery disease: a systematic review of the literature. Clin Physiol Funct Imaging. 2012;32:421–430. doi: 10.1111/j.1475-097X.2012.01152.x. [DOI] [PubMed] [Google Scholar]
- 77.Miyajima I, Kawaguchi T, Fukami A, Nagao Y, Adachi H, Sasaki S, Imaizumi T, Sata M. Chronic HCV infection was associated with severe insulin resistance and mild atherosclerosis: a population-based study in an HCV hyperendemic area. J Gastroenterol. 2013;48:93–100. doi: 10.1007/s00535-012-0610-3. [DOI] [PubMed] [Google Scholar]
- 78.Hsue PY, Lo JC, Franklin A, Bolger AF, Martin JN, Deeks SG, Waters DD. Progression of atherosclerosis as assessed by carotid intima-media thickness in patients with HIV infection. Circulation. 2004;109:1603–1608. doi: 10.1161/01.CIR.0000124480.32233.8A. [DOI] [PubMed] [Google Scholar]
- 79.Thiébaut R, Aurillac-Lavignolle V, Bonnet F, Ibrahim N, Cipriano C, Neau D, Dupon M, Dabis F, Mercié P. Change in atherosclerosis progression in HIV-infected patients: ANRS Aquitaine Cohort, 1999-2004. AIDS. 2005;19:729–731. doi: 10.1097/01.aids.0000166097.46940.35. [DOI] [PubMed] [Google Scholar]
- 80.Jericó C, Knobel H, Calvo N, Sorli ML, Guelar A, Gimeno-Bayón JL, Saballs P, López-Colomés JL, Pedro-Botet J. Subclinical carotid atherosclerosis in HIV-infected patients: role of combination antiretroviral therapy. Stroke. 2006;37:812–817. doi: 10.1161/01.STR.0000204037.26797.7f. [DOI] [PubMed] [Google Scholar]
- 81.Lang S, Mary-Krause M, Simon A, Partisani M, Gilquin J, Cotte L, Boccara F, Costagliola D. HIV replication and immune status are independent predictors of the risk of myocardial infarction in HIV-infected individuals. Clin Infect Dis. 2012;55:600–607. doi: 10.1093/cid/cis489. [DOI] [PubMed] [Google Scholar]
- 82.Currier JS, Kendall MA, Zackin R, Henry WK, Alston-Smith B, Torriani FJ, Schouten J, Mickelberg K, Li Y, Hodis HN. Carotid artery intima-media thickness and HIV infection: traditional risk factors overshadow impact of protease inhibitor exposure. AIDS. 2005;19:927–933. doi: 10.1097/01.aids.0000171406.53737.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart. 2009;95:1826–1835. doi: 10.1136/hrt.2009.177774. [DOI] [PubMed] [Google Scholar]
- 84.Stein JH, Klein MA, Bellehumeur JL, McBride PE, Wiebe DA, Otvos JD, Sosman JM. Use of human immunodeficiency virus-1 protease inhibitors is associated with atherogenic lipoprotein changes and endothelial dysfunction. Circulation. 2001;104:257–262. doi: 10.1161/01.cir.104.3.257. [DOI] [PubMed] [Google Scholar]
- 85.Badiou S, Merle De Boever C, Dupuy AM, Baillat V, Cristol JP, Reynes J. Decrease in LDL size in HIV-positive adults before and after lopinavir/ritonavir-containing regimen: an index of atherogenicity? Atherosclerosis. 2003;168:107–113. doi: 10.1016/s0021-9150(03)00058-3. [DOI] [PubMed] [Google Scholar]
- 86.Asztalos BF, Schaefer EJ, Horvath KV, Cox CE, Skinner S, Gerrior J, Gorbach SL, Wanke C. Protease inhibitor-based HAART, HDL, and CHD-risk in HIV-infected patients. Atherosclerosis. 2006;184:72–77. doi: 10.1016/j.atherosclerosis.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 87.Duffy P, Wang X, Lin PH, Yao Q, Chen C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J Surg Res. 2009;156:257–264. doi: 10.1016/j.jss.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hsue PY, Hunt PW, Schnell A, Kalapus SC, Hoh R, Ganz P, Martin JN, Deeks SG. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS. 2009;23:1059–1067. doi: 10.1097/QAD.0b013e32832b514b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Desvarieux M, Boccara F, Meynard JL, Bastard JP, Mallat Z, Charbit B, Demmer RT, Haddour N, Fellahi S, Tedgui A, et al. Infection duration and inflammatory imbalance are associated with atherosclerotic risk in HIV-infected never-smokers independent of antiretroviral therapy. AIDS. 2013;27:2603–2614. doi: 10.1097/QAD.0b013e3283634819. [DOI] [PubMed] [Google Scholar]
- 90.Jackson L, Britton J, Lewis SA, McKeever TM, Atherton J, Fullerton D, Fogarty AW. A population-based epidemiologic study of Helicobacter pylori infection and its association with systemic inflammation. Helicobacter. 2009;14:108–113. doi: 10.1111/j.1523-5378.2009.00711.x. [DOI] [PubMed] [Google Scholar]
- 91.Rogha M, Dadkhah D, Pourmoghaddas Z, Shirneshan K, Nikvarz M, Pourmoghaddas M. Association of helicobacter pylori infection with severity of coronary heart disease. ARYA Atheroscler. 2012;7:138–141. [PMC free article] [PubMed] [Google Scholar]
- 92.Schiavoni G, Di Pietro M, Ronco C, De Cal M, Cazzavillan S, Rassu M, Nicoletti M, Del Piano M, Sessa R. Chlamydia pneumoniae infection as a risk factor for accelerated atherosclerosis in hemodialysis patients. J Biol Regul Homeost Agents. 2010;24:367–375. [PubMed] [Google Scholar]
- 93.Pejcic A, Kesic LJ, Milasin J. C-reactive protein as a systemic marker of inflammation in periodontitis. Eur J Clin Microbiol Infect Dis. 2011;30:407–414. doi: 10.1007/s10096-010-1101-1. [DOI] [PubMed] [Google Scholar]
- 94.Campbell LA, Yaraei K, Van Lenten B, Chait A, Blessing E, Kuo CC, Nosaka T, Ricks J, Rosenfeld ME. The acute phase reactant response to respiratory infection with Chlamydia pneumoniae: implications for the pathogenesis of atherosclerosis. Microbes Infect. 2010;12:598–606. doi: 10.1016/j.micinf.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kebschull M, Demmer RT, Papapanou PN. “Gum bug, leave my heart alone!”--epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res. 2010;89:879–902. doi: 10.1177/0022034510375281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roivainen M, Viik-Kajander M, Palosuo T, Toivanen P, Leinonen M, Saikku P, Tenkanen L, Manninen V, Hovi T, Mänttäri M. Infections, inflammation, and the risk of coronary heart disease. Circulation. 2000;101:252–257. doi: 10.1161/01.cir.101.3.252. [DOI] [PubMed] [Google Scholar]
- 97.Okada T, Ayada K, Usui S, Yokota K, Cui J, Kawahara Y, Inaba T, Hirohata S, Mizuno M, Yamamoto D, et al. Antibodies against heat shock protein 60 derived from Helicobacter pylori: diagnostic implications in cardiovascular disease. J Autoimmun. 2007;29:106–115. doi: 10.1016/j.jaut.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 98.Choi J, Lee SY, Kim K, Choi BK. Identification of immunoreactive epitopes of the Porphyromonas gingivalis heat shock protein in periodontitis and atherosclerosis. J Periodontal Res. 2011;46:240–245. doi: 10.1111/j.1600-0765.2010.01339.x. [DOI] [PubMed] [Google Scholar]
- 99.Kreutmayer S, Csordas A, Kern J, Maass V, Almanzar G, Offterdinger M, Öllinger R, Maass M, Wick G. Chlamydia pneumoniae infection acts as an endothelial stressor with the potential to initiate the earliest heat shock protein 60-dependent inflammatory stage of atherosclerosis. Cell Stress Chaperones. 2013;18:259–268. doi: 10.1007/s12192-012-0378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andrié RP, Bauriedel G, Braun P, Höpp HW, Nickenig G, Skowasch D. Prevalence of intimal heat shock protein 60 homologues in unstable angina and correlation with anti-heat shock protein antibody titers. Basic Res Cardiol. 2011;106:657–665. doi: 10.1007/s00395-011-0171-2. [DOI] [PubMed] [Google Scholar]
- 101.Ausiello CM, Palazzo R, Spensieri F, Fedele G, Lande R, Ciervo A, Fioroni G, Cassone A. 60-kDa heat shock protein of Chlamydia pneumoniae is a target of T-cell immune response. J Biol Regul Homeost Agents. 2005;19:136–140. [PubMed] [Google Scholar]
- 102.Ford P, Gemmell E, Walker P, West M, Cullinan M, Seymour G. Characterization of heat shock protein-specific T cells in atherosclerosis. Clin Diagn Lab Immunol. 2005;12:259–267. doi: 10.1128/CDLI.12.2.259-267.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ayada K, Yokota K, Hirai K, Fujimoto K, Kobayashi K, Ogawa H, Hatanaka K, Hirohata S, Yoshino T, Shoenfeld Y, et al. Regulation of cellular immunity prevents Helicobacter pylori-induced atherosclerosis. Lupus. 2009;18:1154–1168. doi: 10.1177/0961203309106600. [DOI] [PubMed] [Google Scholar]
- 104.Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;265:391–394. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- 105.Dengler TJ, Raftery MJ, Werle M, Zimmermann R, Schönrich G. Cytomegalovirus infection of vascular cells induces expression of pro-inflammatory adhesion molecules by paracrine action of secreted interleukin-1beta. Transplantation. 2000;69:1160–1168. doi: 10.1097/00007890-200003270-00022. [DOI] [PubMed] [Google Scholar]
- 106.Rahbar A, Söderberg-Nauclér C. Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J Virol. 2005;79:2211–2220. doi: 10.1128/JVI.79.4.2211-2220.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Naghavi M, Wyde P, Litovsky S, Madjid M, Akhtar A, Naguib S, Siadaty MS, Sanati S, Casscells W. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation. 2003;107:762–768. doi: 10.1161/01.cir.0000048190.68071.2b. [DOI] [PubMed] [Google Scholar]
- 108.Naghavi M, Barlas Z, Siadaty S, Naguib S, Madjid M, Casscells W. Association of influenza vaccination and reduced risk of recurrent myocardial infarction. Circulation. 2000;102:3039–3045. doi: 10.1161/01.cir.102.25.3039. [DOI] [PubMed] [Google Scholar]
- 109.Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circ Res. 2001;89:763–771. doi: 10.1161/hh2101.099270. [DOI] [PubMed] [Google Scholar]
- 110.Petty GW, Duffy J, Houston J. Cerebral ischemia in patients with hepatitis C virus infection and mixed cryoglobulinemia. Mayo Clin Proc. 1996;71:671–678. doi: 10.4065/71.7.671. [DOI] [PubMed] [Google Scholar]
- 111.Capra F, De Maria E, Lunardi C, Marchiori L, Mezzelani P, Beri R, Gabrielli GB. Serum level of soluble intercellular adhesion molecule 1 in patients with chronic liver disease related to hepatitis C virus: A prognostic marker for responses to interferon treatment. J Infect Dis. 2000;181:425–431. doi: 10.1086/315265. [DOI] [PubMed] [Google Scholar]
- 112.Cacoub P, Ghillani P, Revelen R, Thibault V, Calvez V, Charlotte F, Musset L, Youinou P, Piette JC. Anti-endothelial cell auto-antibodies in hepatitis C virus mixed cryoglobulinemia. J Hepatol. 1999;31:598–603. doi: 10.1016/s0168-8278(99)80337-7. [DOI] [PubMed] [Google Scholar]
- 113.Shrestha S, Irvin MR, Grunfeld C, Arnett DK. HIV, inflammation, and calcium in atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:244–250. doi: 10.1161/ATVBAHA.113.302191. [DOI] [PubMed] [Google Scholar]
- 114.Khor CC, Hibberd ML. Host-pathogen interactions revealed by human genome-wide surveys. Trends Genet. 2012;28:233–243. doi: 10.1016/j.tig.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 115.Georges JL, Rupprecht HJ, Blankenberg S, Poirier O, Bickel C, Hafner G, Nicaud V, Meyer J, Cambien F, Tiret L. Impact of pathogen burden in patients with coronary artery disease in relation to systemic inflammation and variation in genes encoding cytokines. Am J Cardiol. 2003;92:515–521. doi: 10.1016/s0002-9149(03)00717-3. [DOI] [PubMed] [Google Scholar]
- 116.Westhorpe CL, Maisa A, Spelman T, Hoy JF, Dewar EM, Karapanagiotidis S, Hearps AC, Cheng WJ, Trevillyan J, Lewin SR, et al. Associations between surface markers on blood monocytes and carotid atherosclerosis in HIV-positive individuals. Immunol Cell Biol. 2014;92:133–138. doi: 10.1038/icb.2013.84. [DOI] [PubMed] [Google Scholar]