

Abstract
Aggressive natural killer cell leukemia (ANKL) is a rare hematological malignancy that is particularly common among the Asian population. In the current study, we retrospectively evaluated six Chinese ANKL patients, including five males and one female, with a median age of 42 years (range 22 to 50 years). A number of unusual pathogenic manifestations were found in these ANKL patients, such as isolated extraocular muscle involvement, and hemophagocytic syndrome (HPS) with acute renal failure and multiple cavity effusion. Four of the patients died between two and six months after the diagnosis; however, there were two ANKL cases whose clinical behavior differed from the typical clinical course. One survived for over 30 months after splenectomy and chemotherapy treatment, and another ANKL case derived from chronic lymphoproliferative disorders of NK-cells (CLPD-NK) was treated with allogeneic bone marrow transplant (allo-BMT) and survived over 18 months. In conclusion, four cases experienced an aggressive clinical course whereas two demonstrated an indolent manifestation of their disease. New therapeutic regimens including allo-BMT should be optimized in order to improve outcomes of this disease.
Keywords: Aggressive NK-cell leukemia, lymphoproliferative disorders of NK-cells, hemophagocytic syndrome, retrospective study
Introduction
Lymphoproliferative disorders of natural killer cells (LPD-NK) are rare and accounts for less than 5% of all lymphoid neoplasms [1-6]. In the updated World Health Organization (WHO) classification for hematopoietic tumors and lymphoid tissues in 2008, three diseases originating from mature NK-cells were proposed based on their distinct clinical and pathological features, including: two aggressive mature NK-cell neoplasms, the extranodal NK/T cell lymphomanasal type (NK/TCL-NT), the aggressive NK-cell leukemia (ANKCL), and one provisional entity-chronic lymphoproliferative disorders of NK-cells (CLPD-NK) [7]. Aggressive NK-cell leukemia shares many features with the NK/TCL-NT, such as a high incidence among Asians, strong association with Epstein-Barr virus (EBV), and a similar immunophenotype [6,8-10]. Since the NK/TCL-NT also presents with bone marrow invasion and often associates with a hemophagocytic syndrome like ANKCL, some researchers believe that ANKCL represents a leukemic performance of the NK/TCL-NT, akin to the relationship between lymphoblastic lymphoma and leukemia [3].
ANKL is the rarest and poorest characterized malignancy among lymphoid neoplasms and the clinical features have been described only in a few case reports [10-16]. The common symptoms and signs include fever, B symptoms, hepatosplenomegaly, disseminated intravascular coagulopathy (DIC), and hemophagocytosis [17]. Acute leukemia complicated with orbital involvement is rare and has a very poor survival prognosis, even following treatment with radiotherapy and chemotherapy [18]. Until now, only one ANKL case in the world reported ocular involvement but there was a lack of pathological evidence due to the poor general condition of the patient [19]. Primary manifestation of malignant lymphoma and/or leukemia rarely invades in the kidney; however, it can be the cause of a hitherto unexplained acute renal failure that is detected incidentally [20]. NK-cell malignancies generally show poor response to chemotherapy through high expression of multidrug resistance associated p-glycoproteins [21]. There is no standard treatment currently available for advanced-stage patients. Patients with advanced stage NK-cell neoplasms have poor prognosis, with a reported survival between two and six months. To address the above issues, we performed a retrospective survey of mature NK-cell leukemia in our department. The current case study reports information on six patients diagnosed with ANKL who presented with hemophagocytosis and acute renal failure.
Patients
Between 1990 and 2012, six patients diagnosed with aggressive NK-cell leukemia in the Department of Hematology, Nanjing Drum Tower Hospital was enrolled in this retrospective study. The data retrieved include patients’ ages at diagnosis, gender, type of leukemia, the prominent presenting sign, imaging features, histopathological findings of the bone marrow, spleen and orbital specimen if available, bone marrow and/or peripheral blood immunophenotype, EBV status, treatment regimen and outcome. All available archival imaging records of the patients were re-examined.
This study was approved by the institutional review board of Nanjing Drum Tower hospital. All patients signed the informed consent.
Case reports
Case 1: A 37-year-old woman presented with fever, fatigue and abdominal distension for two months and was referred to the department of infectious disease. Two months prior she had experienced an incomplete abortion, and later became febrile. Although antibiotics were used, her fever remained high and so additional surgery was performed to clear the remaining uterine contents. She then developed jaundice with splenomegaly, and multiple abdominal lymphadenopathies. Blood counts showed pancytopenia (hemoglobin: 109 g/dl; white blood cell count: 4.5×109/L, lymphocytes: 76%, neutrophils: 7%, monocytes: 17%, and platelets: 37×109/L). The peripheral blood smear revealed 76% abnormal blasts. Blood chemistry showed high bilirubin (total bilirubin: 47.3 μM/L, direct bilirubin: 43.3 μM/L), elevated aspartate aminotransferase (911.3 IU/L), and alanine aminotransferase (840.7 IU/L). Blood urea nitrogen and creatinine were normal. Lactate dehydrogenase was 1453 IU/L. Routine blood coagulation tests were performed. Prothrombin time (PT) was 28.60 s (10-16 s normal), PT/INR was 2.46, and aPTT was 73.80 s (20-40 s normal), TT was 38.00 s (13-21 s normal), and fibrinogen: 0.9 (2-4 g/L normal). A bone marrow examination showed large granular lymphocytic leukemia (Figure 1A). Combined with the bone marrow immunophenotype and clinical presentation, a final diagnosis of ANKL was indicated.
Figure 1.

Bone marrow morphology of aggressive NK leukemia. A. A typical type III NK cell exhibited basophilic cytoplasm and bizarre nucleus. B. A typical type I NK cell exhibited large granular lymphocyte appearance in bone marrow smear. C. Dyserythropoiesis was shown in the erythroid cells. D. A typical type II NK cell with one hematophagocytic cells were found in the bone marrow smear.
Case 2: A 47-year-old man presented with fever, abdominal distension and pain. Physical examination showed hepatosplenomegaly, ultrasound examination revealed multiple lymphadenopathies in abdomen, bilateral pleural effusions and ascites. Serum ferritin was 647 ng/ml. Routine blood coagulation tests showed fibrinogen: 1.1 (2-4 g/L); PT, aPTT, and TT were normal. Blood chemistry revealed high bilirubin (total bilirubin: 28.7 μM/L, direct bilirubin: 10.9 μM/L) and elevation of aspartate aminotransferase (194.8 IU/L) and alanine aminotransferase (279.1 IU/L). Blood urea nitrogen and creatinine were normal. Lactate dehydrogenase was 307 IU/L. Blood counts showed pancytopenia (WBC: 0.7×109/L, Hb: 99 g/dL, PLT: 17×109/L). This patient had a history of nasal peripheral T-cell lymphoma (PTCL) six years prior and received six cycles of CHOP (cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisone) and radiation therapy. Chemotherapy was discontinued due to myocardial infarction. Upon admission, ENT examination and CT scan found no evidence of any abnormality in the nasal cavity. Bone marrow examination showed 37% lymphoblastic leukemic cells. Combined with the patient’s bone marrow immunophenotype of CD3-, CD4-, CD8+, CD56+, HLA-, DR+, IgH and TCR rearrangement (-), the diagnosis was determined to be compatible ANKL transformed from PTCL.
Case 3: The third case was a 50-year-old man who presented with complaints of left abdominal region discomfort, showed massive splenomegaly upon CT scan with uneven density. Blood counts showed WBC: 1.2×109/L, Hb: 101 g/dl, and PLT: 22×109/L. Blood chemistries were almost normal, but LDH level was as low as 76 IU/L (109-245 IU/L normal). Bone marrow examination showed 57.50% lymphomatoid cell infiltration, and large granular lymphocyte was seen in the bone marrow smear (Figure 1B); however, the immunophenotype was only positive for CD7+ and CD56+, but other markers were all negative. Various treatment regimens, including cyclophosphamide, oncovin, prednisone (COP), methotrexate (MTX), and EPOCH (etoposide, prednisone, oncovin, cyclophosphamide, and hydroxydaunorubicin) were used intermittently. Cyclosporines (CsA) were taken orally for approximately two years and the therapeutic effect showed partial remission (bone marrow: 14% leukemic cells). In order to improve the pancytopenia, splenectomy was performed. Spleen histopathology showed red pulp and medullary sinus infiltrated by lymphomatoid cells in clusters, white pulp withered or disappeared, focal spleen necrosis, and EBV (+). After splenectomy, pancytopenia was improved to near normal blood count level for approximately one year; however, pancytopenia deteriorated again and the patient died of sepsis 30 months after diagnosis.
Case 4: A 46-year-old male patient was referred to our hospital with a one month history of exophthalmos, one week history of dizziness, and sudden onset of vision loss. Physical examination showed right eye proptosis and restricted abduction movement. ENT examination showed no neoplasm in the nasal cavity or paranasal sinuses. CT scan indicated that the tendon and belly of the right eye lateral recti muscles were irregularly enlarged. Incisional biopsy histopathology showed tumor infiltration by undifferentiated, large cells with irregular shaped nuclei, finely dispersed nuclear chromatin and clear cytoplasm (Figure 1C). Immunohistochemistry was positive for CD56, TIA-1, perforin; and negative for CD3, CD4, CD8, CD19, CD20, CD22, MPO and cytokeratin. These findings indicated a NK-cell lineage origin for infiltration cells. Bone marrow immunophenotype were consistent with the diagnosis of aggressive NK-cell leukemia. The final diagnosis of this patient was aggressive NK-cell leukemia.
Case 5: A 37-year-old male patient was referred to our hospital with a 10 day history of fever and sputum-producing cough. Initial physical examination showed no lymphadenopathy or hepatosplenomegaly. Systemic examination was unremarkable. Laboratory workup revealed the following levels: WBC: 19.2×109/L, neutrophils: 77%, lymphocytes: 16%, monocytes: 0.06%, eosinophils: 0.01%, Hb: 123 g/L, PLT: 189×109/L. The peripheral blood lymphocyte immunophenotype was normal: CD3+: 76.2% (59.4-84.6% normal), CD19+: 7.7% (6.4-22.6% normal), CD3+CD4+: 33.7% (28.5-60.5% normal), CD3+CD8+: 33.4% (11.1-38.3% normal), CD56+: 12.6% (5.6-30.9% normal). Serum urea nitrogen, serum creatinine, calcium, phosphorous, and uric acid levels were within normal limits; lactate dehydrogenase was 1,292 U/L, and Serum ferritin >1,500 mg/L. Bone marrow smear disclosed hypercellularity with 1% hemophagocytic cells, normal immunophenotype findings in bone marrow by FCS analysis. The hemophagocytic syndrome was first diagnosed. Meropenan and vancomycin were used empirically because of high fever. One day later, hemorrhage of the upper digestive tract occurred. On the second day the patient lost consciousness and became anuric and hyperkalemic and hemodialysis was performed. At this time the initial blood culture revealed staphylococcemia MRSE (+); vancomycin drug concentration was within safety levels. After hemodialysis, renal function improved, however WBC increased to 32.0-54.5×109/L, pleural effusion and ascites occurred with hepatosplenomegaly (liver below costal margin 5.9 cm, spleen just below costal margin). Abnormal cells with atypical nuclear morphology were found in pleural effusion smear and in the end (Figure 1D), flow cytometry (FCM) of peripheral blood indicated aggressive NK-cell leukemia phenotype, bone marrow IgH and TCR rearrangement (-). The patient died two days later after the final diagnosis of ANKL, due to multiple organ failure and disseminated intravascular coagulopathy (DIC).
Case 6: A 50-year-old male patient with splenomegaly and lymphocytosis were found during routine physical examination. Later, small neck and cervical lymphadenopathies were found, and biopsy pathology revealed lymph node reactive proliferation. PET-CT showed enlarged spleen; however, the splenic metabolic activity as indicated by F-18 FDG uptake was within normal range (Figure 2). Initial EBV-DNA copy was within normal range (<1,000 copies/ml), however during following up, the EBV load increased from 3.81×106 copies/ml to 5.60×106 copies/ml and the clinical course gradually became aggressive. Finally diagnoses of aggressive NK-cell leukemia and hemophagocytic syndrome (HPS) were made. After investigational device exemption [22] regimen, bone marrow examination showed no response. The patient was then treated according to the HLH 2004 protocol, after which fever decreased and bone marrow transplantation was performed from an HLA-matched sibling donor (patient’s sister). The conditioning regimen was BUCY2, the infused MNC: 6.31×108/kg, CD34+: 7.81×106/kg, CD3+: 72.3%. Thereafter, hepatosplenomegaly quickly resolved, and GLs and EBV DNA were consistently undetectable. FISH with probes for the X and Y chromosomes revealed no recipient-derived cells in the peripheral blood and bone marrow. The patient sustained complete response, with stage II graft-versus-host disease (GVHD) affecting the skin and liver. His general condition remained stable for 1.5 years.
Figure 2.
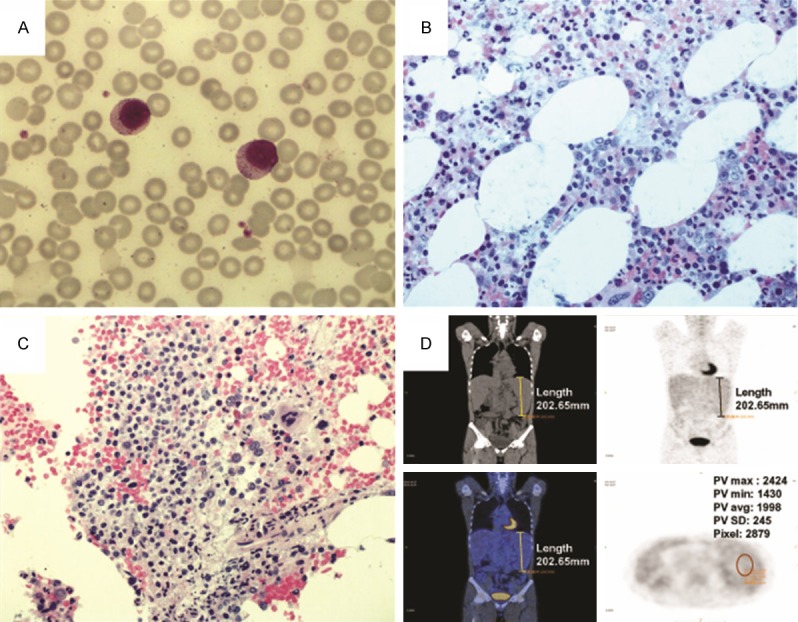
Bone marrow morphology and PET-CT of the case 6. A. Two large granular lymphocytes in bone marrow smear. B. Interstitial infiltration of large granular lymphocytes in bone marrow tissue. C. Focal aggregates of large granular lymphocytes in bone marrow tissue. D. Enlarged spleen with the metabolic activity as indicated by F-18 FDG uptake was within normal range.
Bone marrow morphological findings
We classified the leukemic NK-cells into three types according to the nuclear-cytoplasmic ratio, nuclear morphology, and color of the cytoplasm, nucleoli number and size of the cells with May-Giemsa staining of peripheral blood or bone marrow smear specimens (Figure 1). In brief, Type I has large granular lymphocyte (LGL) appearance. In type III, the cells exhibit pleomorphic-like appearance, together with basophilic cytoplasm and a bizarre nucleus containing one or two nucleoli. Type II is a mixture of type I and type III in each patient, or intermediate characteristics, such as monocyte-like features. Case #6 (NK-CLPD) initially had typical Type I NK-cell morphology but latter transformed to aggressive NK-cell leukemia. The NK-cell morphology varied from Type I to Type II morphology predominantly. The morphology of other cases were 1 type III and 4 type II (Figures 1A and 2B).
Bone marrow cellularity ranged from 40% to 100% (median, 60%), and the bone marrow stromal damages - including edema, serous fat atrophy and focal reticulum fibrosis - were easily seen. In four patients, the NK-cell infiltration was interstitial and in smaller aggregates, the other two showed diffuse infiltration. There were no striking dysplastic changes in granulocytic precursors or megakaryocytes, dyserythropoiesis were occasionally seen in case number three (Figure 1C). Hemophagocytosis was found in four patients (Figure 1D).
Immunophenotype analysis
The results of immunophenotype analysis of this group of patients are summarized as following Table 1. In the present series, the neoplastic cells from all six patients were positive for CD7 and CD56; no case was positive for CD3. Positive results were also found for HLA-DR in five cases, for CD5 in one case, and for CD8 in two cases. All were negative for CD4, CD10, CD19, CD20, and CD22. CD45 positivity was found in all six cases with the intensity ranging from moderate to bright.
Table 1.
Clinical features of six patients with aggressive NK cell leukemia
| Case No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Gender | F | M | M | M | M | M |
| Age (years) | 37 | 47 | 50 | 46 | 37 | 50 |
| Fever | + | + | + | + | + | - |
| Lymphadenopathy | + | + | - | + | + | - |
| Splenomegaly | + | + | + | + | + | + |
| Hepatomegaly | - | + | - | + | - | + |
| WBC (×109/L) | 4.5 | 0.7 | 1.2 | 6.4 | 19.2 | 1.3 |
| Hemoglobin | 109 | 99 | 101 | 139 | 123 | 109 |
| PLT (×109/L) | 37 | 17 | 22 | 46 | 189 | 46 |
| LDH index | 5.93 | 1.25 | 2.62 | 5.27 | 1.41 | 0.11 |
| EBV | - | + | + | - | + | NA |
| Hemophagocytic syndrome | - | + | + | + | + | - |
Note: NA, not available.
Discussion
Aggressive NK-cell leukemia (ANKL) is an uncommon disease, but occurs with higher frequency in Asians and Central/South Americans than in other ethnic groups [23]. The geographic distribution is likely to be due to both genetic and environmental factors and the disease is almost always associated with EBV. NK-cell leukemia occurs almost exclusively in younger adults (median age 39 years) and is slightly more common in males. Presentation is usually acute with B symptoms (particularly fever), jaundice, lymphadenopathy, hepatosplenomegaly, circulating leukemic cells and cytopenias. Disseminated intravascular coagulation, HPS, liver dysfunction and multi-organ failure may occur. Besides the above-mentioned presentations, in our case series we found one case of orbital involvement as an initial manifestation of ANKL in a 46-year-old male. The patient presented with proptosis and sudden vision loss. Orbital CT revealed isolated extraocular muscle involvement. Histopathology of incisional biopsy disclosed natural killer cell infiltration in lateral rectus muscle. Orbital involvement, which was found in 8% of myeloid leukemias and in 12% of lymphoid leukemias, is very rare compared with intraocular leukemia. Isolated extraocular muscle (EOM) involvement is a highly unusual presentation of orbital leukemia. Kiratli reported five such cases [24]. Acute myeloid leukemia was diagnosed in two patients while chronic lymphocytic, myeloid and biphenotypic acute leukemias were detected in each of the patients. One of these cases had bilateral involvement. Four patients were affected in the lateral rectus muscle, while the other one was affected in the superior rectus muscle. In one patient who had no prior history of leukemia, an incisional biopsy established the diagnosis [25]. Isolated EOM involvement is extremely rare. Kincaid and Green found EOM infiltration only in 1.3% of leukemia patients in an autopsy study [26]. Even large, prospective clinical studies relate EOM palsies to cranial nerve involvements rather than to direct muscular leukemic infiltrations. Most orbital leukemias are believed to arise from the bone marrow. The higher bone marrow density in the greater wing of the sphenoid and zygoma may explain the propensity for leukemias to develop in the lateral orbital structures [25]. Aydin hypothesized that circulating leukemia cells predominantly invade the lateral rectus muscle, as the lacrimal artery is larger than the muscular branches and arises from the ophthalmic artery just after the central retinal artery [25]. Until now, only Tsubokura reported one case with ocular palsy associated with aggressive NK-cell leukemia [19]. Magnetic resonance imaging of orbital cavity showed marked enlargement, and diffused high intensity signals in the right superior rectus muscle and left inferior rectus muscle on fat-saturated T2-weighted image. The authors could not perform biopsy of the lesions due to the poor general condition of the patient, and the exact etiology of ocular manifestation remains to be elucidated. Since the patient’s ocular symptoms occurred in parallel to the progression of the leukemia, tumor infiltration, infection, or paraneoplastic myositis can all be the possible reasons. To the best of our knowledge, our report is the first one in which isolated extraocular muscle involvement was the ophthalmic manifestation of ANKL, which was further identified by histopathologic evidence.
Acute kidney injury (AKI) is a dreaded complication in patients with hematological malignancies and may be caused by the disease itself or by its treatment. Of the many factors that can cause AKI in this setting, sepsis and hypoperfusion have been reported to be the most common. Other causes include metabolic complications (tumor lysis syndrome; TLS and hypercalcemia), renal infiltration by neoplastic cells, obstructive nephropathy, glomerulonephritis, and treatment-induced nephrotoxicity. The primary manifestation for one patient in our case series was HPS without any evidence of peripheral blood or bone marrow NK cell involvement. Later, upper gastric bleeding and acute renal failure occurred and led to pleural effusion, ascites, splenomegaly and increased NK leuke mic cells in peripheral blood. In acute leukemia, renal complications occur due to several factors including leukemic infiltration of the kidneys, therapy-related side effects such as tumor lysis syndrome, nephrotoxic drugs, and septicemias. The reason in our case may be multifactorial and the above mentioned factors should all be considered as multiorgan infiltration manifestations are impressive in this patient. Although ANKL has been well described in the previous case reports, this particular case suggests that a rapid and early diagnosis sometimes remains difficult in clinical practice because most cases present with hemophagocytic syndrome and neoplastic cells for laboratory analysis are extremely difficult. One reason is that peripheral adenopathy is usually absent even if the clinical presentation is clear. A second possibility is that flow cytometry detection was also limited and difficult, particularly in cases where there were a low number of malignant cells because neoplastic cells exist in various sizes and have varied cytoplastic granules. The patient experienced an exacerbation of clinical conditions although more than one aspiration and bone marrow biopsies were done but no evidence were gained from the early samples. From this patient’s presentation we hypothesized that aggressive NK-cell leukemia may represent a leukemic form of NK/TCL-NT, akin to the relationship between lymphoblastic lymphoma and leukemia.
Aggressive NK-cell leukemia is a catastrophic disease with an almost uniform mortality. Here, one unusual case first presented with the splenomegaly and cytopenia, bone marrow NK-cell infiltration >50%, LDH was lower than normal low range, and the patient was EBV (-). The clinical course of the case was relatively longer (30 months) than typical ANKL. Our observations suggested that the clinical behavior of some ANKL patients differs from the typical clinical course. Therefore, patients with indolent clinical courses, similar to the patient in this study, can be found among ANKL patients. Additional studies should be performed to explore the presence of an indolent subtype among ANKL patients.
Another case included in our review evolved from chronic lymphoproliferative disorder of NK-cell and which later became aggressive. The PET-CT was performed three times during follow up. The results showed an enlarged spleen twice; however, the metabolic activity of all organs as identified by F-18 FDG uptake was within normal range even when disease transformed to ANKL with HPS. Derlin and Park both reported one case of aggressive NK-cell leukemia detected by F-18 fluorodeoxyglucose PET-CT, and suggest that PET-CT might be a valuable tool enabling diagnosis and comprehensive staging of NK-cell leukemia [27,28].
The prognosis of ANKL is very poor and the median survival time from diagnosis is approximately 58 days. Refractoriness to chemotherapy may be attributed to the expression of multidrug resistance associated with P-glycoprotein. Allo-SCT has been found to improve the prognosis of NK-cell neoplasms. Up to this point no study has reported patients with ANKL who have sustained long-term complete remission without undergoing allo-SCT. It is extremely difficult to plan allo-SCT for ANKL in some candidate patients due to the rapid disease progression and deteriorating condition. Therefore, early planning of allo-HCT, from the beginning of care as soon as the diagnosis of ANKL is made, is essential. Although L-asparaginase and gemcitabine-containing regimens have shown some therapeutic effect, optimal chemotherapy for ANKL is another concern because preceding chemotherapy to control the disease enables more patients to have a chance for allo-HCT. Our case had a HLA-matched sibling donor and myeloablative stem cell transplantation which was performed soon after diagnosis, from which the patient achieved complete response and maintained stable remission for more than 18 months.
In conclusion, ANKL presented with isolated extraocular muscle involvement as the ophthalmic manifestation is extremely rare and the clinical behavior of some ANKL may differ from the typical clinical course. Allo-HCT might be a promising therapy for ANKL with curative potential.
Disclosure of conflict of interest
None.
References
- 1.Aozasa K, Takakuwa T, Hongyo T, Yang WI. Nasal NK/T-cell lymphoma: epidemiology and pathogenesis. Int J Hematol. 2008;87:110–117. doi: 10.1007/s12185-008-0021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung MM, Chan JK, Wong KF. Natural killer cell neoplasms: a distinctive group of highly aggressive lymphomas/leukemias. Semin Hematol. 2003;40:221–232. doi: 10.1016/s0037-1963(03)00136-7. [DOI] [PubMed] [Google Scholar]
- 3.Hasserjian RP, Harris NL. NK-cell lymphomas and leukemias: a spectrum of tumors with variable manifestations and immunophenotype. Am J Clin Pathol. 2007;127:860–868. doi: 10.1309/2F39NX1AL3L54WU8. [DOI] [PubMed] [Google Scholar]
- 4.Oshimi K. Progress in understanding and managing natural killer-cell malignancies. Br J Haematol. 2007;139:532–544. doi: 10.1111/j.1365-2141.2007.06835.x. [DOI] [PubMed] [Google Scholar]
- 5.Semenzato G, Marino F, Zambello R. State of the art in natural killer cell malignancies. Int J Lab Hematol. 2012;34:117–128. doi: 10.1111/j.1751-553X.2011.01374.x. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki R, Suzumiya J, Nakamura S, Aoki S, Notoya A, Ozaki S, Gondo H, Hino N, Mori H, Sugimori H, Kawa K, Oshimi K. Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia. 2004;18:763–770. doi: 10.1038/sj.leu.2403262. [DOI] [PubMed] [Google Scholar]
- 7.Dearden CE, Johnson R, Pettengell R, Devereux S, Cwynarski K, Whittaker S, McMillan A. Guidelines for the management of mature T-cell and NK-cell neoplasms (excluding cutaneous T-cell lymphoma) Br J Haematol. 2011;153:451–485. doi: 10.1111/j.1365-2141.2011.08651.x. [DOI] [PubMed] [Google Scholar]
- 8.Akashi K, Mizuno S. Epstein-Barr virus-infected natural killer cell leukemia. Leuk Lymphoma. 2000;40:57–66. doi: 10.3109/10428190009054881. [DOI] [PubMed] [Google Scholar]
- 9.Au WY, Weisenburger DD, Intragumtornchai T, Nakamura S, Kim WS, Sng I, Vose J, Armitage JO, Liang R. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood. 2009;113:3931–3937. doi: 10.1182/blood-2008-10-185256. [DOI] [PubMed] [Google Scholar]
- 10.Ruskova A, Thula R, Chan G. Aggressive Natural Killer-Cell Leukemia: report of five cases and review of the literature. Leuk Lymphoma. 2004;45:2427–2438. doi: 10.1080/10428190400004513. [DOI] [PubMed] [Google Scholar]
- 11.Castelli R, Molteni M, Gianelli U, Cro L, Grimoldi MG, Cortelezzi A. Aggressive natural killer cell leukaemia with a complex karyotype: a case report. Ann Hematol. 2006;85:66–68. doi: 10.1007/s00277-005-0001-4. [DOI] [PubMed] [Google Scholar]
- 12.Chan JK, Sin VC, Wong KF, Ng CS, Tsang WY, Chan CH, Cheung MM, Lau WH. Nonnasal lymphoma expressing the natural killer cell marker CD56: a clinicopathologic study of 49 cases of an uncommon aggressive neoplasm. Blood. 1997;89:4501–4513. [PubMed] [Google Scholar]
- 13.Guerrero AM, Lira VP, Bertin CP, Galleguillos VM, Ocqueteau TM. Natural killer cell leukemia. Case report. Rev Med Chil. 2005;133:457–460. doi: 10.4067/s0034-98872005000400010. [DOI] [PubMed] [Google Scholar]
- 14.Kagami Y, Suzuki R, Taji H, Yatabe Y, Takeuchi T, Maeda S, Kondo E, Kojima M, Motoori T, Mizoguchi Y, Okamoto M, Ohnishi K, Yamabe H, Seto M, Ogura M, Koshikawa T, Takahashi T, Kurita S, Morishima Y, Suchi T, Nakamura S. Nodal cytotoxic lymphoma spectrum: a clinicopathologic study of 66 patients. Am J Surg Pathol. 1999;23:1184–1200. doi: 10.1097/00000478-199910000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Murdock J, Jaffe ES, Wilson WH, McManus DT, Alexander HD, Morris TC. Aggressive natural killer cell leukemia/lymphoma: case report, use of telesynergy and review of the literature. Leuk Lymphoma. 2004;45:1269–1273. doi: 10.1080/10428190310001646879. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi E, Asano N, Li C, Tanaka T, Shimada K, Shimada S, Yoshino T, Kojima M, Hara K, Eimoto T, Nakamura S. Nodal T/NK-cell lymphoma of nasal type: a clinicopathological study of six cases. Histopathology. 2008;52:585–596. doi: 10.1111/j.1365-2559.2008.02997.x. [DOI] [PubMed] [Google Scholar]
- 17.Song SY, Kim WS, Ko YH, Kim K, Lee MH, Park K. Aggressive natural killer cell leukemia: clinical features and treatment outcome. Haematologica. 2002;87:1343–1345. [PubMed] [Google Scholar]
- 18.Maka E, Lukats O, Toth J, Fekete S. Orbital tumour as initial manifestation of acute myeloid leukemia: granulocytic sarcoma: case report. Pathol Oncol Res. 2008;14:209–211. doi: 10.1007/s12253-008-9028-x. [DOI] [PubMed] [Google Scholar]
- 19.Tsubokura M, Yamashita T, Kageyama S, Endo I, Tsuda H, Akiyama H. Ocular palsy associated with aggressive NK-cell leukemia. Int J Hematol. 2011;93:687–688. doi: 10.1007/s12185-011-0874-z. [DOI] [PubMed] [Google Scholar]
- 20.Ishida F, Ko YH, Kim WS, Suzumiya J, Isobe Y, Oshimi K, Nakamura S, Suzuki R. Aggressive natural killer cell leukemia: therapeutic potential of L-asparaginase and allogeneic hematopoietic stem cell transplantation. Cancer Sci. 2012;103:1079–1083. doi: 10.1111/j.1349-7006.2012.02251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi M, Kita K, Miwa H, Nishii K, Oka K, Ohno T, Shirakawa S, Fukumoto M. Frequent expression of P-glycoprotein/MDR1 by nasal T-cell lymphoma cells. Cancer. 1995;76:2351–2356. doi: 10.1002/1097-0142(19951201)76:11<2351::aid-cncr2820761125>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 22.Mattheolabakis G, Rigas B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine (Lond) 2012;7:1577–1590. doi: 10.2217/nnm.12.128. [DOI] [PubMed] [Google Scholar]
- 23.Perkovic S, Basic-Kinda S, Gasparovic V, Krznaric Z, Babel J, Ilic I, Aurer I, Batinic D. Epstein-Barr virus-negative aggressive natural killer-cell leukaemia with high P-glycoprotein activity and phosphorylated extracellular signal-regulated protein kinases 1 and 2. Hematol Rep. 2012;4:e16. doi: 10.4081/hr.2012.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiratli H, Balci KE, Himmetoglu C, Uner A. Isolated extraocular muscle involvement as the ophthalmic manifestation of leukaemia. Clin Experiment Ophthalmol. 2009;37:609–613. doi: 10.1111/j.1442-9071.2009.02099.x. [DOI] [PubMed] [Google Scholar]
- 25.Aydin A, Cakir A, Ersanli D. Isolated extraocular muscle involvement as the ophthalmic manifestation of leukaemia: an alternative explanation. Clin Experiment Ophthalmol. 2010;38:651. doi: 10.1111/j.1442-9071.2010.02345.x. [DOI] [PubMed] [Google Scholar]
- 26.Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol. 1983;27:211–232. doi: 10.1016/0039-6257(83)90123-6. [DOI] [PubMed] [Google Scholar]
- 27.Derlin T, Mester J, Klutmann S. F-18 FDG PET/CT findings of aggressive NK-cell leukemia. Clin Nucl Med. 2011;36:932–933. doi: 10.1097/RLU.0b013e318219b38f. [DOI] [PubMed] [Google Scholar]
- 28.Park JA. 18F-FDG PET for Diagnosis and Response Assessment for Aggressive NK Cell Leukemia. Clin Nucl Med. 2014;39:e281–2. doi: 10.1097/RLU.0b013e3182952976. [DOI] [PubMed] [Google Scholar]