

Abstract
Background
We recently reported an increase in N-methyl-d-aspartate (NMDA) receptor subunit expression and CaMKII-dependent phosphorylation of NR2B in the rostral cingulate cortical (rCC) neurons following esophageal acid exposure in rats. As α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors mediate the fast excitatory transmission and play a critical role in synaptic plasticity, in this study, we investigated the effect of esophageal acid exposure in rats on the expression of AMPA receptor subunits and the involvement of these molecular alterations in acid-induced sensitization of neurons in the anterior cingulate (ACC) and midcingulate (MCC) cortices.
Methods
In molecular study, we examined GluA1 and GluA2 expression and phosphorylation in membrane preparations and in the isolated postsynaptic densities (PSDs) from rats receiving acute esophageal exposure of either saline (control group) or 0.1 NHCl (experimental group). In electrophysiological study, the effect of selective AMPA receptor (Ca2+ permeable) antagonist IEM-1460 and CaMKII inhibitor KN-93 was tested on responses of cortical neurons during acid infusion to address the underlying molecular mechanism of acid-induced sensitization.
Key Results
The acid exposure significantly increased expression of GluA1, pGluA1Ser831, and phosphorylated CaMKIIThr286, in the cortical membrane preparations. In isolated PSDs, a significant increase in pGluA1Ser831 was observed in acid-treated rats compared with controls. Microinjection of IEM-1460 or KN-93 near the recording site significantly attenuated acid-induced sensitization of cortical neurons.
Conclusions & Inferences
The underlying mechanism of acid-induced cortical sensitization involves upregulation and CaMKII-mediated phosphorylation of GluA1. These molecular changes of AMPA receptors subunit GluA1 in the cortical neurons might play an important role in acid-induced esophageal hypersensitivity.
Keywords: AMPA receptor subunits, anterior cingulate cortex (ACC), CaMKII, GluA1, midcingulate cortex (MCC), postsynaptic densities (PSDs)
INTRODUCTION
Visceral hypersensitivity is thought to be due to a complex interaction between peripheral and spinal pathways, midbrain descending modulatory pathways, and higher cortical networks triggered by painful stimuli.1,2 In the past, several attempts have been made to elucidate the mechanism of visceral hypersensitivity using brain imaging techniques.3 In human fMRI (functional magnetic resonance imaging) study, esophageal acid exposure demonstrates cortical sensitization to subliminal and liminal non-painful mechanical stimulations.4 Studies have also reported a selective activation of anterior cingulate cortex (ACC) and insular cortex (IC) under various painful visceral stimuli.5,6 In anesthetized rabbits, cortical recordings following colonic distension demonstrate that viscero-nociceptive neurons are most frequent in ACC (39%) and MCC (midcingulate cortex; 36%) and less frequent in retrospenial cortex (RSC 12%).7 Although several brain imaging studies in humans and electrophysiological recordings from animals indicate the activation of cortical and insular neurons to visceral stimuli, the exact neurochemical changes that occur during this activation are not clearly delineated.
In the brain, ionotropic glutamate receptors are primarily involved in excitatory neurotransmission and synaptic plasticity. Among ionotropic glutamate receptors, AMPA receptors mediate the majority of fast excitatory synaptic transmission, whereas NMDA receptors are involved in the induction of long-term synaptic plasticity.8 An enhancement of NMDA receptor-mediated excitatory transmission in ACC neurons correlates with hyperalgesia and allodynia in rats.9,10 A long-term molecular alteration of NMDA receptor subunits (NR2A and NR2B) expression and phosphorylation of these subunits in the rostral cingulate cortex has been reported recently in rats following early-in-life esophageal acid exposure.11 Several studies have also implicated AMPA receptor-mediated synaptic activity during acute as well as persistent pain.12-18 However, the dynamic distribution of AMPA receptor subunits in cortical neurons in the setting of noxious visceral hypersensitivity has not been well defined.
This study was undertaken to test the hypothesis that the esophageal acid exposure induces AMPA receptor subunit upregulation and phosphorylation in the cingulate cortex and this molecular alteration contributes to sensitization of cortical neurons. As AMPA receptor subunits GluA1 and GluA2 are ubiquitously present in the central nervous system, we investigated the expression of these two subunits in ACC and MCC following acute esophageal acid exposure in rats. As the role of CaMKII is critical in glutamate receptor phosphorylation and synaptic plasticity,19 we have also studied the expression of CaMKII and pCaMKIIThr286 in cortical neurons following esophageal acid infusion. To validate the implication of our molecular findings, we examined the effect of esophageal acid infusion on the sensitization of ACC neurons. We further studied the pharmacological intervention of selective AMPA receptor subunit (Ca2+ permeable) antagonist IEM-1460 and CaMKII inhibitor KN-93 on acid-induced cortical sensitization.
MATERIALS AND METHODS
Animals
A total of 85 adult male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA) were used for this study. All procedures were performed in strict accordance to the guidelines laid down by the NIH and the Office of Laboratory Animal Welfare (OLAW) of Public Health Service (PHS). The Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin approved all experimental procedures (AUA2297) in accordance to the guidelines of the International Association for Study of Pain.
Experimental procedures
Surgical Procedures for esophageal acid infusion in adult rats were carried out as described earlier.11 Briefly, rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and a 15 cm long acid infusion catheter (PE-10) was inserted into the esophagus through an incision made 2 cm below the UES (upper esophageal sphincter). The tip of the catheter was positioned near the mid-thoracic area and the catheter was tied near the incision to prevent backflow of the acid into the pharynx. A second catheter (drainage catheter, 4 cm length, PE-160) was placed 1 cm rostral to the incision with the tip directed towards the pharynx. This catheter was used to remove accumulated saliva to prevent aspiration. For acute treatment, rats received 2 mL of either saline or 0.1 N HCl, pH 1.2 at 0.1 mL/min for 20 min and the brains were removed as described in the following section. Based on our previous study, we selected 4 h post infusion as the optimum time for tissue harvesting, since this time point exhibited a significant changes in NMDA receptor subunits in the rostral cingulate cortex following acute esophageal acid exposure in rats.11
Tissue harvest
For procuring brain tissues, rats were deeply anesthetized (sodium pentobarbitol, 50 mg/kg, i.p.) and decapitated. The skull bone was removed and the brain was placed in a cooled metal slicing block (Zivic rat brain slicer, Cat # BSRS005.1) for coronal sectioning (two 3 mm slices between 6–9 mm and 9–12 mm anterior to the cortical-cerebellar junction). In each slice, we used a 2 mm diameter tissue punch to bilaterally sample the cingulate cortices. The posterior slice (6–9 mm) carried MCC and the anterior slice (9–12 mm) carried the ACC. Punched out tissues were placed in liquid nitrogen for molecular studies and remaining slices were fixed for histological verification of location from where tissues were punched.
Western blot analysis of cortical membrane preparations
Cortical membrane preparations were carried out as previously described.20 Various antibodies used for western blot analysis were rabbit anti-GluA1 (1 : 2500, Alomone Labs, Israel), rabbit anti-GluA2 (1 : 5000; Alomone), rabbit anti-pGluA1Ser831 (1 : 1000; Millipore, Billerica, MA, USA), rabbit anti-pGluA1Ser845 (1 : 1000; Millipore), rabbit anti-pGluA2Ser880 (1 : 1000; Millipore), rabbit anti-CaMKII (1 : 1000; Cell Signaling, Danvers, MA, USA), rabbit anti-pCaMKIIThr286 (1 : 1000; Cell Signaling) and mouse anti-β actin (1 : 5000; Sigma, St Louis, MO, USA). The intensity of protein expression for experimental and housekeeping gene (mouse anti β-actin) for individual tissue sample was measured by densitometric scanning using Alpha Image software program (Cell Biosciences Inc., Santa Clara, CA, USA).
Isolation of PSDs by unltracentrifugation
For postsynaptic densities (PSDs) isolation, ACC and MCC were combined from each animal, and tissues from three animals receiving same treatment were pooled together for isolating sufficient PSDs for further analysis. Three sets of PSDs (n = 9/group) were prepared from animals receiving either acid or saline. PSDs isolation were carried out using density gradient ultracentrifugation as described previously.21 Briefly, the streak-like cloudy bands between 2.0 M/1.5 M sucrose was removed carefully in a microfuge tube and re-suspended in an equal amount of 75 mM KCl with 0.5% Triton X-100 and centrifuged at 50 000 rpm for 30 min at 4 °C. The resulting pellet carrying the final PSD product was resuspended in solubilization buffer containing 1% SDS and incubated at 37 °C for 45 min and centrifuged at 14 000 rpm for 15 min. The protein concentration of isolated PSDs was estimated by BCA method.
Immunohistochemical analysis of synaptic pGluA1Ser831 and PSD-95 expression in cortical neurons
We have followed the method as described previously.20 In brief, ACC tissues were embedded in HistoPrep (Fisher Scientific, Pittsburgh, PA, USA), and serial sections of 25-μm thickness were taken on a cryostat. To unmask synaptic receptors, the floating sections were then subjected to antigen retrieval using sodium citrate buffer (10 mM, pH 8.5) at 80 °C for 30 min. This was followed by incubation with blocking buffer (0.1 M PBS+0.25% Triton X-100+10% NGS) for 1 h at room temperature. The sections were then incubated with mouse anti-PSD-95 (1 : 100; BD Biosciences, San Jose, CA, USA) and rabbit anti-pGluA1Ser831 (1 : 100) antibodies for 48 h at 4 °C. After five washes of 15 min each, sections were incubated with goat anti-rabbit Alexa 568 and goat anti-mouse Alexa 488 antibodies (Life Technologies, Carlsbad, CA, USA) for 2 h at room temperature. The sections were mounted on clean slides using Vectashield mounting medium (Vector Laboratory, Burlingame, CA, USA). Both antibodies exhibited specific binding with cortical extracts and pGluA1Ser831 appeared as a single band at 100 kDa (in the present study) and PSD-95 exhibited a distinct band at 95 kDa in our previous report.11 The fluorescent images were captured using Leica TCS NT scanning laser confocal microscope equipped with argon (Ar, 488 nm), krypton (kr, 568) and helium-neon (He-Ne, 633) lasers. For simultaneous imaging of Alexa-488 and Alexa-568, a dual channel fluorescence arrangement was used. To study the immunostaining pattern of ACC neurons at low magnification, we used 20× Plan Apo objective lens. To resolve individual immunostained puncta, we used a 60× oil immersion Plan Apo objective lens. For improved signal to noise ration, up to eight scans were averaged at each optical sections and stacks of 8–15 optical sections (1024 × 1024 pixel array) yielded voxel dimensions between 0.15 and 0.4 for the X, Y and Z planes.
Neuronal recording from ACC and pharmacological intervention
Fourteen rats were anesthetized with a mixture of α-chloralose (80 mg/kg, i.p.) + urethane (80 mg/kg, i.p.). Femoral vein and artery were cannulated for infusion of saline and monitoring blood pressure, respectively. The trachea was intubated below the larynx for free breathing. A small drainage catheter was placed into the gastro-esophageal (GE) junction through the stomach and tied securely to prevent acid entering the stomach. The anesthesia was maintained with a supplemental dose (1/4th of initial dose) every hour. The head was fixed on a stereotaxic head-holder and a craniotomy was performed to access the ACC (bregma: +1.0–5.0 mm, 0.1–2.0 mm lateral). Single barrel carbon fiber microelectrodes (10 MΩ, Carbostar-1, Catalog #: E1011, Kation Scientific, MN, USA) were used for extracellular recordings from ACC neurons (bregma: +1.7–3.7 mm, 0.3–1.0 mm lateral, 1.3–3.5 mm dorso-ventral). The infusion of 0.1 mL of saline (pH 5.6) or 0.1 N HCl (pH 1.2) was given at mid esophagus, 2 cm caudal to the upper esophageal sphincter at every 4 min interval to avoid acid-induced desensitization. A guide cannula (20GA, Plastic One Inc., Roanoke, VA, USA) was inserted close to the proximity (5–10 μm from the recording electrode) of recording site. The drug was injected through an injector (24GA, C311I-SPC, Plastic One Inc) with the tip extending 0.5 mm from the guide cannula. IEM-1460 or KN-93 was microinjected (0.5 μL of KN93 [100 μg/mL] or IEM-1460 [100 μM]) 120 s before the acid infusion. All dosings were chosen based on previous publications by us11 and others22-24. Responses of neurons to intra-esophageal saline or acid were recorded before and after the injection of the drugs. The recovery from the drug effect was checked 45 min after the microinjection. A 15% change in firing frequency over the baseline was considered as an effective response.
Data analysis
Results are represented as mean ± SD. Student’s t-test was used for the comparison between two groups and a p < 0.05 was considered significant. Since most of the cortical recording yielded multiunit recording, we used signal waveform analysis (spike 2, v4.01) to identify each neuron in each recording session. Neuron that reliably matched the template was selected for further analysis. The total number of action potentials during 60 s of resting period prior to the esophageal acid infusion was considered as baseline activity represented as impulses/sec. The total number of action potentials within 60 s following acid infusion was counted as the effect of acid. A 15% increase in firing frequency during acid infusion was considered as effective response. The statistical analysis was performed using sigmastat (V2.03, SPSS, Chicago, IL, USA). Statistical comparison was performed using Student’s t-test. All values are expressed as mean ± SEM. The value of p < 0.05 was considered statistically significant.
RESULTS
Acid-induced alteration of AMPA receptor subunits expression in ACC and MCC
The technique used for punching out brain regions representing ACC and MCC are shown in Fig. 1. In the first set of experiments, membrane preparations from ACC and MCC regions were examined. We observed a high level of GluA1 constitutively expressed as a 100 kDa protein in both ACC and MCC regions. However, acid treatment resulted in further increase in its expression both in ACC and MCC (Fig. 1B, *p <0.05, **p <0.01 vs saline-treated controls). As phosphorylation of AMPA receptor subunits is an important posttranslational modification that determines many aspects of channel function,25,26 we further investigated the effect of acid on GluA1 phosphorylation at its C-terminal end amino acids (Ser831 and Ser845) in cortical membrane extracts. As shown in Fig. 1B, a low level of expression of pGluA1Ser831 was observed in saline-treated animals. However, esophageal acid exposure resulted in a significant increase in membrane expression of pGluA1Ser831 both in ACC and MCC tissue extracts (Fig. 1C, **p < 0.01 vs controls). We did not observe significant difference in pGluA1Ser845 expression between acid- and saline-treated groups (Fig. 1C), but the level of pGluA1Ser845 expression was constitutively high in both regions. We further analyzed the relative expression of pGluA1Ser831 against GluA1 in ACC and MCC for both acid- and saline-treated rats (Fig. S1). In ACC, the relative expression of pGluA1Ser831 against GluA1 is significantly higher in acid-treated rats compared to saline controls (Fig. 1, *p ≤ 0.05, Data S1). Whereas, MCC samples from acid-treated rats failed to exhibit a significant increase in the ratio pGluA1Ser831 to GluA1 expression compared to saline controls (Fig. S1). We also examined the expression and phosphorylation of GluA1 in insular cortex, the brain region also reported to be involved in cortical sensitization; however, no significant difference in GluA1 expression and phosphorylation was observed in insular cortices following esophageal acid exposure (see Fig. S2). Although there is a high level of constitutively expressed GluA2 (100 kDa) in cortical regions, esophageal acid treatment failed to induce any significant difference in expression of GluA2 and pGluA2Ser880 (Fig. S3). The specificity of the antibody bindings was examined in Western blots using respective blocking peptides (see Fig. S3).
Figure 1.

(A) Typical examples of Thionin-stained sections used to identify the acquired anterior cingulate cortex (ACC) and midcingulate cortex (MCC) from the rat brain. The removed areas of the brain sections include most of the rat ACC and MCC regions. Asterisks (*) represent the area of the tissues extracted for evaluation. The sections depicted are close to the most rostral and caudal sections of the brain sampled. MC, Motor cortex; SSC, Somatosensory cortex; cg, Cingulum; cc, Corpus collosum; ac, Anterior commissure; ec, External capsule; CL, Claustrum; LV, Lateral ventricle; HP, Hippocampus; fi, Fimbria of the hippocampus; Hb, Habenula; St, Striatum (caudate putamen). The regions of ACC and MCC are referred to Bregma. (B) GluA1, pGluA1Ser831, and pGluA1Ser845 expression in crude cortical membrane preparations from rats following acute esophageal exposure to either saline or acid (n = 5/group). (C) Bar graphs in lower panel represent relative intensity of expression against β-actin and presented as mean ± SD (*p < 0.05, **p < 0.01 vs saline controls).
Upregulation of phosphorylated CaMKIIThr286 in cortical membrane
As reported data suggest the involvement of CaMKII and PKC in phosphorylation of GluA1 at Ser831 and Ser845 in response to noxious stimulations,25,26 we analyzed the expression of CaMKII and its activated form pCaMKIIThr286 in ACC and MCC following esophageal acid or saline treatment. CaMKII expressed as a 50 kDa protein (alpha form) in both ACC and MCC. No significant difference in CaMKII expression was observed between groups. However, acid treatment resulted in a significant upregulation of pCaMKIIThr286 both in ACC and MCC neurons in acid-treated rats (Fig. 2, *p < 0.01 vs controls).
Figure 2.

pCaMKIIThr286 and CaMKII expression in crude cortical membrane preparations from rats following acute esophageal exposure of either acid or saline. Upper panels show the expression in individual animals (n = 5/group). The bar graphs in lower panel represent relative intensity of pCaMKII expression against CaMKII and presented as mean ± SD (*p < 0.05 vs saline controls).
Acid-induced upregulation of pGluA1Ser831 in the isolated PSDs
Figure 3A illustrates the isolated PSDs from the gradient between 1.5 and 2.0 M sucrose following density gradient ultracentrifugation of the cortical membrane preparation with a high enrichment of various synapse-associated proteins. The isolated PSDs were examined for the expression of pGluA1Ser831 and pGluA2Ser880. A significant increase in pGluA1Ser831 was observed in the PSDs isolated from acid-treated animals compared to saline-treated controls (Fig. 3B and C, **p < 0.001), whereas no significant difference in the expression of pGluA2Ser880 was observed between these two groups of rats.
Figure 3.
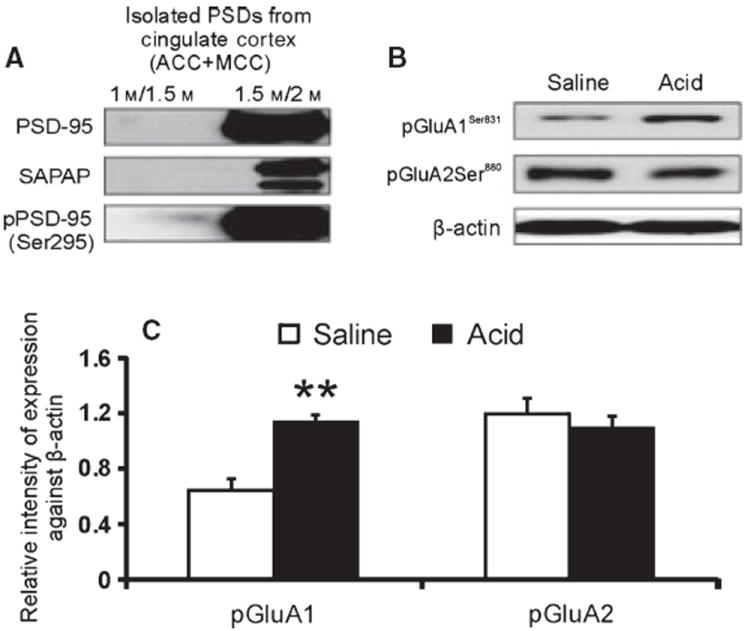
(A) Expression profile of synapse-associated proteins in the isolated PSDs. (B) Western blots of pGluA1ser831 and pGluA1Ser880 expression in the PSDs isolated from saline- and acid-treated animals. (C) Three sets of PSDs were isolated each from saline- and acid-treated animals and data presented as mean ± SD (**p < 0.001 vs saline controls).
Upregulation of PSD-95 and pGluA1Ser831 expression in ACC
In immunohistochemistry (IHC) experiments, double immunostaining of cortical neurons in layer II/III of ACC from acid- and saline-treated rats is shown in Fig. 4A. As GluA1 and PSD-95 are mainly associated at the PSDs through their binding to another synaptic protein stargazine, we did not anticipate their exact colocalization in the cortical membrane. However, in merged images, especially in sections from acid-treated groups, the higher level of expression of both GluA1 and PSD-95 in very close proximity appeared as coexpression with yellow punctas on the membrane as shown by arrows (Fig. 4A). In acid-treated rats, PSD-95 and pGluA1Ser831 expression were significantly high compared with saline-treated controls (Fig. 4B, **p < 0.001 vs saline control). We further evaluated PSD-95-positive puncta around the cortical membrane for the coexpression of pGluASer831 (Fig. 4C). In acid-treated rats, association of pGluA1Ser831 expression with PSD-95 positive puncta is significantly higher compared to saline -treated controls (Fig. 4C, *p < 0.01 vs saline control). In Western blots, both anti-pGluA1Ser831 and anti-PSD-95 antibodies exhibited single bands at molecular weight 100 and 95 kDa, respectively, indicating the specificity of antibody binding with cortical tissues (Fig. 4E). To examine further specificity of antibody binding, we carried out immunostaining without primary antibody. No significant immunostaining was observed in tissue sections without primary antibody incubation (Fig. 4D).
Figure 4.

(A) Immunostaining of pGluA1Ser831 (red) and PSD-95 (green) in layer II/III anterior cingulate cortex (ACC) neurons following acute esophageal acid or saline exposure in rats. Arrows in bottom row indicate the colocalization of pGluA1Ser831 and PSD-95. The scale bar is 10 μm. (B) It shows significantly higher expression of pGluA1Ser831 and PSD-95 in acid-treated rats compared with saline-treated controls (**p <0.001 vs saline control). (C) It shows percentage of PSD-95-positive puncta associated with pGluA1Ser831 expression (*p ≤ 0.01 vs saline control). (D) It shows cortical sections immunostained without primary antibodies. (E) It shows the specific binding (a single band) for both pGluA1Ser831 and PSD-95 antibodies with ACC extract in Western blot.
Effect of selective AMPA receptor (Ca++ permeable) antagonist IEM-1460 and CaMKII inhibitor KN-93 on acid-induced ACC neuron sensitization
Extracellular microelectrode recording from ACC neurons exhibited progressive increase in firing to repeated infusion of acid into the esophagus (Fig. 5). Microinjection of selective calcium permeable AMPA receptor antagonist IEM-1460 (1 μL of 100 μm) to close proximity (5–10 μm away) of the recording site completely blocked the subsequent acid-induced excitation of these neurons (Fig. 5A). IEM-1460 did not change the resting firing of ACC neurons following microinjection. The excitation of these neurons by esophageal acid was retested 45 min after IEM-1460 injection. Neurons exhibited full recovery of response to acid infusion, suggesting a reversible effect of the antagonist (Fig. 5B). Figure 5C summarizes the analytical data of eight neurons that were tested to intraesophageal acid followed by IEM-1460 microinjection. Esophageal saline infusion did not produce excitation of ACC neurons (Fig. 5D).
Figure 5.

Illustration of the effect of selective AMPA receptor (Ca2+ permeable) antagonist IEM-1460 on responses of ACC neurons following intraesophageal acid infusion. (A) It shows an example of recording from ACC. The top trace represents frequency histogram and the bottom trace shows the action potentials of ACC neurons. Neurons exhibited progressive increase in firing to repeated acid (0.1 mL of 0.1 N HCl) infusion. IEM-1460 was injected (down arrow) following third acid infusion. Neurons stopped firing to subsequent acid infusion. (B) Responses of these neurons recovered when tested after 45 min of IEM-1460 injection. (C) It shows analytical data of eight neurons tested to microinjection of IEM-1460. Intraesophageal acid infusion significantly (*p < 0.05 vs baseline 30–60 s) increased the firing frequency of the neuron. Following IEM-1460 injection, responses of these neurons to subsequent acid infusion were significantly (#p < 0.05 vs third acid infusion 540–570 s) inhibited. Responses of neurons recovered significantly (+p < 0.05 vs baseline 3750–3780 s) 45 min after IEM-1460 injection. (D) intraesophageal saline infusion did not produce excitation of cortical neurons (n = 14).
Similarly, acid-induced excitation of ACC neurons was significantly (p < 0.05 vs acid) blocked after microinjecting CaMKII inhibitor KN-93 (Fig. 6A), suggesting that CaMKII-mediated receptor phosphorylation may be involved in the excitation of these neurons. Like IEM-1460, KN-93 did not change the resting firing of ACC neurons following microinjection. Responses of these neurons to acid recovered when tested 45 min after KN-93 injection (Fig. 6B). Figure 6C summarizes the analytical data of 13 neurons that were tested to intraesophageal acid followed by KN-93 microinjection. In three separate experiments, KN-93 was injected to the contralateral side of ACC and intraesophageal acid infusion was carried out in similar fashion. This microinjection of KN-93 to contralateral side did not block the acid-induced excitation of ACC neurons, which validates the focal blocking effect of the antagonist following microinjection of the drug to close proximity of recording electrode (data not shown).
Figure 6.

Illustration of the effect of CaMKII inhibitor KN-93 on responses of ACC neurons following intraesophageal acid infusion. (A) It shows an example of recording from ACC neuron. KN-93 was injected (down arrow) following third acid infusion. The top trace represents frequency histogram and the bottom trace shows the action potentials of ACC neurons. (B) Responses of these neurons recovered when tested after 45 min of KN-93 injection. (C) It shows analytical data of 13 neurons tested to microinjection of KN-93. Intraesophageal acid infusion significantly (*p < 0.05 vs baseline 30–60 s) increased the firing frequency of the neuron. Following KN-93 injection, responses of these neurons to subsequent acid infusion were significantly (#p <0.05 vs third acid infusion 540–570 s) inhibited. Responses of neurons recovered significantly (+p <0.05 vs baseline 3750–3780 s) 45 min after KN-93 injection. (D) The effect of KN-93 (CaMKII inhibitor) microinjection on esophageal acid–induced pGluA1Ser831 expression in ACC. Upper panel represents pGluA1Ser831 expression in individual animals (n = 4/group) receiving either vehicle DMSO or KN-93 before acute esophageal acid exposure. Lower panel represents relative intensity of expression against β-actin (*p <0.05 vs vehicle controls).
KN-93 downregulates acid-induced GluA1Ser831 phosphorylation in ACC
To confirm the involvement of CaMKII in acid-induced phosphorylation of GluA1 in cortical neurons, ACC extracts from rats receiving intracortical application of either KN-93 or DMSO prior to esophageal acid exposure were examined by Western blots. A significant downregulation in the expression of phosphorylated GluA1Ser831 was observed in ACC extracts from rats receiving KN-93 compared with DMSO-treated rats (Fig. 6D, p < 0.05 vs DMSO).
DISCUSSION
In this study, we have demonstrated an upregulation of AMPA receptor subunit GluA1 in cortical neurons in rats following acute esophageal acid exposure. This upregulation is also accompanied by an increase in phosphorylation of GluA1 subunit at Ser831 (pGluA1Ser831) in ACC and MCC neurons. Moreover, intracortical application of highly selective AMPA receptor antagonist IEM-1460 resulted in blocking of acid-induced cortical neuron sensitization indicating the functional implication of this receptor subunit upregulation. We have also demonstrated an activation of the enzyme CaMKII in the cortical neurons with significant increase in phosphorylated CaMKII (pCaMKIIThr286) in acid-treated rats compared to saline-treated controls. Moreover, microinjection of CaMKII inhibitor KN-93 into the ACC resulted in significant inhibition of acid-induced excitation of cortical neurons as well as downregulation of pGluA1Ser831 expression. We observe a high level of constitutively expressed GluA2 in cortical membrane preparation from both ACC and MCC and acid exposure did not change the expression of GluA2 and pGluA2Ser880 suggesting that the esophageal acid–induced cortical sensitization mainly involved in activation of GluA1, but not GluA2 subunit. Recently, we have documented CaMKII-mediated upregulation of cortical NR2B subunit phosphorylation (pNR2BSer1303) of NMDA receptors following acute esophageal acid exposure.11 Taken together, these findings emphasize the involvement of both AMPA and NMDA receptor subunits upregulation and phosphorylation in cortical sensitization following esophageal acid exposure.
We also demonstrate an upregulation of pGluA1Ser831 expression in the isolated postsynaptic densities (PSD) indicating acid-induced accumulation of pGluA1Ser831 in the postsynaptic membrane of the cortical tissues. We have previously shown an upregulation of PSD-95 (a highly abundant receptor anchoring protein of PSDs) in the rCC following esophageal acid exposure.11 Consistent with this finding, our IHC study also exhibits an upregulation of PSD-95 in the cortical membrane, and also its coexpression with pGluA1Ser831 in acid-treated rats. This observation further confirms postsynaptic accumulation of phosphorylated AMPA receptor subunits GluA1 in the cortical neurons following esophageal acid exposure.
AMPA receptor subunit expression at the excitatory synapses
In the central nervous system, NMDA and AMPA receptors mediate most of the synaptic function and highly concentrated at the PSDs of excitatory synapses. While AMPA receptors mediate rapid synaptic transmission, the activation of NMDA receptors induces synaptic plasticity.27-30 Several reports support the involvement of AMPA receptor subunits in spinal nociception and also in the induction of LTP in hippocampal and cortical neurons.19,30-32 In mice model of visceral hyperalgesia, a significant trafficking of GluA1 from cytosol to plasma membrane has been observed following painful visceral stimuli.33 At supraspinal level, CFA-induced peripheral inflammation significantly enhances the synaptic insertion of GluA1 subunits in the ACC neurons, which subsequently increases the central excitatory transmission during chronic pain.18
The involvement of GluA1 in the inflammation-induced synaptic potentiation and activation of ERK signaling pathway in the ACC has been documented using GluA1 knockout mice.32 Moreover, a close association between NMDA and AMPA receptor subunits are evident in recent studies, suggesting that the surface expression of GluA1 is dependent on subunit-specific interaction with NR2A subunit of NMDA receptors, and this interaction is involved in many forms of synaptic plasticity.34,35 We recently documented a significant upregulation of NR2A subunit of NMDA receptor in cortical neurons following esophageal acid exposure.11 Moreover, we have also demonstrated a long-lasting upregulation of NR2A subunits in the cortex of rats that were subjected to esophageal acid early in life indicating a critical role for NR2A subunits in acid-induced cortical sensitization. Moreover, this report and our present findings together suggest a possible involvement of NR2A-NMDA receptor subunit in GluA1 surface expression and activation following acid-induced cortical sensitization.
AMPA receptor subunit phosphorylation and neuronal sensitization
Distinct roles of AMPA receptor subunits phosphorylation in neuronal sensitization have been documented in recent studies.25,26,36,37 The phosphorylated GluA1Ser845 has been reported to regulate the open channel probability and GluA1Ser831 regulates the channel conductance, whereas GluA1Ser818 phosphorylation is critical for LTP-driven incorporation of AMPA receptors into the postsynaptic membrane. Studies also suggest the role of phosphorylated GluA1Ser831 and GluA1Ser845 in inflammatory pain, but not in neuropathic pain.25,26,38 In this study, esophageal acid only induced upregulation of phosphorylated GluA1ser831, but not GluA1Ser845 in the cingulate cortex. This could be due to distinct stimulus-dependent response of esophageal acid and in agreement with a recent study demonstrating a selective upregulation of GluA1ser831, but not GluA1Ser845 in the rat spinal dorsal horn following postoperative pain.39 Moreover, pharmacological blockade of the acid-induced cortical neuron sensitization using highly specific Ca2+ permeable AMPA subunit antagonist IEM-1460 also indicates the possible involvement of GluA1 in synaptic transmission and downstream signaling in cortical neurons. We further demonstrated that this acid-induced alteration in AMPA receptor subunit is specific for cingulate cortex, as no significant change in expression and phosphorylation of GluA1 expression was observed in the insular cortex; the region also reported to be involved in acid-induced cortical activation in human fMRI studies.4
AMPA receptors are mainly heterotetrameric channels assembled from subunits GluA1-GluA4.30 However, GluA1 and GluA2 are the subunits predominantly expressed in the brain. In this study, both GluA2 and phosphorylated GluA2ser880 in cortical neurons are constitutively expressed in high levels in control rats and esophageal acid exposure failed to induce a significant alteration in the expression as well as phosphorylation of GluA2. In contrast to GluA1, GluA2 subunit is Ca2+ impermeable and its phosphorylation at Ser880 has been reported to involve in disengaging AMPA receptors from synaptic location and also facilitates AMPA receptor endocytosis.40,41 Studies also suggest that in the dorsal horn neurons, disruption of GluA2 binding to its synaptic anchoring protein (glutamate receptor interacting protein) can result in a switch of GluA2-containing AMPA receptors to GluA2 lacking ones and further may increase AMPA receptor Ca2+ permeability at the synapses.42 The activity-dependent insertion of GFP (green fluorescence protein)-tagged AMPA receptor subunits at synapses has also shown that the GluA2 subunit is constitutively delivered to synapses, whereas the GluA1 subunit requires high-frequency stimulation or the presence of CaMKII for significant synaptic delivery.43,44 Consistent with these findings, we also observe a significant inhibition of cortical sensitization by selective Ca2+ permeable AMPA receptor antagonist IEM-1460 indicating a possible involvement of GluA1 over GluA2 in esophageal sensitization.
In the isolated PSDs, we have demonstrated a significant increase in pGluA1Ser831 following esophageal acid exposure and immunostaining of cortical tissues also demonstrates a significantly higher expression of both pGluA1Ser831 and PSD-95 indicating postsynaptic trafficking of AMPA receptor subunits. Interestingly, C-terminal region of the GluA1 has been reported to interact with PDZ domain of another synapse-associated protein SAP97, but not PSD-95.45 However, we could not investigate the interaction of GluA1 with its synaptic anchoring protein because the anti-SAP7 antibody used in our study failed to show specific immunoreactivity with the cortical membrane preparations. The logical explanation of synaptic coexpression of pGluA1Ser831 and PSD-95 could be due to the presence of another PDZ domain-containing protein stargazine at the synapses that interacts with both AMPA receptor subunits and PSD-95.46 The interaction between stargazine and AMPA receptor subunits appears to be important for the surface delivery of the receptors, whereas interaction between stargazine and PSD-95 is important for the synaptic targeting of AMPA receptors.
Involvement of CamKII in cortical sensitization
A critical role for CaMKII phosphorylation and its interaction with NMDA receptor subunits at the PSDs has recently been established in viscerally hypersensitive rats.47 Studies have also shown that the translocation of CaMKII at the PSDs and its phosphorylation via Ca2+ influx through NMDA receptor stabilizes the enzyme in its active state, which in turn phosphorylates nearby AMPA receptor subunits at the synapses.35 We have recently demonstrated an increase in phosphorylated NR2B Ser1303 at the cortical PSDs following esophageal acid exposure in rats, and further established that this phosphorylation was CaMKII-mediated.11 In a recent study, inhibition of CaMKII activation in cultured hippocampal neurons prevented NMDA receptor-dependent delivery of GluA1 to the cell surface indicating that the CaMKII activation is important for the activity-dependent recruitment of GluA1-containing AMPA receptors.31 Moreover, intrathecal application of a CaMKII inhibitor KN-93 in spinal neurons before the painful visceral stimulus apparently inhibits GluA1 insertion into the plasma membrane fraction.33 Similar to these reports, we have demonstrated that acid-induced cortical sensitization as well as phosphorylation of GluA1Ser831 could be inhibited by intracortical application of KN-93 suggesting that CaMKII-mediated signaling mechanism is certainly involved in esophageal hypersensitivity. Overall, our findings suggest that there are two distinct mechanisms involved in enhancement of cortical AMPA receptor function during esophageal hypersensitivity. The first mechanism is the CaMKII-mediated GluA1 Ser831 phosphorylation resulting in increase in channel conductance and neuronal sensitization, and second is the CaMKII-dependent synaptic delivery of GluA1-containing AMPA receptors and long-term synaptic plasticity in cortical neurons.
In conclusion, this study demonstrates an increase in AMPA receptor subunit GluA1 expression and its phosphorylation in neurons of the cingulate cortex after acute esophageal acid exposure. This study along with our previous findings strongly suggest the involvement of CaMKII-mediated AMPA and NMDA receptor subunits activation and phosphorylation as the underlying mechanisms of esophageal acid–induced cortical sensitization. Further study of the synaptic AMPA and NMDA receptor interactions in long-term cortical sensitization may provide a better understanding of the esophageal pain mechanism of patients with GERD.
Supplementary Material
Relative expression of pGluA1Ser831 against GluA1 membrane preparations from ACC and MCC following acute esophageal exposure of either acid or saline in rats.
GluA1 and pGluA1Ser831 expression in membrane preparations from insular cortex following acute esophageal exposure of either acid or saline in rats.
GluA2 and pGluA2Ser880 expression in cortical membrane preparations from rats following acute esophageal exposure of either acid or saline.
Key Messages.
The findings of this study provide a better understanding of the esophageal pain mechanisms for patients with GERD.
The main objectives were to examine the effect of esophageal acid in Sprague Dowley rats on AMPA receptor subunits expression and phosphorylation in neurons from anterior and mid-cingulate cortices.
The underlying mechanism of cortical sensitization was studied by pharmacological interventions during electrophysiological recordings from cortical neurons following esophageal acid exposures.
Esophageal acid resulted in upregulation and CaMKII-mediated phosphorylation of AMPA receptor subunit GluA1 in cortical neurons.
Acknowledgments
FUNDING
This study was supported in part by NIH grants R01DK025731 and R56DK089493-01.
Footnotes
AUTHOR CONTRIBUTION
BB involved in manuscript writing, study design, data acquisition, and analysis; BKM performed surgical procedures, undertaking electrophysiology experiments, and data acquisition; SP provided assistance in molecular experiments and data acquisition; PK contributed to data analysis, graphing, critical reading of the manuscript; IML involved in tissue collection; JNS involved in supervision of electrophysiology experiments, interpretation of functional data, and critical reading of the manuscript; RS provided scientific concept, critical revision of the manuscript, and support of the work.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
DISCLOSURE
All authors agreed to participate in this study without any potential conflict of interest.
References
- 1.Fass R, Tougas G. Functional heartburn: the stimulus, the pain, and the brain. Gut. 2002;51:885–92. doi: 10.1136/gut.51.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer EA, Collins SM. Evolving pathophysiologic models of functional gastrointestinal disorders. Gastroenterology. 2002;122:2032–48. doi: 10.1053/gast.2002.33584. [DOI] [PubMed] [Google Scholar]
- 3.Mayer EA, Naliboff BD, Craig AD. Neuroimaging of the brain-gut axis: from basic understanding to treatment of functional GI disorders. Gastroenterology. 2006;131:1925–42. doi: 10.1053/j.gastro.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Lawal A, Kern M, Sanjeevi A, et al. Neurocognitive processing of esophageal central sensitization in the insula and cingulate gyrus. Am J Physiol Gastrointest Liver Physiol. 2008;294:G787–94. doi: 10.1152/ajpgi.00421.2007. [DOI] [PubMed] [Google Scholar]
- 5.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–84. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Hobson AR, Aziz Q. Modulation of visceral nociceptive pathways. Curr Opin Pharmacol. 2007;7:593–7. doi: 10.1016/j.coph.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Sikes RW, Vogt LJ, Vogt BA. Distribution and properties of visceral nociceptive neurons in rabbit cingulate cortex. Pain. 2008;135:160–74. doi: 10.1016/j.pain.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu LJ, Steenland HW, Kim SS, et al. Enhancement of presynaptic glutamate release and persistent inflammatory pain by increasing neuronal cAMP in the anterior cingulate cortex. Mol Pain. 2008;4:40. doi: 10.1186/1744-8069-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu X, Gao J, Yan J, Fan J, Owyang C, Li Y. Role for NMDA receptors in visceral nociceptive transmission in the anterior cingulate cortex of viscerally hypersensitive rats. Am J Physiol Gastrointest Liver Physiol. 2008;294:G918–27. doi: 10.1152/ajpgi.00452.2007. [DOI] [PubMed] [Google Scholar]
- 10.Cao Z, Wu X, Chen S, et al. Anterior cingulate cortex modulates visceral pain as measured by visceromotor responses in viscerally hypersensitive rats. Gastroenterology. 2008;134:535–43. doi: 10.1053/j.gastro.2007.11.057. [DOI] [PubMed] [Google Scholar]
- 11.Banerjee B, Medda BK, Schmidt J, Lang IM, Sengupta JN, Shaker R. Neuronal plasticity in the cingulate cortex of rats following esophageal acid exposure in early life. Gastroenterology. 2011;141:544–52. doi: 10.1053/j.gastro.2011.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartmann B, Ahmadi S, Heppenstall PA, et al. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–50. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 13.Jones TL, Sorkin LS. Calcium-permeable alpha-amino-3-hydroxy-5-methyl -4-isoxazolepropionic acid/kainate receptors mediate development, but not maintenance, of secondary allodynia evoked by first-degree burn in the rat. J Pharmacol Exp Ther. 2004;310:223–9. doi: 10.1124/jpet.103.064741. [DOI] [PubMed] [Google Scholar]
- 14.Pogatzki EM, Niemeier JS, Sorkin LS, Brennan TJ. Spinal glutamate receptor antagonists differentiate primary and secondary mechanical hyperalgesia caused by incision. Pain. 2003;105:97–107. doi: 10.1016/s0304-3959(03)00169-6. [DOI] [PubMed] [Google Scholar]
- 15.Sorkin LS, Yaksh TL, Doom CM. Pain models display differential sensitivity to Ca2 + -permeable non-NMDA glutamate receptor antagonists. Anesthesiology. 2001;95:965–73. doi: 10.1097/00000542-200110000-00028. [DOI] [PubMed] [Google Scholar]
- 16.Park JS, Yaster M, Guan X, et al. Role of spinal cord alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in complete Freund’s adjuvant-induced inflammatory pain. Mol Pain. 2008;4:67. doi: 10.1186/1744-8069-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu H, Wu LJ, Wang H, et al. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci. 2008;28:7445–53. doi: 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bie B, Brown DL, Naguib M. Increased synaptic GluR1 subunits in the anterior cingulate cortex of rats with peripheral inflammation. Eur J Pharmacol. 2011;653:26–31. doi: 10.1016/j.ejphar.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Wu J, Wu Z, Lin Q, Yue Y, Fang L. Regulation of AMPA receptors in spinal nociception. Mol Pain. 2010;6:5. doi: 10.1186/1744-8069-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee B, Medda BK, Schmidt J, et al. Altered expression of P2X3 in vagal and spinal afferents following esophagitis in rats. Histochem Cell Biol. 2009;132:585–97. doi: 10.1007/s00418-009-0639-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villasana LE, Klann E, Tejada-Simon MV. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods. 2006;158:30–6. doi: 10.1016/j.jneumeth.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopach O, Kao SC, Petralia RS, Belan P, Tao YX, Voitenko N. Inflammation alters trafficking of extrasynaptic AMPA receptors in tonically firing lamina II neurons of the rat spinal dorsal horn. Pain. 2011;152:912–23. doi: 10.1016/j.pain.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniels BA, Wood L, Tremblay F, Baldridge WH. Functional evidence for D-serine inhibition of non-N-methyl-D-aspartate ionotropic glutamate receptors in retinal neurons. Eur J Neurosci. 2012;35:56–65. doi: 10.1111/j.1460-9568.2011.07925.x. [DOI] [PubMed] [Google Scholar]
- 24.Fortin DA, Srivastava T, Dwarakanath D, et al. Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors. J Neurosci. 2012;32:8127–37. doi: 10.1523/JNEUROSCI.6034-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang L, Wu J, Zhang X, Lin Q, Willis WD. Increased phosphorylation of the GluR1 subunit of spinal cord alpha-amino-3-hydroxy-5-methyl-4-is oxazole propionate receptor in rats following intradermal injection of capsaicin. Neuroscience. 2003;122:237–45. doi: 10.1016/s0306-4522(03)00526-8. [DOI] [PubMed] [Google Scholar]
- 26.Fang L, Wu J, Lin Q, Willis WD. Protein kinases regulate the phosphorylation of the GluR1 subunit of AMPA receptors of spinal cord in rats following noxious stimulation. Brain Res Mol Brain Res. 2003;118:160–5. doi: 10.1016/j.molbrainres.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 28.Collingridge GL, Bliss TV. Memories of NMDA receptors and LTP. Trends Neurosci. 1995;18:54–6. [PubMed] [Google Scholar]
- 29.Wisden W, Seeburg PH. Mammalian ionotropic glutamate receptors. Curr Opin Neurobiol. 1993;3:291–8. doi: 10.1016/0959-4388(93)90120-n. [DOI] [PubMed] [Google Scholar]
- 30.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 31.Appleby VJ, Correa SA, Duckworth JK, et al. LTP in hippocampal neurons is associated with a CaMKII-mediated increase in GluA1 surface expression. J Neurochem. 2011;116:530–43. doi: 10.1111/j.1471-4159.2010.07133.x. [DOI] [PubMed] [Google Scholar]
- 32.Toyoda H, Zhao MG, Ulzhofer B, et al. Roles of the AMPA receptor subunit GluA1 but not GluA2 in synaptic potentiation and activation of ERK in the anterior cingulate cortex. Mol Pain. 2009;5:46. doi: 10.1186/1744-8069-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–23. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for post-synaptic plasticity. Annu Rev Neurosci. 2006;29:325–62. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–25. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Lu Y, Sun YN, Wu X, et al. Role of alpha-amino-3-hydroxy-5-methyl-4-is oxazolepropionate (AMPA) receptor subunit GluR1 in spinal dorsal horn in inflammatory nociception and neuropathic nociception in rat. Brain Res. 2008;1200:19–26. doi: 10.1016/j.brainres.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Mu X, Wu J, et al. Differential roles of phosphorylated AMPA receptor GluR1 subunits at Serine-831 and Serine-845 sites in spinal cord dorsal horn in a rat model of post-operative pain. Neurochem Res. 2011;36:170–6. doi: 10.1007/s11064-010-0288-y. [DOI] [PubMed] [Google Scholar]
- 40.Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–67. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19:2765–74. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park JS, Voitenko N, Petralia RS, et al. Persistent inflammation induces GluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. J Neurosci. 2009;29:3206–19. doi: 10.1523/JNEUROSCI.4514-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–7. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 44.Shi SH, Hayashi Y, Petralia RS, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–6. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 45.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–24. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 46.Chen L, Chetkovich DM, Petralia RS, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–43. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Zhang X, Liu H, et al. Phosphorylated CaMKII post-synaptic binding to NR2B subunits in the anterior cingulate cortex mediates visceral pain in visceral hypersensitive rats. J Neurochem. 2012;121:662–71. doi: 10.1111/j.1471-4159.2012.07717.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative expression of pGluA1Ser831 against GluA1 membrane preparations from ACC and MCC following acute esophageal exposure of either acid or saline in rats.
GluA1 and pGluA1Ser831 expression in membrane preparations from insular cortex following acute esophageal exposure of either acid or saline in rats.
GluA2 and pGluA2Ser880 expression in cortical membrane preparations from rats following acute esophageal exposure of either acid or saline.