

Summary
Post-tetanic potentiation (PTP) is a widely observed form of short-term plasticity lasting for tens of seconds after high-frequency stimulation. Here we show that although protein kinase C (PKC) mediates PTP at the calyx of Held synapse in the auditory brainstem before and after hearing onset, PTP is produced primarily by an increased probability of release (p) before hearing onset, and by an increased readily-releasable pool of vesicles (RRP) thereafter. We find that these mechanistic differences, which have distinct functional consequences, reflect unexpected differential actions of closely related calcium-dependent PKC isoforms. Prior to hearing onset, when PKCγ and PKCβ are both present, PKCγ mediates PTP by increasing p and partially suppressing PKCβ actions. After hearing onset, PKCγ is absent and PKCβ produces PTP by increasing RRP. In hearing animals, virally expressed PKCγ overrides PKCβ to produce PTP by increasing p. Thus, two similar PKC isoforms mediate PTP in distinctly different ways.
Introduction
Many forms of synaptic plasticity regulate neurotransmitter release by a combination of increasing the probability of release (p) and the size of the readily-releasable pool of vesicles (RRP) (Pan and Zucker, 2009; Regehr et al., 2009; Zucker and Regehr, 2002). Whether increases in p or RRP underlie synaptic enhancement is important from a functional point of view, because these two mechanisms have very different effects on responses to stimulus trains (Pan and Zucker, 2009; Thanawala and Regehr, 2013). An increase in the RRP simply scales up the size of synaptic responses evoked by repetitive activation, and the effect is similar in many ways to increasing the number of postsynaptic receptors. In contrast, increasing p also increases use-dependent depression, and as a result for repetitive activation the initial synaptic response is more strongly enhanced than subsequent responses. Consequently, overall neurotransmitter release evoked by high frequency stimulation is doubled if RRP doubles, but is essentially unchanged if p doubles. It is particularly controversial whether changes in p or RRP underlie post-tetanic potentiation (PTP), a form of short-term plasticity lasting tens of seconds to minutes following tetanic stimulation (Alle et al., 2001; Bao et al., 1997; Griffith, 1990; Magleby, 1979; Magleby and Zengel, 1975; Zucker and Regehr, 2002).
PTP is thought to be a neural mechanism that contributes to short-term memory, synaptic filtering, and information processing (Abbott and Regehr, 2004; Klug et al., 2012; Silva et al., 1996). High-frequency (tetanic) stimulation induces PTP by transiently increasing presynaptic calcium, which in turn activates downstream molecular effectors that elevate neurotransmitter release (Delaney and Tank, 1994; Delaney et al., 1989; Habets and Borst, 2006; Korogod et al., 2005; Regehr et al., 1994; Zucker and Regehr, 2002). A growing body of evidence supports a critical role for protein kinase C (PKC) in PTP (Alle et al., 2001; Beierlein et al., 2007; Brager et al., 2003; Fioravante et al., 2011; Korogod et al., 2007; Lee et al., 2008; Wierda et al., 2007). The role of PKC in PTP has been most extensively studied at the calyx of Held synapse (Fioravante et al., 2011; Korogod et al., 2007; Lee et al., 2008), where it was established that in postnatal day (P)11-14 animals PTP is mediated primarily by PKCβ (Fioravante et al., 2011), one of the “classical” calcium-dependent isoforms (PKCα, PKCβ and PKCγ, as opposed to the many calcium-insensitive “novel” and “atypical” isoforms (Newton, 2001; Newton, 1995; Steinberg, 2008). There is, however, considerable debate regarding whether PKC enhances release at the calyx of Held by increasing p or RRP. Most studies suggest that PKC mainly increases p (Habets and Borst, 2005; Habets and Borst, 2006; Korogod et al., 2007; Lou et al., 2005; Turecek and Trussell, 2001; Wu and Wu, 2001), but others suggest that PKC prominently increases RRP (Chu et al., 2012; Fioravante et al., 2011; Habets and Borst, 2007).
Because the calyx of Held synapse undergoes age-dependent anatomical and functional changes (Borst and Soria van Hoeve, 2012; Nakamura and Cramer, 2011; Rodriguez-Contreras et al., 2008; Taschenberger et al., 2002; von Gersdorff and Borst, 2002), we compared the properties of PTP before and after the onset of hearing, and we assessed the roles of the calcium-dependent PKC isoforms. We find that PKCγ produces PTP by increasing p before hearing onset, and that PKCβ produces PTP by increasing RRP afterwards. In pre-hearing PKCγ ko animals, PTP persists but is mainly due to PKCβ increasing RRP. This indicates that the key to whether PTP is due to an increase in p or RRP is whether PKCγ or PKCβ mediates synaptic enhancement.
When both PKCγ and PKCβ are present, PTP is mediated by an increase in p, suggesting that PKCγ suppresses PKCβ-dependent regulation of the RRP. Thus, even though PKCγ and PKCβ are highly similar calcium-dependent isoforms that both mediate PTP, they do so by different synaptic mechanisms with different functional consequences.
Results
Age-dependent properties of PTP
We initially determined whether the mechanisms of PTP are developmentally regulated by comparing the properties of synaptic plasticity before (P8-10) and after (P16-19) the onset of hearing (~P12) (Sonntag et al., 2009; Sonntag et al., 2011). A widely-used approach to assess whether changes in p contribute to changes in neurotransmitter release is to determine whether synaptic changes are accompanied by alterations in the paired-pulse ratio (PPR) of two closely spaced stimuli. Typically, low p synapses facilitate, due to an accumulation of residual calcium, and high p synapses depress, likely as a result of significant depletion of the RRP during the first stimulus. If PTP reflects an increase in p it is expected to be accompanied by a decrease in PPR. e stimulated with pairs of pulses (Δt = 10 ms) every 5 s prior to and following tetanic stimulation (4s, 100 Hz) (Figure 1A, top). On average there was a significant reduction in PPR (−32 ± 3%; n=6; p<0.01) in P8-10 animals (Figure 1A, bottom) during PTP (Figure 1A, middle). In P16-19 animals, even though the basal properties of synaptic transmission were different from P8-10 animals, as described previously (Iwasaki and Takahashi, 2001; Taschenberger et al., 2002), the magnitude and time course of PTP were comparable to that observed in P8-10 animals (Table S1). However, only a minimal reduction in PPR accompanied PTP (−3.1±1.3%; p=0.36) in P16-19 animals (n=8) (Figure 1B). These findings suggest that changes in p make a larger contribution to PTP in P8-10 animals, but they cannot be used to precisely quantify the contributions of p and RRP to plasticity.
Figure 1. PTP is accompanied by a large decrease in paired-pulse plasticity before the onset of hearing but not after hearing onset.

The calyx of Held was stimulated every 5 seconds with a pair of pulses separated by 10 ms, PTP was induced at t=0 using a 4 s, 100 Hz train and stimulation with pairs of pulses resumed. Results are shown for P8-10 animals (A) and P16-19 animals (B). Representative traces show the average baseline paired-pulse EPSC (A, B, top left, gray), the potentiated response during the peak of PTP (A, B, top middle, black), and traces normalized to the first EPSCs (A, B, top right). The EPSC amplitudes (A, B, middle) and the PPR are plotted as a function of time (A, B, bottom). Error bars are SEM.
It is possible, however, to quantify the contributions of p and RRP to PTP from synaptic responses evoked by action-potential trains in the presence of cyclothiazide and kynurenate to prevent postsynaptic receptor desensitization and saturation (Fioravante et al., 2011; Korogod et al., 2005; Lee et al., 2008; Schneggenburger et al., 1999). In the case of PTP, the responses to a stimulus train used to induce PTP and to a stimulus train 10 seconds later (at the peak of PTP), are compared. Such an approach is illustrated for pre-hearing and hearing animals (Figure 2). The amplitudes of the EPSCs can then be used to determine the size of the RRP in several different ways. A plot of the cumulative EPSC versus the stimulus number can be used to determine the size of the RRP (Moulder and Mennerick, 2005; Pan and Zucker, 2009; Stevens and Williams, 2007; Thanawala and Regehr, 2013). The key to this approach is that the EPSC amplitude eventually reaches a steady-state level, and under these conditions the RRP is depleted and the remaining release is due to replenishment from a reserve pool (Schneggenburger et al., 1999; Thanawala and Regehr, 2013). Extrapolation is then used to determine the size of the RRP (RRPtrain). Although this approach is widely used, it is known to overestimate the amount of replenishment that occurs, which leads to an underestimate of the size of the RRP (Lee et al., 2008; Schneggenburger et al., 1999; Thanawala and Regehr, 2013). It is possible to refine this approach by correcting the estimate of the amount of replenishment that occurs early in the train to obtain a corrected estimate (RRPtrainC) (Thanawala and Regehr, 2013). Lastly, we estimated RRP using an approach that was first introduced by Elmqvist and Quastel (Elmqvist and Quastel, 1965; Grande and Wang, 2011; Taschenberger et al., 2002; Taschenberger et al., 2005; Thanawala and Regehr, 2013). This method is based on the assumption that during the train the synaptic currents get progressively smaller as the RRP depletes. A plot of the amplitude of the EPSCs in the train as a function of the cumulative EPSC is then used to estimate RRP (RRPEQ). The amplitudes of the EPSCs were then used to determine the amount of synaptic enhancement, and p was computed from the equation EPSC=RRP*p. The use of these three approaches to quantify RRP and p at the calyx of Held has been addressed previously (Thanawala and Regehr, 2013). Our estimates of the RRP size were not confounded by large increases in the postsynaptic neurotransmitter sensitivity because there was only a very small increase in the size of quantal responses after PTP was induced (7.5 ± 6.5 %, n=12 in P8-10 wildtype animals; 6.4 ± 6.6%, n=10 in P16-19 wildtype mice for the interval 6-16 s post-tetanus).
Figure 2. Assessing the contributions of p and RRP to PTP before (A–D) and after (E–H) hearing onset.

Mechanisms of PTP were examined using trains in the presence of kynurenate and CTZ to prevent receptor saturation and desensitization. (A, E) Synaptic currents evoked by the first 40 stimuli of 4s, 100 Hz train (top) and by a 40 pulse 100 Hz train (bottom) 10 s after tetanic stimulation (at the peak of PTP) are shown. The change in pool size and p were determined using the cumulative EPSC method, the corrected EPSC method (B, F) and the EQ method (C,G). (D, H) Summary of the changes in synaptic strength, RRP and p determined using the three different quantification methods. Paired t-tests were used to compare the changes in EPSC, RRPtrain, ptrain, RRPtrainC, ptrainC, RRPEQ, and pEQ during PTP to baseline, as indicated (*p<0.05, **p<0.01, ***p<0.001). Error bars are SEM.
We used all three of these approaches to determine the contributions of RRP and p to PTP in P8-10 animals (Figure 2A–D). The responses to 40 stimuli at 100 Hz during the initial conditioning train (a total of 400 stimuli at 100 Hz) that is used to induce PTP (Figure 2A, top) and during a second conditioning train 10 seconds after the end of initial train (Figure 2A, bottom) are shown for a representative experiment (Figure 2A–C). As shown in this example, the responses to both trains showed prominent use-dependent depression, pool size increases were small (RRPtrainC 4%, RRPEQ 2%) and the enhancement of the EPSC was primarily a consequence of an increase in p (ptrainC 41%, pEQ 45%). On average, in P8-10 wildtype animals PTP was 67 ± 16 %, RRPtrainC 18 ± 6%, RRPEQ 11 ± 5%, ptrainC 42 ± 12%, and pEQ 52 ± 15% (Figure 2D, Table S2).
We used the same approaches to determine the contributions of RRP and p to PTP in P16-19 animals (Figure 2E–H). In comparison to the responses observed in pre-hearing animals the initial EPSCs were larger, the depression was less pronounced, and the steady-state EPSCs were larger (Figure 2E, Figure S1). The magnitude of PTP was comparable to that observed in pre-hearing animals, but the changes in RRP played a much larger role. In the illustrated example, the initial EPSC was enhanced by 64%, and there was a large increase in the size of the readily releasable pool (RRPtrainC 57%, RRPEQ 52%; Figure 2F,G). A summary of the contributions of p and RRP, regardless of the method used to estimate them, revealed that the enhancement of the EPSC was primarily a consequence of an increase in RRP in P16-19 animals (Figure 2H, Table S2). These findings support the hypothesis that prior to the onset of hearing PTP is mediated primarily by an increase in p, but after hearing onset it is mediated predominantly by an increase in RRP.
Age-dependent differences in the decrease of PPR that accompanied PTP were also observed when cyclothiazide and kynurenate were included in the bath (Fucile et al., 2006; Taschenberger et al., 2002). Under these conditions there was a significant reduction in PPR in P8-10 mice (−42 ± 8 %, n=10, p<0.05) and in P16-19 mice (−14 ± 5%, n=8, p<0.05) but the reduction was larger in pre-hearing mice (p<0.05).
Contributions of different PKC isoforms to PTP
To elucidate the mechanisms underlying PTP in pre-hearing and hearing animals, we performed a series of experiments using pharmacological and genetic manipulations. We characterized the basal properties of transmission and found that for all conditions tested there were no differences in the initial amplitudes of the EPSC, initial PPRs, the initial sizes of RRP and the initial probability of release (Figure S1). These data suggest that the basal properties of synaptic transmission among wildtype and knockout groups are not different within each age group of animals. For a subset of experimental conditions we also performed experiments in the presence of cyclothiazide and kynurenate to quantify contributions of RRP and p to PTP, and to determine the changes in PPR that accompany PTP (Figure S1 and Table S2). We found that cyclothiazide and kynurenate did not significantly alter the amplitude of PTP (Tables S1 and S2). For the remainder of the paper we also determined RRP and p by the three methods shown in Figure 2, but as there was good agreement between the corrected train method and the Elmqvist and Quastel method (Thanawala and Regehr, 2013) we present only RRPtrainC and ptrainC for simplicity.
We have previously shown that in P11-14 mice PKCα and PKCβ are both present in the calyx of Held, but PTP is mediated mainly by PKCβ-dependent increases in RRP (Fioravante et al., 2011). The age-dependence of the properties of PTP could arise from differences in PKC signaling. There are two leading hypotheses by which PKC could account for the observed differences between pre-hearing and hearing animals: (1) PTP is mediated primarily by PKCβ at all ages but it enhances transmission through different mechanisms throughout development, or (2) in pre-hearing animals PTP is not mediated by PKCβ.
The role of PKC isoforms in pre-hearing and hearing animals was assessed with knockout animals and with an isoform-specific inhibitor. Whereas PTP was greatly reduced in P16-19 PKCαβ knockout animals (8.3 ± 6.4%; p<0.05; Figure 3A), it was largely intact in pre-hearing PKCαβ knockout animals (40 ± 9.0%; p=0.15; Figure 3B). We also used a newly available PKCβ inhibitor to examine the role of PKCβ (Tanaka et al., 2004). Such a pharmacological approach compliments the use of knockout animals by allowing acute inhibition of PKCβ in a manner that is free from potential developmental complications that could occur in global knockout animals. This compound inhibits PKCα, PKCβI, PKCβII, and PKCγ, with Kms of 330 nM, 21 nM, 5 nM, and >1 μM, respectively. These properties suggest that this inhibitor may be well-suited to our experiments in which we need to inhibit PKCβ without affecting PKCγ (although it is possible that this inhibitor could also partially inhibit PKCα, which plays a very minor role in PTP at the calyx of Held (Fioravante et al., 2011). We found that at a concentration of 250 nM this drug reduced the magnitude of PTP from 57 ± 11% (n=10) to 12 ± 1.5 % (n=7, p<0.01) in P16-19 animals (Figure 3C). Intriguingly, in P8-10 widltype animals the PKCβ inhibitor did not attenuate PTP (59 ± 15%; n=10; p=0.97; Figure 3D). Moreover, the inhibitor did not disrupt PTP in P8-10 PKCαβ ko mice (54 ± 15%; n=3; p=0.50; Figure 3G) suggesting that it does not have off-target effects. These findings indicate that PTP is dependent on PKCβ in hearing animals, but not in pre-hearing animals.
Figure 3. Age-dependent differences in the roles of calcium-dependent PKC isoforms in PTP.

PTP as a function of time for P16-19 animals (A, C) and P8-10 animals (B, D, G, H). (A, B) PTP from wildtype and PKCαβ dko animals are compared. (C, D) The effects of the PKCβ inhibitor (Calbiochem 539654) on PTP in wildtype animals are shown for the corresponding age groups. (E, F) Summary plots of RRP and p contributions to PTP in pre-hearing animals. Experiments were performed in the presence of CTZ and kynurenate as shown in Supplementary Figure S2. (G, H) The effects of the PKCβ inhibitor (G) and a broad spectrum PKC inhibitor (GF109203X) (H) on PTP in P8-10 PKCαβ dko animals are shown. *p<0.05, **p<0.01. Error bars are SEM.
We went on to examine the contribution of p and RRP to synaptic enhancement in pre-hearing animals in which PKCβ was absent or inhibited. In PKCαβ ko animals PTP is mediated exclusively by an increase in p and the small increase in pool size that is apparent in age-matched wildtype animals (Figure 2D) is absent (ptrainC 45 ± 8%, n=9; Figure 3E, Figure S2A). For wildtype animals in the presence of a PKCβ inhibitor there was a small but non-significant increase in RRP, and PTP was mediated by an increase in p (ptrainC 47 ± 16%, n= 14; Figure 3F, Figure S2B).
These results suggest that prior to the onset of hearing, PTP is mediated by a mechanism that is independent of PKCα and PKCβ. Alternatively, there could be a compensatory adaptation that only mediates PTP in the absence of PKCα and PKCβ. For example, at the granule cell to Purkinje cell synapse in the cerebellum, PTP is mediated by PKCα and PKCβ in wildtype animals, but it is mediated by a PKC-independent mechanism in PKCαβ ko mice (Fioravante et al., 2012). Although the observation that PTP is strongly attenuated in PKCαβ ko mice indicates that such a compensatory mechanism is not present at the calyx of Held in hearing animals, it is possible that a compensatory mechanism is present before the onset of hearing.
PTP in P8-10 PKCαβ ko animals could be mediated either by a mechanism that involves other PKC isoforms, or one that is completely PKC-independent. It is possible to distinguish between these possibilities by testing the effects of a broad spectrum PKC inhibitor on PTP in these animals. Previous studies have shown that in slices from wildtype mice a broad spectrum PKC inhibitor eliminates most of the PTP, and a small component remains that is mediated at least in part by myosin light chain kinase (Fioravante et al., 2011; Lee et al., 2008). We found that in P8-10 PKCαβ ko mice the pan-PKC inhibitor GF109203X (GF) greatly attenuates most PTP (14 ± 3%; n=11; p<0.05; Figure 3H). The small remaining enhancement is comparable to the enhancement mediated by myosin light chain kinase in P11-14 wild type mice (Fioravante et al., 2011). This observation suggests that the PKCαβ-independent PTP observed in pre-hearing mice could be mediated by other PKC isoform(s).
There are many PKC isoforms, but PKCα, PKCβ and PKCγ are the only calcium-dependent ones (Newton, 2001; Newton, 1995; Steinberg, 2008). PKCγ therefore seemed like a reasonable candidate to mediate PTP in P8-10 animals. Although previous studies suggested that PKCγ is not present at calyx of Held synapses in P13-15 rats (Saitoh et al., 2001), the possibility that it could be present before hearing onset had not been assessed. We therefore used immunohistochemical techniques to determine whether PKCγ is present in P8-10 animals. We co-labeled glutamatergic calyces with an antibody against vesicular glutamate transporter 1 (vGlut1; red) and an anti-PKCγ antibody (green) (Figure 4). Confocal images through the center of MNTB neurons show a characteristic ring of vGlut1 labeling that demarcates the calyceal presynaptic terminals surrounding the cell bodies of MNTB neurons. In brainstem slices from a P10 representative animal, the PKCγ labeling showed a similar distribution, consistent with it being expressed presynaptically, and this labeling was absent in P8-10 PKCγ ko animals (Figure 4, top and middle). These findings establish that PKCγ is indeed present at the calyx of Held in pre-hearing animals.
Figure 4. Immunohistochemical localization of PKCγ at the calyx of Held in animals before and after hearing onset.
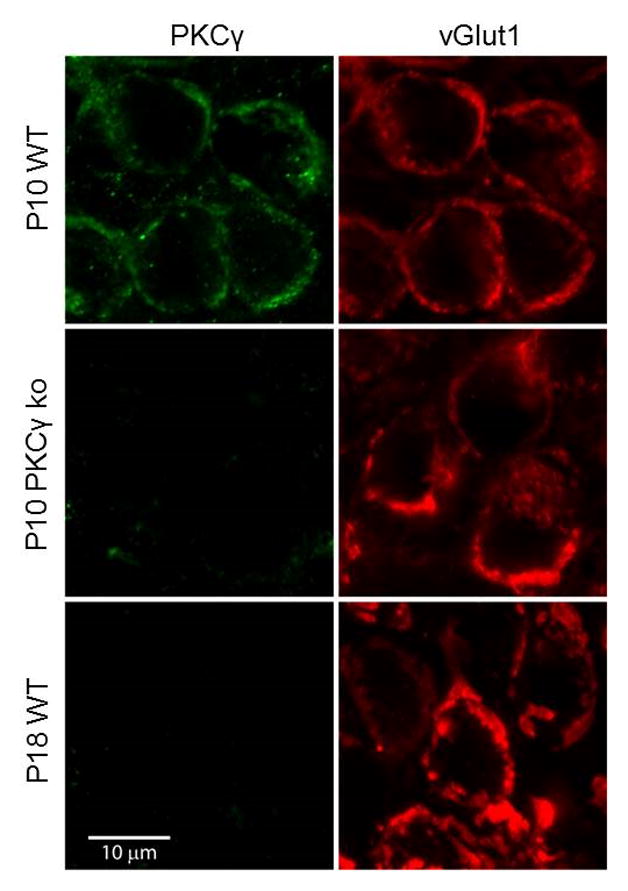
Brain slices containing the MNTB region from wildtype and PKCγ ko animals prior to and after hearing onset were co-labeled with antibodies to PKCγ (green) and an antibody to the presynaptic marker vGlut1 (red). Representative images are shown for P10 and P18 slices from wildtype (WT) and PKCγ ko animals.
The lack of a contribution of PKCγ to PTP in hearing animals could arise either because PKCγ expression is downregulated during development, or because it is still present but no longer able to produce PTP. To distinguish between these possibilities, we tested for the presence of PKCγ in P16-19 mice. We observed intense vGlut1 labeling with a pattern that was consistent with the morphology of adult calyces, but PKCγ was absent (Figure 4, bottom). This suggests that PKCγ cannot contribute to PTP in animals after hearing onset because its expression is significantly downregulated.
The finding that PKCγ is present at the calyx of Held in pre-hearing animals prompted us to test the role of this isoform in PTP. In P8-10 PKCγ ko mice, PTP is present (54 ± 8%; n=14) at a magnitude that is comparable to that observed in wildtype animals (p=0.65; Figure 5A). This indicates that in pre-hearing animals PKCγ is not essential, and it suggests other PKC isoforms can mediate PTP in the absence of PKCγ. At first sight, the similarity in the amplitude and time course of PTP between pre-hearing wildtype and PKCγ ko animals does not seem to support an important role for PKCγ in PTP at this age. But an examination of the contribution of p and RRP to PTP indicates that PTP in PKCγ ko mice differs markedly from that observed in wildtype mice, and indicates that PKCγ plays an important role in PTP prior to hearing onset. Remarkably, in P8-10 PKCγ ko mice the mechanism of PTP is very different from that seen in wildtype or in PKCαβ ko mice, and PTP is primarily a result of an increase in the size of the RRP (RRPtrainC 37 ± 13%; n=8) rather than an increase in p (ptrainC 20 ± 11%; Figure 5B, Figure S2C). Indeed, the contributions of RRP and p are similar to those seen after the onset of hearing in wildtype animals (Figure 2H). The elimination of PKCγ from the synapses of pre-hearing animals has essentially transformed the properties of PTP to those typically observed only after the onset of hearing.
Figure 5. PKCγ mediates PTP in pre-hearing animals at the calyx of Held synapse.

(A) PTP in P8-10 wildtype and PKCγ ko animals. (B) Summary of the contributions of RRP and p to synaptic enhancement in P8-10 PKCγ ko animals. Paired t-tests were used to compare the changes in EPSC, RRPtrainC, ptrainC during PTP to baseline, as indicated (*p<0.05). (C) PTP in P8-10 wildtype and PKCαβγ triple ko animals. (D) PTP in PKCαγ ko animals in the absence and the presence of a PKCβ inhibitor (250 nM). Error bars are SEM.
The important role of PKCγ in PTP prior to hearing onset is further supported by the strong attenuation of PTP when all three calcium-dependent isoforms are either genetically eliminated or inhibited. In contrast to PKCαβ ko animals in which PTP was present, PTP is strongly attenuated in P8-10 PKCαβγ triple ko animals (12 ± 9%; n=17; p<0.01; Figure 5C). A PKCβ inhibitor attenuated the PTP in PKCαγ mice to a similar extent (17 ± 5%; n=12; p<0.01; Figure 5D). These observations indicate that before hearing onset a combination of PKCβ and PKCγ mediate PTP and that PKCγ plays a prominent role in wildtype animals.
Assessing the contribution of changes in calcium influx to PTP
Prior to the onset of hearing PTP is primarily produced by PKCγ increasing p. One possible mechanism is that PKCγ could regulate calcium entry to increase p, because increases in calcium entry are known to contribute to some forms of short-term synaptic plasticity (Catterall and Few, 2008). Previously we found that increases in calcium entry do not account for PKCβ-mediated PTP at the calyx of Held in P11-14 animals (Fioravante et al., 2011), but at that age PKCβ-mediates PTP by increasing the RRP, whereas increasing action-potential-evoked calcium influx is expected to act primarily by increasing p (although see (Thanawala and Regehr, 2013)). Thus, PTP mediated by PKCγ is a stronger candidate for the involvement of increases in calcium influx. Moreover, at the calyx of Held in pre-hearing animals, it is thought that increases in calcium influx contribute to PTP evoked by prolonged stimulation (Habets and Borst, 2006), and may also contribute following 4s at 100 Hz (Korogod et al., 2007).
We therefore tested the hypothesis that PTP in pre-hearing animals is mediated by PKCγ-dependent increases in calcium influx in wildtype animals. We measured presynaptic calcium in the calyx of Held from P8-10 animals as we had done previously for older animals (Fioravante et al., 2011, Figure S3). We loaded calyces with Calcium Green-1 dextran and Alexa-594 dextran, and labeled calyces were readily identified (Figure 6A, top). Single stimuli evoked rapid calcium transients of 31 ± 9 nM that decayed with a time constant of 66 ±19 ms in wildtype calyces (representative trace shown in Figure 6A, bottom). Following tetanic stimulation (4s, 100 Hz) the residual calcium increased to over 100 nM and decayed to resting calcium levels with a time constant of 23 ± 7 s (Figure 6C, top). We used the calcium increase evoked by single stimuli as a means of detecting changes in calcium influx. Calcium increases evoked by single stimuli were unaltered by tetanic stimulation, indicating that tetanic stimulation does not result in an increase in calcium entry in pre-hearing animals (Figure 6C, bottom). We performed similar experiments in PKCγ ko animals in which PTP is mediated by an increase in RRP rather than p. Calcium signaling in PKCγ ko animals and wildtype animals was indistinguishable (Figure 6B, C). Thus, we find that PKCγ does not produce PTP by increasing action potential evoked calcium entry, and PKCγ-dependent increases in calcium influx do not account for PTP in pre-hearing animals.
Figure 6. Tetanic stimulation produced similar presynaptic residual calcium signals in wildtype and PKCγ ko animals in pre-hearing animals.
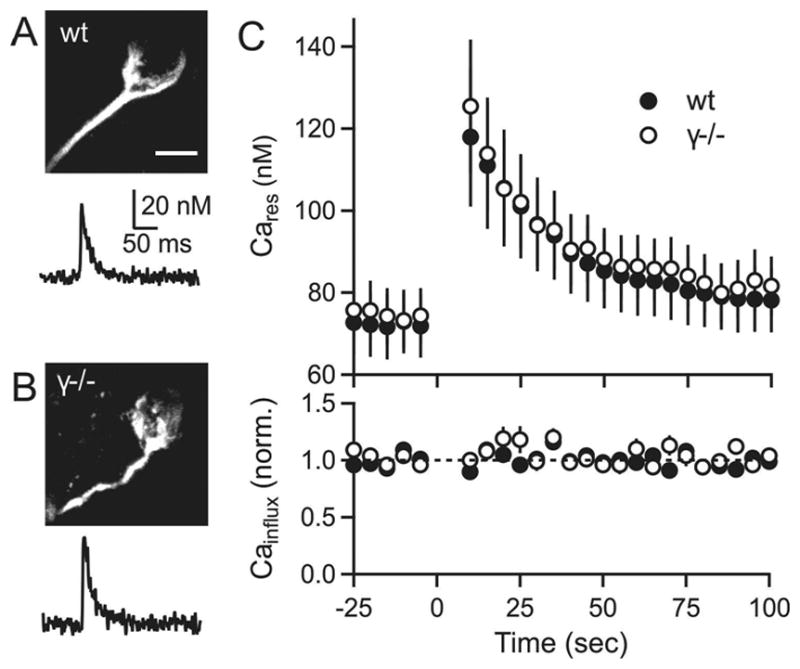
(A) top: two-photon image of a calyx from a wildtype animal filled with Alexa-594 dextran and calcium-green dextran. bottom: Calcium transient evoked by a single stimulus for a wildtype calyx. (B) Same as A, but for a PKCγ ko animal. (C) Plots of residual calcium (Cares, top) and calcium influx (bottom) in slices from wildtype (filled symbols) and PKCγ ko (open symbols) animals. Tetanic stimulation (4 s, 100 Hz) was at time t=0. Scale bar for (A, B) is 10 μm. Error bars are SEM.
PKCγ suppresses the actions of PKCβ to dictate the mechanism of PTP
Based on the prominent contribution of p to PTP in wildtype animals prior to the onset of hearing, it seems that PTP is mediated primarily by PKCγ. However, in pre-hearing PKCγ ko animals PTP is produced by an RRP increase mediated by PKCβ. Why does PKCβ mediate PTP in pre-hearing PKCγ ko animals but play such a minor role in P8-10 wildtype animals?
To begin to address this question, we used immunohistochemistry to examine the expression of PKCβ at the calyx of Held prior to the onset of hearing. We found that PKCβ is present in wildtype animals (Figure 7, top). Immunofluorescence is eliminated in PKCαβ dko animals (Figure 7, middle). In PKCγ knockout animals, PKCβ is still present and there is no obvious increase in expression levels (Figure 7, bottom). These findings indicate that in pre-hearing wildtype animals PKCβ is present, but it does not contribute significantly to PTP. This suggests that PKCγ somehow prevents PKCβ from increasing the RRP.
Figure 7. PKCβ is present at the calyx of Held prior to hearing onset.
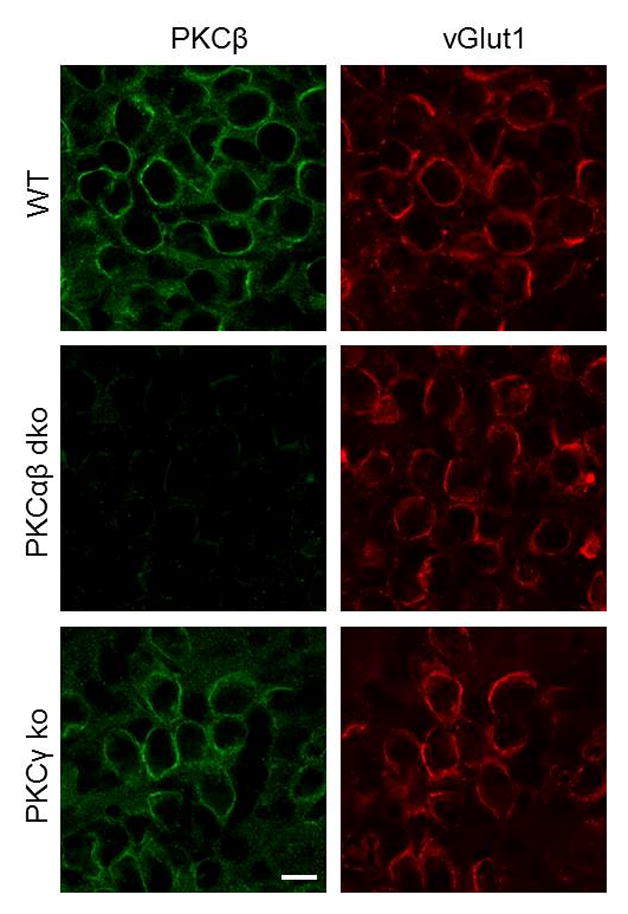
Brain slices from P8-10 animals containing the MNTB region were co-labeled with antibodies to PKCβ (green) and an antibody to the presynaptic marker vGlut1 (red). Representative images are shown for P10 slices from wildtype (WT), PKCαβ ko, and PKCγ ko animals. Scale bar is 10 μm.
If PKCγ does indeed suppress the activity of PKCβ, then the expression of PKCγ in hearing animals should also suppress the increase in the RRP by PKCβ and lead to PTP mediated predominantly by an increase in p. We determined if this is the case by using AAV to express PKCγ-YFP in globular bushy cells that give rise to calyx of Held synapses. Wildtype animals were used for these experiments. Calyces expressing PKCγ-YFP were readily identified by strong fluorescence (Figure 8A). Contributions of RRP and p to PTP were assessed (Figure 8B, C) and synapses expressing PKCγ-YFP showed robust PTP (75 ± 9%; n=9) that was mediated primarily by an increase in p (ptrainC 58 ± 5%), with very little contribution from RRP (11 ± 5%) (Figure 8D). Synapses from the same animals in which PKCγ-YFP was not expressed exhibited PTP (60 ± 7%; n=3) that was mediated mainly by an increase in RRP (RRPtrainC 45 ± 10%) (Figure 8E), which is typical of hearing wildtype animals. Although the magnitude of PTP was slightly larger for calyces infected with PKCγ-YFP, there was no significant difference in the amount of PTP between YFP-expressing and non-expressing cells (p=0.23). These experiments show that the presence of PKCγ prevents PKCβ from contributing to PTP by increasing the RRP.
Figure 8. Viral expression of PKCγ in the calyx of Held of hearing animals alters the mechanism of PTP.
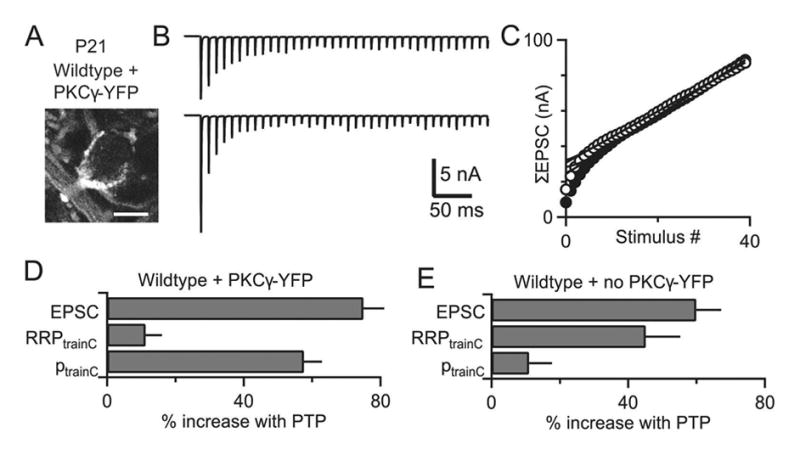
An AAV expressing PKCγ-YFP was injected in the ventral cochlear nucleus at P4. Synaptic properties were examined at postnatal day 19–22 and calyces of Held expressing PKCγ-YFP were identified. Mechanisms of PTP were examined using trains in the presence of kynurenate and CTZ to prevent receptor saturation and desensitization. (A) Representative fluorescence image from a P21 wildtype calyx showing PKCγ-YFP fluorescence. (B) Synaptic currents evoked by the first 40 stimuli of 4s, 100 Hz train (top) and by a 40 pulse, 100 Hz train (bottom) 10 s after tetanic stimulation (at the peak of PTP) are shown. (C) Cumulative EPSCs for the initial train (closed circles) and the second train (open circles) are plotted against stimulus number. (D) Summary of the contributions of RRP and p to PTP for synapses expressing PKCγ-YFP. (E) Summary of the contributions of RRP and p to PTP for synapses not expressing PKCγ-YFP from the same animals as in D. Scale bar for (A) is 10 μm.
The magnitude of PTP and the contributions of RRP and p to this enhancement in different knockout animals and in the presence of PKC inhibitors are summarized in Figure 9A and Table S2 for P8-10 animals. The changes in PPR associated with PTP for all groups are summarized in Table S2. PTP in pre-hearing animals is accompanied by a large decrease in PPR only when PKCγ is present. This is qualitatively consistent with PKCγ mediating PTP by increasing the probability of release.
Figure 9. Summary and schematic showing the effects of different PKC isoforms on PTP before and after hearing onset.

(A) Summary bar graphs are shown for the magnitude of PTP, and the changes in RRP and p that occur following tetanic stimulation in wildtype and knockout mice, and in the presence of pharmacological inhibitors. Bar graphs are color-coded to indicate whether PKCβ, PKCγ or both isoforms were knocked out or pharmacologically inhibited. Green bars indicate summaries of experiments in which PKCβ-YFP was expressed in wildtype animals. (B) A schematic illustration of the differential ways PKCβ and PKCγ contribute to PTP and the functional consequences of their response to a stimulus train.
Discussion
Our primary finding is that PKCβ and PKCγ both mediate PTP, but PKCγ predominantly enhances the probability of release and PKCβ mainly increases the size of the readily releasable pool. These findings indicate that the identity of the calcium-dependent PKC isoform that mediates PTP controls the mechanism and functional consequences of PTP.
After the onset of hearing PTP is mediated largely by PKCβ, which governs plasticity primarily by increasing the size of the RRP. The situation is more complicated before the onset of hearing. Although all three calcium-dependent isoforms of PKC are present at the calyx of Held, PTP is due largely to an increase in p mediated by PKCγ. In PKCγ ko animals, PTP is still observed, but it is due largely to an increase in RRP mediated by PKCβ. In fact, the contributions of RRP and p in pre-hearing PKCγ ko animals are remarkably similar to those observed in wildtype animals after the onset of hearing. The elimination of PKCγ in P8-10 animals essentially transforms the properties of PTP to those of wildtype animals after the onset of hearing, where PKCγ is absent from the calyx of Held. Moreover, the viral expression of PKCγ after the onset of hearing leads to PTP with properties similar to pre-hearing wildtype animals. Thus, it is not simply the age of the animal that determines the properties of PTP; it is the complement of calcium-dependent PKC isoforms available to mediate PTP.
PKC isoform-specific modulation
There is a growing appreciation that different PKC isoforms can perform specialized roles (Harper and Poole, 2007; Heemskerk et al., 2011; Sossin, 2007; Steinberg, 2008). An important factor in isoform-specific actions is that PKC isoforms are expressed differentially throughout the nervous system, and often in a developmentally regulated manner (Huang et al., 1990; Kose et al., 1990; Roisin and Barbin, 1997). But even for cells that express multiple PKC isoforms, differential subcellular compartmentalization and differential activation of substrate targets can allow individual PKC isoforms to perform unique cellular functions (Dekker and Parker, 1994; Hofmann, 1997; Shirai and Saito, 2002; Steinberg, 2008). For example, the calcium-sensitive isoforms PKCα and PKCβ and the novel isoform PKCδ have opposing actions in platelet activation and aggregation (Gilio et al., 2010; Harper and Poole, 2007; Heemskerk et al., 2011; Strehl et al., 2007). Similarly, at Aplysia sensory-motor synapses a calcium-independent PKC isoform mediates serotonin-induced recovery from depression (Manseau et al., 2001), whereas synaptic enhancement lasting for hours after stimulation is mediated by a calcium-dependent PKC isoform (Sossin, 2007; Zhao et al., 2006). As in these examples, isoform-specific actions within a cell often involve different classes of PKCs that are known to be activated by different types of signals. It is also known that different classes of PKCs are differentially effective at certain substrates. For example, calcium-dependent PKCs prefer basic residues N-terminal to the phosphorylation site, whereas novel PKCs prefer hydrophobic residues (Nishikawa et al., 1997; Sossin, 2007). The observation that PKC isoforms from different classes can act on different phosphorylation sites to differentially modulate L-type calcium channels (Yang et al., 2009) raises the possibility that PKC isoforms could act on the same protein but at different phosphorylation sites to produce PTP with different properties.
Although less is known about the ability of closely related calcium-dependent isoforms such as PKCβ and PKCγ to target different substrates, it is possible that anchoring proteins could allow such interactions to occur. PKCs have isoform-specific interactions with receptors for activated C kinase (RACKs), a family of membrane-associated anchoring proteins that function as molecular scaffolds to localize individual PKCs to distinct membrane microdomains so that PKC isoforms are in close proximity to their unique substrates. Cells may express unique RACKs for each PKC isoform and such PKC-RACK interactions could be essential for isoform-specific cellular responses (Csukai et al., 1997; Mackay and Mochly-Rosen, 2001; Schechtman et al., 2004). Different RACKS could localize specific PKC isoforms to different subdomains within the calyx of Held. Another possibility is that PKCβ and PKCγ could prefer different substrates (Nishikawa et al., 1997).
Possible targets of PKCβ and PKCγ
Further studies are required to determine the molecular targets of PTP and the means by which PKCβ and PKCγ produce PTP with different functional properties. We tested and excluded the hypothesis that PKCγ modulates presynaptic calcium entry. This is consistent with the observation that even at synapses where changes in calcium influx contribute to short-lived forms of plasticity, they do not contribute to PTP (Korogod et al., 2007). A previous study (Habets and Borst, 2006) suggested a role for calcium influx modulation in short-term plasticity, but our findings differ from those results, probably because a much different induction protocol was used there to induce a longer lasting form of short-term plasticity.
Munc18-1 remains a leading candidate effector molecule for PTP. For cultured hippocampal neurons it was shown that synaptic enhancement produced by phorbol esters, and a form of use-dependent plasticity following tetanic stimulation, both rely on PKC phosphorylating Munc18-1 (Wierda et al., 2007). Munc18-1 has also been implicated in regulating pool size (Nili et al., 2006; Toonen et al., 2006) and the probability of release, and there are multiple PKC phosphorylation sites on Munc18-1 (Barclay et al., 2003; Fujita et al., 1996). Based on these observations it is possible that PKCβ and PKCγ could both enhance transmission by phosphorylating Munc18-1, perhaps by phosphorylating different sites. At present it is not known if either or both PKCγ and PKCβ enhance transmission by phosphorylating Munc18-1. One or both of these isoforms could also regulate transmission by phosphorylating other targets in the presynaptic bouton including SNAP-25 (Gonelle-Gispert et al., 2002; Houeland et al., 2007; Nagy et al., 2002; Zamponi et al., 1997).
PKCγ suppresses the actions of PKCβ to dictate the mechanism of PTP
We tested the hypothesis that PKCγ increases p and PKCβ increases RRP by independent mechanisms acting on different targets, with multiplicative effects. Our findings indicate that PKCβ-dependent increases in RRP and PKCγ-dependent increases in p are not independent mechanisms. If they were, then when both PKCγ and PKCβ are present, it would be expected that PKCγ would increase p by ~ 40%, PKCβ would increase RRP by ~40% and the overall increase in EPSC amplitude would be ~96% [(1.4*1.4–1)*100]. But when both PKCγ and PKCβ are present, either in pre-hearing wildtype animals or in calyces expressing PKCγ-YFP in hearing wildtype animals, increases in RRP are small, and the magnitude of PTP is smaller than expected if both p and RRP increased. These results indicate that PKCβ and PKCγ do not act through independent mechanisms. We conclude instead that PKCγ suppresses the PKCβ pathway. Further studies are required to determine how this occurs.
Functional consequences of PTP being mediated by increased p prior to hearing onset and by increased RRP thereafter
Whether PTP is induced by an increase in p or RRP is important from a functional perspective, because these properties have very different effects on prolonged responses during firing of action potentials at the calyx of Held (Figure 9B). The properties of use-dependent plasticity at a synapse are likely tailored to the activity patterns of the presynaptic cell. In vivo, before the onset of hearing, neurons in the anteroventral cochlear nucleus (aVCN) fire spontaneous bursts of up to 5 action potentials often at more than 100 Hz (Sonntag et al., 2009; Sonntag et al., 2011). Such activity patterns could be crucial for setting up tonotopic maps prior to hearing onset (Kandler et al., 2009; Kandler and Friauf, 1993; Keuroghlian and Knudsen, 2007). After hearing onset, aVCN neurons fire more regularly and continuously, transmission reliability is high, and the calyx of Held synapse appears well-suited to convey auditory responses driven at high frequencies (Sonntag et al., 2011). It is perhaps not a surprise that the properties of short-term synaptic plasticity are regulated to respond appropriately to such different patterns of presynaptic activity before and after the onset of hearing. Increasing p results in a more rapidly depressing synaptic response, and the overall release by a burst of activity is unaffected. It seems that such a mechanism of PTP would not be particularly effective after hearing onset when sustained activity predominates. Increasing RRP essentially scales up responses and enhances release even for sustained activity and may be a more suitable mechanism after the onset of hearing.
Experimental Procedures
Animals
All animal experiments were completed in accordance with guidelines by the Harvard Medical Area Standing Committee on Animals. PKCαβ double knockout (ko) mice were obtained through breeding of PKCα and PKCβ knockout animals generated by M. Leitges (Leitges et al., 2002; Leitges et al., 1996). PKCγ ko mice (Abeliovich et al., 1993) were obtained from Jackson Labs. Because PKCβ and PKCγ are both located on chromosome 7, we first generated PKCβγ double ko mice through breeding of single PKCβ and PKCγ knockouts. PKCβγ double ko mice were then bred with PKCα ko mice to produce PKCαβγ triple ko animals. To prevent genetic drift in the inbred ko lines, we backcrossed them every second generation to C57BL/6J or 129S2. Because of the low probability of obtaining a double or triple knockout from heterologous crosses, we bred het-knockout animals together to increase the probability of getting desired animals.
Similarly, to increase the probability of obtaining wildtype mice, we crossed PKC het-het or het-wildtype mice to use as wildtype controls. Wildtype mice were derived from the same genetic line describe above. Animals of both sexes were used for experiments. Mice (C57BL/6J) from Charles River were used for all experiments in Figure 1 to test for changes in paired-pulse plasticity during PTP. For all other figures, mice of mixed background were used, and age-matched wildtype, PKCαβ, PKCγ, and PKCαβγ ko mice from our colony were interleaved for all experiments.
Brain Slices and Electrophysiology
Isoflurane was used to anesthetize animals, then their brains were dissected at 4°C using a solution containing the following (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.1 CaCl2, 3 MgCl2, 25 glucose, 3 myo-inositol, 2 Na-pyruvate, 0.4 ascorbic acid, pH 7.4 and continuously bubbled with 95% O2/5% CO2. Transverse slices (190–200 μm thick) containing the MNTB were cut from postnatal day (P)8-10 and P16-19 mice using a Leica vibratome slicer (VT1000S). Slices were then incubated at 32°C for 30 min with a solution of the same composition as the cutting solution above, but altered to have 2 CaCl2 and 1 MgCl2. Electrophysiological recordings were made as previously described (Fioravante et al., 2011) with the external solution containing 25 μM bicuculline and 1 μM strychnine to block inhibitory synaptic activity. Whole-cell patch-clamp recordings of excitatory postsynaptic currents (ESPCs) from MNTB neurons were made with an internal solution containing (in mM): 140 Cs-gluconate, 20 CsCl, 20 TEA-Cl, 10 HEPES, 5 EGTA, 5 Na2-phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na, pH: 7.3, 315–320 mOsm.
A custom-made bipolar electrode was placed near the midline near the MNTB region to stimulate presynaptic calyceal fibers. PTP was induced using a tetanic train (4 s, 100 Hz), and the baseline and post-tetanic EPSCs were measured at 0.2 Hz. To measure RRP and p, a second high-frequency train (0.4 s, 100 Hz) was induced 10 s after the first tetanus after wash-in of 0.1 mM cyclothiazide and 1 mM kynurenate to prevent AMPA receptor desensitization and saturation, respectively. In some experiments, slices were incubated for 30 min in the PKCβ inhibitor (3-(1-(3-Imidazol-1-ylpropyl)-1H-indol-3-yl)-4-anilino-1H-pyrrole-2,5-dione, Calbiochem) or for 60 min in the pan-PKC inhibitor (GF109203X HCl, Abcam).
Presynaptic Calcium Imaging
Calyces of Held were bulk loaded with Calcium Green-1 dextran (0.5%; 10 kDa, potassium salt, anionic, Invitrogen, Eugene, OR) and Alexa-594 dextran (0.025%) as previously described (Beierlein et al., 2004; Fioravante et al., 2011). Briefly, glass pipettes containing the loading dyes were placed next to calyceal fiber bundles arising from the midline. Loading times were 3–5 min at 32 °C, and slices were incubated for 1 h at 32 °C after loading. Fluorescence calcium signals were obtained using a two-photon microscope, and were converted to calcium by determining the Rmax/Rmin ratio (~5) in sealed pipettes. To determine Rmax, we washed on 20 μM ionomycin at the end of the experimental session, and the Rmax in ionomycin was used for all calcium calculations (see Supplemental Figure S3A). The Rtrain determined using a high-frequency train did not reach the Rmax value obtained with ionomycin. Calyces with bright green fluorescence at rest were rejected for further study, because they either had elevated resting calcium levels or were overloaded with calcium indicator.
Data Analysis
Custom written programs in IgorPro (WaveMetrics) were used to analyze all data. PPR was calculated as EPSC2/EPSC1. The contributions of RRP and p to PTP were quantified using the cumulative EPSC method and the Elmqvist and Qaustel method (Elmqvist and Quastel, 1965; Fioravante et al., 2011; Thanawala and Regehr, 2013). Additionally, changes in RRP and p were also calculated using a corrected cumulative EPSC method (Thanawala and Regehr, 2013). Statistical analyses were completed using paired Student’s t-tests, or for multiple comparisons, one-way ANOVAs or Student’s t-tests with Bonferroni corrections. The level of significance was set at p<0.05.
Immunohistochemistry
Transverse brainstem slices (190 μm thick) were obtained with a vibratome from P8-10 and P16-19 mice as described above. Slices were fixed in 2% paraformaldehyde for 1 hr at 4°C, washed three times in phosphate buffered solution (PBS, Sigma-Aldrich, St. Louis, MO), and then incubated for 1 hour at room temperature in PBS with 0.25% TritonX-100 (PBST) and 10% normal goat serum (NGS). Slices were incubated afterwards in primary antibodies (1:500 dilution) in PBST and 10% NGS overnight at 4°C. Slices were rinsed in PBS, incubated in secondary antibodies (1:500 dilution), then rinsed in PBS and mounted on Superfrost glass slides (VWR, West Chester, PA). The antibodies used were: anti-vGlut1 guinea pig polyclonal (Synaptic Systems, Göttingen, Germany), anti-PKCγ rabbit polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-guinea pig rhodamine-conjugated and goat anti-rabbit Alexa Fluor 488-conjugated secondaries (Santa Cruz Biotechnology). Images were obtained using an Olympus FluoViewTM FV1000 laser scanning confocal microscope with a 63x oil objective. Excitation wavelengths were 543 nm for rhodamine (vGlut1) and 488 nm for Alexa Fluor 488 (PKCγ). Emission filters were LP560 for vGlut1 and BP505-530 for PKCγ. Stacked optical sections at 1024 × 1024 were obtained sequentially for each channel.
DNA constructs and viruses
Cloning was performed by Genscript. The adeno-associated virus for PKCγ-YFP was generated by the University of Pennsylvania Vector Core. All constructs were verified by sequencing. Mouse PKCγ was obtained through PCR from Addgene plasmid #21236, using the following primers: 5′-TACAAGGCTGGTACCGAGCTCGGATCCGCGGGTCTGGGCCCTGGCGGAGGCGAC T -3′; 3′-ACCAGTGCCTCGAGCATGACAGGCACGGGCACAGGGCTTGT-5′. To generate the AAV vector, PKCγ was inserted between the 2 YFP sequences of a custom pENN.AAV.CMV.YFP.YFP.RBG cis-plasmid (based on the University of Pennsylvania vector core plasmid pENN.AAV.CMV.TurboRFP.RBG) using SacII and SalI.
Surgery
P4 pups were stereotactically and unilaterally injected under isofluorane anesthesia into the VCN (from lambda: 1.3 mm lateral, 0.9 mm caudal, 3 mm ventral), where globular bushy cells that give rise to calyx of Held synapses in the contralateral MNTB reside. Injections (600 nl at a rate of 1 nl/s) were performed with an UltraMicroPump (UMP3, WPI) and Wiretrol II capillary micropipettes (Drummond Scientific) pulled to a fine tip (10–20 μm diameter). After the injection, pups were allowed to recover on a heating pad prior to returning to the home cage. 14–18 days were allowed for expression prior to slice preparation.
Supplementary Material
Acknowledgments
We thank M. Antal, A. de Jong, S. Jackman, P. Kaeser, S. Rudolph, and L. Witter for comments on the manuscript, M. Thanawala for help with Igor routines and data analysis, K. McDaniels for help with genotyping, and E. Raviola for guidance on immunohistochemistry. This work was supported by NIH NS032405 to WGR, a Howard Hughes Medical Institute Medical Research Fellow grant and a NIH grant F30 DC013716-01 to YXC and DF was supported by NIH grant T32 NS007484.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell. 1993;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- Alle H, Jonas P, Geiger JR. PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc Natl Acad Sci U S A. 2001;98:14708–14713. doi: 10.1073/pnas.251610898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao JX, Kandel ER, Hawkins RD. Involvement of pre- and postsynaptic mechanisms in posttetanic potentiation at Aplysia synapses. Science. 1997;275:969–973. doi: 10.1126/science.275.5302.969. [DOI] [PubMed] [Google Scholar]
- Barclay JW, Craig TJ, Fisher RJ, Ciufo LF, Evans GJ, Morgan A, Burgoyne RD. Phosphorylation of Munc18 by protein kinase C regulates the kinetics of exocytosis. J Biol Chem. 2003;278:10538–10545. doi: 10.1074/jbc.M211114200. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gee KR, Martin VV, Regehr WG. Presynaptic calcium measurements at physiological temperatures using a new class of dextran-conjugated indicators. J Neurophysiol. 2004;92:591–599. doi: 10.1152/jn.00057.2004. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fioravante D, Regehr WG. Differential expression of posttetanic potentiation and retrograde signaling mediate target-dependent short-term synaptic plasticity. Neuron. 2007;54:949–959. doi: 10.1016/j.neuron.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Soria van Hoeve J. The calyx of held synapse: from model synapse to auditory relay. Annu Rev Physiol. 2012;74:199–224. doi: 10.1146/annurev-physiol-020911-153236. [DOI] [PubMed] [Google Scholar]
- Brager DH, Cai X, Thompson SM. Activity-dependent activation of presynaptic protein kinase C mediates post-tetanic potentiation. Nat Neurosci. 2003;6:551–552. doi: 10.1038/nn1067. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann N Y Acad Sci. 1999;868:144–159. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Chu Y, Fioravante D, Thanawala M, Leitges M, Regehr WG. Calcium-dependent isoforms of protein kinase C mediate glycine-induced synaptic enhancement at the calyx of Held. J Neurosci. 2012;32:13796–13804. doi: 10.1523/JNEUROSCI.2158-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csukai M, Chen CH, De Matteis MA, Mochly-Rosen D. The coatomer protein beta′-COP, a selective binding protein (RACK) for protein kinase Cepsilon. J Biol Chem. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- Dekker LV, Parker PJ. Protein kinase C--a question of specificity. Trends Biochem Sci. 1994;19:73–77. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Delaney KR, Tank DW. A quantitative measurement of the dependence of short-term synaptic enhancement on presynaptic residual calcium. J Neurosci. 1994;14:5885–5902. doi: 10.1523/JNEUROSCI.14-10-05885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney KR, Zucker RS, Tank DW. Calcium in motor nerve terminals associated with posttetanic potentiation. J Neurosci. 1989;9:3558–3567. doi: 10.1523/JNEUROSCI.09-10-03558.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist D, Quastel DM. A quantitative study of end-plate potentials in isolated human muscle. J Physiol. 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Chu Y, Myoga MH, Leitges M, Regehr WG. Calcium-dependent isoforms of protein kinase C mediate posttetanic potentiation at the calyx of Held. Neuron. 2011;70:1005–1019. doi: 10.1016/j.neuron.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Myoga MH, Leitges M, Regehr WG. Adaptive regulation maintains posttetanic potentiation at cerebellar granule cell synapses in the absence of calcium-dependent PKC. J Neurosci. 2012;32:13004–13009. doi: 10.1523/JNEUROSCI.0683-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucile S, Miledi R, Eusebi F. Effects of cyclothiazide on GluR1/AMPA receptors. Proc Natl Acad Sci U S A. 2006;103:2943–2947. doi: 10.1073/pnas.0511063103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Sasaki T, Fukui K, Kotani H, Kimura T, Hata Y, Sudhof TC, Scheller RH, Takai Y. Phosphorylation of Munc-18/n-Sec1/rbSec1 by protein kinase C: its implication in regulating the interaction of Munc-18/n-Sec1/rbSec1 with syntaxin. J Biol Chem. 1996;271:7265–7268. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- Gilio K, Harper MT, Cosemans JM, Konopatskaya O, Munnix IC, Prinzen L, Leitges M, Liu Q, Molkentin JD, Heemskerk JW, Poole AW. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. 2010;285:23410–23419. doi: 10.1074/jbc.M110.136176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Costa M, Takahashi M, Sadoul K, Halban P. Phosphorylation of SNAP-25 on serine-187 is induced by secretagogues in insulin-secreting cells, but is not correlated with insulin secretion. Biochem J. 2002;368:223–232. doi: 10.1042/BJ20020896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande G, Wang LY. Morphological and functional continuum underlying heterogeneity in the spiking fidelity at the calyx of Held synapse in vitro. J Neurosci. 2011;31:13386–13399. doi: 10.1523/JNEUROSCI.0400-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith WH. Voltage-clamp analysis of posttetanic potentiation of the mossy fiber to CA3 synapse in hippocampus. J Neurophysiol. 1990;63:491–501. doi: 10.1152/jn.1990.63.3.491. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets RL, Borst JG. An increase in calcium influx contributes to post-tetanic potentiation at the rat calyx of Held synapse. J Neurophysiol. 2006;96:2868–2876. doi: 10.1152/jn.00427.2006. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JG. Dynamics of the readily releasable pool during post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2007;581:467–478. doi: 10.1113/jphysiol.2006.127365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper MT, Poole AW. Isoform-specific functions of protein kinase C: the platelet paradigm. Biochem Soc Trans. 2007;35:1005–1008. doi: 10.1042/BST0351005. [DOI] [PubMed] [Google Scholar]
- Heemskerk JW, Harper MT, Cosemans JM, Poole AW. Unravelling the different functions of protein kinase C isoforms in platelets. FEBS Lett. 2011;585:1711–1716. doi: 10.1016/j.febslet.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Hofmann J. The potential for isoenzyme-selective modulation of protein kinase C. Faseb J. 1997;11:649–669. doi: 10.1096/fasebj.11.8.9240967. [DOI] [PubMed] [Google Scholar]
- Houeland G, Nakhost A, Sossin WS, Castellucci VF. PKC modulation of transmitter release by SNAP-25 at sensory-to-motor synapses in aplysia. J Neurophysiol. 2007;97:134–143. doi: 10.1152/jn.00122.2006. [DOI] [PubMed] [Google Scholar]
- Huang FL, Young WS, 3rd, Yoshida Y, Huang KP. Developmental expression of protein kinase C isozymes in rat cerebellum. Brain Res Dev Brain Res. 1990;52:121–130. doi: 10.1016/0165-3806(90)90227-p. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental regulation of transmitter release at the calyx of Held in rat auditory brainstem. J Physiol. 2001;534:861–871. doi: 10.1111/j.1469-7793.2001.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Clause A, Noh J. Tonotopic reorganization of developing auditory brainstem circuits. Nat Neurosci. 2009;12:711–717. doi: 10.1038/nn.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Friauf E. Pre- and postnatal development of efferent connections of the cochlear nucleus in the rat. J Comp Neurol. 1993;328:161–184. doi: 10.1002/cne.903280202. [DOI] [PubMed] [Google Scholar]
- Keuroghlian AS, Knudsen EI. Adaptive auditory plasticity in developing and adult animals. Prog Neurobiol. 2007;82:109–121. doi: 10.1016/j.pneurobio.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Klug A, Borst JG, Carlson BA, Kopp-Scheinpflug C, Klyachko VA, Xu-Friedman MA. How do short-term changes at synapses fine-tune information processing? J Neurosci. 2012;32:14058–14063. doi: 10.1523/JNEUROSCI.3348-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci. 2005;25:5127–5137. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Posttetanic potentiation critically depends on an enhanced Ca(2+) sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A. 2007;104:15923–15928. doi: 10.1073/pnas.0704603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kose A, Ito A, Saito N, Tanaka C. Electron microscopic localization of gamma- and beta II-subspecies of protein kinase C in rat hippocampus. Brain Res. 1990;518:209–217. doi: 10.1016/0006-8993(90)90974-g. [DOI] [PubMed] [Google Scholar]
- Lee JS, Kim MH, Ho WK, Lee SH. Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci. 2008;28:7945–7953. doi: 10.1523/JNEUROSCI.2165-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV. Knockout of PKC alpha enhances insulin signaling through PI3K. Mol Endocrinol. 2002;16:847–858. doi: 10.1210/mend.16.4.0809. [DOI] [PubMed] [Google Scholar]
- Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Immunodeficiency in protein kinase cbeta-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- Mackay K, Mochly-Rosen D. Localization, anchoring, and functions of protein kinase C isozymes in the heart. J Mol Cell Cardiol. 2001;33:1301–1307. doi: 10.1006/jmcc.2001.1400. [DOI] [PubMed] [Google Scholar]
- Magleby KL. Facilitation, augmentation, and potentiation of transmitter release. Prog Brain Res. 1979;49:175–182. doi: 10.1016/S0079-6123(08)64631-2. [DOI] [PubMed] [Google Scholar]
- Magleby KL, Zengel JE. A quantitative description of tetanic and post-tetanic potentiation of transmitter release at the frog neuromuscular junction. J Physiol. 1975;245:183–208. doi: 10.1113/jphysiol.1975.sp010840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manseau F, Fan X, Hueftlein T, Sossin W, Castellucci VF. Ca2+-independent protein kinase C Apl II mediates the serotonin-induced facilitation at depressed aplysia sensorimotor synapses. J Neurosci. 2001;21:1247–1256. doi: 10.1523/JNEUROSCI.21-04-01247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sorensen JB. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci. 2002;22:9278–9286. doi: 10.1523/JNEUROSCI.22-21-09278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura PA, Cramer KS. Formation and maturation of the calyx of Held. Hear Res. 276:70–78. doi: 10.1016/j.heares.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Nili U, de Wit H, Gulyas-Kovacs A, Toonen RF, Sorensen JB, Verhage M, Ashery U. Munc18–1 phosphorylation by protein kinase C potentiates vesicle pool replenishment in bovine chromaffin cells. Neuroscience. 2006;143:487–500. doi: 10.1016/j.neuroscience.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- Pan B, Zucker RS. A general model of synaptic transmission and short-term plasticity. Neuron. 2009;62:539–554. doi: 10.1016/j.neuron.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 2009;63:154–170. doi: 10.1016/j.neuron.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Contreras A, van Hoeve JS, Habets RL, Locher H, Borst JG. Dynamic development of the calyx of Held synapse. Proc Natl Acad Sci U S A. 2008;105:5603–5608. doi: 10.1073/pnas.0801395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roisin MP, Barbin G. Differential expression of PKC isoforms in hippocampal neuronal cultures: modifications after basic FGF treatment. Neurochem Int. 1997;30:261–270. doi: 10.1016/s0197-0186(96)00095-2. [DOI] [PubMed] [Google Scholar]
- Saitoh N, Hori T, Takahashi T. Activation of the epsilon isoform of protein kinase C in the mammalian nerve terminal. Proc Natl Acad Sci U S A. 2001;98:14017–14021. doi: 10.1073/pnas.241333598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechtman D, Craske ML, Kheifets V, Meyer T, Schechtman J, Mochly-Rosen D. A critical intramolecular interaction for protein kinase Cepsilon translocation. J Biol Chem. 2004;279:15831–15840. doi: 10.1074/jbc.M310696200. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem. 2002;132:663–668. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Rosahl TW, Chapman PF, Marowitz Z, Friedman E, Frankland PW, Cestari V, Cioffi D, Sudhof TC, Bourtchuladze R. Impaired learning in mice with abnormal short-lived plasticity. Curr Biol. 1996;6:1509–1518. doi: 10.1016/s0960-9822(96)00756-7. [DOI] [PubMed] [Google Scholar]
- Sonntag M, Englitz B, Kopp-Scheinpflug C, Rubsamen R. Early postnatal development of spontaneous and acoustically evoked discharge activity of principal cells of the medial nucleus of the trapezoid body: an in vivo study in mice. J Neurosci. 2009;29:9510–9520. doi: 10.1523/JNEUROSCI.1377-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag M, Englitz B, Typlt M, Rubsamen R. The calyx of held develops adult-like dynamics and reliability by hearing onset in the mouse in vivo. J Neurosci. 2011;31:6699–6709. doi: 10.1523/JNEUROSCI.0575-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn Mem. 2007;14:236–246. doi: 10.1101/lm.469707. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Williams JH. Discharge of the readily releasable pool with action potentials at hippocampal synapses. J Neurophysiol. 2007;98:3221–3229. doi: 10.1152/jn.00857.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehl A, Munnix IC, Kuijpers MJ, van der Meijden PE, Cosemans JM, Feijge MA, Nieswandt B, Heemskerk JW. Dual role of platelet protein kinase C in thrombus formation: stimulation of pro-aggregatory and suppression of procoagulant activity in platelets. J Biol Chem. 2007;282:7046–7055. doi: 10.1074/jbc.M611367200. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Sagawa S, Hoshi J, Shimoma F, Matsuda I, Sakoda K, Sasase T, Shindo M, Inaba T. Synthesis of anilino-monoindolylmaleimides as potent and selective PKCbeta inhibitors. Bioorg Med Chem Lett. 2004;14:5171–5174. doi: 10.1016/j.bmcl.2004.07.061. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leao RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Scheuss V, Neher E. Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J Physiol. 2005;568:513–537. doi: 10.1113/jphysiol.2005.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala MS, Regehr WG. Presynaptic calcium influx controls neurotransmitter release in part by regulating the effective size of the readily releasable pool. J Neurosci. 2013;33:4625–4633. doi: 10.1523/JNEUROSCI.4031-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Wierda K, Sons MS, de Wit H, Cornelisse LN, Brussaard A, Plomp JJ, Verhage M. Munc18–1 expression levels control synapse recovery by regulating readily releasable pool size. Proc Natl Acad Sci U S A. 2006;103:18332–18337. doi: 10.1073/pnas.0608507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Borst JG. Short-term plasticity at the calyx of held. Nat Rev Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Wu XS, Wu LG. Protein kinase c increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J Neurosci. 2001;21:7928–7936. doi: 10.1523/JNEUROSCI.21-20-07928.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Doshi D, Morrow J, Katchman A, Chen X, Marx SO. Protein kinase C isoforms differentially phosphorylate Ca(v)1.2 alpha(1c) Biochemistry. 2009;48:6674–6683. doi: 10.1021/bi900322a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Leal K, Abi-Farah C, Martin KC, Sossin WS, Klein M. Isoform specificity of PKC translocation in living Aplysia sensory neurons and a role for Ca2+-dependent PKC APL I in the induction of intermediate-term facilitation. J Neurosci. 2006;26:8847–8856. doi: 10.1523/JNEUROSCI.1919-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.