

Abstract
Rosai-Dorfman disease (RDD) is an uncommon histiocytic disease of unknown etiology. It typically presents as massive lymphadenopathy with a predilection for the cervical lymph nodes of children and young adults. However, extranodal involvement is not uncommon and may cause confusion with other neoplasms or reactive disease. We describe here a unique case of extranodal RDD manifesting as a pericardial mass in a 69-year-old man. The lesion was detected by computed tomography during a periodic examination of the chest. Subsequently positron emission tomography scan showed mild increase of flurodeoxyglucose uptake. Clinically, it was supposed to be a mesothelioma. Histological examination showed the typical features of RDD confirmed by the staining of S100 protein, which highlighted the emperipolesis of the characteristic histiocytes. To the best of our knowledge, pericardial RDD represents an extremely rare condition and should be included in the differential diagnosis of pericardial neoplasms.
Keywords: Rosai-Dorfman disease, pericardial neoplasms, differential diagnosis
Introduction
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy (SHML), is a non-clonal proliferative histiocytic disease which was described as a distinctive entity by Rosai and Dorfman in 1969 [1]. The lesion typically manifests as painless massive lymphadenopathy with a predilection for the cervical lymph nodes [2]. Occurrence outside the lymph node is not uncommon, accounting for approximately 43% of cases. Extranodal RDD may involve a wide variety of anatomic sites, either as isolated or in association with lymphadenopathy. The majority of extranodal RDD are encountered in the skin, nasal cavity and paranasal sinuses, orbit, upper respiratory tract and bone [3]. Less commonly, the disease may also involve the breast, gastrointestinal tract, urogenital tract, soft tissues and central nervous system. Of note, extranodal RDD arising in unusual sites may cause diagnostic pitfalls. We describe here a unique case of primary pericardial RDD which was initially mistaken as an inflammatory myofibroblastic tumor (IMT).
Case report
A 69-year-old man with a 10-year history of hypertension, chronic bronchial asthma and gout was incidentally found to have a pericardial mass during a routine checkup. No symptoms such as chest pain or dyspnea were complained by the patient. On physical examination, no lymphadenopathy was identified. Electrocardiogram showed sinus rhythm and nonspecific T wave change. Cardiac ultrasound detected no obvious abnormality. Chest computed tomographic (CT) scan revealed a 3.0-cm solitary mass of soft tissue density in the left pericardium (Figure 1A). Subsequent CT/positron emission tomography (CT/PET) imaging demonstrated irregular thickening of the pericardium with a mild increase of metabolic activity (Figure 1B). At surgery, the mass was located on the superficial aspect of the left ventricle with partial involvement of the underlying myocardium. A subtotal resection of the mass was performed. The postoperative course was uneventful. The excised mass was initially interpreted as an inflammatory myofibroblastic tumor (IMT). After a final diagnosis of pericardial RDD was established, no additional treatment has been administrated. The patient is well with no signs of disease progression at 5 months of follow up.
Figure 1.
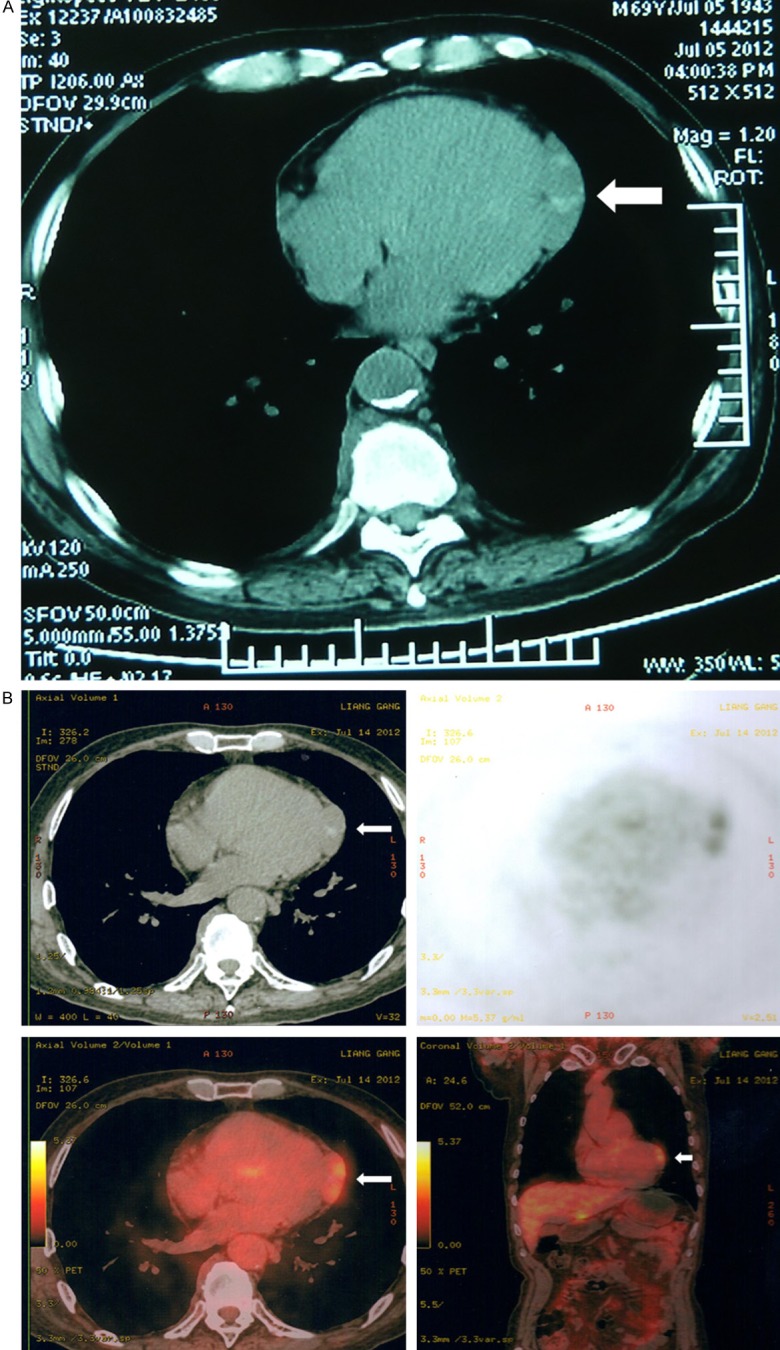
A. Chest computed tomographic (CT) scan shows a 3.0 cm solitary mass in the left visceral pericardium. B. Computed tomography/positron emission tomography imaging demonstrates increased metabolic activity (arrows).
Grossly, the excised mass was described as an unencapsulated nodule measuring 3 × 2 × 1.5 cm in size. On cut section, it appeared as gray-white with a fibrous consistency. On histology, the lesion showed a fibroinflammatory process with prominent fibrosis (Figure 2A). The inflammatory infiltration consisted mostly of lymphocytes and plasma cells with sparse neutrophils and eosinophils. Intermixed with the inflammatory cells and fibrotic collagen fibers were scattered large histiocytes with palely staining cytoplasm. On high power, a few histiocytes exhibited the characteristic feature of emperipolesis, but not obvious on H&E stained slides (Figure 2B). By immunohistochemistry, the histiocytes showed strong expression of S100 protein and CD68 (Figure 3). Other markers included cytokeratin (AE1/AE3), cytokeratin 5/6, D2-40, calretinin, alphasmooth muscle actin, desmin, ALK-1 and CD1a were all negative. There was no increased staining with IgG4 of the background plasma cells. The analysis of in-situ hybridization using ALK1 and EBER probes did not engender positive signals.
Figure 2.
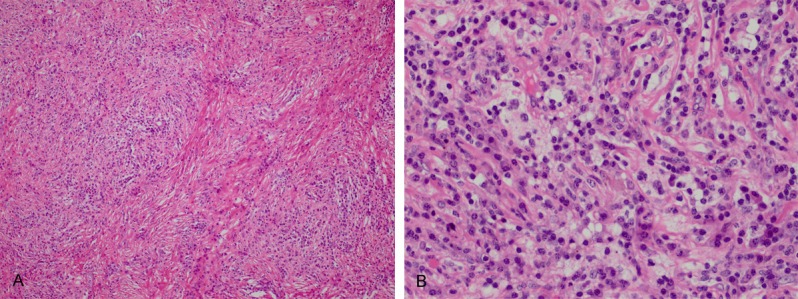
A. Low-power H&E of the pericardial neoplasm shows a fibrohistiocytic lesion with increased fibrosis. B. High-power H&E of the pericardial neoplasm shows the histiocytes with emperipolesis.
Figure 3.
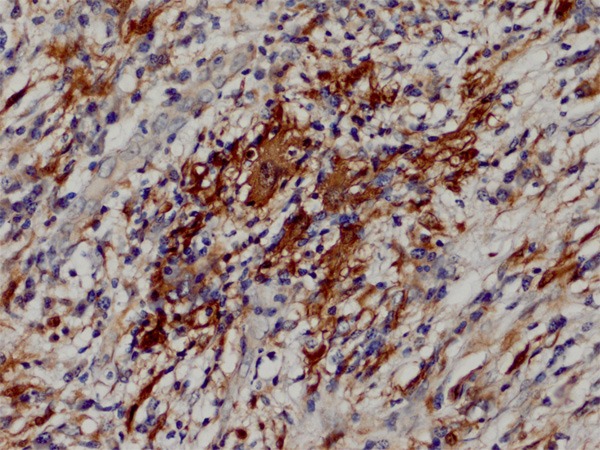
S100 protein immunostain highlights the feature of emperipolesis.
Discussion
Extranodal manifestation of RDD in the heart is very rare. To date, only nine reliable cases have been described in the literature [4-10]. Taking the current case into account, the overall median and average ages of patients with cardiac RDD at diagnosis were 53 years and 45.8 years (range, 12-79 years). There was a male predominance, with male to female ratio of 5:1. Seven cases Extranodal manifestation ed as pure extranodal RDD, whereas three cases were in association with nodal RDD [4,6,10]. The presenting symptoms available in six patients included chest pain (4 cases), fever (2 cases), shortness of breath (1 case), chest distress (1 case), and dyspnea (1 case). Two cases were found incidentally at autopsy [4,5]. The current case was discovered incidentally during a routine checkup. The symptoms in one case were not stated [3]. Reported sites of involvement included the tricuspid and pulmonic valves [3], atrium [4,6,7], visceral pericardium [5], and interartrial septum [8]. In two cases, the lesion presented with multifocal disease, involving the left atrium and ventricle, the ascending aorta and superior vena cava in one [9] and the lungs, pleura, epicardium and colon in the other [10]. Approximately half of the cases arose in the atrium with a predilection for the right sides. To date, RDD presenting as a primary pericardial RDD is extremely rare with only one case has been documented in a German literature [5]. Our case represented the first one being reported in the English literature.
The radiological features of extranodal RDD are nonspecific. However, imaging study is important in the localization of the disease. On CT scan, extranodal RDD typically appears as a homogenous mass isointense to soft tissue. On MRI, the lesion enhances with gadolinium-based contrast agents, exhibiting slightly increased signal density on T1-weighted and variable signal intensity on T2-weighted imaging. As RDD is usually hypermetabolic, increased metabolic activity is common on PET/CT.
The hallmark of extranodal RDD is the presence of scattered large histiocytes with emperipolesis. However, emperipolesis is usually difficult to appreciate in extranodal lesions as there is usually increased fibrosis which may obscure the histiocytes. In such settings, the staining of S100 protein will facilitate in the identification of emperipolesis. Owing to the proliferative spindle cells with intermixed lymphocytes and plasma cells, extranodal RDD may be mistaken as other fibrohistiocytic lesions, especially IMT. IMT is a distinctive myofibroblastic neoplasm that typically occurs in the lung, mesentery and omentum of children and young adults. Although any part of the body can be involved, IMT presenting as a cardiac lesion is exceedingly rare [11]. Histologically, cardiac IMT is composed of smooth muscle actin positive myofibroblasts and characterized by zonal ischemic necrosis and myxoid background. The absence of emperipolesis help distinguish a cardiac IMT from a cardiac RDD. Detection of ALK1 with FISH is less valuable as most cardiac IMTs showed negative results. The increased fibrosis with dispersed plasma cells in an extranodal RDD may resemble IgG4-related sclerosing disease. Although a link between cutaneous RDD and IgG4-related sclerosing disease was suggested by Kuo et al [12], other studies as well as ours did not find increased number of IgG4-positive plasma cells in the background of cardiac RDD. Nevertheless, additional studies are needed to further elucidate the relationship.
The etiology of RDD remains uncertain. Proposed mechanisms include a disorder of immune regulation or potential infectious agents like Herpesvirus 6 (HHV-6) and Epstein-Barr virus (EBV). At present, most authors favor an idiopathic histiocytosis of RDD. The histiocytes in RDD are thought to represent an unusual type of activated marcrophage. It has been supposed that a cytokine-mediated migration of monocytes may be involved in the accumulation and activation of histiocytes.
With respect to the clinical behavior, whereas nodal RDD is a benign, self-limited disease with spontaneous regressing, the outcome of extranodal RDD disease is quite variable. For patients with resectable lesions, surgical excision may bring in complete remission. However, massive involvement of vital organs might cause life-threatening complications. Mortality is primarily due to the disruption of organ function rather than from the disease itself [10]. Surgical approach in patients with multiple disease is usually limited for biopsy. The effectiveness of systemic chemotherapy, corticosteroid treatment or radiotherapy varies. The results may be encouraging or discouraging depending on different patients [6,10]. Generally, these approaches may be applied in cases whenever surgery is not feasible. Most recently, tyrosine kinase inhibitor imatinib and drugs that specifically target cytokines (TNF-α and IL-6) have been found to be effective in a few examples of recurrent, refractory or severe RDD. However, more clinical trials are necessary before drawing a conclusion.
In summary, we describe a rare case of extranodal RDD arising primarily in the pericardium. Due to the unexpected occurrence of RDD in this unusual site and appearance of a fibrohistiocytic lesion, the disease may be easily misdiagnosed as IMT. In challenging cases, immunostaining of S100 protein will facilitate in the differential diagnosis.
Disclosure of conflict of interest
None.
References
- 1.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63–70. [PubMed] [Google Scholar]
- 2.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19–73. [PubMed] [Google Scholar]
- 3.Buchino JJ, Byrd RP, Kmetz DR. Disseminated sinus histiocytosis with massive lymphadenopathy: its pathologic aspects. Arch Pathol Lab Med. 1982;106:13–6. [PubMed] [Google Scholar]
- 4.Maleszewski JJ, Hristov AC, Halushka MK, Miller DV. Extranodal Rosai-Dorfman disease involving the heart: report of two cases. Cardiovasc Pathol. 2009;19:380–4. doi: 10.1016/j.carpath.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Scheffel H, Vogt P, Alkadhi H. Cardiac manifestations of Rosai-Dorfman disease (article in German) Herz. 2006;31:715–6. doi: 10.1007/s00059-006-2872-0. [DOI] [PubMed] [Google Scholar]
- 6.Halabe-Cherem J, Cuan-Perez M, Wacher N, et al. A case of sinusoidal histiocytosis with possible invasion of the heart. Rev Med Inst Mex Seguro Soc. 1988;26:295–8. [Google Scholar]
- 7.Ajise OE, Stahl-Herz J, Goozner B, Cassai N, McRae G, Wieczorek R. Extranodal Rosai-Dorfman disease arising in the right atrium: a case report with literature review. Int J Surg Pathol. 2011;19:637–42. doi: 10.1177/1066896911409577. [DOI] [PubMed] [Google Scholar]
- 8.Yontz L, Franco A, Sharma S, Lewis K, McDonough C. A case of Rosai-Dorfman disease in a pediatric patient with cardiac involvement. Radiology Case. 2012;6:1–8. doi: 10.3941/jrcr.v6i1.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter JT, Strange RG Jr, Fisher SI, Miller DV, Delvecchio DM. Extranodal Rosai-Dorfman disease presenting as a cardiac mass in an adult: report of a unique case and lack of relationship to IgG4-related sclerosing lesions. Hum Pathol. 2010;41:297–301. doi: 10.1016/j.humpath.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Tang H, Li B, Xiu Q. Rosai-Dorfman disease of multiple organs, including the epicardium: An unusual case with poor prognosis. Heart Lung. 2011;40:168–71. doi: 10.1016/j.hrtlng.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Burke A, Li L, Kling E, Kutys R, Virmani R, Miettinen M. Cardiac inflammatory myofibroblastic tumor: a “benign” neoplasm that may result in syncope, myocardial infarction, and sudden death. Am J Surg Pathol. 2007;31:1115–22. doi: 10.1097/PAS.0b013e31802d68ff. [DOI] [PubMed] [Google Scholar]
- 12.Kuo TT, Chen TC, Lee LY, Lu PH. IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease: an additional immunohistochemical feature and possible relationship to IgG4-related sclerosing disease. J Cutan Pathol. 2009;36:1069–73. doi: 10.1111/j.1600-0560.2008.01222.x. [DOI] [PubMed] [Google Scholar]