

SUMMARY
Neurodegenerative diseases represent an increasing burden in our aging society, yet the underlying metabolic factors influencing onset and progression remain poorly defined. The relationship between impaired IGF-1/insulin-like signaling (IIS) and lifespan extension represents an opportunity to investigate the interface of metabolism with age-associated neurodegeneration. Using datasets of established daf-2/IIS-signaling components in Caenorhabditis elegans, we conducted systematic RNAi screens in worms to select for daf-2-assoicated genetic modifiers of α-synuclein misfolding and dopaminergic neurodegeneration, two clinical hallmarks of Parkinson’s disease. An outcome of this strategy was the identification of gpi-1/GPI, an enzyme in glucose metabolism, as a daf-2-regulated modifier that acts independent of the downstream cytoprotective transcription factor, DAF-16/FOXO, to modulate neuroprotection. Subsequent mechanistic analyses using Drosophila and mouse primary neuron cultures further validated the conserved nature of GPI neuroprotection from α-synuclein proteotoxicity. Collectively, these results support glucose metabolism as a conserved functional node at the intersection of proteostasis and neurodegeneration.
INTRODUCTION
Parkinson’s disease (PD) is an age-dependent neurodegenerative disease characterized by the accumulation of α-synuclein (α-syn) within Lewy bodies and the selective loss of dopamine (DA) neurons in the substantia nigra. While human genetic and genomic studies have illuminated various contributors to disease pathology, aging remains the single most definitive risk factor for the development of PD (Amaducci and Tesco, 1994). Therefore, an unresolved issue is the underlying molecular relationship between genetic factors influencing aging-associated metabolic changes and the loss of DA neurons associated with PD.
The IGF-1/insulin-like signaling (IIS) pathway has been implicated in human aging (Suh et al., 2008) and neurodegeneration (Cohen and Dillin, 2008). This pathway has been studied extensively in the nematode, Caenorhabditis elegans (C. elegans), where mutations in DAF-2, the sole worm insulin/IGF-1 receptor, doubles lifespan and protects against a wide variety of cellular stressors. This effect is mediated through activation of DAF-16, the only FOXO/forkhead transcription factor (FOXO) in worms, which regulates the expression of numerous cytoprotective genes. In flies, reduced IIS via mutations in chico/insulin receptor substrate (IRS) extend lifespan and provide stress resistance (Clancy et al., 2001), phenotypes also are mediated by FOXO/FKHR (Yamamoto and Tatar, 2011). Similarly, mutations in Drosophila insulin-like receptor (InR), the homolog of DAF-2, also extend lifespan in adult flies (Tatar et al., 2001). While the IIS pathway is comparably simplified in worms and flies, it is important to note that this signaling cascade is nearly identical to that in mammals (Taguchi and White, 2008). This provides the exciting opportunity to utilize age-modified animals to enrich for gene candidates involved in neurodegenerative disease processes.
Among proteotoxicity models in worms, reduced IIS has demonstrated protection against amyloid-β (Aβ), polyglutamine-repeat (polyQ) proteins, and SOD-1 (Morley et al., 2002; Cohen et al., 2006; Boccitto et al., 2012). Similarly, expression of Insulin Degrading Enzyme in Drosophila suppresses Aβ neurotoxicity (Tsuda et al., 2010). We previously established worm models in which two pathological hallmarks of PD, α-syn-induced DA neurodegeneration (Cao et al., 2005), and α-syn misfolding (Hamamichi et al., 2008) can be visualized and assayed. Similarly, we have established a Drosophila model for DA neurodegeneration, which takes advantage of its greater neuronal complexity to develop neurochemical and behavioral markers of PD-related degeneration (Chaudhuri et al., 2007). Here, we combine these established assay systems with the wealth of existing data on aging in these distinct models, as a synergistic platform to uncover a relationship between metabolic change and neurodegeneration.
In this study, we report that reduced IIS suppresses α-syn toxicity and DA neurodegeneration in Drosophila and C. elegans. We performed a large-scale RNAi screen in C. elegans to reveal 60 distinct genetic modifiers associated with this protection. Subsequent bioinformatic and functional analyses of these candidates identified gpi-1/GPI, a key enzyme in glucose metabolism, as a potent modifier of α-syn misfolding and DA neurodegeneration in C. elegans. We also found that mutation of Pgi/GPI in Drosophila elevated a neuroinflammatory signal, disrupted DA homeostasis, and induced DA neurodegeneration. Lastly, this effect was translated to mammals, as gpi1/GPI knockdown resulted in α-syn accumulation and neurotoxicity in mouse primary cortical neurons. These data from worms, flies, and mice collectively advance our understanding of proteotoxicity, with glucose metabolism as a conserved unifying feature.
RESULTS
Reduced IIS suppresses α-syn-induced DA neurodegeneration in Drosophila
Although the IIS pathway is highly conserved in worms, flies, and mammals, a direct link between the IIS pathway and proteotoxicity or neurodegeneration has not previously been demonstrated in Drosophila. Chico/IRS mutations extend Drosophila lifespan and provide stress resistance (Clancy et al., 2001). To determine whether reduced IIS modulates α-syn toxicity in flies, we evaluated DA neuron in CNS of α-syn-expressing adults heterozyogous for a mutant chico allele. Quantification of these neuron clusters at day 1 (24–48 hrs post-eclosion) revealed no significant variations in the number of DA neurons for any genotype (Figure S1A). At day 20, chico mutants maintain wildtype (WT) levels of DA neurons in all clusters (Figure 1A–1C). In contrast, flies expressing human α-syn showed significant declines in neuron numbers in all DA neuron clusters. However, the presence of the chico mutation suppressed α-syn-induced DA neuron loss (Figure 1A–1E).
Figure 1. chico mutation protects against age-dependent α-syn toxicity in DA neurons.
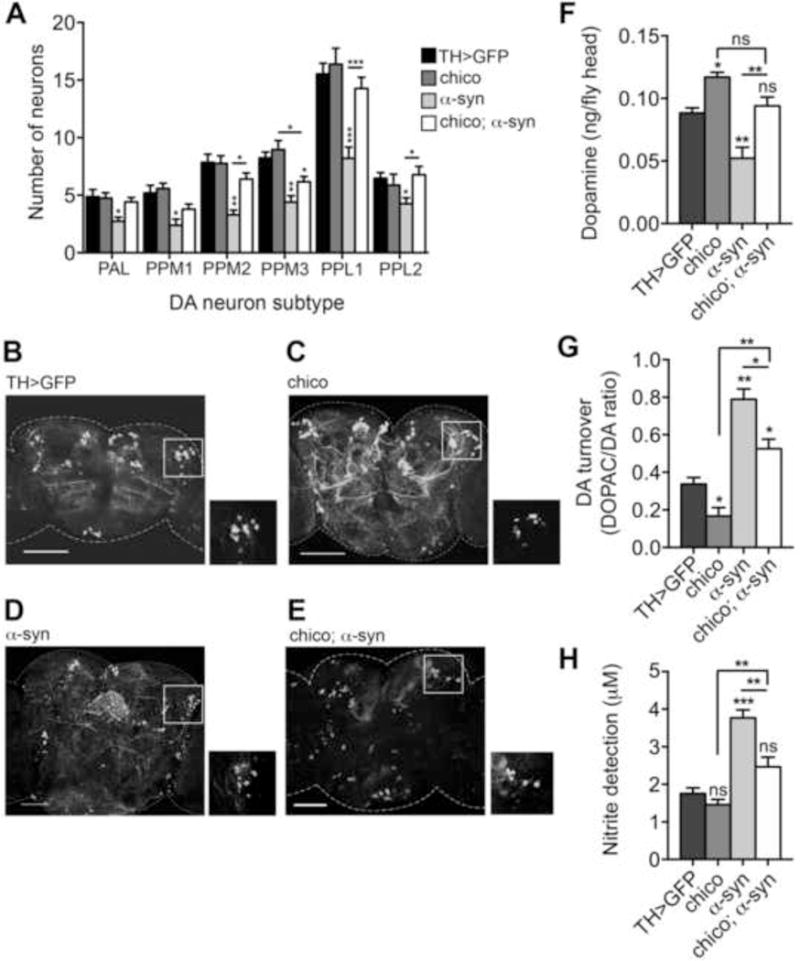
DA neurons in the brains of 20 day-old adult males were visualized by expressing UAS-GFP tyrosine hydroxylase (TH)-Gal4. A) DA neurons were counted in each of the following DA subtype regions: anterior PAL (protocerebral anterolateral), posterior PPM1, PPM2 and PPM3 (protocerebral posterior medial), posterior PPL1 and PPL2 (protocerebral posterolateral). Values are averages of 15 brains per genotype. B–E) Representative images of brains used for neuron counts with insets of the PPL1 region. F–G) DA and DOPAC pools in 20 day-old male heads. Values are averages of assays from three independent head extractions and three technical replicas for each extract, per genotype. Pools are quantified as ng per fly head. H) Nitric oxide synthase activity in brains of 20 day-old males was measured using a modified Griess Reagent assay. Results are displayed as μM concentration of nitrites in incubation medium. Values are averages of three replications of 20 fly brains per replication, and three technical replicas per genotype. Genotypes tested were: TH>GFP (UAS-GFP/+; TH-GAL4/+), chico (UAS-GFP/chico; TH-GAL4/+), α-syn (UAS-GFP/+; TH-GAL4/UAS-α-syn), chico; α-syn (UAS-GFP/chico; TH-GAL4/UAS-α-syn). All values compared to TH>GFP controls unless otherwise indicated. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with a Dunnett’s post-test. Error bars indicate ± SEM.
IIS mediates DA homeostasis and inflammatory signaling in Drosophila
To investigate the effect of chico on DA homeostasis we quantified cellular DA levels and turnover. At day 1, α-syn flies showed no significant change in DA pools relative to the WT control (Figure S1B). Interestingly, chico mutant flies and chico mutant-α-syn flies exhibited elevated levels of DA when compared to either WT or α-syn flies alone (Figure S1B). By day 20, all genotypes except α-syn have experienced a slight, age-dependent reduction in DA (compare Figure 1F and Figure S1B), while the α-syn flies displayed strongly reduced DA, relative to controls. chico mutation rescued the effect of α-syn on DA (Figure 1F).
We then quantified the ratio of DOPAC:DA, which we have shown to be elevated in Drosophila DA neurons prior to degeneration (Chaudhuri et al., 2007). Analysis of 1 day-old flies revealed no significant change in DA turnover in chico or chico;α-syn versus the control, but elevated DA turnover was evident in α-syn flies (Figure S1C). At 20 days post-eclosion, control flies had a slight increase in DA turnover associated with normal aging, while chico mutants remain virtually unchanged (Figure 1G). Thus, flies expressing α-syn exhibit elevated DA turnover, which is partially rescued in the chico; α-syn strain (Figure 1G).
We tested whether there was an accompanying inflammatory response associated with these phenotypes. Using a modified Griess Reagent assay (Ajjuri and O’Donnell, 2013), we determined the levels of secreted nitrites, a stable metabolite of NO, as a measure of relative inflammatory response. At day 1, α-syn flies displayed an increase in NO levels, while chico and chico;α-syn showed little variation from controls (Figure S1D). A strong increase in NO levels was observed in α-syn flies after 20 days of aging, which was significantly reduced in chico;α-syn transgenic flies (Figure 1H). Taken together, these findings suggest the Drosophila IIS pathway can influence α-syn-induced DA neurotoxicity.
IIS pathway modulates α-syn-induced DA neurodegeneration in C. elegans
Next, we investigated whether the link between the IIS pathway and α-syn toxicity represents an evolutionarily conserved mechanism that may be applicable toward understanding PD. We performed genetic crosses to generate daf-2 mutant worms that overexpress human α-syn and GFP in DA neurons. At day 7, we found that only 15% of α-syn worms displayed the WT complement of 6 anterior DA neurons (Figure 2A and 2D) compared to worms expressing GFP alone, which have 100% WT DA neurons (Figure 2C; Hamamichi et al., 2008). Strikingly, 40% of daf-2;α-syn worms exhibited complete retention of DA neurons (Figure 2A, 2E). A well-characterized downstream component of the IIS pathway is DAF-16/FOXO, which functions as a key regulator of numerous cytoprotective genes (Murphy et al., 2003). We examined daf-16 mutants that overexpress α-syn + GFP in DA neurons. As predicted, daf-16 enhanced neurodegeneration (Figure 2A). Surprisingly, we observed an intermediate level of neuroprotection in daf-2; daf-16 double mutants overexpressing α-syn + GFP in DA neurons (Figure 2A). We verified that the genetic markers (unc-75 and dpy-1) used in these mutants did not affect the DA neurodegeneration phenotype by examining the balancers alone (Figure 2A) and by crossing α-syn into the same daf-2 and daf-16 alleles balanced with different genetic markers (Figure S2A). Our findings that daf-2 mutation dramatically suppressed neurodegeneration, daf-16 mutants enhanced neurodegeneration, and daf-2;daf-16 double mutants suppressed neurodegeneration (although not to the same extent as daf-2 mutants), indicate that DAF-16 is not the only genetic component responsible for DA neuroprotection. Additional pathways downstream of DAF-2 must contribute to neuroprotection.
Figure 2. IIS pathway modulates α-syn aggregation and α-syn-induced DA neurodegeneration in C. elegans.
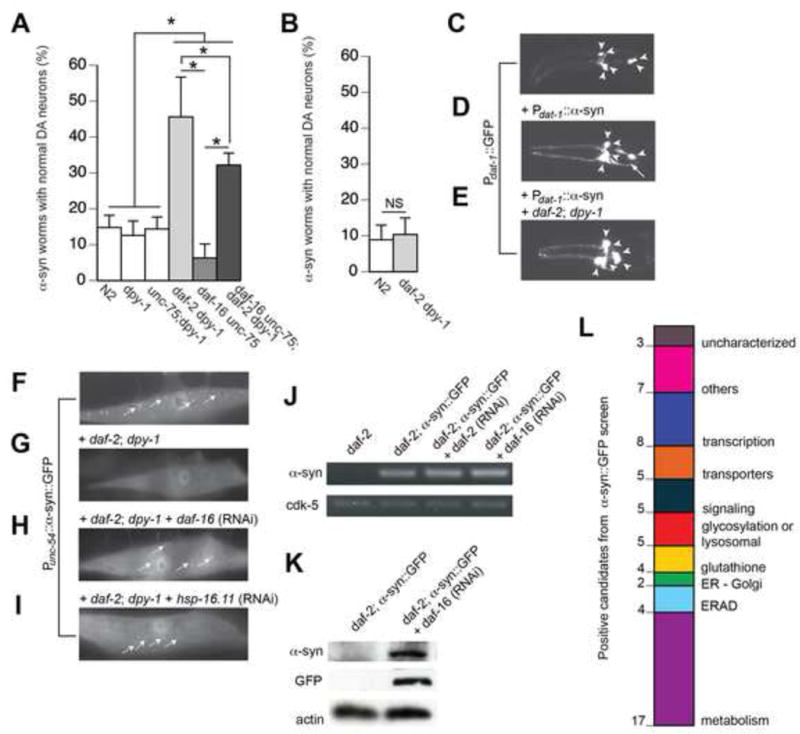
A–E) IIS pathway modulates α-syn-induced DA neurodegeneration. A) IIS pathway modulates α-syn-induced DA neurodegeneration at chronological aging (day 7) in worms. daf-2 mutation dramatically suppressed α-syn-induced DA neurodegeneration while the daf-16 mutation enhanced α-syn-induced DA neurodegeneration. Notably, the daf-2; daf-16 double mutation moderately suppressed α-syn-induced DA neurodegeneration at chronological aging. White bars indicate controls of the genetic backgrounds of the normal (N2), daf-2, and daf-16 mutations. B) The daf-2 mutation did not suppress α-syn-induced DA neurodegeneration at biological aging (mean lifespan: day 20 for WT, day 40 for daf-2 mutants). Three independent trials were performed (n=90 total), and positives were considered significant if p<0.05, Student’s t-test. C–E) Representative images of DA neurons in Pdat-1::GFP (C), Pdat-1::GFP + Pdat-1::α-syn (D), and Pdat-1::GFP + Pdat-1::α-syn + daf-2; dpy-1 mutant (E). Arrowheads denote normal neurons; arrows denote degenerated neurons. F–L) IIS pathway modulates α-syn aggregation. F) In the WT background, α-syn::GFP accumulates in the cytoplasm of muscle cells. G) In the daf-2 mutant background, the α-syn::GFP fusion protein is completely degraded. This daf-2 mutant + α-syn::GFP strain was used in an RNAi screen. H–I) RNAi knockdown of daf-16 and hsp-16.11 resulted in a return of α-syn::GFP aggregates in the daf-2 background. J) Semi-quantitative RT-PCR demonstrates that daf-2 mutation and RNAi do not affect mRNA level of α-syn::GFP. K) Western blot confirms the degradation of the fusion protein in the daf-2 mutant background. L) Summary of the 60 positive candidates that, when knocked-down by RNAi in a daf-2 mutant + α-syn::GFP strain, reproducibly enhanced α-syn::GFP accumulation. Positive candidates were categorized using KOG and/or GO annotations.
DA neurodegeneration at biological vs. chronological aging in worms
Associated with daf-2 mutant lifespan extension is a delayed rate of aging. We questioned whether daf-2-mediated neuroprotection at day 7 (chronological aging) might result from slower development than WT (N2) control animals. We performed lifespan assays to determine biological aging (mean lifespan) and found that it was 20 days for WT; α-syn worms while it was 40 days for daf-2; α-syn worms (data not shown). Interestingly, we found that daf-2 was no longer neuroprotective at biological aging (Figures 2B; S2B). These findings, together with our results demonstrating IIS-mediated neuroprotection at chronological aging in worms (Figures 2A; S2A) and flies (Figure 1), suggest that the metabolic changes associated with reduced IIS are responsible for protection against α-syn proteotoxicity and DA neurodegeneration.
RNAi screen for IIS-mediated protection against α-syn misfolding
We next sought to identify the genetic factors responsible for this IIS pathway-mediated protection against α-syn by generating a daf-2 strain that overexpresses α-syn::GFP in muscle cells. Misfolded α-syn::GFP fusion protein accumulated in the cytoplasm of N2 worms (Figure 2F), while in daf-2 mutants the fusion protein was almost undetectable (Figure 2G). We reasoned that daf-2 mutation likely promoted degradation of the fusion protein rather than affecting the α-syn::GFP expression level (Figure 2J–2K). We then knocked down daf-16 and hsp-16.11 (positive controls) via RNAi in daf-2; α-syn::GFP worms and discovered that GFP inclusions reproducibly returned in these animals (Figure 2H–2I).
To conduct the RNAi screen, we compiled a list of candidate genes and/or proteins with clear human orthologs that are up-regulated in the daf-2 background (Murphy et al., 2003; McElwee et al., 2004; Halaschek-Wiener et al., 2005; Dong et al., 2007) and genes that mediate daf-2 lifespan-extension (Samuelson et al., 2007). We included genes up-regulated upon pan-neuronal overexpression of α-syn (Vartiainen et al., 2006) and previously identified genetic modifiers of α-syn misfolding and toxicity in worms (Hamamichi et al., 2008; Kuwahara et al., 2008; van Ham et al., 2008). In total we assayed 625 targets in triplicate and identified 60 genes that, when knocked-down by RNAi, reproducibly enhanced α-syn::GFP accumulation in the daf-2 mutant background (Tables S1–S2; Figure 3A). These positive hits were categorized into functional groups using KOG and GO annotations (Figure 2L, Table S2).
Figure 3. Secondary screening of positive candidates.
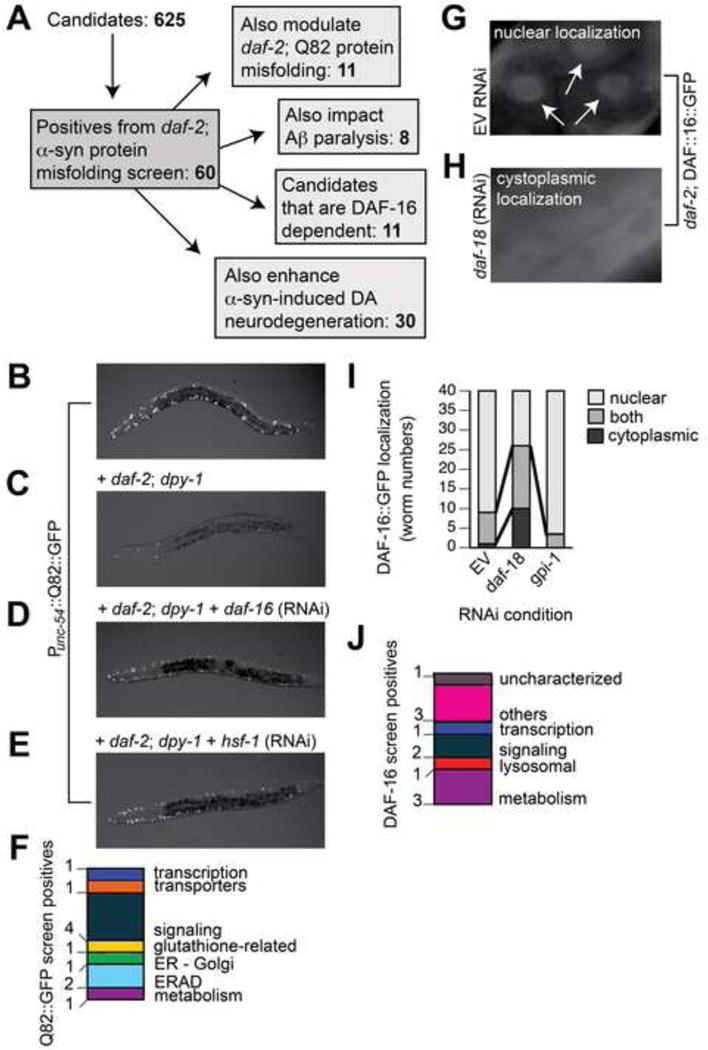
A) Summary of secondary RNAi screens. B) In the WT background, Q82::GFP accumulates in the cytoplasm of muscle cells. C) Q82::GFP aggregation is suppressed in daf-2 mutants, with approximately 9 ± 2 aggregates per worm. D–E) RNAi knockdown of daf-16 or hsf-1 in daf-2 mutant background caused a return of the formation of Q82::GFP aggregates, with averages of 18 ± 1 and 23 ± 6 aggregates per worm, on average. We used the daf-2 + Q82::GFP worms (C) to screen for modifiers of polyglutamine aggregation. F) Summary of the candidates that, when knocked-down in daf-2 mutant background, significantly enhanced polyQ aggregation. Two independent trials (n=40 total) were performed, and positives were determined as significant if *p<0.05, one-way ANOVA with Dunnett post test. G) In daf-2 mutants treated with EV control RNAi, DAF-16::GFP is localized to the nucleus (arrowheads). H) In daf-2 mutants treated with daf-18 RNAi, DAF-16::GFP is distributed evenly throughout the cytoplasm. I) Graph showing quantification of DAF-16::GFP localization in worms. Localization was scored as “nuclear” (light gray), “cytoplasmic” (black), or “both” nuclear and cytoplasmic (dark gray). In daf-2 mutants fed EV RNAi, DAF-16::GFP is predominantly localized to the nucleus. In daf-2 mutants exposed to the RNAi clone targeting daf-18 there is increased cytoplasmic distribution of DAF-16::GFP. We performed RNAi against all 60 positive candidates in daf-2; DAF-16::GFP worms and examined the effect of candidate knockdown on DAF-16 localization vs. the DAF-16::GFP pattern obtained with mock, EV, RNAi knockdown. One candidate from our screen, gpi-1 (RNAi), is displayed in this graph. J) Summary of candidates that, when knocked-down, altered DAF-16::GFP localization. Two independent trials (n=40 total) were performed, and positives were determined as significant if p<0.05, Chi-Square test.
Examination of IIS-mediated α-syn modifiers in additional proteostasis models
We next determined whether the 60 candidates were specific to α-syn misfolding or are more general effectors of proteostasis. A previous study identified genetic modifiers of polyQ aggregation (Nollen et al., 2004) none of those reported were identified in the daf-2 background. We generated daf-2 worms that overexpress Q82::GFP in muscle cells, and consistent with a previous report (Morley et al., 2002), found that reduced IIS suppressed polyQ aggregation (Figure 3B–C). We then performed RNAi of the 60 candidates to identify genes that, when knocked-down in the daf-2 + Q82::GFP background, enhanced polyQ aggregation (Figure 3A–E). We discovered that 11/60 modifiers of α-syn misfolding also affected polyQ aggregation (Figure 3F, Table S2).
We further investigated whether any of the 60 candidates modified amyloid-beta (Aβ) peptide toxicity. To evaluate these targets, an established worm model that utilizes temperature-sensitive induction of Aβ-induced paralysis (Dostal and Link, 2011) was employed. We found that 8/60 targets enhanced Aβ-induced paralysis (Figure S3A–B, Table S2).
In total we found that 18/60 α-syn effectors also modified polyQ or Aβ misfolding; one gene (Y45F10B.9, an uncharacterized zinc-finger protein) impacted misfolding in all three models. Thus, many of these genes appear to be general age-associated modifiers of proteostasis that may affect susceptibility to a variety of protein misfolding diseases.
The majority of α-syn modifiers act independent of DAF-16/FOXO
Since DAF-16 is a key regulator of numerous cytoprotective genes and was found to mediate α-syn misfolding (Figure 2H) and DA neurodegeneration (Figure 2A), we investigated whether the 60 identified effectors were DAF-16-dependent. We performed genetic crosses to generate daf-2; DAF-16::GFP worms to visualize the distribution of DAF-16 within cells. Consistent with previous reports (Lin et al., 2001), we observed predominantly nuclear localization of DAF-16 in untreated and empty vector (EV) control RNAi-fed animals and increased cytoplasmic distribution of DAF-16 in daf-18 RNAi-fed animals, in accordance with daf-18 negative regulation of DAF-16 activity (Figure 3G–3I). We performed RNAi against each of the 60 targets and examined the knockdown effect on DAF-16 localization. Surprisingly, we found that only 11/60 candidates altered the distribution of DAF-16 within worms, including 3/17 metabolism-categorized candidates (Figure 3A, 3J, Table S2). For example, a metabolic candidate, gpi-1, when knocked down, did not have an overt effect on DAF-16::GFP localization in the daf-2 background (Figure 3I and Table S2). Thus, while some candidates may function as DAF-16-dependent effectors of α-syn misfolding, the majority of these modifiers appear to modulate proteotoxicity through distinct mechanisms.
In order to further define mechanistic pathways that may be enriched in our screens, we analyzed our 60 α-syn modifiers using IPA, which relies on the Ingenuity Pathway Knowledge Base (IPKB). Table S3 shows the five most significant functional networks of α-syn modifiers identified by IPA analysis. The leading network included functional categories of energy production and nucleic acid metabolism. We further investigated the most significant functional network by visualizing its interaction with daf-2/IGFR and daf-16/FOXO (Figure S4). We found that 21 proteins from our initial 60 α-syn modifiers interact within this network, revealing an important role for metabolism in age-associated α-syn toxicity. We hypothesize this subset of functional modifiers mediate metabolic changes connected to PD.
Examination of IIS-mediated α-syn modifiers in C. elegans DA neurons
Next we assessed whether our 60 candidate genes modify α-syn-induced DA neurodegeneration independent of the daf-2 mutation. Our reasoning was two-fold: 1) most candidates were not daf-16 dependent, and 2) as described in Figure 2A, daf-2; daf-16 double mutants only decrease α-syn-induced DA neurodegeneration by 13%, compared to a 30% decrease in the WT background, suggesting there are additional pathways impacting neuroprotection in parallel to DAF-16. We utilized a worm strain that enables DA neuron-selective RNAi with transgenic animals expressing α-syn (and GFP) in DA neurons to knockdown the 60 IIS-related candidates (Harrington et al., 2012). We found that 30/60 candidates upon RNAi treatment significantly enhanced DA neurodegeneration (Figure 4A–E, Table S2). Interestingly, 10/30 positives were involved in the energy production and metabolism IPA network (Figure S3), including glucose-6-phosphate isomerase (gpi-1/GPI), a key enzyme in glycolysis. Interestingly, when GPI-1 is secreted by cancer cells it can also serve as a cytokine to activate autocrine motility factor (AMF) signaling (Haga et al., 2000). It was therefore significant that the receptor for AMF (hrdl-1/AMFR) was also identified in this screen. Accordingly, both gpi-1 and hrdl-1 RNAi enhanced α-syn-induced DA neurotoxicity (Figures 2L, 4A–D, Table S2). Conversely, overexpression of gpi-1 and hrdl-1 in DA neurons resulted in significant protection from α-syn-induced neurotoxicity (Figure 4F–I).
Figure 4. IIS-associated modifiers of α-syn misfolding also affect α-syn-induced DA neurodegeneration.
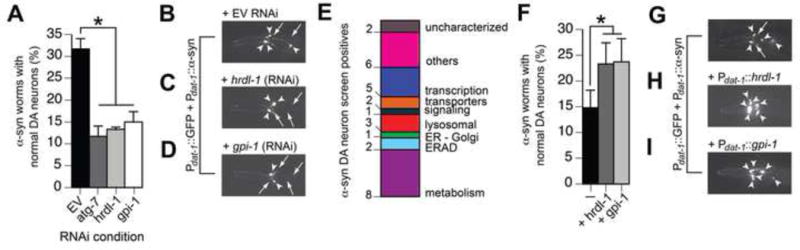
A) Graph showing percentage of 6 day-old worms with normal DA neurons for two RNAi targeting controls (EV and atg-7) and two positive candidates from RNAi screen (gpi-1 and hrdl-1). RNAi targeting each of the 60 candidates was performed in sid-1 mutant worms overexpressing Punc-119::SID-1 + Pdat-1::GFP + Pdat-1::α-syn. B–D) Representative images of Punc-119::SID-1 + Pdat-1::GFP + Pdat-1::α-syn worms treated with EV RNAi (B), hrdl-1 RNAi (C), and gpi-1 RNAi (D). Arrowheads show intact DA neuron cell bodies. Arrows indicate areas where DA neurons have degenerated. E) Summary of candidates that, when knocked-down, enhanced DA neurodegeneration in transgenic worms expressing Punc-119::SID-1 + Pdat-1::GFP + Pdat-1::α-syn. F) Graph showing the percentage of 7 day-old worms with normal DA neurons for Pdat-1::GFP + Pdat-1::α-syn worms overexpressing either hrdl-1 or gpi-1. G–I) Representative images of Pdat-1::GFP + Pdat-1::α-syn worms (G), Pdat-1::GFP + Pdat-1::α-syn + Pdat-1::HRDL-1 worms (H), and Pdat-1::GFP + Pdat-1::α-syn + Pdat-1::GPI-1 (I).*p<0.05, one-way ANOVA with Dunnett post-test.
GPI-1 protects DA neurons in parallel to DAF-16
To investigate the mechanism for GPI-1-mediated neuroprotection, we performed combinatorial RNAi against daf-2 + gpi-1 and daf-2 + hrdl-1 to determine whether daf-2-mediated neuroprotection is dependent on these gene products. Combinatorial RNAi eliminated the neuroprotection phenotype conferred by daf-2 RNAi (Figure 5A). Notably, neuroprotection decreased 79% in daf-2; gpi-1 double RNAi vs. 30% in daf-2; daf-16 double mutants (Figures 5A vs. 2A), suggesting the cellular events involving gpi-1 provide greater protection than the DAF-16 pathway. We also found that combinatorial RNAi against daf-16 + gpi-1 enhances DA neurodegeneration compared to gpi-1 RNAi alone (Figure 5B), providing further evidence that gpi-1 is functioning independently of daf-16. We validated this result with quantitative RT-PCR and found that gpi-1 RNAi did not significantly impact the expression level of two known endogenous DAF-16 transcriptional targets, sod-3 and mtl-1 (Robida-Stubbs et al., 2012). Similar results were observed in the WT genetic background (Figure 5C).
Figure 5. GPI-1 mechanistic analyses.
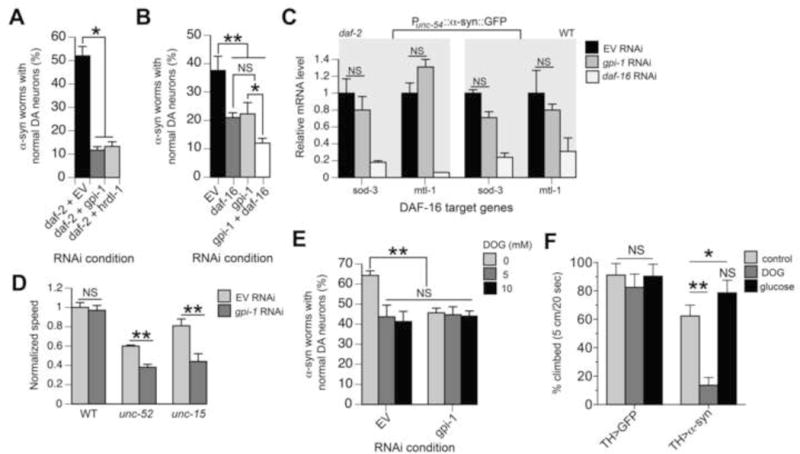
A–B) Graphs depicting percentage of 6-day old worms with normal neurons following combinatorial RNAi of daf-2 + EV control, gpi-1, or hrdl-1 (A) and combinatorial RNAi of daf-16 + EV control or gpi-1 (B) in Punc-119::SID-1 + Pdat-1::GFP + Pdat-1::α-syn C. elegans. *p<0.05; **p<0.01, one-way ANOVA with Dunnett post-test. C) No induction of endogenous DAF-16 target genes in response to gpi-1 RNAi. Quantitative RT-PCR was performed with young adult daf-2 mutant or N2 WT worms expressing Punc-54::α-syn::GFP, treated with EV control RNAi, gpi-1 RNAi, or daf-16 RNAi (positive control). For each daf-16 target gene, its mRNA levels were normalized to the expression in worms treated with EV control RNAi. Values are means ± SD (n=3 independent biological samples with 100 worms in each). P value was calculated by nonparametric one-way ANOVA. NS, not significant, compared with worms treated with EV control RNAi. D) gpi-1 RNAi at permissive temperature (16°C throughout the experiment) significantly reduced the motility of young adult unc-15(e1402) and unc-52(e669su250) worms. Animals were exposed to EV or gpi-1 RNAi from embryos. The movement of young adult worms were recorded and analyzed by WormLab3.0 with the crawling mode. Values are means ± SD (n = 3 independent experiments with 15–20 worms in each). ** p<0.01, unpaired Student’s two tailed t-test with Welch correction. E) Graph showing percentage of 4 day-old worms with normal DA neurons. Punc-119::SID-1 + Pdat-1::GFP + Pdat-1::α-syn worms fed EV control RNAi or gpi-1 RNAi were treated with 0 mM, 5 mM, and 10 mM 2-deoxyglucose (DOG), then analyzed 24 hours after exposure. **p<0.01, one-way ANOVA with Dunnett post-test. F) Mobility of α-syn expressing flies aged to 10 days and then fed 200 mM DOG or 1 M glucose for 5 days. * p<0.05, ** p<0.01, one-way ANOVA with Bonferroni post-test.
GPI-1 influences protein homeostasis through glycolysis
We tested whether GPI-1 could regulate the folding of endogenous metastable proteins by using worm strains carrying temperature-sensitive (ts) mutations unc-15(e1402) and unc-52(e669su250), encoding paramyosin and perlecan, respectively. At permissive temperature these ts metastable proteins fold and function properly, while at elevated temperature the proteins misfold and induce motility defects (Gidalevitz et al., 2006). Figure 5D shows that gpi-1 RNAi at permissive temperature significantly (P<0.01) reduced the motility of young adult unc-15 and unc-52 worms by 46% and 37% respectively, suggesting a role for gpi-1 in maintaining global proteostasis.
To study the underlying mechanism impacting proteostasis, we inquired whether GPI-1 neuroprotection involved glycolysis, since this enzyme is involved in its initial steps. Treatment with 2-deoxyglucose (DOG), a glucose analog that blocks glycolysis, enhanced α-syn-induced DA neurodegeneration in worms (Figure 5E) and α-syn-induced mobility defects in flies (Figure 5F). Importantly, combinatorial treatment of worms with DOG + gpi-1 RNAi did not enhance α-syn-induced DA neurodegeneration compared to gpi-1 RNAi alone (Figure 5E). In addition, excess glucose suppressed α-syn-induced mobility defects in Drosophila (Figure 5F, Movies S1–S3). Lastly, we quantified ATP levels in worms treated with gpi-1 RNAi, and found these were unaffected (Figure S5), implying certain glycolytic metabolites may be responsible neuroprotection. Along with prior studies (Schulz et al., 2007; Tauffenberger et al., 2012), these data provide strong evidence that gpi-1 mediates proteotoxicity via glycolysis.
Pgi/GPI mutation induces DA neurodegeneration in Drosophila
We sought to investigate whether a relationship between glucose metabolism, proteostasis, and neurodegeneration is conserved in other species. To this end, we first examined the link between IIS and the gpi-1 fly ortholog, pgi. While heterozygous chico mutants had no deficiency in climbing behavior, heterozygous pgi mutants displayed a significant reduction in mobility relative to WT flies (Figure 6A). The presence of the mutant allele of chico in double heterozygotes was unable to rescue the climbing deficits of heterozygous pgi mutants. This result suggests that pgi acts downstream of chico or that the two genes function in genetically separate pathways.
Figure 6. Pgi/GPI mutation enhances α-syn-induced DA neuron loss.
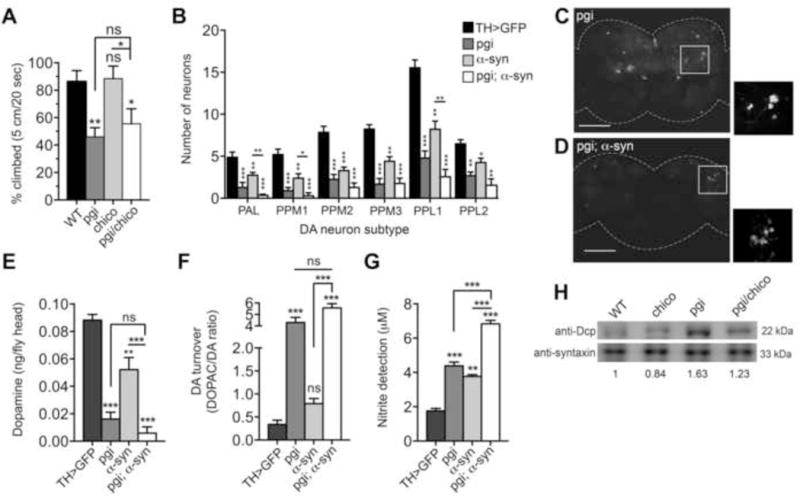
A) Mobility assays of heterozygous chico and pgi mutant alleles, individually and in combination, relative to WT flies. B–D) DA neurons in the brains of 20 day old adult males, visualized by TH-Gal4 driven GFP. B) Neurons were counted in anterior and posterior DA neuron clusters. Values are averages of 15 brains per genotype. C–D) Representative images of brains used for neuron counts with insets of the PPL1 region. E–F) DA and DOPAC pools were measured in heads of 20-day old male flies. Assay values were determined using three independent head extracts and three technical replicas for each genotype and calculated as ng per fly head. G) Nitric oxide synthase activity in brains of 20 day-old males was measured using a modified Griess Reagent assay. Results are displayed as μM concentration of nitrites in incubation medium. Values are averages of 3 replications of 20 fly brains per replication, and three technical replicas per genotype. Genotypes for strains tested are: TH>GFP (UAS-GFP/+; TH-GAL4/+), Pgi (UAS-GFP/Pgi; TH-GAL4/+), α-syn (UAS-GFP/+; TH-GAL4/UAS-α-syn), Pgi; α-syn (UAS-GFP/Pgi; TH-GAL4/UAS-α-syn). These assays were performed simultaneously with those in Figure 1 and employed the same TH>GFP controls. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Dunnett’s post-test analysis. Error bars indicate ± SEM. H) Western blot reveals that levels of the apoptosis factor DCP-1 (death caspase-1) in 20 day-old adult male heads was elevated 1.6 fold in the pgi mutant flies and reduced slightly in chico alone (0.83:1). Expression of chico and pgi together only slightly ameliorated DCP-1 protein expression relative to wild type (1.23: 1).
Next we examined whether pgi modulates α-syn-induced DA neurodegeneration. A heterozygous pgi mutation was introduced into flies expressing α-syn and GFP in DA neurons, under the control of a TH-GAL4 driver. To investigate the effect of pgi on α-syn toxicity, we compared DA neuron numbers for the WT and heterozygous pgi mutant genotypes in GFP-expressing neurons, in the absence and presence of α-syn. No significant changes were observed DA neuron numbers for any genotype at day 1 post-eclosion (Figure S6A). However, we found a significant decline at day 20 in DA neuron numbers in pgi heterozygous mutant brains in the absence of α-syn (Figure 6B–6D). Similarly, α-synx, pgiWT flies exhibited significant degeneration of DA neurons (Figure 6B). The combination of the pgi mutant allele with α-syn significantly reduced neuron numbers further in nearly all DA neuron clusters (Figure 6B–6D).
Pgi/GPI mutation exacerbates the effects of α-syn on DA metabolism and nitric oxide production in Drosophila
Since glucose restriction increases oxidative stress in worms (Schulz et al., 2007), we investigated whether the pgi mutation would result in cytosolic accumulation and oxidation of DA in fly brains. HPLC analysis of DA and DOPAC levels in fly heads revealed that even at day 1 post-eclosion pgi mutant flies possessed significantly lower DA levels (Figure S6B) and dramatically elevated DOPAC:DA ratios (Figure S6C). While α-syn alone did not result in significant diminution of DA pools or elevated DA turnover at day 1, it enhanced the effects of the pgi mutant allele on both DA loss and DOPAC production (Figure S6B–S6C). After aging to 20 days, DA turnover in pgi mutants increased nearly 2.5-fold (Figure 6F). At this age, α-syn flies displayed significantly reduced DA, further loss in combination with the pgi mutation and an enhanced DOPAC:DA ratio (Figure 6E–6F). These findings reveal a strong age-associated effect of pgi on DA regulation.
We assayed NO production in heterozygous pgi mutants alone and in the α-syn background. Both pgi mutants and α-syn-expressing flies individually displayed strong inflammatory responses as measured by NO production and, in combination, nearly a 4-fold increase in NO relative to control flies was observed at day 1 and day 20 post-eclosion (Figures S6D and 6G). Consistent with these data, levels of the apoptosis factor DCP-1, encoded by death caspase-1, were significantly elevated in pgi mutant flies (Figure 6H). While chico alone showed a minor decrease in DCP-1 levels, compared to WT flies, expressing both chico and pgi together only slightly ameliorated DCP-1 protein expression triggered by the mutant pgi allele alone. In summary, α-syn expression and pgi mutation individually cause deleterious effects on DA neurons, and the combination of these two modifications is synergistic in their damage to the adult Drosophila brain.
Knockdown of gpi1/GPI in mouse primary cortical neurons
To determine if the effect of gpi1 knockdown translated to a mammalian system, mouse neuronal cortical cultures were transduced with lentiviral constructs expressing a short hairpin RNA (shRNA) sequence targeted against gpi1. Infection of neurons at a multiplicity of infection (MOI) of 1 resulted in over 90% knock-down of gpi1 protein compared to cells infected with a scrambled shRNA sequence (scrb), when assessed at 7 days post infection (dpi) (Figure 7A). Neurotoxicity was determined by immunofluorescence analysis of neurofilament protein, a sensitive assay that measures the degeneration of neurites. Infection with lenti-gpi1-shRNA at MOI 0.5 resulted in significant neurotoxicity compared to both non-transduced (N.T.) and scrb infected neurons, when assessed at both 7 and 9 dpi (Figure 7B). Increasing the viral titer to an MOI of 1 further enhanced the neurotoxic effect (Figure 7C).
Figure 7. Knockdown of Gpi1/GPI in mouse cortical neurons causes neurotoxicity and α-syn accumulation.
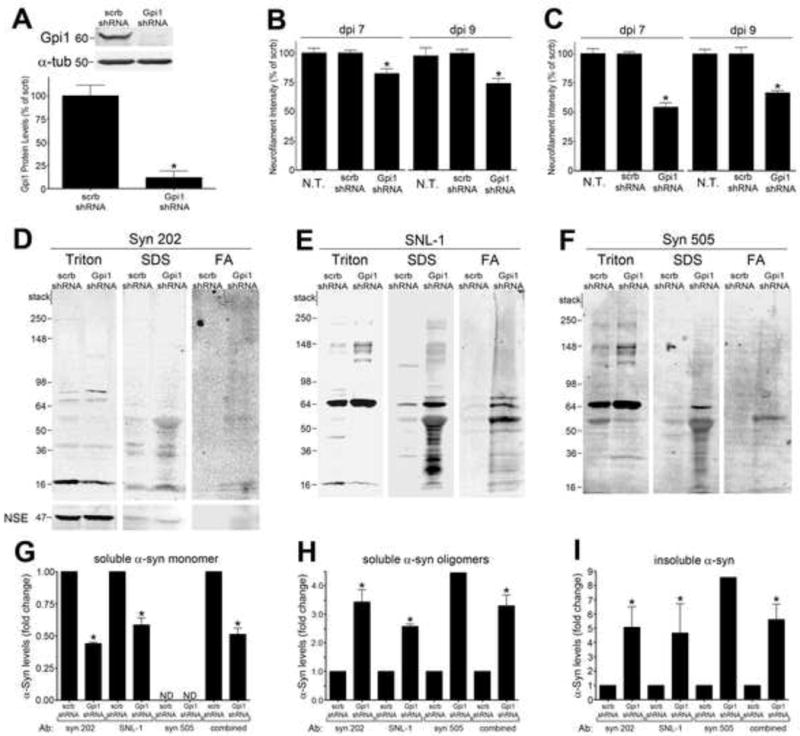
A) Neurons were transduced with lenti-gpi1 shRNA or scrambled shRNA (scrb) at MOI 1, harvested at 7 days post infection (dpi), and gpi1 protein levels were determined by western blot and densitometry. α-tubulin was used as a loading control (n=4). B) Neurotoxicity was assessed by neurofilament measurement at dpi 7 and 9 in neurons infected at MOI 0.5. C) Neurotoxicity assessment at MOI 1 infection. (n=4). D–F) Sequential extraction analysis of dpi 7 neurons infected at MOI 0.5 using anti-α-syn antibodies syn 202, SNL-1, and syn 505. Reactivity with syn 505 reveals the presence of misfolded, oxidized/nitrated α-syn. Neural specific enolase (NSE) was used as a loading control. G–I) Densitometric quantification of various biochemical forms of α-syn (n=2 for syn 202, n=2 for SNL-1, n=1 for syn 505). Values are the mean SEM, *p<0.05).
We subsequently characterized the effect of gpi1 knockdown on α-syn solubility in neuronal cultures by sequential biochemical extraction. Neurons from gpi1 shRNA transduced neurons were sequentially extracted in 1% Triton X-100, 2% SDS, then 70% formic acid (FA), and compared to extracts from scrb shRNA infected neurons. Western blot analysis revealed that gpi1 knockdown significantly reduced the levels of Triton X-100-soluble monomeric α-syn migrating at 18kDa by 50%, while increasing the levels of soluble high molecular weight (HMW) oligomers by 3-fold (Figures 7D–7H). Additionally, gpi1 knockdown appeared to cause a dramatic accumulation of Triton X-100 insoluble α-syn (~5 fold increase) extracted in SDS and FA-containing fractions (Figures 7D–7F, 7I). The solubility changes of α-syn were confirmed with two antibodies that were generated against unmodified α-syn (syn 202 and SNL-1), as well mAb syn 505, which was generated against oxidized/nitrated α-syn (Duda et al., 2002) and recognizes crosslinked species of the protein; thus syn 505 preferentially detects misfolded forms of α-syn (Waxman et al., 2008). These results indicate that gpi1 knockdown results in neurotoxicity and concomitantly causes the accumulation of soluble α-syn oligomers and insoluble species.
DISCUSSION
The wealth of information that has accumulated from decades of elegant research in model systems has led to lists of genetic factors influencing lifespan and healthspan (Kenyon, 2010; Gems and Partridge, 2013). Likewise, human genetic studies have also generated an expanding catalog of heritable and candidate susceptibility factors for PD. We set out to better define the molecular intersection of aging and PD in the context of established genetic modifiers, by taking advantage of pre-existing datasets and the respective strengths of select model systems. Without question, the IIS pathway is central to understanding the organismal control of metabolism. We show here that reduced IIS suppresses α-syn misfolding and neurotoxicity in flies and worms and identify a conserved DAF-16/FOXO-independent mechanism through which the IIS pathway integrates metabolic regulation with proteotoxicity and DA neurodegeneration.
GPI is up-regulated in daf-2 mutants (Dong et al., 2007) and functions independently of DAF-16/FOXO to modulate neurodegeneration (Figure 4). Interestingly, GPI RNAi/mutation extends lifespan in C. elegans (Schulz et al., 2007) and influences lifespan in Drosophila (Lai et al., 2007). While this relationship is seemingly paradoxical, it highlights the distinction between chronological aging and neuronal dysfunction and health. An expression level change in GPI-1 in daf-2 mutant worms does not necessarily contribute to the further lifespan extension and may reflect a compensatory mechanism activated by a reduction in DAF-2 signaling. Indeed, this was implied by our result that gpi-1 + daf-2 combinatorial RNAi abolished the protective effect of daf-2 RNAi (Figure 5A) and was echoed by the climbing assay in flies (Figure 6A). We also determined that GPI RNAi/mutation enhances DA metabolism (Figures 6F and S6C), induces an inflammatory response and apoptosis (Figure 6G–6H and S6D), causes widespread protein aggregation and neurodegeneration in worms, flies, and mammalian primary neurons (Figures 4–7; Table S2), but does not affect ATP production (Figure S5). Furthermore, we found that increased glucose metabolism via overexpression of GPI and glucose surplus suppressed α-syn-induced DA neurodegeneration in worms (Figure 4F) and flies (Figure 5F), respectively. Our data, together with Tauffenberger et al (2012), support a role for gpi-1/GPI in mediating proteotoxicity and neurodegeneration via glucose metabolism.
In addition to the role as a cytoplasmic enzyme, GPI has an alternative role in cancer cells were it acts as a ligand (gpi-1/AMF) in the AMF pathway. Its receptor, HRDL-1/AMFR, was also identified in our screen to modify α-syn misfolding and neurodegeneration (Figure 4). AMF/AMFR attenuate ER stress and apoptosis in cancer cells (Fu et al., 2011). However, HRDL-1 also functions as a homolog of HRD family of E3 ligases that regulates HMG-CoA reductase degradation. The identification of HRDL-1/AMRF activity in neuroprotection suggests a potential relationship between cholesterol biosynthesis and α-syn toxicity in PD. This is underscored by the results of Scherzer et al. (2003), in which changes in the expression of lipid metabolism genes represented a major response to α-syn expression in Drosophila. Albeit (presumably) mechanistically distinct, these data are intriguing given the reported efficacy of statins in PD models (Bar-On et al., 2008). How an alteration of lipid metabolism contributes to α-syn misfolding and neurotoxicity remains to be determined. Nevertheless, glucose metabolism may impact α-syn toxicity through a mechanism involving the elevation of certain lipids that bind to α-syn and facilitate the process of toxic oligomerization.
Importantly, three positive candidates from this study are associated with neurodegenerative disease in patients: C01A2.4/CHMP2B [frontotemporal dementia and degeneration; (Isaacs et al., 2011)]; gpd-2/GAPDH [Parkinson’s, Alzheimer’s, and Huntington’s (Mazzola and Sirover, 2001)]; and C54D10.10/TFPI [Alzheimer’s (Piazza et al., 2012)]. Notably, we did not identify C. elegans orthologs of other known heritable PD genes among these modifiers. While this may seem surprising, in the context of reduced IIS, these findings were not completely unexpected. Indeed, patients with monogenic forms of PD do not develop the disease until later in life, demonstrating a key role for the metabolic changes associated with aging in disease onset. Consistent with this point is the observation that energy metabolism-associated genes are a key class of genes whose expression is modified as α-syn-induced neurodegeneration progresses (Scherzer et al., 2003). Likewise, our screening paradigm involved analysis of different phenotypes (aggregation; degeneration) in distinct cell types (bodywall muscles; dopamine neurons). Thus, contextual differences in expression may account for an absence of select modifiers. These collective results highlight the utility of model systems in deciphering the integrated consequences of imbalances in protein management and metabolic networks on neuronal survival.
EXPERIMENTAL PROCEDURES
Basic C. elegans genetic and biochemical methods
Strain maintenance, genetic crosses, creation of transgenic nematodes and supporting biochemical procedures were carried out using standard methods (see Extended Experimental Procedures for details).
C. elegans RNAi screening
Screening was performed using RNAi feeding clones (Geneservice, Cambridge, UK). Twenty age-synchronized young-adult F1 animals were analyzed per clone. The RNAi clones resulting in significant α-syn aggregation (80% of worms with increased quantity and size of aggregates) were scored as positive, and all positives were tested in triplicate. RNAi feeding procedures, assays, and worm strains are described in the Extended Experimental Procedures.
C. elegans secondary screening of candidate genes from RNAi screen
Nematode models of polyglutamine aggregation in bodywall muscle cells, Aβ paralysis, DAF-16::GFP localization, and α-syn-induced DA neurodegeneration were examined as secondary screening assays. These models were interrogated using RNAi of the 60 gene candidates identified from the screen by exploring by the impact of gene knockdown on F1 offspring (vs. EV RNAi). Polyglutamine aggregation and Aβ paralysis worm models (daf-2; Punc-54::Q82::GFP and Pmyo-3::Aβ1–42) were scored at the L3-stage for total number of aggregates or numbers of worms paralyzed, respectively. DA neurons were also examined for α-syn-induced neurodegeneration in 7-day old animals (sid-1; Punc-119::sid-1; Pdat-1::α-syn; Pdat-1::GFP) that had been exposed to RNAi since embryonic stages. A worm was considered rescued when all six anterior DA neurons were intact had no visible signs of degeneration. Statistical analyses for RNAi experiments with Q82 aggregation, Aβ paralysis, and DA neuron degeneration were performed using the oneway ANOVA with Dunnett post-hoc test (p<0.05) to compare control worms (fed RNAi bacteria not targeting any gene) with experimental worms (fed RNAi bacteria targeting candidate gene). C. elegans scored for DAF-16::GFP localization were L3 staged and had been exposed to RNAi since the embryonic stage (daf-2; Pdaf-16a/b::GFP). For each worm, GFP localization was scored as “completely nuclear”, “both nuclear and cytoplasmic”, or “completely cytoplasmic”. Two independent trials (n=40 total) were performed, and positives were determined as significant if p<0.05, Chi-Square test. Extended Experimental Procedures provide more details for all of these procedures.
C. elegans ts mutant phenotype assays
The nematode mutants, unc-15 and unc-52, were treated with EV or gpi-1 (RNAi) and maintained at permissive temperature. Behavioral analysis was recorded and analyzed using WormLab3.0 (MBF Bioscience) with Kalman smoothing. Three independent trials with 15–20 worms; unpaired Student’s two-tailed t-test with Welch correction (see Extended Experimental Procedures for details).
DOG analysis in C. elegans
DA neurodegeneration assays were performed on Pdat-1::α-syn, Pdat-1::GFP animals that were age-synchronized, exposed to 5 or 10 mM DOG for 24 hours, and analyzed at day 4. Three trials were performed (n = 90 animals/treatment); one-way ANOVA with Dunnett post-hoc test.
Ingenuity Pathway Analysis
The Ingenuity Pathway Analysis (IPA) software (Ingenuity® Systems, www.ingenuity.com) was used for distributing the 60 positive candidate genes from our primary screen into biological networks and for evaluation of functional significance.
Basic Drosophila genetic and biochemical methods
Strain maintenance, genetic crosses, and supporting biochemical procedures were carried out using standard methods (see Extended Experimental Procedures for details).
Drosophila DA neuron quantification
DA neurons from adult males were dissected for analysis either one or twenty days post-eclosion. These neurons were visualized by expression of GFP under the control of the DA neuron driver TH-Gal4 and samples were quantified following confocal microscopy imaging. Additional details are outlined in the Extended Experimental Procedures section.
Drosophila DA neuron HPLC and NO synthase analyses
Monoamines from male heads were separated using HPLC as described in Chaudhuri et al. (2007). A modified Griess reagent assay described in Ajjuri and O’Donnell (2013) was used to quantify nitric oxide synthase activity. Male heads were also examined in this assay. For all experiments, genotypes were analyzed in triplicate and statistics were analyzed using one-way ANOVA followed by Dunnett post-hoc test. See Extended Experimental Procedures for more details.
Drosophila DOG and glucose feeding
Male flies were aged to 10 days post-eclosion and then fed either standard corn syrup food or standard food supplemented with either 200 mM 2-deoxyglucose or glucose with a final concentration of 1M continuously for 5 days. Statistics were analyzed using oneway ANOVA followed by Bonferroni post-test.
Drosophila climbing assay
Flies were anesthetized using cold coma, transferred in groups of 10 to climbing vials and allowed 45 min to recuperate. Vials were gently tapped before each trial, and climbing was scored by calculating the number of flies to climb 6cm in 20 seconds. The results represent 10 trials with 3 repetitions per trial. Statistical analyses were performed using one-way ANOVA with Bonferroni post-test.
Mouse cortical cultures, lentiviral infection, and neurotoxicity analysis
Murine neuronal cortical cells were obtained at embryonic day 17 as previously described (Tsika et al., 2010). Cells were seeded in 96 well plates at 50,000 cells/well, infected at 5 days in vitro (DIV) with lentiviral particles containing shRNA against gpi1 at either MOI 0.5 or 1. The cells were then fixed in 4% paraformaldehyde at the indicated time points. The staining and analysis procedures have been described in detail previously (Tsika et al., 2010). Neurotoxicity analysis was performed with 2 separate culture preparations, with 4 replicates for each preparation. One-way ANOVA with Tukey’s post-hoc test was used to determine statistical significance, and p<0.05 was considered significant. See Extended Experimental Procedures for details.
Sequential biochemical extraction of mouse neuronal cultures
6,000,000 cells/condition were extracted for SDS-PAGE analyses using the methods described in the Extended Experimental Procedures section. The subsequent gels were transferred to polyvinylidene difluoride membranes and probed with anti-α-synuclein antibodies (syn 202, dilution 1:500, Covance; SNL-1, dilution 1:500, gift of Benoit I. Giasson, University of Pennsylvania; or syn 505, dilution 1:500, Invitrogen). Anti-Neural specific enolase and anti-alpha-tubulin were used as loading controls. Primary antibodies were detected with anti-mouse or rabbit IgG conjugated to IRDye 680 or 800. For controls, blots were scanned after the blocking step to determine autofluorescent bands, and also after the addition of secondary Ab alone. Any nonspecific bands detected were not included in densitometric analyses. Quantification of the Triton-soluble band migrating at 18kDa was used for monomer measurements (syn 202 and SNL-1), quantification of aggregated forms of α-synuclein was done with syn 202, SNL-1, and syn 505, and repeated with separated culture preparations. A Student’s t-test was used to determine statistical significance; p<0.05 was considered significant.
Supplementary Material
HIGHLIGHTS.
Insulin signaling modulates neurodegeneration in fly and worm Parkinson’s models
Reduced insulin-signaling screen reveals metabolic modifiers of protein misfolding
A glycolytic enzyme, GPI, is neuroprotective across worms, flies and mouse neurons
GPI functions independently of DAF-16/FOXO to modulate proteostasis via glycolysis
Acknowledgments
We are grateful to all members of the Caldwell and O’Donnell labs for their collegiality and collective teamwork. Special thanks to Laura Berkowitz for her invaluable contributions and Chris Link and Richard Morimoto for the Aβ paralysis and Q82::GFP strains, respectively. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This research was funded by grants from the National Institutes of Health (R15 NS075684-01 to GAC and R15 NS078728 to JMO). Other support came from a Howard Hughes Medical Institute Undergraduate Science Program Grant to The University of Alabama (ARB, SKL, SMD), as well as the Parkinson’s Support Group of Huntsville (LRR) and the Parkinson’s Association of Alabama (ALK, MWZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajjuri RR, O’Donnell JM. Novel whole-tissue quantitative assay of nitric oxide levels in Drosophila neuroinflammatory response. J Vis Exp. 2013;82 doi: 10.3791/50892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaducci L, Tesco G. Aging as a major risk for degenerative diseases of the central nervous system. Curr Opin Neurol. 1994;7:283–286. doi: 10.1097/00019052-199408000-00001. [DOI] [PubMed] [Google Scholar]
- Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, Masliah E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J Neurochem. 2008;105:1656–1667. doi: 10.1111/j.1471-4159.2008.05254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccitto M, Lamitina T, Kalb RG. Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS ONE. 2012;7:e33494. doi: 10.1371/journal.pone.0033494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen K. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Broughton S, Partridge L. Insulin/IGF-like signalling, the central nervous system and aging. Biochem J. 2009;418:1–12. doi: 10.1042/BJ20082102. [DOI] [PubMed] [Google Scholar]
- Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin-Mediated Protection from Cellular Stress in the Dopaminergic Neurons of Caenorhabditis elegans. Journal of Neuroscience. 2005;25:3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A, Bowling K, Funderburk C, Inamdar A, O’Donnell J. Interaction of genetic and environmental factors in a Drosophila parkinsonism model. J Neurosci. 2007;27:2457–2467. doi: 10.1523/JNEUROSCI.4239-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nature Publishing Group. 2008;9:759–767. doi: 10.1038/nrn2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing Activities Protect Against Age-Onset Proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Dong MQ, Venable JD, Au N, Xu T, Park SK, Cociorva D, Johnson JR, Dillin A, Yates JR. Quantitative Mass Spectrometry Identifies Insulin Signaling Targets in C. elegans. Science. 2007;317:660–663. doi: 10.1126/science.1139952. [DOI] [PubMed] [Google Scholar]
- Dostal V, Link CD. Assaying β-amyloid Toxicity using a Transgenic C. elegans Model. J Vis Exp. 2010;44:e2252. doi: 10.3791/2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM-Y, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Fu M, Li L, Albrecht T, Johnson JD, Kojic LD, Nabi IR. Autocrine mobility factor/phosphoglucose isomerase regulates ER stress and cell death through control of ER calcium release. Cell Death Diff. 2011;18:1057–1070. doi: 10.1038/cdd.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013;75:621–644. doi: 10.1146/annurev-physiol-030212-183712. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine disease. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Haga A, Niinaka Y, Raz A. Phosphohexose isomerase/autocrine motility factor/neuroleukin/maturation factor is a multifunctional phosphoprotein. Biochim Biophys Acta. 2000;1480:235–244. doi: 10.1016/s0167-4838(00)00075-3. [DOI] [PubMed] [Google Scholar]
- Halaschek-Wiener J, Khattra JS, McKay S, Pouzyrev A, Stott JM, Yang GS, Holt RA, Jones SJM, Marra MA, Brooks-Wilson AR, et al. Analysis of long-lived C. elegans daf-2 mutants using serial analysis of gene expression. Genome Research. 2005;15:603–615. doi: 10.1101/gr.3274805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc Natl Acad Sci USA. 2008;105:728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AJ, Yacoubian TA, Slone SR, Caldwell KA, Caldwell GA. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J Neurosci. 2012;32:2142–2153. doi: 10.1523/JNEUROSCI.2606-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signaling in brain and fat body. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- Isaacs AM, Johannsen P, Holm I, Nielsen JE, FReJA consortium Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheim Res. 2011;8:246–251. doi: 10.2174/156720511795563764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Koyama S, Yoshina S, Ren CH, Kato T, Mitani S, Iwatsubo T. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in α-synuclein transgenic C. elegans. Human Molecular Genetics. 2008;17:2997–3009. doi: 10.1093/hmg/ddn198. [DOI] [PubMed] [Google Scholar]
- Labandeira-Garcia JL, Rodriguez-Pallares J, Villar-Cheda B, Rodríguez-Perez AI, Garrido-Gil P, Guerra MJ. Aging, Angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis. 2011;2:257–74. [PMC free article] [PubMed] [Google Scholar]
- Lai C-Q, Parnell LD, Lyman RF, Ordova JM, Mackay TFC. Candidate genes affecting Drosophila life span identified by integrating microarray gene expression analysis and QTL mapping. Mech Aging Devel. 2007;128:237–249. doi: 10.1016/j.mad.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- Mazzola JL, Sirover MA. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J Neurochem. 2001;76:442–449. doi: 10.1046/j.1471-4159.2001.00033.x. [DOI] [PubMed] [Google Scholar]
- McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. Shared transcriptional signature in Caenorhabditis elegans dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J Biol Chem. 2004;279:44533–44543. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Nollen EAA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RHA. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci USA. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza F, Galimberti G, Conti E, Isella V, Perlangeli MV, Speranza T, Borroni B, Pogliani EM, Padovani A, Ferrarese C. Increased tissue factor pathway inhibitor and homocysteine in Alzheimer’s disease. Neurobiology of Aging. 2012;33:226–233. doi: 10.1016/j.neurobiolaging.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson AV, Carr CE, Ruvkun G. Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes & Development. 2007;21:2976–2994. doi: 10.1101/gad.1588907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer CR, Jensen RV, Gullans SR, Feany MB. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2003;12:2457–2466. doi: 10.1093/hmg/ddg265. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metabolism. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Slack C, Werz C, Wieser D, Alic N, Foley A, Stocker H, Withers DJ, Thornton JM, Hafen E, Partridge L. Regulation of lifespan, metabolism, and stress responses by the Drosophila SH2B protein, Lnk. PLoS Genet. 2010;6:e1000881. doi: 10.1371/journal.pgen.1000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh Y, Atzmon G, Cho M-O, Hwang D, Liu B, Leahy DJ, Barzilai N, Cohen P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A, White MF. Insulin-Like Signaling, Nutrient Homeostasis, and Life Span. Annu Rev Physiol. 2008;70:191–212. doi: 10.1146/annurev.physiol.70.113006.100533. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu M-P, Yin C-M, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Tauffenberger A, Vaccaro A, Aulas A, Velde CV, Parker JA. Glucose delays age-dependent proteotoxicity. Aging Cell. 2012;11:856–866. doi: 10.1111/j.1474-9726.2012.00855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsika E, Moysidou M, Guo J, Cushman M, Gannon P, Sandaltzopoulos R, Giasson BI, Krainc D, Ischiropoulos H, Mazzulli JR. Distinct Region-Specific α-Synuclein Oligomers in A53T Transgenic Mice: Implications for Neurodegeneration. Journal of Neuroscience. 2010;30:3409–3418. doi: 10.1523/JNEUROSCI.4977-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Kobayasi T, Matsuo T, Aigaki T. Insulin-degrading enzyme antagonizes insulin-dependent tissue growth and Aβ-induced neurotoxicity in Drosophila. FEBS Lett. 2010;584:2916–2920. doi: 10.1016/j.febslet.2010.05.010. [DOI] [PubMed] [Google Scholar]
- van Ham TJ, Thijssen KL, Breitling R, Hofstra RMW, Plasterk RHA, Nollen EAA. C. elegans Model Identifies Genetic Modifiers of α-Synuclein Inclusion Formation During Aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen S, Pehkonen P, Lakso M, Nass R, Wong G. Identification of gene expression changes in transgenic C. elegans overexpressing human α-synuclein. Neurobiology of Disease. 2006;22:477–486. doi: 10.1016/j.nbd.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Duda JE, Giasson BI. Characterization of antibodies that selectively detect alpha-synuclein in pathological inclusions. Acta Neuropathol. 2008;116:37–46. doi: 10.1007/s00401-008-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Tatar M. Insulin receptor substrate chico acts with the transcription factor FOXO to extend Drosophila lifespan. Aging Cell. 2011;10:729–732. doi: 10.1111/j.1474-9726.2011.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi SA. Mitochondrial Superoxide Signal Triggers Increased Longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.