

Abstract
Leptin mimics many of the anti-diabetic actions of insulin in insulin-deficient diabetes but the mechanism is controversial. Fujikawa et al. (2013) reveal that leptin receptors in γ-aminobutyric acid (GABA)-ergic and pro-opiomelanocortin (POMC) neurons in the central nervous system are sufficient to mediate the life-saving and anti-diabetic actions of leptin in insulin-deficient mice.
The discovery of insulin in 1921-22 and its profound, life-saving effect on people with type 1 diabetes (T1DM) was one of the miracles of modern medicine. Normal glucose control without insulin seemed impossible. Yet, recent studies demonstrate that the adipocyte-secreted, anorexigenic hormone, leptin, also has potent anti-diabetic actions in insulin-deficient diabetes. In this issue, Fujikawa et al elucidate the underlying mechanisms by identifying specific neuronal subtypes that can mediate the glucose-lowering effects of leptin in the absence of insulin (Fujikawa et al., 2013).
Although insulin therapy is effective for T1DM, diabetic complications including retinopathy, nephropathy, cardiovascular disease and lower hindlimb amputation are still major problems. Insulin therapy lacks the precision necessary to mimic the finely-tuned dynamics of insulin secretion from pancreatic ß-cells. Intensive insulin treatment reduces the incidence and progression of diabetic complications but increases the frequency of severe hypoglycemia and may contribute to increased adiposity, hepatic steatosis, and adverse plasma lipoprotein profiles in T1DM. Leptin could be a potential adjunctive therapy for T1DM because of its beneficial effects on glucose metabolism and its anti-steatotic and appetite-suppressive effects (Wang et al, 2010; Morton and Schwartz, 2011). Leptin increases glucose utilization in peripheral tissues, suppresses hepatic glucose production, and reverses hyperglucagonemia in insulin-deficient rodents (German et al., 2011, Wang et al, 2010) (Figure 1). These effects are independent of leptin’s anorexic effect, and appear to be mediated through the hypothalamus although the neuronal circuits are unknown.
Figure 1.
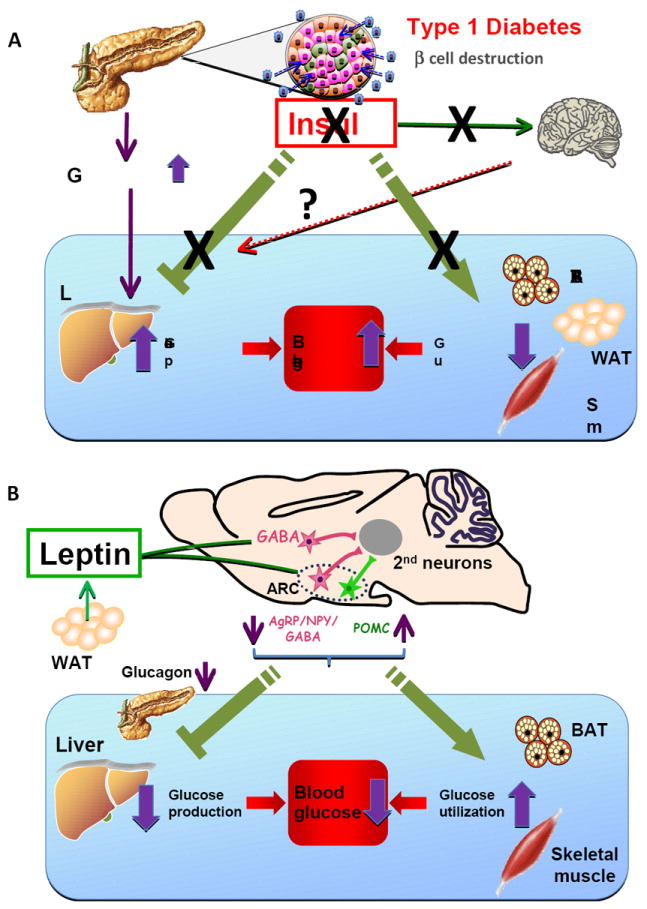
Metabolic effects of insulin deficiency and CNS leptin in Type 1 diabetes A. Alterations of glucose metabolism in T1DM. Autoimmune destruction of pancreatic ß cells markedly reduces insulin secretion, which increases glucagon secretion from pancreatic α cells. Hyperglucagonemia drives HGP resulting in hyperglycemia. Insulin deficiency reduces glucose utilization in peripheral tissues such as BAT, white adipose tissue (WAT) and skeletal muscle, further contributing to hyperglycemia. Insulin also regulates peripheral metabolism via the CNS, including inhibition of HGP and lipid metabolism in WAT. Whether impairment of these central effects of insulin contributes to the metabolic abnormalities in T1DM is unknown.
B. Anti-diabetic actions of leptin. Leptin is released mainly from white adipose tissue and acts through LEPR on GABAergic as well as POMC neurons, to induce anti-diabetic actions in the insulin-deficient state. Leptin inhibits the activity of AgRP/NPY/GABA neurons, while it activates that of POMC neurons in the ARC. The 2nd order neurons for leptin’s anti-diabetic actions may be located in the paraventricular hypothalamus (PVH), lateral hypothalamus (LH) VMH or brainstem. LEPR in GABAergic neurons in other hypothalamic regions such as the dorsomedial hypothalamus (DMH) may also mediate leptin’s actions on glycemia. Through its effects on GABAergic and POMC neurons, leptin suppresses HGP and increases glucose utilization in peripheral tissues such as BAT and skeletal muscle. Suppression of the hyperglucagonemia that is characteristic of insulin deficiency appears to be critical for the suppression of HGP by leptin, whereas the mechanism for the leptin-induced increase in glucose utilization is unknown.
Fujikawa et al. used two complementary models of insulin-deficient diabetes induced by 1) the pancreatic ß-cell toxin, streptozotocin or 2) diphtheria toxin treatment in mice expressing the diphtheria-toxin receptor driven by a rat insulin promoter (RIP), which ablated nearly all pancreatic ß-cells. To determine which neuronal population(s) mediate the effects of intracerebroventricular (icv) leptin – leptin administered directly into the brain ventricles - in insulin-deficient mice, the authors deleted or re-expressed the leptin receptor (LEPR) in a neuron type-specific manner.
LEPR ablation selectively in POMC neurons marginally blunted the hyperglycemia-lowering action of icv leptin in insulin-deficient mice and LEPR ablation in steroidgenic factor 1 (SF1) neurons (enriched in the ventromedial hypothalamus, VMH), had no effect. Furthermore, LEPR re-expression in POMC neurons alone in insulin-deficient mice lacking LEPR in all other tissues failed to improve hyperglycemia or survival in response to icv leptin. In contrast, LEPR re-expression selectively in GABAergic neurons, increased survival of insulin-deficient mice treated with icv leptin, partially improved hyperglycemia, and fully reversed hyperglucagonemia. LEPR re-expression in both GABAergic and POMC neurons had additive metabolic effects and was sufficient to mediate the life-saving and anti-diabetic effects of leptin in insulin-deficient mice. The major input was from GABAergic neurons.
Fujikawa et al.’s paper raises important questions. First, which GABAergic neurons mediate the anti-diabetic effects of leptin and what are the target neurons? GABAergic neurons expressing LEPRs are present in the arcuate, dorsomedial hypothalamus and lateral hypothalamus. Although leptin injection into VMH can normalize diabetic hyperglycemia, LEPR in SF1-expressing neurons is not necessary for these effects (Meek et al., 2013, Fujikawa et al. 2013). Leptin may act on presynaptic GABAergic neurons projecting to the VMH or other regions. In the arcuate, neuropeptide Y (NPY) and agouti-related peptide (AgRP)-expressing neurons co-release GABA, which potently inhibits POMC neuronal activity. Leptin suppresses the neuronal activity of NPY/AgRP/GABAergic neurons, thereby activating POMC neurons (Morton and Schwartz, 2011). Loss of GABAergic signaling in a subset of arcuate AgRP/NPY/GABAergic neurons that project to the parabrachial nucleus leads to starvation (Wu et al., 2009). Furthermore, mice lacking GABA release in arcuate RIP-expressing neurons have decreased energy expenditure and become obese via a circuit involving paraventricular hypothalamic neurons (Kong et al., 2012), underscoring the importance of hypothalamic GABAergic neurons in energy balance.
Second, what are the mechanism(s) for leptin’s actions in peripheral tissues (Figure 1) and is suppression of hyperglucagonemia critical? Glucagon appears to be essential for development of hyperglycemia in insulin-deficient mice (Wang et al., 2010). But the anti-diabetic actions of icv leptin are partial in diabetic mice in which LEPR is re-expressed in GABAergic neurons alone, while hyperglucagonemia is fully reversed. Thus, suppression of hyperglucagonemia is not sufficient for the full anti-diabetic actions of leptin. Furthermore, suppression of hyperglucagonemia does not explain the leptin-induced glucose uptake in peripheral tissues. ß-adrenergic receptors mediate leptin-stimulated glucose uptake in brown fat and muscle in lean non-diabetic rodents (Haque et al., 1999) but Fujikawa et al. showed that the anti-diabetic actions of icv leptin were intact in ß1,ß2,ß3-adrenergic receptor-knockout mice, indicating that different pathways are engaged by hypothalamic neurons to regulate glucose homeostasis in the presence and absence of insulin. Insulin-like growth factor binding protein 2 is induced in liver by leptin (Hedbacker et al., 2010) and overexpression improves hyperglycemia in insulin-deficient mice. Whether it mediates the anti-diabetic effects of icv leptin is unknown.
Third, how do the CNS effects of leptin on glucose homeostasis integrate with the peripheral and CNS effects of insulin? Insulin inhibits hepatic glucose production both directly and through the hypothalamus (Figure 1) (Pocai et al., 2005). The relative importance of these central effects of insulin in normal physiology and whether impairment of these effects contributes to metabolic abnormalities in T1DM is unknown. Furthermore, whether CNS insulin acts through the same GABAergic neurons as leptin is unknown.
The most fundamental question is whether leptin therapy will be effective in T1DM people. Independent of leptin’s insulin-mimicking effects, its potential appetite-suppressing effects could be beneficial for glucose control in T1DM. T1DM rodents are leptin-deficient due to reduced fat mass resulting from uncontrolled diabetes, while T1DM people who are controlled with insulin therapy have normal or increased fat mass. Thus, leptin supplementation in leptin-sufficient T1DM humans may not have the same glycemic effects as leptin replacement in leptin-deficient T1DM mice. Furthermore, potential adverse effects of leptin such as hypertension and pro-inflammatory and pro-coagulatory effects may occur when leptin levels are raised above normal in T1DM humans. If used in combination with insulin, leptin’s effect to potently suppress glucagon may increase the risk for severe hypoglycemia in T1DM people.
Collectively, recent studies provide convincing evidence that leptin has beneficial effects on glucose homeostasis in insulin-deficient (and leptin-deficient) mice via the CNS. Fujikawa et al. found that the vast majority of leptin’s anti-diabetic effects may be mediated by CNS GABAergic neurons. While further studies are needed to determine the full neuronal circuitry, these findings extend our understanding of CNS control of glucose metabolism. Clinical studies are needed to determine whether leptin or molecules targeting the GABAergic pathways mediating leptin’s glucose-lowering effects could be effective and safe adjunctive therapies for T1DM and whether, by lowering the insulin dose, these could diminish some adverse effects of intensive insulin therapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Fujikawa T, Berglund ED, Patel VR, Giorgio Ramadori G, Vianna CR, Vong L, Thorel F, Chera S, Herrera PL, Lowell BB, Elmquist JK, Pierre Baldi P, Roberto Coppari R. Cell Metab. 2013 doi: 10.1016/j.cmet.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German JP, Thaler JP, Wisse BE, Oh-I S, Sarruf DA, Matsen ME, Fischer JD, Taborsky GJ, Jr, Schwartz MW, Morton GJ. Endocrinology. 2011;152:394–404. doi: 10.1210/en.2010-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedbacker K, Birsoy K, Wysocki RW, Asilmaz E, Ahima RS, Farooqi IS, Friedman JM. Cell Metab. 2010;11:11–22. doi: 10.1016/j.cmet.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Kong D, Tong Q, Ye C, Koda S, Fuller PM, Krashes MJ, Vong L, Ray RS, Olson DP, Lowell BB. Cell. 2012;151:645–657. doi: 10.1016/j.cell.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek TH, Matsen ME, Dorfman MD, Guyenet SJ, Damian V, Nguyen HT, Taborsky GJ, Jr, Morton GJ. [10.1210/en.2013-1328];Endocrinology. 2013 doi: 10.1210/en.2013-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minokoshi Y, Haque MS, Shimazu T. Diabetes. 1999;48:287–291. doi: 10.2337/diabetes.48.2.287. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Schwartz MW. Physiol Rev. 2011;91:389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L. Nature. 2005;434:1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- Wang MY, Chen L, Clark GO, Lee Y, Stevens RD, Ilkayeva OR, Wenner BR, Bain JR, Charron MJ, Newgard CB, Unger RH. Proc Natl Acad Sci USA. 2010;107:4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD. Cell. 2009;137:225–234. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]