

Abstract
To determine the effects of adiponectin on human placenta during gestational diabetes mellitus (GDM) and on high glucose (HG)-induced BeWo cell proliferation. We examined the expression levels of adiponectin in control and GDM placenta using quantitative real-time PCR, Western blot, and immunohistochemistry (IHC). Cell proliferation and viability were assessed using a colorimetric assay (cell counting kit-8), PCNA immunocytochemical staining, and Western blot analysis of cyclin D1. Transfection of siRNA against c-jun was performed using Lipofectamine 2000. Cell cycle analysis was performed using propidium iodide staining and flow cytometry. Results show a decreased expression of adiponectin and an increased degree of trophoblast cell proliferation in GDM placenta compared to the normal placenta. Similarly, HG can promote BeWo cell proliferation that is associated with adiponectin down-regulation. This proliferation could be depressed by addition of exogenous adiponectin, i.e. adiponectin exerts antiproliferative effects on HG-induced trophoblast cells. Adiponectin suppresses the HG-induced BeWo cell proliferation by inhibiting the activation of JNK/c-jun. In conclusion, adiponectin inhibits HG-induced proliferation of BeWo cells through down-regulation of JNK/c-jun phosphorylation.
Keywords: Adiponectin, gestational diabetes mellitus, placenta, JNK/c-Jun
Introduction
Pregnancy, a state of insulin resistance, is associated with elevated levels of various cytokines resulting in profound alterations in the maternal metabolism-geared towards ensuring adequate supply of nutrition to the fetus. Placental functions are regulated by hormones and cytokines that are produced either locally or distally which together coordinate normal placental development and fetal growth [1-3].
Adiponectin, a 30-kDa protein, is an adipocytokine that has been shown to have anti-atherogenic, anti-inflammatory, and anti-diabetic roles [4,5]. In contrast to the elevated levels of cytokines such as interleuckin-6 (IL-6), tumor necrosis factor (TNF-α), and leptin, serum adiponectin levels are lower in insulin-resistant states [6]. Adiponectin is also known to have antiproliferative effects in various human cells [7] such as hepatic stellate cells [8], colon cancer cell [9]. Adiponectin orchestrates its effects by activating two seven-transmembrane domain receptors: adiponectin receptor type 1 (ADIPOR1) and adiponectin receptor type 2 (ADIPOR2) [10]. Binding of adiponectin to these receptors activates various signal transduction pathways such as the AMP-activated protein kinase (PRKA), phosphoinositide-3-kinase (PIK3), mitogen-activated protein kinase (P38/P42/P44 MAPK), and the JUN kinase pathways [11,12].
It has been reported that the plasma adiponectin levels are reduced in women with GDM [13,14]. Furthermore, umbilical vein serum adiponectin levels were three-fold higher than maternal serum adiponectin levels in normal pregnancies [15]. It has also been established that the human placenta is a source of adiponectin [16]. According to the new American Diabetes Association (ADA) criteria made after the HAPO study, GDM is defined as the glucose intolerance that first occurs or is first identified during pregnancy-a condition that complicates 15-20% of all pregnancies [17]. GDM is a state of increased insulin resistance that can lead to complications during pregnancy. Although the etiology of GDM has not been identified, increasing body of evidence suggests that the placenta, the organ connecting the developing fetus to the maternal body, plays an important role in the process [18,19]. In turn, metabolic dysfunctions in GDM, such as hyperglycemia, hyperinsulinemia, and dyslipidemia can induce changes in the cytokines leading to the abnormalities of the placenta and trophoblast cells [20].
Previous studies have reported that GDM can induce apoptosis of trophoblast cells. However, large for gestational babies (macrosomia) are much more common than small for gestational babies (fetal growth restriction) delivered by GDM mothers in clinics. In light of these findings, we hypothesized that there are changes which can be found in GDM placenta and adiponectin is likely playing a role in this process. Therefore, we determined the expression level of adiponectin and the proliferation of trophoblast cells in the placenta with different glucose tolerance states-GDM versus normal healthy controls. Furthermore, we examined if adiponectin is involved in the molecular mechanisms controlling proliferation of trophoblastic cells. We further explored the antiproliferative effect of adiponectin, modulated via the JNK/c-Jun signal transduction pathway. Finally, we studied the direct effects of adiponectin on growth capacities of BeWo choriocarcinoma cell line, which is commonly used as model system for trophoblast cells.
Materials and methods
Ethics statement
This study was approved by the Research Ethical Committee of the First Affiliated Hospital of Sun Yat-sen University. Written informed consent was obtained from all volunteers.
Subjects
Thirty pregnant women with GDM were recruited for the GDM group. The diagnosis of GDM was based on the new ADA criteria made after the HAPO study. To prevent potential interference from insulin or metformin, only diet controlled GDM subjects were recruited for the study. The control group consisted of thirty healthy pregnant women with normal glucose tolerance (NGT) during pregnancy.
Sample collection
To improve homogeneity and comparability, placentas were selected from subjects undergoing Caesarean sections for breech presentation, maternal request, uterine scar, or possible fetal distress. Placenta villi of approximately 3 cm3 were obtained from five different intact cotyledons immediately following the delivery. After the maternal decidual layer was removed, the tissue was quickly washed with ice-cold PBS, snap-frozen in liquid nitrogen, and stored at -80°C until needed for analysis. Samples for immunohistochemistry were fixed in 4% paraformaldehyde for three days and embedded in paraffin before sectioning.
Protein isolation and Western blotting
Expression levels of adiponectin in normal and GDM placentas were evaluated by Western blot. Placental tissue was first washed with cold PBS and homogenized in 1 ml of RIPA lysis buffer per 100 mg of tissue using a homogenizer. Samples were centrifuged at 10,000 × g for 10 minutes at 4°C. Protein concentration was determined using Bradford protein assay (Forevergen bioscience, China). Western blot was performed according to routine procedures.
Expression of adiponectin/PCNA in normal and GDM placenta by IHC
Immunohistochemical staining of placental tissue using adiponectin/PCNA antibodies was performed to examine the level of adiponectin and cell proliferation respectively. Human placental tissue was first washed with cold PBS, fi xed with 4% paraformaldehyde, embedded in paraffi n and sliced into sections (5 micron thickness). Deparaffinized and hydrated tissue sections were subjected to microwave heating in 0.1 M sodium citrate buffer (pH 6.0) for antigen retrieval. The endogenous peroxidase activity was quenched and non-specific binding was blocked with non-immune sera as described earlier. Visualization was achieved using Vectastain ABC Peroxidase Elite Kit (Vector Laboratories, Burlingame, CA, USA) and freshly prepared 3,3’-diaminobenzidine (DAB) tetrahydrochloride (Sigma Chemical Company, St Louis, MO, USA) as substrate. Expression of adiponectin was evaluated by IPP (version 6.0; Media Cybernetics, Silver Spring, Maryland, USA). Ten digital images at 1360 × 1024 pixel resolution, 200 × magnification, were captured by the BX51WI microscope (Olympus, Japan) for every section. Optical density was calibrated, the image was converted to gray scale, and the integral optical density (IOD) was counted. Number of cells expressing PCNA was counted from 10 representative fields.
RNA isolation and qPCR
Transcript levels of adiponectin in normal and GDM placenta by quantitative real-time PCR (qRT-PCR). To determine mRNA levels of adiponectin in normal and GDM placenta, we performed qRT-PCR experiments. Placental tissue was washed with cold PBS and homogenized in 1 ml of TRIZOL reagent per 100 mg of tissue using a homogenizer. RNA was isolated according to the manufacturer’s instructions. Reverse transcription was performed using the Revert Aid First Strand cDNA Synthesis Kit (Fermentas, USA). The real-time PCR amplification was performed on MiniOpticon™ Real-Time PCR Detection System with the following primers and probe SYBR Green adiponectin primers: Forward primer 5’-TATGATGGCTCCACTGGTA-3’ Reverse primer 5’-GAGCATAGCCTTGTCCTTCT-3’ GAPDH primers: Forward primer 5’-GAGTCAACGGATTTGGTCGT-3’ Reverse primer 5’-GACAAGCTTCCCGTTCTCAG-3’.
Cell proliferation analysis
BeWo cells were seeded in 96-well plates at the density of 1000 cells per well in 100 μl of complete culture medium. After treatments with glucose (25 mM) and adiponectin [20 μg/ml] [8], three methods were utilized for cell proliferation analysis. Cell Counting Kit-8 (CCK-8) analysis: 10 ul of CCK-8 was added to each well. The culture plates were shaken for 10 min and the optical density (OD) values were read at 450 nm. To investigate protein expression levels of Cyclin D1 in BeWo cells, we carried out Western blot analysis on the control and treated cells. Proliferating cell nuclear antigen (PCNA) is an evolutionarily well-conserved protein found in all eukaryotic species. We performed an immunocytochemical assay examining PCNA to analyze BeWo cell proliferation.
Analysis of cell cycle by propidium iodide staining and flow cytometry
BeWo cells were seeded in 96-well plates at the density of 1000 cells per well in 100 μl of complete culture medium. After treatments with glucose (25 mM) and adiponectin, cells were harvested and fixed in cold 70% ethanol overnight at -20°C. Cells were then washed with PBS, treated with RNaseA (50 μg/ml), and stained with propidium iodide (25 μg/ml) for 30 min at 37°C. Samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and distribution of the cell-cycle phases was determined using Modfit Software (BD Biosciences).
Effect of adiponectin on BeWo choriocarcinoma cell lines
Human choriocarcinoma BeWo cell line, obtained from American Type Culture Collection (Manassas, VA), was maintained under standard culture conditions of 5% CO2 atmosphere at 37°C. Cells were cultured in phenol red-free DMEM/F12 medium supplemented with 15% fetal calf serum (FCS) and antibiotics [streptomycin (0.1 mg/ml) and penicillin (100 U/ml)]. Control and GDM BeWo cells were treated with high glucose and adiponectin.
Signal transduction pathway of adiponectin
BeWo cells were cultured in 6-well plates and then treated with glucose (25 mM), adiponectin, and with one of the various inhibitors tested. Total protein concentration was determined and Western blot analyses were performed to investigate protein levels involved in p-JNK/JNK pathway.
Statistical analysis
Data were analyzed by SPSS19.0 database. The results were expressed as mean ± SEM and statistical analysis was carried out using independent student’s t-test or one-way analysis of variance (ANOVA). P values of < 0.05 were considered significant.
Results
Baseline characteristics of the research populations
Clinical and laboratory data were compared between groups of 30 pregnant women with either GDM or NGT. As shown in Table 1, no statistically significant differences can be found between GDM and NGT groups in parity, gravidity, pre-gravidity and pre-partum BMI, gestational age, SBP (systolic blood pressure) and DBP (diastolic blood pressure) at admission, newborn gender, birth length, neonatal head circumference, shoulder circumference, upper arm circumference, placental diameter, and rate of admission to neonate intensive care unit (NICU). Pregnant women with GDM were older, had higher fasting glucose levels in early pregnancy, and had higher glucose levels at each time point of the oral glucose tolerance test (OGTT). They also had a higher glycated hemoglobin (HbA1c) level than women in the NGT group. GDM mothers had heavier fetuses (at birth) and placentas as compared to NGT mothers. Although statistically insignificant, the GDM group had 3 macrosomias compared to none in the NGT group.
Table 1.
Baseline clinical characteristics and biochemical parameters of the research population (Mean ± SD)
| Gestational diabetes Mellitus (N = 30) | Normal Glucose Tolerance (N = 30) | P | |
|---|---|---|---|
| Maternal parameters | |||
| Age (year) | 32.30 ± 4.43 | 29.23 ± 4.11 | 0.007 |
| Gravidity | 2.13 ± 1.36 | 1.67 ± 0.84 | 0.115 |
| Parity | 1.33 ± 0.61 | 1.10 ± 0.31 | 0.065 |
| Pre-gravidity BMI (kg/m2) | 21.87 ± 2.48 | 20.87 ± 2.68 | 0.138 |
| Pre-partum BMI (kg/m2) | 26.89 ± 2.65 | 26.81 ± 3.20 | 0.921 |
| FBG in 1 st trimester (mmol/l) | 4.64 ± 0.72 | 4.27 ± 0.31 | 0.012 |
| OGTT-fast (mmol/l) | 4.58 ± 0.55 | 4.20 ± 0.31 | 0.002 |
| OGTT-1 h (mmol/l) | 9.81 ± 1.28 | 6.93 ± 1.32 | 0.000 |
| OGTT-2 h (mmol/l) | 9.10 ± 1.14 | 6.39 ± 1.04 | 0.000 |
| HbA1c (%) | 5.14 ± 0.49 | 4.91 ± 0.32 | 0.036 |
| SBP (mmHg) | 114.57 ± 10.24 | 117.37 ± 7.70 | 0.236 |
| DBP (mmHg) | 70.93 ± 7.13 | 72.73 ± 6.14 | 0.299 |
| Fetal Parameters | |||
| Gestational Age (week) | 38.67 ± 1.06 | 39.14 ± 1.04 | 0.090 |
| Gender | |||
| Male (n, %) | 17, 56.7 | 15, 50 | 0.605 |
| Female (n, %) | 13, 43.3 | 15, 50 | 0.605 |
| Birth Weight (kg) | 3.40 ± 0.36 | 3.20 ± 0.30 | 0.026 |
| Birth Length (cm) | 50.00 ± 1.60 | 49.80 ± 1.94 | 0.664 |
| Macrosomia (n, %) | 3, 10 | 0, 0 | 0.236 |
| Head Circumference (cm) | 34.49 ± 1.30 | 34.46 ± 1.56 | 0.950 |
| Shoulder Circumference (cm) | 36.18 ± 2.54 | 35.79 ± 2.11 | 0.523 |
| Upper Arm Circumference (cm) | 11.39 ± 0.94 | 11.68 ± 0.87 | 0.209 |
| Placental diameter (cm) | 20.13 ± 3.23 | 20.10 ± 2.22 | 0.963 |
| Placental weight (kg) | 0.66 ± 0.08 | 0.56 ± 0.06 | 0.000 |
| NICU admission rate (n, %) | 6, 20 | 3, 10 | 0.470 |
BMI = body mass index; FBG = fasting blood glucose; OGTT = oral glucose tolerance test; HbA1c = Glycated hemoglobin; SBP = systolic blood pressure; DBP = diastolic blood pressure; NICU = neonate intensive care unit.
Human adiponectin expression in GDM and normal placental tissue
Previous findings showed that the human placenta is a source of adiponectin. We employed RT-PCR, Western blotting, and immunohistochemistry to quantify the expression of adiponectin in human placenta of different glucose tolerance states; GDM versus NTG. To analyze the mRNA expression level of adiponectin, we carried out qRT-PCR experiments with corresponding cDNA generated from total RNA and results normalized to the GAPDH expression. The results demonstrate that the level of adiponectin mRNA was significantly decreased in GDM patients compared with the healthy controls (Figure 1A). In agreement with the quantitative RT-PCR results, Western blotting also showed a decreased expression of adiponectin in GDM placenta (Figure 1B). Strengthening the findings further, IHC also showed decreased expression level of adiponectin in GDM placenta, while immunoreactivity showed cytoplasmic expression (Figure 1C).
Figure 1.
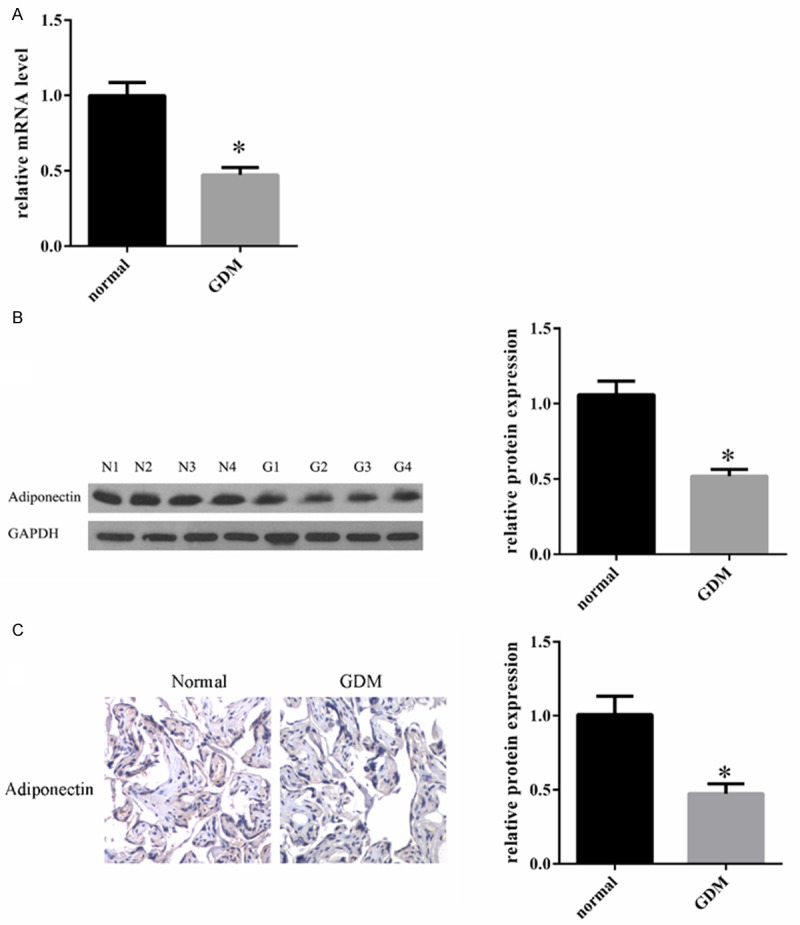
Human adiponectin expression in GDM and normal placental tissue. A. Quantitative RT-PCR analysis of adiponectin mRNA in GDM and normal placental tissue. Transcript level of adiponectin in GDM was relatively low as compared to the normal placental tissues. B. Western blot analysis for the detection of adiponectin. One hundred micrograms of total protein was loaded and separated on a 10% SDS-PAGE gel. To confirm equal loading, the same blot was stripped and probed with a monoclonal GAPDH antibody. N: normal group; G: GDM group. C. Immunohistochemical analysis for the expression of adiponectin in GDM and normal placental tissue. Immunohistochemical adiponectin staining was performed. Adiponectin expression was lower in the GDM group compared to the normal group. Qualified statistical data are the mean ± SE of at least 3 separate experiments. *, P < 0.05.
Cell proliferation in human placenta trophoblast cells of different glucose tolerance states; normal and GDM
The IHC (Figure 2A) and statistics (Figure 2B) results showed a significantly increased degree of trophoblast cell proliferation in the GDM placenta in comparison to the normal controls.
Figure 2.
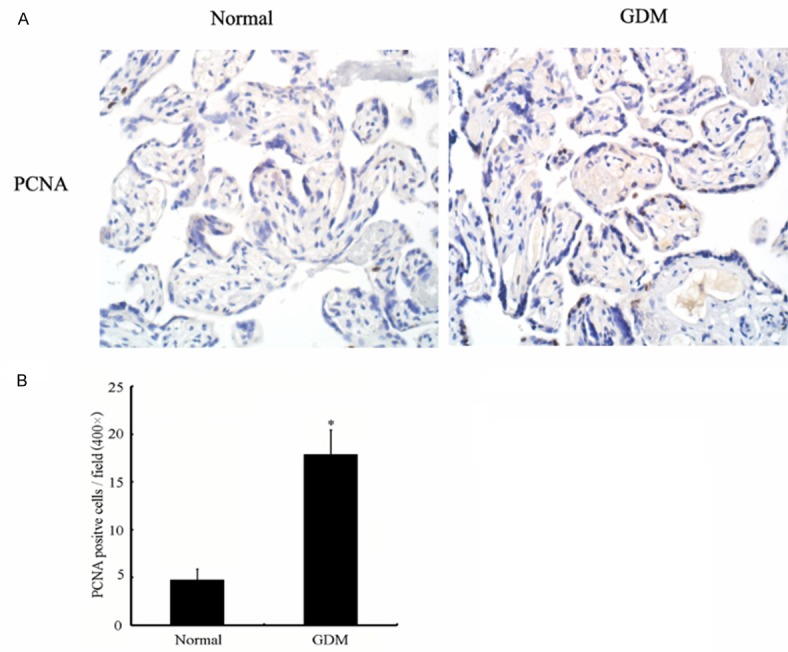
Cell proliferation in human placenta trophoblast cells of different glucose tolerance states; normal and GDM. A. Histological sections were immunostained for PCNA expression (see text). The number of trophoblasts in 10 representative fields was counted. Cell proliferation was quantified by the percentage of trophoblasts stained positive for PCNA. B. There are a significantly higher percentage of PCNA positive cells in GDM than in normal group. Qualified statistical data are the mean ± SE of at least 3 separate experiments. *, P < 0.05.
Effect of HG on BeWo cells and adiponectin expression. Our previous studies showed that in GDM patients, proliferation of human placenta trophoblast cells increased while expression of adiponectin decreased. Next we tested the effect of HG on the growth potential of BeWo cells–a placental cell line that has been widely used as an in vitro model for the placenta and for the study of adiponectin expression. Figure 3A shows the results of CCK-8 assays where high glucose markedly enhanced the proliferation potential of BeWo cells in contrast with control or high mannitol treated cells. In a time course CCK-8 assay, high glucose promoted cell growth for up to the analysis time of five day. Compared with the high mannitol treated cells, the protein levels of Cyclin D1 increased significantly in high glucose treated cells (Figure 3B). To understand the relationship between BeWo cell proliferation and adiponectin, we investigated the effect of high glucose on adiponectin expression. Interestingly, compared to the control high mannitol treated cells, expression of adiponectin significantly decreased as the proliferation of BeWo cells increased (Figure 3C). These results indicate that high glucose can promote BeWo cell proliferation that is associated with adiponectin down-regulation.
Figure 3.
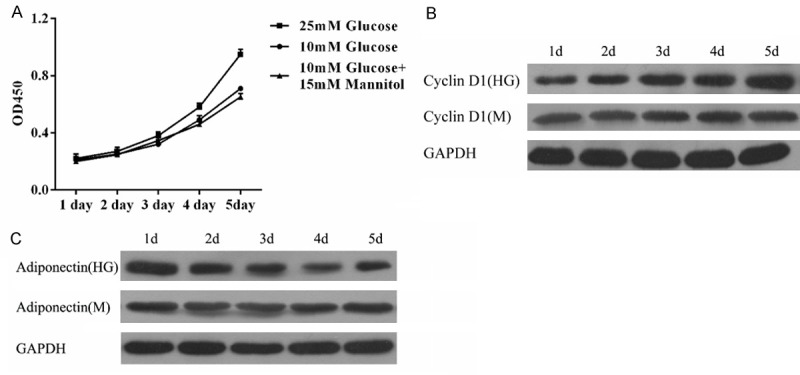
Effect of HG on BeWo cells and adiponectin expression A. Proliferation potential of control, HG, and high mannitol treated BeWo cells as determined by the CCK-8 assay. The data represent means ± S.D. of three independent experiments each performed in triplicates. Mannitol was used as an osmotic control. B. HG increases the expression of Cyclin D1 as detected by Western blot analysis. C. Western blot analysis of control, HG, and high mannitol treated BeWo cells for adiponectin protein expression.
Effect of adiponectin on BeWo cell proliferation Our previous data suggested a crosstalk between adiponectin and different glucose tolerance states. We hypothesized that adiponectin exerts a regulatory role on the proliferation of BeWo cells. Therefore, we examined the anti-proliferative effect of adiponectin on BeWo cells. As shown in Figure 4A, adiponectin + high glucose treatment of BeWo cells resulted in an inhibition of cell growth compared with the high glucose treatment alone. The anti-proliferative action of adiponectin was also confirmed by the cell cycle analysis performed by staining DNA with propidium iodide followed by flow cytometry. Results showed a statistically significant increase in the percentage of cells in the G0/G1 phase and a decrease in the percentage of cells in the G2/M phase after adiponectin + high glucose treatment compared with the high glucose treatment alone (Figure 4B). Finally, we estimated proliferative activity of adiponectin by immunostaining for PCNA. The number of PCNA-positive cells was significantly lower with adiponectin + high glucose treatment than with the high glucose treatment alone (Figure 4C and 4D), indicating an inhibitory role of adiponectin on BeWo cell proliferation.
Figure 4.
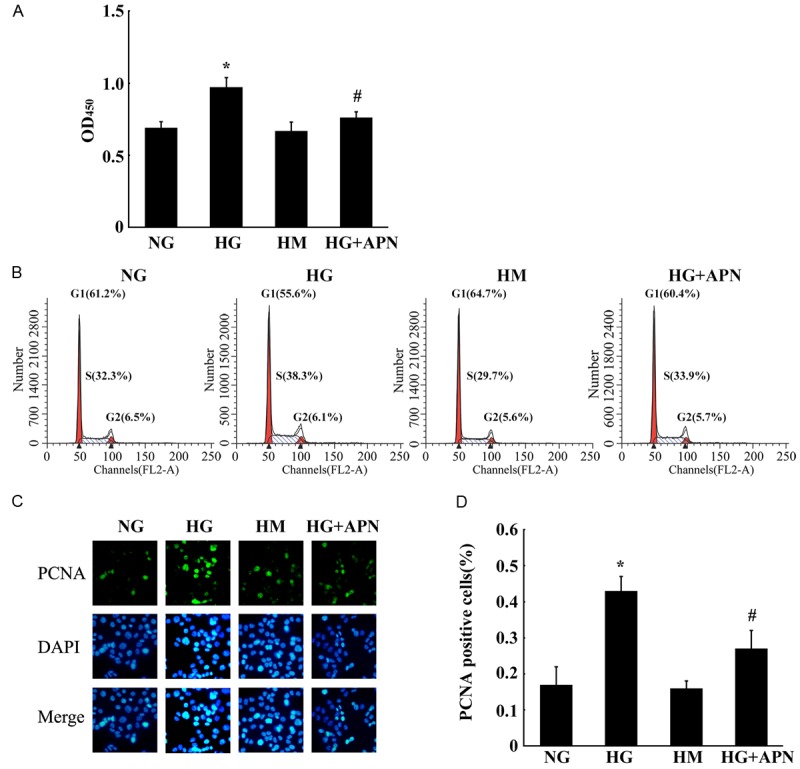
Effect of adiponectin on BeWo cell proliferation. A. Adiponectin inhibited proliferation of BeWo cells as determined by the CCK-8 assay. Bar graph shows the extent of BeWo cell proliferation upon various treatments. No significant change was found for the high mannitol compared with the normal glucose control groups. The proliferation of the high glucose group was increased markedly compared with the controls. The proliferation of the high glucose + adiponectin group was decreased significantly compared with the high glucose group. B. The cell cycle distribution of BeWo cells after different treatments. Statistically significant differences between samples were observed using the Student’s t-test (P < 0.05). C. Adiponectin down regulates the expression of PCNA. Immunocytochemistry for PCNA was performed at the end of the incubation period. DAPI staining was used to visualize the nuclei. D. Image analysis was performed to quantify the percentage of positive cells (blue nuclear staining). Mean ± SEM of 3 independent experiments done in triplicates is presented *, #, P < 0.05.
Effects of adiponectin on HG-triggered phosphorylation of JNK and c-Jun in proliferation of BeWo cells
The JNK/c-Jun pathway regulates proliferation and/or death in several cell types [21,22]. Therefore, we tested whether HG stimulation of BeWo cell proliferation was mediated by the JNK/c-Jun pathway. As shown in Figure 5A, JNK1/2 and c-Jun phosphorylations were upregulated in high glucose treated cells. We blocked JNK activity using SP600125, a selective inhibitor of JNK1 and JNK2. SP reduced the proliferation of BeWo cells to levels comparable to those in controls (Figure 5B). We observed similar effects using another MLK inhibitor, CEP11004. These findings demonstrate that the increased activation of the JNK/c-Jun pathway is essential for HG-induced proliferation of BeWo cells. To confi rm that the elevated c-jun levels contributed to the increased proliferation of BeWo cells, we knocked-down c-Jun by transient expression of c-Jun siRNA in BeWo cells. The knockdown was confi rmed by the Western blot analysis of c-jun expression, while the non-targeting control siRNA had no effect (Figure 5C). When we measured cell proliferation, we observed that the numbers of c-jun siRNA-expressing cells increased at a signifi cantly lower rate when compared with cells expressing control siRNA (Figure 5D). This result confi rms that elevated c-jun expression is suffi cient to increase HG-induced proliferation of BeWo cells. Our previous study demonstrated that HG-induced proliferation of BeWo cells was mediated by JNK/c-jun signaling. We evaluated if adiponectin can inhibit HG-induced JNK/c-jun phosphorylation in BeWo cells exposed to high concentrations of glucose. Western blot analysis showed that adiponectin inhibited the HG-induced phosphorylation of JNK and c-Jun (Figure 5E). These results suggest that adiponectin suppresses the HG-induced BeWo cell proliferation by inhibiting the activation of JNK/c-jun.
Figure 5.
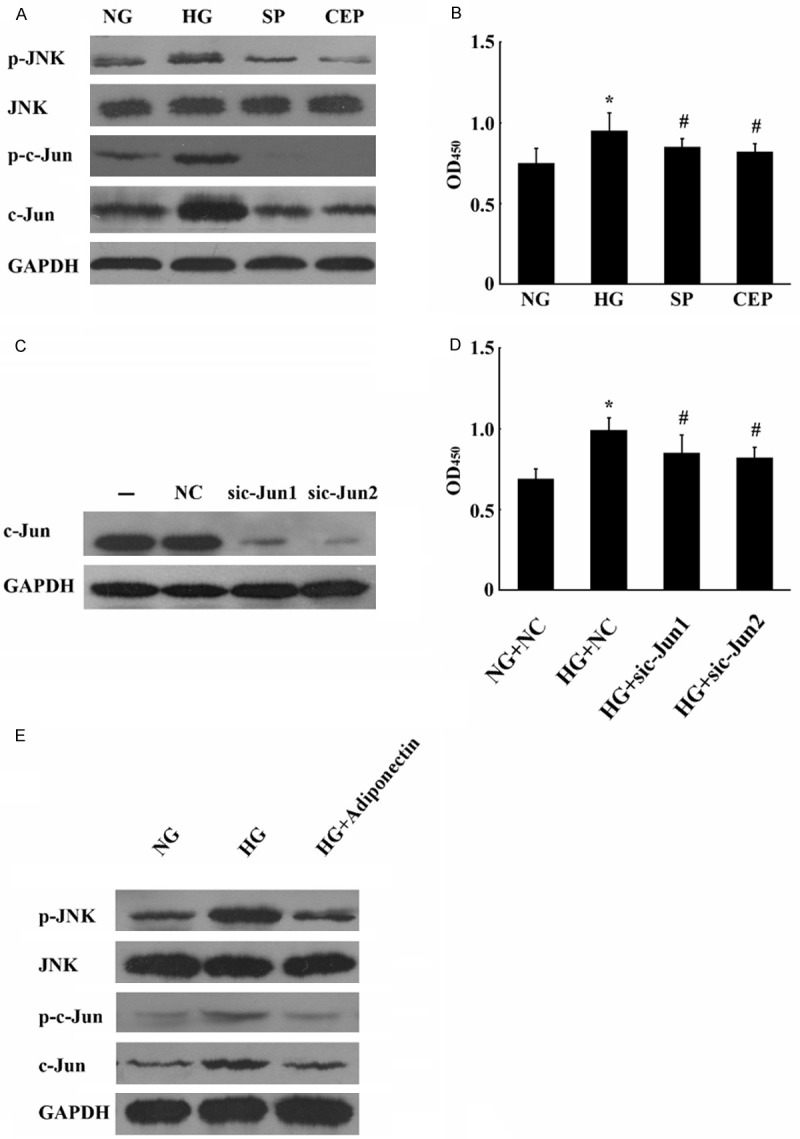
Effects of adiponectin on HG-triggered phosphorylation of JNK and c-Jun in proliferation of BeWo cells. JNK/c-jun mediates the up-regulation of HG-induced proliferation of BeWo cells. A. Western blots of JNK1/2 and c-Jun show increased phosphorylation in HG-induced BeWo cell proliferation. GAPDH was used as an internal control. B. SP and CEP inhibit glucose-induced c-Jun phosphorylation causing reduced proliferation of BeWo cells. C. BeWo cells were cultured in high glucose conditions, transfected with c-Jun siRNA (200 pmol) , and subsequently analyzed by Western blotting for c-Jun and GAPDH as control. Mock transfections without siRNA or with non-targeting siRNA served as controls. D. c-Jun is required for BeWo cell proliferation. E. Effects of adiponectin on JNK/c-Jun pathway in the HG-induced proliferation of BeWo cells. BeWo cells were cultured in either normal glucose (NG), HG, or HG + adiponectin. The cells were harvested and Western blots were probed for JNK/c-Jun and their activated phospho-counterparts. GAPDH was used as an internal control. Mean ± SEM of 3 independent experiments done in triplicates is presented. *, #, P < 0.05.
Discussion
Gestational diabetes mellitus (GDM) has negative effects on the mother and on the offspring in both short- and long-terms. Women with GDM have a higher risk for developing additional complications during pregnancy, and are more likely to suffer from type-2 diabetes following the pregnancy. Offsprings of GDM mothers not only have higher morbidity and adverse infant outcomes, but also have an increased risk of diabetes, obesity, and cardiovascular disease in their adult lives.
The placenta plays an important role in the development of GDM. Hyperglycemia, hyperinsulinemia, and other metabolic dysfunctions in GDM patients in turn induce changes in cytokines causing abnormalities of the placenta and trophoblast cells.
In the present study, we demonstrate that the expression of adiponectin decreased in the GDM placenta. Furthermore, we present novel data showing that the proliferation of trophoblast cells increased in GDM placenta. Taken together, these findings strongly suggest that adiponectin has antiproliferative effects on human placenta.
Cytokines such as TNF-α and IL-6, through their ability to interfere with insulin signalling, have been implicated in insulin resistance in type 2 diabetes. In humans, a significant negative correlation exists between plasma adiponectin levels and insulin resistance states such as the type 2 diabetes and obesity, suggesting crosstalk between adiponectin and different glucose tolerance states.
Recently, it has been shown that plasma adiponectin levels are reduced in pregnancy-induced insulin resistance and the reduced plasma adiponectin correlates negatively with neonatal weight. When adiponectin levels were measured in cord vein, a striking increase in plasma adiponectin concentrations with gestational age was noted; concentrations at term were more than 20-fold higher compared with concentrations at 24 weeks of gestation. Based on our present data and the fact that adiponectin is elevated during pregnancy, it is tempting to speculate that during pregnancy-induced insulin resistance, adiponectin exert regulatory roles that wield change of placenta. Interestingly, we noted significantly reduced adiponectin expression in GDM placenta, in stark contrast to elevated levels of other cytokines, which may potentially contribute to the relatively lower plasma adiponectin levels seen in the GDM patients. In agreement with a previous study in which adiponectin was shown to depress the proliferation of cells, we report similar effects of adiponectin on placental trophoblast cells. We speculate that due to increased proliferation of trophoblast cells caused by the decreased level of adiponectin in GDM placenta, the size of the placenta was larger, the weight of the placenta was heavier, the neonatal birth weight was heavier, and the incidence of macrosomia was higher in the GDM group as compared to the NGT group.
As expected, similar phenomenon can also be observed in BeWo choriocarcinoma cell lines when treated with a high glucose concentration-confirming high glucose level can increase proliferation of trophoblast cells and depress expression of adiponectin. Moreover, we found that the increased proliferation of trophoblast cells may be caused by adiponectin in the GDM placenta. Treatment of BeWo cells with a high glucose concentration resulted in a dramatic decrease in the expression of adiponectin. Additionally, adiponectin levels decreased gradually with time while the proliferation of BeWo cells increased.
To gain further insight, we compared cell numbers, cell cycles and proliferation of BeWo cells in four different culture conditions: High glucose concentration (HG), normal glucose concentration (NG), high mannitol concentration (HM) and high glucose concentration with human recombinant adiponectin (HG + APN). The results showed that there were more cells in total, more cells in S and G2 phases, and higher cell proliferation occured in the HG group than in any other group. On the other hand, the proliferation could be depressed by addition of exogenous adiponectin. These data provides novel findings that adiponectin exerts antiproliferative effects on trophoblast cells treated with high glucose concentrations.
The molecular mechanism for these effects of adiponectin is not entirely clear. What is evident from other biological systems, including human aortic smooth muscle cells and pig skeletal muscle cells for example, is that adiponectin influences the proliferation by binding to its cognate receptors ADIPOR1 and ADIPOR2 activating various signal transduction pathways, such as PRKA, PIK3, P38/P42/P44 MAPK, and JUN kinase pathways.
To identify the signaling pathway involved in adiponectin’s effect in reducing trophoblast cell proliferation, we investigated JUN kinase pathway-known to mediate the effects of adiponectin. SP and CEP (inhibitors of JUN kinase pathway), and siRNA1 and siRNA2 against c-jun, all suppressed the proliferation of cells treated with a high glucose concentration, demonstrating the importance of JUN kinase pathway in antiproliferative effects of adiponectin on BeWo cells.
In conclusion, we demonstrate that adiponectin acts as an antiproliferative factor in human trophoblastic cells. In GDM placenta, JNK/c-Jun signal transduction pathway plays an important role which is the target of modulated by adiponectin. Our findings indicate that adiponectin should be considered as a newly identified regulator of placental development along with its potential role in fetal overgrowth in GDM-complicated pregnancies.
Acknowledgements
This paper is part of the research project Influence of change of adiponectin-mediated placental and trophoblast cells on fetal growth supported by the Grant “Science and Technology Projects of Guangzhou, 2011”, No. 2011J4100110.
Disclosure of conflict of interest
None.
References
- 1.Castro-Rendon WA, Castro-Alvarez JF, Guzman-Martinez C, Bueno-Sanchez JC. Blastocyst-endometrium interaction: intertwining a cytokine network. Braz J Med Biol Res. 2006;39:1373–1385. doi: 10.1590/s0100-879x2006001100001. [DOI] [PubMed] [Google Scholar]
- 2.Mardon HJ, Bagchi MK, Bagchi IC, Peng C, Karpovich N, Wang Y. Hormonal and paracrine regulation of embryonic implantation: a workshop report. Placenta. 2007;28(Suppl A):S82–84. doi: 10.1016/j.placenta.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 3.Campos DB, Palin MF, Bordignon V, Murphy BD. The ‘beneficial’ adipokines in reproduction and fertility. Int J Obes (Lond) 2008;32:223–231. doi: 10.1038/sj.ijo.0803719. [DOI] [PubMed] [Google Scholar]
- 4.Yamauchi T, Hara K, Kubota N, Terauchi Y, Tobe K, Froguel P, Nagai R, Kadowaki T. Dual roles of adiponectin/Acrp30 in vivo as an anti-diabetic and anti-atherogenic adipokine. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:243–254. doi: 10.2174/1568008033340090. [DOI] [PubMed] [Google Scholar]
- 5.Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes. 2001;50:1126–1133. doi: 10.2337/diabetes.50.5.1126. [DOI] [PubMed] [Google Scholar]
- 6.Pellme F, Smith U, Funahashi T, Matsuzawa Y, Brekke H, Wiklund O, Taskinen MR, Jansson PA. Circulating adiponectin levels are reduced in nonobese but insulin-resistant first-degree relatives of type 2 diabetic patients. Diabetes. 2003;52:1182–1186. doi: 10.2337/diabetes.52.5.1182. [DOI] [PubMed] [Google Scholar]
- 7.Dieudonne MN, Bussiere M, Dos Santos E, Leneveu MC, Giudicelli Y, Pecquery R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem Biophys Res Commun. 2006;345:271–279. doi: 10.1016/j.bbrc.2006.04.076. [DOI] [PubMed] [Google Scholar]
- 8.Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–685. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- 9.Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, Kim SE, Lee GY, Lee JW, Jung SA, Chung HY, Jeong S, Kim JB. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol. 2010;24:1441–1452. doi: 10.1210/me.2009-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 11.Luo XH, Guo LJ, Yuan LQ, Xie H, Zhou HD, Wu XP, Liao EY. Adiponectin stimulates human osteoblasts proliferation and differentiation via the MAPK signaling pathway. Exp Cell Res. 2005;309:99–109. doi: 10.1016/j.yexcr.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki T, Bub JD, Uzuki M, Iwamoto Y. Adiponectin activates c-Jun NH2-terminal kinase and inhibits signal transducer and activator of transcription 3. Biochem Biophys Res Commun. 2005;333:79–87. doi: 10.1016/j.bbrc.2005.05.076. [DOI] [PubMed] [Google Scholar]
- 13.Cseh K, Baranyi E, Melczer Z, Kaszas E, Palik E, Winkler G. Plasma adiponectin and pregnancy-induced insulin resistance. Diabetes Care. 2004;27:274–275. doi: 10.2337/diacare.27.1.274. [DOI] [PubMed] [Google Scholar]
- 14.Ranheim T, Haugen F, Staff AC, Braekke K, Harsem NK, Drevon CA. Adiponectin is reduced in gestational diabetes mellitus in normal weight women. Acta Obstet Gynecol Scand. 2004;83:341–347. doi: 10.1111/j.0001-6349.2004.00413.x. [DOI] [PubMed] [Google Scholar]
- 15.Kajantie E, Hytinantti T, Hovi P, Andersson S. Cord plasma adiponectin: a 20-fold rise between 24 weeks gestation and term. J Clin Endocrinol Metab. 2004;89:4031–4036. doi: 10.1210/jc.2004-0018. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Tan B, Karteris E, Zervou S, Digby J, Hillhouse EW, Vatish M, Randeva HS. Secretion of adiponectin by human placenta: differential modulation of adiponectin and its receptors by cytokines. Diabetologia. 2006;49:1292–1302. doi: 10.1007/s00125-006-0194-7. [DOI] [PubMed] [Google Scholar]
- 17.American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;32(Suppl 1):S62–67. doi: 10.2337/dc09-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desoye G, Hauguel-de Mouzon S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care. 2007;30(Suppl 2):120–126. doi: 10.2337/dc07-s203. [DOI] [PubMed] [Google Scholar]
- 19.Kirwan JP, Hauguel-De Mouzon S, Lepercq J, Challier JC, Huston-Presley L, Friedman JE, Kalhan SC, Catalano PM. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes. 2002;51:2207–2213. doi: 10.2337/diabetes.51.7.2207. [DOI] [PubMed] [Google Scholar]
- 20.Leach L, Taylor A, Sciota F. Vascular dysfunction in the diabetic placenta: causes and consequences. J Anat. 2009;215:69–76. doi: 10.1111/j.1469-7580.2009.01098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- 22.Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol. 2000;60:1015–1021. doi: 10.1016/s0006-2952(00)00372-5. [DOI] [PubMed] [Google Scholar]