

Abstract
We report a rare autopsy case of early infantile-onset vanishing white matter disease, with a submicroscopic deletion of 14q24.3, which included EIF2B2 and a missense mutation of EIF2B2 (V85E) of the remaining allele. The patient was a 4-year-old boy, who was found to have suddenly died during sleep. Physical and mental development began to deteriorate after convulsions at 10 month of age, and did not recover to baseline measurements. At autopsy, the brain showed a marked decrease in volume of white matter, with no typical cystic rarefaction. Histopathologically, the affected white matter showed diffuse loss of myelin fibers, meager astrogliosis with dysmorphic astrocytes, and loss of oligodendrocytes. Proliferative and apoptotic markers were negative for oligodendrocytes in the severely affected area. These findings may be related to the severity of the disease, and might be a feature of the EIF2B2 mutation pattern of the patient. Additionally, unusual fatty infiltration of both ventricles of the heart was found. These findings were suspected as early pathology of arrhythmogenic right ventricular cardiomyopathy due to characteristic gene mutation in the present case. In the present case, the defect EIF2B2 caused by hemizygosity may be related to early onset of the disease and the unusual pathological changes with vulnerability of oligodendrocytes and astrocytes, as well as cardiac abnormalities and sudden unexpected death.
Keywords: Vanishing white matter disease, EIF2B2 mutation, hemizygosity, sudden unexpected death, arrhythmogenic right ventricular cardiomyopathy
Introduction
Vanishing white matter disease (VWMD) (OMIM number 603896) is a childhood leukoencephalopathy with autosomal recessive traits. However, this disease has an extremely wide phenotypic variation and may affect people of all ages, including neonates and adults [1,2]. A characteristic clinical feature of VWMD is that neurological deterioration follows a chronic progressive course, with additional episodes of rapid deterioration by stresses, such as fever and minor head trauma. Neuropathological features generally show increasing white matter rarefaction and cystic degeneration, absence of inflammatory signs, diffuse loss of myelin fibers, conservation of grey matter structures, meager astrogliosis with dysmorphic astrocytes, and abnormal, foamy oligodendrocytes [2]. VWMD is caused by mutations in each of the five genes encoding for the five subunits (EIF2B1-5) of eukaryotic translation initiation factor 2B (eIF2B), which is essential for protein synthesis [1,2]. Because of the fact that EIF2B1-5 are housekeeping genes, many questions remain unanswered with respect to how perturbation of these ubiquitously expressed translation initiation factors should result in a highly selective neurological phenotype. We report the interesting pathological findings of a boy with VWMD for a missense mutation of EIF2B2 (V85E) caused by hemizygosity due to a de novo deletion.
Case report
A 4-year-old boy was found to have suddenly died during sleep. The clinical summary of this child up to 2 years, including magnetic resonance imaging (MRI) findings and chromosomal microarray testing results, has been reported previously [3]. He was born to healthy parents, and experienced clonic convulsions with facials twitches on the ipsilateral side lasting a few minutes at 10 months of age. Brain MRI revealed progressive dilatation of the lateral ventricles and vanishing white matter. After the onset of seizures, he began to deteriorate and did not recover to previous baseline measurements. He was then diagnosed with VWMD by identification of a submicroscopic deletion of 14q24.3, which occurred de novo on the paternal allele, and a maternally inherited missense mutation of EIF2B2 (V85E). Physical and mental development had begun to deteriorate after a bout of convulsive seizures, and he became worse than original levels of development. Subsequently, the developmental deficiencies continued to 4 years of age, without improvement. In view of his persistent neurological disease, we decided that he would benefit from percutaneous endoscopic gastrostomy, which was considered to be superior to gastric feeding 2 months before death. Soft palate movement disorders and mild central apnea were also observed. Cardiac abnormalities were not observed by a routine examination. He was discharged 10 days after surgery and then remained in a stable condition for approximately 1.5 months.
Autopsy findings
The deceased was 94 cm tall and weighed 10.1 kg (body mass index, 11.4). The brain weighed 564 g. Histopathological and immunohistochemical findings were evaluated by comparing the findings with a healthy control (4-year-old boy, died by accidental drowning). The external aspect of the present case revealed significant atrophy of the cerebrum and cerebellum, but there was little or no swelling of the brain. On coronal sections of the cerebrum, almost the entire deep cerebral white matter was grayish in color with severe rarefaction. However, no cystic or vacuolar degeneration was evident in the white matter. Moderately dilated lateral ventricles and thin atrophy of the corpus callosum and anterior commissure were also observed. Gray matter structures, including the cerebral cortex, basal ganglia, cerebellum, and brainstem nuclei, were relatively well preserved (Figure 1).
Figure 1.
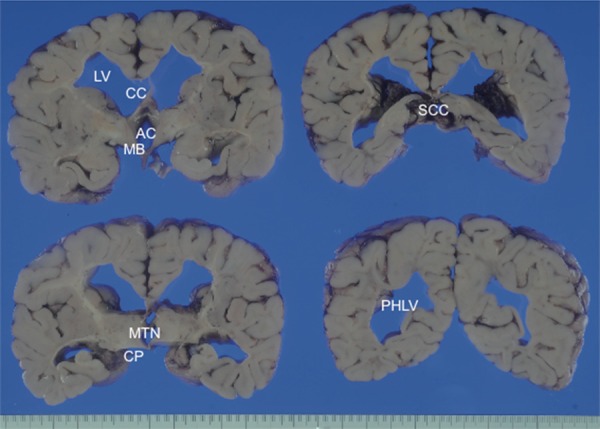
Coronal sections of the cerebral hemisphere through the mammillary, thalamus, cerebral peduncle, splenium of the corpus callosum, and posterior horn of the lateral ventricle. The white matter is grayish and clearly shows rarefaction. Abbreviations: AC anterior commissure; CC corpus callosum; CP cerebral peduncle; LV lateral ventricle; MB mammillary body; MTN medial thalamic nucleus; PHLV posterior horn of the lateral ventricle; SCC splenium of the corpus callosum.
Microscopically, near total absence of myelin in the deep cerebral white matter was found, and myelin fibers of subcortical U-fibers, the corpus callosum, the cerebral peduncles, the lower third part of the internal capsules, the fornix, red nuclei, pyramid tracts of medulla, and folia of the cerebellum were also severely affected. Additionally, myelin fibers running through the upper internal and external capsules, intrinsic fibers of the thalami, the optic tract, white matter surrounding the deep cerebellar nuclei, the raphe, transverse pontine fibers, as well as the spinocerebellar tract, anterior corticospinal tract, and anterior and lateral fascicles containing the spinoreticular and spinothalamic tract of the spinal cord, were relatively preserved (Figure 2A-E). The grey matter was mostly preserved, and six-layer lamination and neuronal density were completely preserved. Only mild loss of neuronal cells in CA1 of the hippocampus, Purkinje cells in the vermis of the cerebellum, and cerebellar granular cells was found as a result of recurrent hypoxia in the clinical course.
Figure 2.

Pathological findings in the cerebrum, cerebellum, and brainstem of VWMD. The stains used were LFB-H&E (A-E) and Holzer staining (F). (A) Coronal section of the left cerebral hemisphere at the level of the frontal lobe. The white matter is reduced in volume, showing a complete absence of myelin. (B) Coronal section of the left cerebral hemisphere through the thalamus and the cerebral peduncle. Temporal lobes of this patient show widespread loss of myelin and rarefaction of the white matter. Thalami are macroscopically preserved. (C) Transverse section of the midbrain. (D) Transverse section of the medulla oblongata. (E) Transverse section of the cerebellum, including the pons. Myelin fibers running through the cerebral peduncles are severely affected. (F) Gliosis of white matter in the present case was inconspicuous. Abbreviations: CN caudate nucleus; CP: cerebral peduncle; DN: dentate nucleus; EC: external capsule; HC: hippocampus; IC: internal capsule; ICP: inferior cerebellar peduncle; IO: inferior olive; MCP: middle cerebellar peduncle; MRN: medial reticular nucleus; MTN: medial thalamic nucleus; PY: pyramid tract (corticospinal tract); RN: red nucleus; Rn: raphe nucleus; TL: temporal lobe; TPF: transverse pontine fibers; Scale bar for (A-F) 10 mm.
In affected white matter, Holzer staining (Figure 2F) and immunohistochemistry for GFAP showed a lack of or mild gliosis. The density of oligodendrocytes was also decreased in severely affected white matter. Additionally, some of the oligodendrocytes showed an abnormal foamy appearance and an eccentric location of the nuclei (Figure 3A, 3B). Some astrocytes were dysmorphic with large blunt processes (Figure 3C, 3D).
Figure 3.
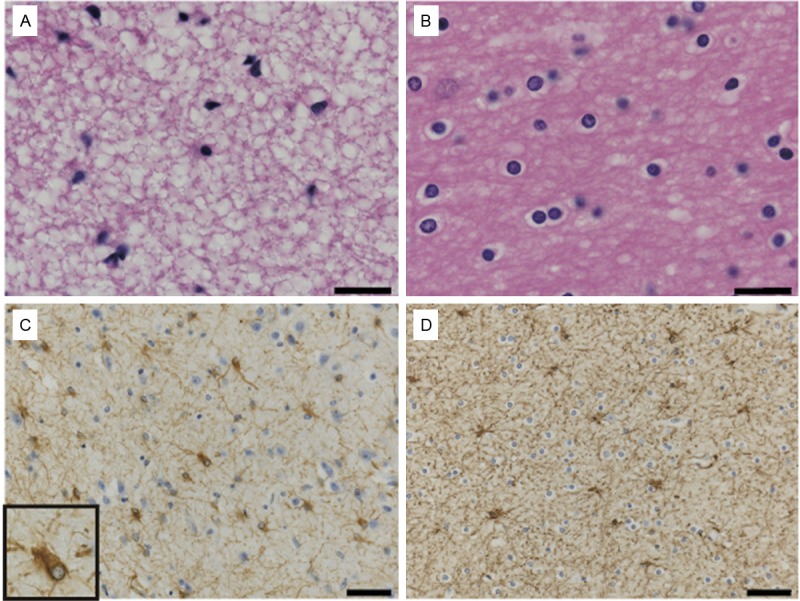
Astrocytes and oligodendrocytes in the white matter from the middle temporal lobe of a VWMD patient (A, C) and normal control (B, D). The stains used were LFB-H&E (A, B) and immunohistochemistry for GFAP (C, D). (A, B) The density of oligodendrocytes was lower in VWMD (A) compared with the control (B). Oligodendrocytes in our VWMD patient show an abnormal appearance. (LFB-H&E staining). A significant increase in astrocytes was not found in the severely affected temporal lobe of the VWMD (C) compared with the control (D). Astrocytes display abnormal morphology with coarse blunt processes in the inset of (C). (Immunohistochemistry for anti-GFAP). Scale bar for (A, B): 40 μm; (C, D): 20 μm.
Immunohistochemistry for Oligo2 exhibited no oligodendrocyte progenitors in severely affected lesions, such as white matter of the temporal lobe. Some positive cells for Oligo2 were found in white matter that was not considerably affected, but the incidence of positive labeling was not significantly different to the control case.
In the severely affected white matter, positive labeling for DNA damage-associated markers (anti-ssDNA and anti-histone H2AX [Ser139]) was not evident in astrocytes or oligodendrocytes. There were few Ki67-positive cells (cell proliferative marker), and the labeling index was low in all areas of white matter (<1%) and identical with that of the control.
The heart weighed 60 g. The heart was examined as previously reported [4]. The heart showed slightly fatty infiltration of the apex and left ventricle. The thickness of the anterior wall of the left ventricle, ventricular septum, and right ventricle was 0.7 cm, 0.7 cm, and 0.1 cm, respectively. The coronary arteries were normal in appearance. Histologically, mild fibrous and fatty tissue replacement of the myocardium was visible by microscopy (Figure 4A, 4B), in the presence of inflammation in the right ventricle (Figure 4C). Replacement of fibrous tissue of the myocardium was slightly visible by microscopy in the left ventricle (Figure 4D). The sinoatrial node and atrioventricular conduction systems were normal in appearance. No other clinically significant lesions were found in other organs.
Figure 4.

Microscopic appearance of the heart. (A, B, D) Elastica-Masson staining. (C) H&E staining. (A) Low-power view of the right ventricle. Moderate fibrous replacement of the myocardium is present. (B) High-power view of the right ventricle. (C) Higher magnification of the right ventricle. Mild fatty tissue replacement of the myocardium is present. (D) High-power view of the left ventricle. Moderate fibrous replacement of the myocardium is present. Scale bar for (A): 1 mm; (B): 50 μm; (C): 20 μm; and (D): 100 μm.
Discussion
The period of clinical onset of VWMD is divided into the prenatal/congenital form, subacute infantile form (onset age <1 year), an early childhood onset form (onset age of 1-5 years), a late childhood/juvenile onset form (onset age of 5-15 years), and an adult onset form. The clinical course of prenatal/congenital or early-infantile onset forms is more severe than that of the adult form [5-8]. In the present case, besides commonly affected areas, such as the deep cerebral area, U-fibers, spinocerebellar tract, lateral and anterior corticospinal tract, and anterolateral fascicles containing the spinoreticular and spinothalamic tract [2,6,7,9-12], areas that are reported to be relatively less severely affected were severely involved in the present case. These severely affected areas included the corpus callosum, cerebral peduncles, red nucleus, anterior commissure, fornix, and folia of the cerebellum. We consider that the cause of the clinical severity and other unusual pathological features of the present case may be related to the characteristic gene abnormality of the patient.
Although the volume of deep white matter of the present case was markedly decreased, the cystic rarefaction of white matter was not obvious. This finding is an interesting feature of the case, which may be different to previously presented cases [9-13]. Myelination occurs regionally, beginning with the brain stem at 29 weeks of gestation. A period of rapid myelination starts after the 5th month of delivery, and reaches a mature appearance by the 18th month. Most major tracts are significantly myelinated by early childhood, except in axons within the cortex, and in some regions, such as the arcuate fasciculus, a white matter bundle near the temporal lobe, which continues to myelinate into the second and third decades of life [14]. In cases that show typical cystic rarefaction of VWMD, the volume of white matter may develop in advance of starting the pathological process. However, in the present case with an extremely low volume of deep white matter, the process of abnormal myelination may have been caused in a different manner compared with typical cases.
Morphological and numerical changes in glial cells have been considered to be related to the pathogenesis of VWMD. The cellular pathology of astrocytes and oligodendrocytes in the present case is identical to that in previous reports [10,15]. A reduction in the number of astrocytes without significant gliosis in affected areas in our case is also similar to previously reported cases [15]. Bugiani et al. considered that the lack of significant gliosis in VWMD is caused by dysfunction of astrocytes. They also showed that there is a significant increase in the number of premyelinating oligodendrocyte pregenitors and paucity of myelin in VWMD [15]. However, while proliferation markers were entirely negative in severely affected lesions in the present case, the proliferative activity of relatively spared areas was not different from that in the control case. Additionally, although it has been suggested that apoptotic loss of oligodendrocytes is more frequently seen in infants and younger children in early lesions, there were no apoptotic cells in affected white matter in the present case. Immunohistochemistry results of the present case suggested that proliferative and apoptotic activity was low in affected areas. We speculate that the pathological course of the present case was already near termination at the period of death. Additionally, an extremely low volume of deep white matter without cystic degeneration observed in our patient may have been caused by congenital loss of myelination or initiation of the pathological process in the early stages of myelination.
Our patient had a submicroscopic deletion of 14q24.3, which occurred de novo on the paternal allele, and a maternally inherited missense mutation of EIF2B2 (V85E) [3]. To the best of our knowledge, the patient described in this paper is the first to be reported with a hemizygous EIF2B2 mutation. A homozygous mutation of V85 in EIF2B2 (V85E) has also been reported in adult-onset VWMD, and guanine nucleotide-exchange factor activity of eIF2B containing this mutant subunit is decreased by approximately 20% [16]. Because our patient had a submicroscopic deletion of 14q24.3, which included EIF2B2, the remaining allele with EIF2B2 (V85E) was the only copy available to produce EIF2B2. Although the EIF2B2 (V85E) allele may result in a mild decrease in guanine nucleotide-exchange factor activity in the eIF2B complex, the remaining EIF2B2 (V85E) allele may not provide enough protection from this severe phenotype. A hemizygous EIF2B2 mutation could explain the pathogenesis of disease severity in our patient.
Reports of other organ involvement of patients with VWMD have included hepatosplenomegaly, renal hypoplasia, pancreatitis [17], ovarian dysgenesis [18], and peripheral neuropathy [19]. Although our patient did not clinically exhibit symptoms of cardiac disease, fibrous and fatty tissue replacement of the myocardium with minimal lymphocytic infiltration was found. This pathological finding has not been previously reported in patients with VWMD. These findings in the heart were not so severe, but are similar to the findings of arrhythmogenic right ventricular cardiomyopathy (ARVC) [20,21]. We consider that these ARVC-like abnormalities might have caused his sudden unexpected death. However, the deletion on chromosome 14q24.3 was 2.0 Mb in size and gene-rich, containing 25 RefSeq genes [3]. Among 25 RefSeq genes, some are related to human disorders. The transforming-growth factor beta 3 gene (TGFβ3) is associated with ARVC, and is inherited as an autosomal dominant disorder [22], while controversy still remains about a clear relation between TGFβ3 and ARVC [23]. The mechanism underlying the phenotypic effects of copy number alterations involving 24 RefSeq genes, except for EIF2B2 at 14q24.3, has not clearly been established. However, TGFβ3 haploinsufficiency might be a candidate responsible for ARVD-like changes in the heart of the present case.
We demonstrated pathological findings of VWMD in our case, including different findings to previous reports. The defect EIF2B2 caused by hemizygosity might be due to early infantile-onset and pathogenesis of disease severity, related to the vulnerability of oligodendrocytes and astrocytes. Additionally, the characteristic mutation pattern of EIF2B2 in the present case might be related to the unusual cardiac pathology and sudden death.
Acknowledgements
The authors thank Ms. Syuko Kitora, Ms. Tamae Sasakura, Mr. Noboru Onozuka, and Mr. Osamu Yamamoto for their technical assistance.
Disclosure of conflict of interest
None.
References
- 1.Van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol. 2006;5:413–423. doi: 10.1016/S1474-4422(06)70440-9. [DOI] [PubMed] [Google Scholar]
- 2.Bugiani M, Boor I, Powers JM, Scheper GC, Van der Knaap MS. Leukoencephalopathy with vanishing white matter: A review. J Neuropathol Exp Neurol. 2010;69:987–996. doi: 10.1097/NEN.0b013e3181f2eafa. [DOI] [PubMed] [Google Scholar]
- 3.Shimada S, Miya K, Oda N, Watanabe Y, Kumada T, Sugawara M, Shimojima K, Yamamoto T. An unmasked mutation of EIF2B2 due to submicroscopic deletion of 14q24.3 in a patient with vanishing white matter disease. Am J Med Genet A. 2012;158A:1771–1777. doi: 10.1002/ajmg.a.35431. [DOI] [PubMed] [Google Scholar]
- 4.Hata Y, Mori H, Tanaka A, Fujita Y, Shimomura T, Tabata T, Kinoshita T, Yamaguchi Y, Ichida F, Kominato Y, Ikeda N, Nishida N. Identification and characterization of a novel genetic mutation with prolonged QT syndrome in an unexplained postoperative death. Int J Legal Med. 2014;128:105–115. doi: 10.1007/s00414-013-0853-4. [DOI] [PubMed] [Google Scholar]
- 5.Schiffmann R, Fogli A, Van der Knaap MS, Boespflug-Tanguy O. GeneReviews™ [Internet] Seattle (WA): University of Washington, Seattle; 2003. Childhood Ataxia with Central Nervous System Hypomyelination/Vanishing White Matter; pp. 1993–2013. http://www.ncbi.nlm.nih.gov/books/NBK1258/ [PubMed] [Google Scholar]
- 6.Francalanci P, Eymard-Pierre E, Dionisi-Vici C, Boldrini R, Piemonte F, Virgili R, Fariello G, Bosman C, Santorelli FM, Boespflug-Tanguy O, Bertini E. Fatal infantile leukodystrophy: a severe variant of CACH/VWM syndrome, allelic to chromosome 3q27. Neurology. 2001;57:265–270. doi: 10.1212/wnl.57.2.265. [DOI] [PubMed] [Google Scholar]
- 7.Fogli A, Wong K, Eymard-Pierre E, Wenger J, Bouffard JP, Goldin E, Black DN, Boespflug-Tanguy O, Schiffmann R. Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann Neurol. 2002;52:506–510. doi: 10.1002/ana.10339. [DOI] [PubMed] [Google Scholar]
- 8.Fogli A, Dionisi-Vici C, Deodato F, Bartuli A, Boespflug-Tanguy O, Bertini E. A severe variant of childhood ataxia with central hypomyelination/vanishing white matter leukoencephalopathy related to EIF21B5 mutation. Neurology. 2002;59:1966–1968. doi: 10.1212/01.wnl.0000041666.76863.47. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez D, Gelot A, della Gaspera B, Robain O, Ponsot G, Sarlieve LL, Ghandour S, Pompidou A, Dautigny A, Aubourg P, Pham-Dinh D. Increased density of oligodendrocytes in childhood ataxia with diffuse central hypomyelination (CACH) syndrome: neuropathological and biochemical study of two cases. Acta Neuropathol. 1999;97:469–480. doi: 10.1007/s004010051016. [DOI] [PubMed] [Google Scholar]
- 10.Wong K, Armstrong RC, Gyure KA, Morrison AL, Rodriguez D, Matalon R, Johnson AB, Wollmann R, Gilbert E, Le TQ, Bradley CA, Crutchfield K, Schiffmann R. Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta Neuropathol. 2000;100:635–646. doi: 10.1007/s004010000234. [DOI] [PubMed] [Google Scholar]
- 11.Brück W, Herms J, Brockmann K, Schulz-Schaeffer W, Hanefeld F. Myelinopathia centralis diffusa (vanishing white matter disease): evidence of apoptotic oligodendrocyte degeneration in early lesion development. Ann Neurol. 2001;50:532–536. doi: 10.1002/ana.1227. [DOI] [PubMed] [Google Scholar]
- 12.Van Haren K, van der Voorn JP, Peterson DR, van der Knaap MS, Powers JM. The life and death of oligodendrocytes in vanishing white matter disease. J Neuropathol Exp Neurol. 2004;63:618–630. doi: 10.1093/jnen/63.6.618. [DOI] [PubMed] [Google Scholar]
- 13.van der Knaap MS, Barth PG, Gabreels FJ, Franzoni E, Begeer JH, Stroink H, Rotteveel JJ, Valk J. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48:845–855. doi: 10.1212/wnl.48.4.845. [DOI] [PubMed] [Google Scholar]
- 14.Lenroot RK, Giedd JN. Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci Biobehav Rev. 2006;30:718–729. doi: 10.1016/j.neubiorev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Bugiani M, Boor I, van Kollenburg B, Postma N, Polder E, van Berkel C, van Kesteren RE, Windrem MS, Hol EM, Scheper GC, Goldman SA, van der Knaap MS. Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol. 2011;70:69–82. doi: 10.1097/NEN.0b013e318203ae74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsukawa T, Wang X, Liu R, Wortham NC, Onuki Y, Kubota A, Hida A, Kowa H, Fukuda Y, Ishiura H, Mitsui J, Takahashi Y, Aoki S, Takizawa S, Shimizu J, Goto J, Proud CG, Tsuji S. Adult-onset leukoencephalopathies with vanishing white matter with novel missense mutations in EIF2B2, EIF2B3, and EIF2B5. Neurogenetics. 2011;12:259–261. doi: 10.1007/s10048-011-0284-7. [DOI] [PubMed] [Google Scholar]
- 17.van der Knaap MS, van Berkel CG, Herms J, van Coster R, Baethmann M, Naidu S, Boltshauser E, Willemsen MA, Plecko B, Hoffmann GF, Proud CG, Scheper GC, Pronk JC. eIF2B-related disorders: antenatal onset and involvement of multiple organs. Am J Hum Genet. 2003;73:1199–1207. doi: 10.1086/379524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fogli A, Rodriguez D, Eymard-Pierre E, Bouhour F, Labauge P, Meaney BF, Zeesman S, Kaneski CR, Schiffmann R, Boespflug-Tanguy O. Ovarian failure related to eukaryotic initiation factor 2B mutations. Am J Hum Genet. 2003;72:1544–1550. doi: 10.1086/375404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Federico A, Scali O, Stromillo ML, Di Perri C, Bianchi S, Sicurelli F, De Stefano N, Malandrini A, Dotti MT. Peripheral neuropathy in vanishing white matter disease with a novel EIF2B5 mutation. Neurology. 2006;67:353–355. doi: 10.1212/01.wnl.0000225077.40532.a5. [DOI] [PubMed] [Google Scholar]
- 20.Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases? Circulation. 1998;97:1571–1580. doi: 10.1161/01.cir.97.16.1571. [DOI] [PubMed] [Google Scholar]
- 21.Chimenti C, Pieroni M, Maseri A, Frustaci A. Histologic findings in patients with clinical and instrumental diagnosis of sporadic arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2004;43:2305–2313. doi: 10.1016/j.jacc.2003.12.056. [DOI] [PubMed] [Google Scholar]
- 22.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Campuzano O, Alcalde M, Allegue C, Iglesias A, García-Pavía P, Partemi S, Oliva A, Pascali VL, Berne P, Sarquella-Brugada G, Brugada J, Brugada P, Brugada R. Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2013;50:280–289. doi: 10.1136/jmedgenet-2013-101523. [DOI] [PubMed] [Google Scholar]