

Abstract
Inward rectifier potassium (Kir) channels play fundamental roles in cardiac and renal function and may represent unexploited drug targets for cardiovascular diseases. However, the limited pharmacology of Kir channels has slowed progress toward exploring their integrative physiology and therapeutic potential. Here, we review recent progress toward developing the small-molecule pharmacology for Kir2.x, Kir4.1, and Kir7.1 and discuss common mechanistic themes that may help guide future Kir channel-directed drug discovery efforts.
Introduction
Inward rectifier potassium (Kir) channels play key roles in cardiac excitation-contraction coupling, renal water and solute transport, and other vital physiological and pathophysiological processes [1]. In mammals, the channel superfamily is comprised of at least 16 genes (KCNJx) and 7 sub-families (Kir1.x–7.x) that share a common molecular structure [2]. Kir channels are tetramers of identical (homomeric) or similar (heteromeric) subunits assembled around an aqueous membrane-spanning pore. They lack regulatory voltage-sensing domains, but are gated by polyvalent cations (e.g. polyamines and Mg2+ [3–5]) that occlude the pore at cell potentials more positive than the K+ equilibrium potential (EK). Kir channels thus function as biological diodes by limiting the extent of outward, but not inward, K+ current, a property is termed inward rectification. Strong rectifiers exhibit a sharp cutoff of outward current due to the presence of negatively charged pore-lining residues that stabilize electrostatic interactions with pore-blocking cations (Fig. 1), whereas weak rectifiers, have uncharged amino acids at some of the equivalent positions that weaken block and enable the channels to remain open well above EK [1].
Figure 1. Putative small-molecule binding sites in a model Kir channel.
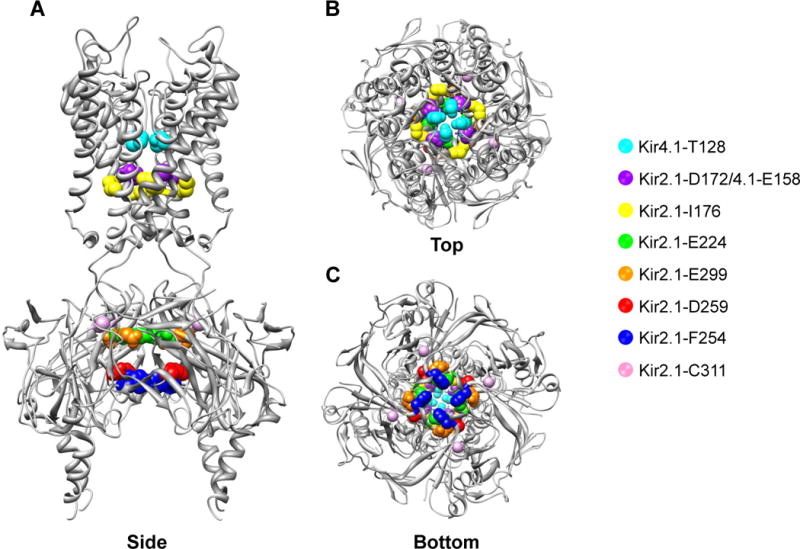
A) Side B) Top and C) Bottom view of chicken Kir2.2 crystal structure with equivalent residues implicated in small-molecule binding highlighted with sphere. Channel-specific residues are indicated in the legend. See text and Table 1 for details.
Kir channels are also gated through interactions with the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2). X-ray structures of Kir2.2 revealed that binding of a PIP2-derivative induces conformational changes in the cytoplasmic domain (Fig. 1) that opens the pore [6]. Growing evidence suggests that some cationic amphiphilic drugs (e.g. carvedilol, mefloquone, thiopental) inhibit Kir channels by interfering with channel-PIP2 interactions [7–9]. These have been discussed recently elsewhere [10] and will not be considered further.
As discussed below, emerging physiological, genetic, and pharmacological data point to specific Kir channels as novel drug targets for cardiac and renal diseases. With few exceptions, the rudimentary pharmacology has mired efforts to explore their integrative physiology and therapeutic potential of Kir channels. This review provides a snapshot of the current state of the field, highlighting recent progress and opportunities for developing the pharmacology of cardiac and renal Kir channels.
Cardiac Kir2.x channels
The ability of the heart to function as a pump requires that atrial and ventricular chambers contract in a highly stereotyped and synchronized fashion. Action potentials (AP) originating in the sinoatrial node (SAN) spread through the atria and then ventricles to initiate contractions. Three Kir channel sub-families contribute to cardiac excitability. Heteromeric Kir3.1/3.4 channels comprising the muscarinic M2 receptor-activated IKACh current slows SAN pacemaker discharge and heart rate in response to parasympathetic nerve stimulation. Activation IKATP carried by heteromeric Kir6.2/SUR channels during metabolic stress contributes to ischemic preconditioning that protects heart function during prolonged ischemia. The molecular physiology, pathophysiology, and pharmacology of IKACh and IKATP have been reviewed extensively [1,11–14] and will not be discussed here. The major focus of this review is on the molecular pharmacology of the third group of cardiac Kir channels: the strong rectifiers Kir2.1, Kir2.2, and Kir2.3. Homomeric and heteromeric assemblies of Kir2.x subunits underlie the IK1 current that dominates the resting K+ conductance and shapes late-phase action potential (AP) repolarization in cardiac myocytes [12]. Genetic loss- and gain-of-function mutations in Kir2.1 (KCNJ2) prolong and shorten, respectively, the AP duration and increase the susceptibility to lethal ventricular arrhythmias [15,16]. No disease-causing mutations in Kir2.2 (KCNJ12) and Kir2.3 (KCNJ4) have been reported.
It is clear that Kir2.x channels play important roles in heart pump function, but their species and regional heterogeneity and lack of specific pharmacological probes has slowed efforts to develop a comprehensive understanding of their integrative physiology and druggability in cardiac diseases. Below we discuss recent progress toward developing these critically needed tools for overcoming this barrier.
Chloroquine
The 4-aminoquinoline derivative chloroquine (Table 1) is used widely as an anti-malarial drug in developing countries. However, prolonged treatment or overdose can induce lethal ventricular arrhythmias through inhibition of various cardiac ion channels [17]. Sanchez-Chapula and colleagues [18] found that chloroquine blocks Kir2.1 at clinically relevant doses (half-maximal inhibitor concentration [IC50] = 8.7 μM) and in a voltage-dependent manner consistent with ‘knock-off’ of the drug from the intracellular pore [19]. Direct application of chloroquine to the cytoplasmic face of Kir2.1 results in channel inhibition that is much faster (~15 sec) than that observed when applied extracellularly (~8 min), suggesting chloroquine must cross the plasma membrane to reach an intracellular binding site. Indeed, alanine-scanning mutagenesis revealed that mutation of four residues in the cytoplasmic domain of Kir2.1 led to progressive loss of chloroquine sensitivity with the following rank-order: E224 > D259 > E299 > F254 (Fig. 1). Molecular modeling identified an energetically favorable docking pose between chloroquine and the channel involving electrostatic interactions between E224, D259, and E299 as well as aromatic pi-stacking with P254. Identifying the putative chloroquine binding site creates opportunities for designing safer analogs exhibiting reduced Kir2.1 activity and cardiotoxicity.
Table 1. Chemical diversity of Kir channel modulators.
| Compound/PubChem CID | Structure | Action | Selectivity (IC50/EC50)* | Channel/Residues | References |
|---|---|---|---|---|---|
| Chloroquine/2719 |
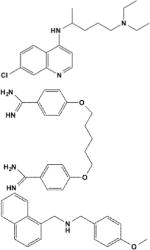
|
Pore blocker | IKACh (0.4) > IKATP (0.5) > IK1 (0.7) | Kir2.1/E224, D259, E299, F254 | [18,22] |
| Pentamidine/4735 | Pore blocker | Kir2.1 (0.2) > 2.2 > Kir2.3 | Kir2.1/E224, D259, E299 | [25] | |
| ML133/781301 | Pore blocker | Kir2.1 (0.3†, 1.9) > 2.6 (2.8) > 2.2 (2.9) > 2.3 (4.0) > 6.2 (7.7) > 7.1 (32.9) > 4.1 (76) > 1.1 (>300) | Kir2.1/D172, I176 | [28] | |
| Flecainide/3356 |
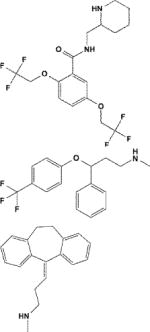
|
Potentiator | Kir2.1 (0.4†† − 0.8‡) > 2.2 ~ 2.3 ~ 2.1/2.2 ~ 2.1/2.3 | Kir2.1/E224, D259, E299, C311 | [33] |
| Fluoxetine/62857 | Pore blocker | Kir4.1 (15) > 2.1 ~ 1.1 | Kir4.1/T128, L151, E158 | [46,48] | |
| Nortriptyline/4543 | Pore blocker | Kir4.1 (28) > 2.1 (245) | Kir4.1/T128, L151, E158 | [47,48] | |
| VU590/4536383 |
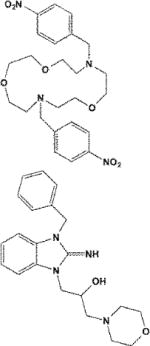
|
Pore blocker | Kir1.1 (0.3) > 7.1 (8) > 2.1 ~ 4.1 | Unknown | [39] |
| VU573/44199324 | Unknown | Kir3.1/3.2 (2) > 2.3 (5) > 7.1 (5) | Unknown | [52] |
See text for details.
μM,
at pH 8.5;
at 50 mV,
at −120 mV
Pharmacological Kir2.1inhibition may provide therapeutic benefits in the setting of certain cardiac pathologies. Gain-of-function mutations in KCNJ2 underlying Short QT Syndrome-Type 3 (SQT3) increase the risk of lethal atrial and ventricular arrhythmias [15]. These mutations (D172N, E299V, and M301K) reduce pore block by Mg2+/polyamines and produce larger outward currents that shorten AP duration. Heterologous expression and in silico studies have suggested that inhibition of the SQT3 mutant Kir2.1-D172N by chloroquine may normalized the AP waveform and improve cardiac function in SQT3 patients [20,21]. This may not bet true for patients carrying E299V and M301K mutations since these residues are near the putative chloroquine binding site (Table 1).
Kir2.1 inhibition may also restore sinus rhythm in the setting chronic atrial fibrillation, where upregulation of Kir2.1 expression and function contributes to arrhythmia recurrence and maintenance. Jalife and colleagues demonstrated that chloroquine treatment terminates atrial or ventricular fibrillation in mice, rabbit, and sheep hearts. At the doses used, however, chloroquine also inhibits IKACh and IKATP [22]. Additional studies are needed to determine if specific Kir2.1 inhibition is sufficient to restore rhythmogenesis.
Pentamidine
Pentamidine is an aromatic diamidine (Table 1) used in the treatment of pneumocystis pneumonia, trypanosomiasis, leishmaniasis, and fungal infections. Intravenous administration in some patients delays ventricular repolarization and induces ventricular tachycardia, consistent with hERG block. Although pentamidine does indeed inhibit hERG function, this requires prolonged drug treatment and is mediated through inhibition of channel trafficking [23], a mechanism that is too slow to explain the more rapid cardiac effects of the drug [24]. Van der Hayden and colleagues [25] reasoned that some of the effects could be due to inhibition of IK1 current carried by members of the Kir2.x subfamily. Indeed, they found that acute pentamidine application at clinically relevant doses inhibits homotetrameric Kir2.x channels with the rank-order potency Kir2.1 (IC50 = 190 nM) > Kir2.2 > Kir2.3. Similar to chloroquine (see above), pentamidine inhibited Kir2.1 much more rapidly when applied to the cytoplasmic side of the channel. In silico ligand docking into the Kir2.1 cytoplasmic domain crystal structure [26] identified a putative pentamidine binding site involving E224, D259, and E299 (Fig. 1), the same residues implicated in chloroquine block (Table 1). Mutations of these residues reduced pentamidine sensitivity, lending support to their model predictions.
In an effort to develop more specific Kir2.1 probes, the investigators analyzed a series of pentamidine analogs for activity toward Kir2.x and several other cardiac ion channels [27]. One compound termed PA-6 exhibited ~15 nM affinity toward Kir2.1, Kir2.2, and Kir2.3, but no discernible activity toward cardiac sodium, calcium, and potassium currents. In terms of potency, PA-6 represents the state-of-the-art in Kir2.1 inhibitors. PA-6 was active on native IK1 in cardiomyocytes and predictably lengthened the action potential duration. Interestingly, and unlike pentamidine, PA-6 had no effect on the hERG biosynthesis. Provided the pharmacokinetic behavior is amenable, PA-6 should provide a useful in vivo probe of Kir2.x function. Furthermore, and given the apparent lack of effect on hERG, PA-6 may provide a safer alternative to pentamidine for the treatment protozoal infections.
ML133
Min Li and colleagues took a modern drug discovery approach to develop a potent and selective small-molecule inhibitor of Kir2.1 [28]. Using a fluorescence-based thallium (Tl+) flux assay [29,30], the investigators screened approximately 300,000 structurally diverse small molecules from the NIH Molecular Libraries Small-Molecule Repository for chemical modulators of Kir2.1. Of 320 confirmed hits, ML133 (N-(4-methoxybenzyl)-1-(naphthalene-1-yl)methanamine) was selected for development because of its drug-like structure, potency toward Kir2.1, and clean ancillary pharmacology. At physiological pH, ML133 exhibits the following rank-order potency: Kir2.1 (IC50 = 2 μM) > 2.6 > 2.2 > 2.3 > 6.2 > 7.1 > 4.1 > >1.1 (Table 1). Interestingly, due to the presence of a protonatable nitrogen in the linker of ML133, its potency toward Kir2.1 is pH-dependent: IC50 = 300 nM at pH 8.5 and 10 μM at pH 6.5 (Table 1). Lead-optimization efforts to improve its potency were unsuccessful. Similar to chloroquine (see above), block of Kir2.1 by ML133 was dependent on the K+ electrochemical driving force, suggesting the binding site is located in ion conduction pathway. In an effort to identify the binding site, the investigators took advantage of the >50-fold selectivity of ML133 for Kir2.1 over Kir1.1 and analyzed a series of Kir1.1-Kir2.1 chimeras and point mutants for sensitivity to ML133. Swapping out incrementally larger regions of Kir2.1 for Kir1.1 localized a potential binding site to the Kir2.1 pore. This region of Kir2.1 and Kir1.1 is highly conserved between the two channels, with the exception of two conspicuous residues: D172 and I176 (Fig. 1). Mutation of these residues to those found in Kir1.1 either alone (i.e. Kir2.1-D172N) or together (Kir2.1-D172N/I176C) reduces ML133 potency by 8- and 23-fold, whereas the reverse mutations in Kir1.1 (i.e. Kir1.1-N171D and Kir1.1-N171D/C175I) confers weak sensitivity to the channel. This putative binding site is different from that of chloroquine and pentamidine (Fig. 1). In terms of selectivity, and based on published data, ML133 represents the state-of-the-art among Kir2.1 channel probes. However, similar to chloroquine and pentamidine, ML133 is not selective for members of the Kir2.x. A future challenge will be designing compounds that can be used to dissect the individual functions and druggability of Kir2.x homo- and heteromeric channels in the heart and other organs.
Flecainide
Flecainide is used widely as a class Ic antiarrhythmic drug that, in addition to inhibiting cardiac sodium channels, displays class III antiarrhythmic activity due to inhibition of Kv4.3 and hERG [31,32]. In an elegant study, Caballero and colleagues reported [33] that clinical doses of flecainide increase ventricular IK1 through potentiation of Kir2.1, but not of Kir2.2, Kir2.3, Kir2.1/2.2 or Kir2.1/2.3 channels. Molecular modeling, mutagenesis, and patch clamp electrophysiology supports a mechanism in which flecainide binding to C311 of Kir2.1 reduces spermine-dependent pore block and increases outward current amplitude. Remarkably, prolonged flecainide treatment partially rescues the function of Kir2.1 channels carrying an R67W LOF mutation identified in Andersen syndrome (AS) patients by increasing the cell surface density of the channel. It is tempting to speculate that this mechanism underlies the propensity of flecainide to suppress ventricular arrhythmias in some AS patients [34,35].
Renal Kir channels
Of the nearly 70 million Americans with hypertension, an insidious disorder which increases the risk of heart disease, stroke, and kidney damage, only about half manage their blood pressure correctly. Diuretics represent the first-line therapy for hypertension and work by reducing sodium and osmotically obliged water reabsorption in the kidney tubule, leading to increased urine output and lowering of blood volume and pressure. The development of new structural classes of diuretic drugs with different mechanisms of action would provide clinicians with greater therapeutic options for managing their patients’ blood pressure, particularly in the settings of diuretic resistance and harmful drug-drug interactions [36]. Below, we discuss recent progress in developing the molecular pharmacology of renal Kir channels that could represent targets for novel-mechanism diuretics.
Kir1.1 (ROMK, KCNJ1)
Genetic LOF mutations in KCNJ1 lower blood pressure by inhibiting renal sodium and water reabsorption [37,38], thus validating Kir1.1 as a diuretic target. Recent advances in the small-molecule pharmacology by our group [39,40] and Merck [41] are providing critically needed tools for exploring Kir1.1 as a therapeutic target for hypertension. This work has been reviewed recently by our group [42] as well as by Garcia and Kaczorowski (2013) in this issue of Current Opinion in Pharmacology and will not be discussed further.
Kir4.1 (KCNJ10)
Inactivating mutations in the gene encoding Kir4.1 (KCNJ10) give rise to SeSAME (or EAST) syndrome, a complex disease presenting with seizures, sensorineural deafness, ataxia, intellectual disability, and electrolyte imbalance [43,44]. The renal consequences of SeSAME syndrome are consistent with impaired NaCl reabsorption in the distal convoluted tubule (for review see [42]). More recently, Zaika et al. (2013) found that dopamine inhibits Na+ reabsorption in the cortical collecting duct (CCD) through inhibition of Kir4.1 homomeric and Kir4.1/5.1 heteromeric channels [45]. Taken together, these data raise the intriguing possibility that renal Kir4.1-containing channels represent novel diuretic targets.
The molecular pharmacology of Kir4.1 is poorly developed and consists of selective serotonin reuptake inhibitors (SSRI), such as fluoxetine (IC50 = 15 μM, [46]) and tricyclic antidepressants (TCA), such nortriptyline (IC50 = 28 μM; [47]). Within the Kir channel family, these drugs appear to be selective for Kir4.1 since they exhibit very limited activity toward Kir1.1 and Kir2.1 [46,47]. Both drugs exhibit voltage-dependent inhibition of Kir4.1, consistent with a pore-blocking mechanism. Kurachi and colleagues found through alanine-scanning mutagenesis that T128, L151, and E158 are required for fluoxetine- and nortriptyline-dependent inhibition of Kir4.1 [48]. Mutation to D or E of N171 in Kir1.1 (N171D/E), which is equivalent to E158 in Kir4.1, confers fluoxetine and nortriptyline sensitivity to Kir1.1. Molecular docking simulations suggested that both drugs can assume energetically favorable interactions with E158 and T128. L151 is located outside of the pore and likely affects drug binding through stabilization of channel tertiary structure. Interestingly, their modeling suggests that both drugs interact with E158 and T128 on diagonally apposed subunits of the tetramer. This raises the possibility that SSRIs and TCAs are specific for Kir4.1 homomeric vs. Kir4.1/5.1 heteromeric channels since Kir5.1 contains an asparagine residue (N161) at the equivalent position of Kir4.1-E158. The development of small-molecule probes targeting Kir4.1 and Kir4.1/5.1 channels would create exciting opportunities for exploring the therapeutic potential of these channels in hypertension and other disorders.
Kir7.1 (KCNJ13)
As one of the newest members of the Kir channel family, there is little known about the function Kir7.1 in the organs known to express the channel, including eye, intestine, stomach, thyroid, spinal cord, brain, and kidney. Studies of Kir7.1 in retinal pigmented epithelial (RPE) cells have begun to shed light on the physiology of the channel in the eye, where it participates in transepithelial ion and water transport required for vision. A LOF mutation in KCNJ13 is associated with Snowflake Vitreoretinal Degeneration (SVD), indicating Kir7.1 function is indeed essential in the eye [49].
The function of Kir7.1 in the kidney is currently a matter of speculation based on its localization and observation that Kir7.1 expression is regulated by serum K+ concentration [50,51]. In rat, Kir7.1 is expressed in the basolateral membrane of several nephron segments, including principal cells of the CCD, which plays fundamental roles in regulated Na+ reabsorption and K+ excretion. Transepithelial K+ excretion in the CCD is mediated by a two-step process, whereby serum K+ is initially pumped across the basolateral membrane by the Na+-K+-ATPase and then secreted into the renal tubule fluid via apical Kir1.1 channels. By hyperpolarizing Vm, K+ secretion enhances the electrochemical driving force favoring Na+ reabsorption across the apical membrane through the K+-sparing diuretic target ENaC (for Epithelial Na+ Channel). Reabsorbed Na+ is transported by the Na+-K+-ATPase across the basolateral membrane before returning to the blood. Although there is no experimental evidence in support of this model, some investigators have postulated that Kir7.1 recycles K+ across the basolateral membrane to maintain robust Na+-K+-ATPase activity needed for transepithelial K+ secretion and Na+ reabsorption. If this model is correct, a Kir7.1 antagonist should 1) slow Na+-K+-ATPase activity, 2) reduce apical K+ secretion, 3) depolarize the apical Vm, 4) reduce ENaC-mediated Na+ reabsorption, and 5) lower blood volume and pressure. In principle, Kir7.1 inhibitors acting in the CCD would mimic the effects of the K+ sparing diuretic amiloride, but may also reduce Na+ reabsorption in other nephron segments such as the DCT, where Kir7.1 function may overlap with that of Kir4.1 (see above). To our knowledge, no studies have reported renal tubule pathologies in SVD patients carrying a loss-of-function mutation in KCNJ13. This could indicate that Kir7.1 is dispensable for renal function or that the loss of channel function leads to subclinical changes in blood pressure.
The small-molecule pharmacology of Kir7.1 is in its infancy and limited to two inhibitors developed by our group (Table 1). VU590 is a 300 nM inhibitor of Kir1.1 that also inhibits Kir7.1 with an IC50 of ~8 μM [39]. The other is the mixed Kir channel inhibitor VU573, which inhibits Kir7.1 with an IC50 of ~5 μM [52]. In an effort to expand the pharmacology, we recently developed a Tl+ flux assay of Kir7.1 that takes advantage of a pore mutation (M125R) that increases the unitary K+ conductance of the channel by ~20-fold [53]. Unlike the WT channel, Kir7.1-M125R mediates robust Tl+ flux that can be measured in a 384-well plate format that is compatible with HTS [52]. We anticipate that this assay will enable the development of small-molecule tool compounds for exploring Kir7.1 physiology and druggability in the kidney tubule and other organ systems.
Conclusions
In conclusion, a growing body of physiological, genetic and pharmacological evidence has implicated some Kir channel as drug targets for cardiac and renal diseases. The last 5 years have seen more progress in the development of the Kir channel pharmacology than in the previous 20 years since the founding member was cloned [54,55]. The identification of clinically used drugs (e.g. chloroquine [18], pentamidine [25,27], flecainide [33], fluoxetine [46,56], and nortriptyline [47]; Table 1) exhibiting Kir channel-directed activity creates opportunities for engineering more potent and selective Kir channel modulators from these chemical scaffolds (e.g. [27]). Chemically unique Kir channel inhibitors have been discovered in publically funded, large-scale screens of diverse chemical libraries, the results of which are deposited in well-annotated databases (http://pubchem.ncbi.nlm.nih.gov/). Counterscreens of focused libraries containing modulators of one Kir channel have uncovered modulators of other Kir channel members [52,57]. Despite this important progress, there are many hurdles to overcome before a comprehensive pharmacological “toolkit” for cardiac and renal inward rectifiers will be realized. These include the development of 1) subtype-selective modulators of the Kir2.x family, including compounds that discriminate between homo- and heteromeric channels, 2) modulators of Kir4.1 homomeric and Ki4.1/5.1 channels, and 3) modulators of Kir7.1. It is noteworthy that most known Kir channel pore blockers studied to date appear to target residues in the membrane-spanning or cytoplasmic pore domain (Fig. 1, Table 1). The development of comparative molecular models of these pore regions could enable in silico screening of “virtual libraries” containing millions of compounds at a fraction of the time, labor, and cost.
Highlights.
Some Kir channels are putative drug targets, but their pharmacology is limited
Newly discovered inhibitors or activators of Kir2.x, 4.1, and 7.1 are discussed
Mechanisms of action involve pore-lining residues that control rectification
This work is a critical first step toward developing more specific modulators
Acknowledgments
This work was supported through National Institutes of Health Grants 1R21NS073097-01 and 1R01DK082884 to J.S.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90(1):291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 2.Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science. 2009;326(5960):1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Z, MacKinnon R. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 1994;371(6494):243–246. doi: 10.1038/371243a0. [DOI] [PubMed] [Google Scholar]
- 4.Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 1994;266(5187):1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- 5.Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+ channels 1localized to a single negatively charged residue. Nature. 1994;371(6494):246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- 6.Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2activation of the classical inward rectifier K+channel Kir2.2. Nature. 477(7365):495–498. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrer T, Ponce-Balbuena D, Lopez-Izquierdo A, Arechiga-Figueroa IA, de Boer TP, van der Heyden MA, Sanchez-Chapula JA. Carvedilol inhibits Kir2.3 channels by interference with PIP2-channel interaction. Eur J Pharmacol. 2011;668(1–2):72–77. doi: 10.1016/j.ejphar.2011.05.067. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Izquierdo A, Ponce-Balbuena D, Moreno-Galindo EG, Arechiga-Figueroa IA, Rodriguez-Martinez M, Ferrer T, Rodriguez-Menchaca AA, Sanchez-Chapula JA. The antimalarial drug mefloquine inhibits cardiac inward rectifier K+ channels: evidence for interference in PIP2-channel interaction. J Cardiovasc Pharmacol. 2011;57(4):407–415. doi: 10.1097/FJC.0b013e31820b7c03. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Izquierdo A, Ponce-Balbuena D, Ferrer T, Rodriguez-Menchaca AA, Sanchez-Chapula JA. Thiopental inhibits function of different inward rectifying potassium channel isoforms by a similar mechanism. Eur J Pharmacol. 2010;638(1–3):33–41. doi: 10.1016/j.ejphar.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 10.van der Heyden MA, Stary-Weinzinger A, Sanchez-Chapula JA. Inhibition of cardiac inward rectifier currents by cationic amphiphilic drugs. Curr Mol Med. 2013;13(8):1284–1298. doi: 10.2174/15665240113139990043. [DOI] [PubMed] [Google Scholar]
- 11.Olson TM, Terzic A. Human KATP channelopathies: diseases of metabolic homeostasis. Pflugers Arch. 2010;460(2):295–306. doi: 10.1007/s00424-009-0771-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anumonwo JM, Lopatin AN. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010;48(1):45–54. doi: 10.1016/j.yjmcc.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Flagg TP, Nichols CG. Cardiac sarcolemmal KATP channels: Latest twists in a questing tale! J Mol Cell Cardiol. 2010;48(1):71–75. doi: 10.1016/j.yjmcc.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coetzee WA. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol Ther. 2013 doi: 10.1016/j.pharmthera.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96(7):800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 16.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105(4):511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Chapula JA, Salinas-Stefanon E, Torres-Jacome J, Benavides-Haro DE, Navarro-Polanco RA. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J Pharmacol Exp Ther. 2001;297(1):437–445. [PubMed] [Google Scholar]
- 18**.Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T, Rupp J, Sachse FB, Tristani-Firouzi M, Sanchez-Chapula JA. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc Natl Acad Sci U S A. 2008;105(4):1364–1368. doi: 10.1073/pnas.0708153105. Using heterologous expression systems, the authors demonstrate that chloroquine interacts with charged residues in the cytoplasmic pore to induce Kir2.1 block. This is the first study to elucidate the mechanism of action of drug-induced inhibition of an inward rectifier potassium channel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin HG, Lu Z. Mechanism of the voltage sensitivity of IRK1 inward-rectifier K+ channel block by the polyamine spermine. J Gen Physiol. 2005;125(4):413–426. doi: 10.1085/jgp.200409242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.El Harchi A, McPate MJ, Zhang Y, Zhang H, Hancox JC. Action potential clamp and chloroquine sensitivity of mutant Kir2.1 channels responsible for variant 3 short QT syndrome. J Mol Cell Cardiol. 2009;47(5):743–747. doi: 10.1016/j.yjmcc.2009.02.027. Using action potential waveform voltage clamp electrophysiology, the authors demonstrate that heterologously expressed Kir2.1 channels carrying the Short QT Syndrome 3 mutation D172N retains sensitivity to block by chloroquine. This study raises the possibility that chloroquine-dependent inhibition of Kir2.1 may correct cardiac rhythmogenesis in SQT3 patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Lopez-Izquierdo A, Ponce-Balbuena D, Ferrer T, Sachse FB, Tristani-Firouzi M, Sanchez-Chapula JA. Chloroquine blocks a mutant Kir2.1 channel responsible for short QT syndrome and normalizes repolarization properties in silico. Cell Physiol Biochem. 2009;24(3–4):153–160. doi: 10.1159/000233241. Using mathematical simulations of the ventricular action potential (AP), the authors demonstrate that chloroquine-dependent inhibition of Kir2.1 channels carrying the SQT3 mutation D172N corrects the AP waveform. This work raises the possibility that chloroquine may correct heart function in SQT3 patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22**.Noujaim SF, Stuckey JA, Ponce-Balbuena D, Ferrer-Villada T, Lopez-Izquierdo A, Pandit S, Calvo CJ, Grzeda KR, Berenfeld O, Chapula JA, Jalife J. Specific residues of the cytoplasmic domains of cardiac inward rectifier potassium channels are effective antifibrillatory targets. FASEB J. 2010;24(11):4302–4312. doi: 10.1096/fj.10-163246. The authors demonstrate that chloroquine treatment terminates atrial or ventricular fibrillation in mice, rabbit, and sheep hearts, raising the possibility that chloroquine-dependent Kir2.1 inhibition may restore sinus rhythm in the setting of chronic cardiac arrhythmias. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuryshev YA, Ficker E, Wang L, Hawryluk P, Dennis AT, Wible BA, Brown AM, Kang J, Chen XL, Sawamura K, Reynolds W, et al. Pentamidine-induced long QT syndrome and block of hERG trafficking. J Pharmacol Exp Ther. 2005;312(1):316–323. doi: 10.1124/jpet.104.073692. [DOI] [PubMed] [Google Scholar]
- 24.Yokoyama H, Nakamura Y, Iwasaki H, Nagayama Y, Hoshiai K, Mitsumori Y, Sugiyama A. Effects of acute intravenous administration of pentamidine, a typical hERG-trafficking inhibitor, on the cardiac repolarization process of halothane-anesthetized dogs. J Pharmacol Sci. 2009;110(4):476–482. doi: 10.1254/jphs.09071fp. [DOI] [PubMed] [Google Scholar]
- 25*.de Boer TP, Nalos L, Stary A, Kok B, Houtman MJ, Antoons G, van Veen TA, Beekman JD, de Groot BL, Opthof T, Rook MB, et al. The anti-protozoal drug pentamidine blocks KIR2.x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel. Br J Pharmacol. 2010;159(7):1532–1541. doi: 10.1111/j.1476-5381.2010.00658.x. The authors demonstrate that pentamidine inhibits native IK1 and Kir2.x channels by interacting with charged residues in the channel cytoplasmic domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8(3):279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 27**.Takanari H, Nalos L, Stary-Weinzinger A, de Git KC, Varkevisser R, Linder T, Houtman MJ, Peschar M, de Boer TP, Tidwell RR, Rook MB, et al. Efficient and specific cardiac IK1 inhibition by a new pentamidine analogue. Cardiovasc Res. 2013;99(1):203–214. doi: 10.1093/cvr/cvt103. The authors identify pentamidine analogs exhibiting greater potency and selectivity toward Kir2.x channels over several other cardiac ion channels. This work demonstrates how analog screening or medicinal chemistry can be used to improve the pharmacological properties of Kir channel modulators. [DOI] [PubMed] [Google Scholar]
- 28**.Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, et al. Selective inhibition of the Kir2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol. 2011;6(8):845–856. doi: 10.1021/cb200146a. The authors employ high-throughput screening and medicinal chemistry to engineer a potent and specific inhibitor of Kir2.x channels. Using a series of Kir1.1–2.1 chimeras and point mutations, they further localize the putative binding site to residues in the membrane-spanning pore. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen. 2004;9(8):671–677. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 30.Raphemot R, Weaver CD, Denton JS. high-throughput screening for small-molecule modulators of inward rectifier potassium channels. J Vis Exp. 2013;(71) doi: 10.3791/4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radicke S, Vaquero M, Caballero R, Gomez R, Nunez L, Tamargo J, Ravens U, Wettwer E, Delpon E. Effects of MiRP1 and DPP6 beta-subunits on the blockade induced by flecainide of Kv4.3/KChIP2 channels. Br J Pharmacol. 2008;154(4):774–786. doi: 10.1038/bjp.2008.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Himmel HM, Bussek A, Hoffmann M, Beckmann R, Lohmann H, Schmidt M, Wettwer E. Field and action potential recordings in heart slices: correlation with established in vitro and in vivo models. Br J Pharmacol. 2012;166(1):276–296. doi: 10.1111/j.1476-5381.2011.01775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Caballero R, Dolz-Gaiton P, Gomez R, Amoros I, Barana A, Gonzalez de la Fuente M, Osuna L, Duarte J, Lopez-Izquierdo A, Moraleda I, Galvez E, et al. Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc Natl Acad Sci U S A. 2010;107(35):15631–15636. doi: 10.1073/pnas.1004021107. The authors demonstrate that flecainide increases currents through Kir2.1 channels in part by interacting with C311 and reducing polyamine-dependent block of outward currents. They further show that prolonged incubation of Kir2.1 channels carrying the Andersen syndrome mutation R67W partially rescues channel function by increasing channel density in the membrane. This study raises the possibility that flecainide may be useful in the treatment of some Andersen syndrome patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellizzon OA, Kalaizich L, Ptacek LJ, Tristani-Firouzi M, Gonzalez MD. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol. 2008;19(1):95–97. doi: 10.1111/j.1540-8167.2007.00910.x. [DOI] [PubMed] [Google Scholar]
- 35.Bokenkamp R, Wilde AA, Schalij MJ, Blom NA. Flecainide for recurrent malignant ventricular arrhythmias in two siblings with Andersen-Tawil syndrome. Heart Rhythm. 2007;4(4):508–511. doi: 10.1016/j.hrthm.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 36.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51(6):1403–1419. doi: 10.1161/HYPERTENSIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
- 37.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet. 1996;14(2):152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 38.Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40(5):592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol. 2009;76(5):1094–1103. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhave G, Chauder BA, Liu W, Dawson ES, Kadakia R, Nguyen TT, Lewis LM, Meiler J, Weaver CD, Satlin LM, Lindsley CW, et al. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol Pharmacol. 2011;79(1):42–50. doi: 10.1124/mol.110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang H, Walsh SP, Yan Y, de Jesus RK, Shahripour A, Teumelsan N, Zhu Y, Ha S, Owens KA, Thomas-Fowlkes BS, Felix JP, et al. Discovery of Selective Small Molecule ROMK Inhibitors as Potential New Mechanism Diuretics. ACS Medicinal Chemistry Letters. 2012;3(5):367–372. doi: 10.1021/ml3000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denton JS, Pao AC, Maduke M. Invited Review - Novel Diuretic Targets. Am J Physiol Renal Physiol. 2013 doi: 10.1152/ajprenal.00230.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360(19):1960–1970. doi: 10.1056/NEJMoa0810276. The authors identify loss-of-function in the gene encoding Kir4.1 in patients with SeSAME/EAST syndrome, demonstrating that Kir4.1 plays important physiological and pathophysiological roles in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106. The authors identify loss-of-function in the gene encoding Kir4.1 in patients with SeSAME/EAST syndrome, demonstrating that Kir4.1 plays important physiological and pathophysiological roles in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45**.Zaika O, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol. 2013 doi: 10.1152/ajprenal.00363.2013. The authors demonstrate that dopamine receptor-dependent inhibition of Kir4.1 and Kir4.1/5.1 channels reduces sodium reabsorption in the collecting duct of the kidney tubule. This study raises the possibility that inhibition of these channels in genetically normal individuals may induce natriuresis, diuresis, and lower blood pressure in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007;1178:44–51. doi: 10.1016/j.brainres.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 47.Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther. 2007;320(2):573–580. doi: 10.1124/jpet.106.112094. [DOI] [PubMed] [Google Scholar]
- 48*.Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol. 2009;75(6):1287–1295. doi: 10.1124/mol.108.052936. The authors employ alanine-scanning mutagenesis and in silico docking to localize the putative fluoxetine and nortriptyline binding site to the membrane-spanning domain of Kir4.1. [DOI] [PubMed] [Google Scholar]
- 49.Hejtmancik JF, Jiao X, Li A, Sergeev YV, Ding X, Sharma AK, Chan CC, Medina I, Edwards AO. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am J Hum Genet. 2008;82(1):174–180. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ookata K, Tojo A, Suzuki Y, Nakamura N, Kimura K, Wilcox CS, Hirose S. Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J Am Soc Nephrol. 2000;11(11):1987–1994. doi: 10.1681/ASN.V11111987. [DOI] [PubMed] [Google Scholar]
- 51.Derst C, Hirsch JR, Preisig-Muller R, Wischmeyer E, Karschin A, Doring F, Thomzig A, Veh RW, Schlatter E, Kummer W, Daut J. Cellular localization of the potassium channel Kir7.1 in guinea pig and human kidney. Kidney Int. 2001;59(6):2197–2205. doi: 10.1046/j.1523-1755.2001.00735.x. [DOI] [PubMed] [Google Scholar]
- 52**.Raphemot R, Lonergan DF, Nguyen TT, Utley T, Lewis LM, Kadakia R, Weaver CD, Gogliotti R, Hopkins C, Lindsley CW, Denton JS. Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for kir2.3, kir3.x, and kir7.1. Front Pharmacol. 2011;2:75. doi: 10.3389/fphar.2011.00075. The authors employ high-throughput screening to interrogate a Kir channel antagonist “focused library” to identify preferential inhibitors of other Kir channels. Medicinal chemistry is used to develop a panel of active and inactive analogs that can be used to probe the function of Kir2.x, Kir3.x, and Kir7.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron. 1998;20(5):995–1005. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 54.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362(6415):31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 55.Zhou H, Tate SS, Palmer LG. Primary structure and functional properties of an epithelial K+ channel. Am J Physiol. 1994;266(3 Pt 1):C809–824. doi: 10.1152/ajpcell.1994.266.3.C809. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac) Br J Pharmacol. 2003;138(6):1119–1128. doi: 10.1038/sj.bjp.0705172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raphemot R, Rouhier MF, Hopkins CR, Gogliotti RD, Lovell KM, Hine RM, Ghosalkar D, Longo A, Beyenbach KW, Denton JS, Piermarini PM. Eliciting renal failure in mosquitoes with a small-molecule inhibitor of inward-rectifying potassium channels. PLoS One. 2013;8(5):e64905. doi: 10.1371/journal.pone.0064905. [DOI] [PMC free article] [PubMed] [Google Scholar]