

Abstract
Matrix metalloproteinases (MMPs) are important enzymes of extracellular matrix (ECM) degradation for creating the cellular environments required during development and morphogenesis. MMPs, collectively called matrixins, regulate also the biological activity of non matrix substrates such as cytokines, chemokines, receptors, growth factors and cell adhesion molecules. Enzymatic activity is regulated at multiple levels. Endogenous specific inhibitors of metalloproteinases (TIMPs) participate in controlling the local activities of MMPs in tissues. The pathological effects of MMPs and TIMPs are involved in cardiovascular disease (CVD) processes, including atherosclerosis and in a number of renal pathophysiologic alterations, both acute and chronic, linking them to acute kidney injury, glomerulosclerosis and tubulointerstitial fibrosis. This review presents an overview of the place of MMPs in atherosclerosis, proteinuria and kidney disease as a subject of considerable interest, given the differentiated and ambiguous role of MMPs in the progression of these diseases.
Keywords: Matrix metalloproteinases, atherosclerosis, proteinuria, kidney disease, glomerulonephritis, glomerulosclerosis, tubulointerstitial fibrosis
Introduction
Matrix metalloproteinases (MMPs) are a gradually multiplying family of zinc-containing and zinc-dependent endopeptidases. To date, 28 different MMPs have been found in humans1. MMPs were thought originally to cleave only extracellular matrix (ECM) proteins, mostly type-IV collagen (col-IV), the main component of vessels and basement membrane (BM), which plays a central role in the induction, progression and repair of kidney disease, just as in atherosclerosis2.
Under normal physiological conditions, the activities of MMPs are precisely regulated at four levels: 1) transcription, 2) activation of the precursor zymogens, 3) interaction with specific ECM components, and 4) inhibition by endogenous tissue inhibitors (TIMPs). Their substrate specificity, sequence similarity and domain organization, classify them into six groups: collagenases, gelatinases, stromelysins, matrilysins, membrane type, and other MMPs3.
MMPs regulate the biological activity of other non matrix substrates, as is now known, such as TNF-a, growth factors, angiotensin-II (AII), transforming growth factor-β1 (ΤG-F-β1) and their receptors, plasminogen and its activators and endothelin2-5. MMPs play an important role, although still vague, in the regulation of cytokine and chemokine activities via proteolytic processing, particularly the gelatinases (MMP-2 and -9). Gelatinases are also the key regulators of leukocyte function6. MMPs play a central role in many biological processes, such as embryogenesis, normal tissue remodeling, wound healing, angiogenesis, embryonic development, blastocyst implantation and organ morphogenesis. Loss of their activity control may result in diseases such as atheroma-atherosclerosis, arthritis, multiple sclerosis, tissue ulceration, cancer, inflammation, bone turn over, nephritis and fibrosis7.
In vitro studies with cultured cells and histological observations of normal and diseased human and experimental blood vessels showed that vascular and inflammatory cells secrete MMPs. Immunocytochemical studies suggest that non diseased human and experimental animal arteries uniformly express, across the wall, MMP-2, TIMPs-1 and -2. Human endothelial cells (ECs) and smooth muscle cells (SMCs), as the major cellular constituents of normal blood vessels, produce constitutively in vitro MMP-2, TIMP-1 and -25. The BM is the part of the ECM that is associated with the vascular endothelium. Many studies have suggested that MMP-9 increased levels play a role in atherosclerosis8-12. Increased expression and activity of several MMPs, mainly MMP-1, -2, -3, and -9, were observed from data related in diseased human arteries and in arterial experimental models of atherosclerosis and restenosis. The expression of MMPs and TIMPs in the kidney is complex, may be species dependent and their localization has not been completely characterized. MMP -1, -2, -3, -9, -13, -14 (MT-1 –MMP), -24, -25, -27, -28, and TIMP-1, -2, -3, -4 are all expressed in renal compartments, but as currently known only MMP-1, -2, -3, -9 and TIMP-1, -2, -3 have been extensively studied in the kidney1,13.
This review focuses on the role of MMPs in atherosclerosis, proteinuria and kidney disease from the side of renal damage progression in glomerular inflammatory diseases and tubulointerstitial fibrosis.
MMPs (-2,-9), TIMPs and ECM receptors
MMP-2, called gelatinase A, of 72KD, located on chr 16q13, degrades mostly collagens IV, fibronectin and laminin, aggrecan, gelatin, elastin and non matrix substrates, as latent TGFβ9, MCP-3 (monocyte chemoattractant protein-3), FGFR-1 (fibroblast growth factor receptor 1), big endothelin-1 and plasminogen. This gelatinase is important in humans for osteogenesis.
MMP-9, called gelatinase B, of 92KD, located on chr 20q11.2-q13.1, degrades mostly col-IV, aggrecan, gelatin, from ECM components and carboxymethylated – transferrin, ΑI, ΑII, plasminogen, pro TGFβ2, IL-2Rα, as non ECM substrates. Both gelatinases (A,B) are able to form proenzyme complexes with TIMPs and use α2- macroglobulin, tissue factor protease inhibitor-2, thrombospondin, as natural inhibitors. MMP-2, mostly constitutively expressed, is activated at the cell surface by a membrane type MMP, whereas MMP-9 is activated through plasmin (ogen)- dependent or - independent mechanisms and is highly regulated by cytokines and growth factors.
TIMPs (21-28KD) are specific endogenous inhibitors that bind MMPs. Four members (TIMP-1, -2, -3 and -4) have been identified in humans and their expression is regulated during development and tissue remodeling. TIMPs inhibit all MMPs, except that TIMP-1 fails to inhibit MT1-MMP, by a mechanism still unknown3.
Activation of ADAMs (a disintegrin and metalloproteinase family) may result in discrete cellular responses14. Integrins, as ECM receptors, regulate several MMPs’ expression and activation in different cell types. MMP-2 is upregulated in part by α2β1-integrin15. Intracellular integrin interaction with cytokines by integrin – linked kinases participate in epithelial-to-mesenchymal transition (EMT) and progression of renal disease16,17. Tissue transglutaminase-2 (TG-2) contributes to the stabilization of ECM and TG inhibition fragilezed matrix to degradation18. Discoidin domain receptor-1 (DDR1) represents another collagen receptor that may interact with MMPs in renal disease19. Hyaluronan receptors in kidney disease seem to influence the TGF-β effect20. The role of EMMPRIN (ECM MMP inducer), fleetingly observed decreasing in tubulointerstitial nephritis expressed in kidney and vessels, remains to be elucidated21.
MMPs in atherosclerosis
Today, experimental in vitro and in vivo investigations are showing that MMPs’ expression and activity are involved in vascular remodeling, hemodynamics, injury, inflammation, oxidative stress and cardiovascular diseases (CVD), including atherosclerosis, restenosis and aneurysms. The participation of MMPs in vascular remodeling and CVD is distinctively conducted from gene expression, in as much as genetic variations may contribute to heterogeneity in the presentation and natural history of atherosclerosis, but enzymatic activities unleash their biological effects5.
The roles of MMPs in subclinical atherosclerosis are early migration and proliferation of SMC, infiltration of leucocytes, intimal thickening and growth of atherosclerotic lesions (intima-media thickness, IMT) and delay of flow-limiting stenosis by expansive remodeling of plaques. The roles of MMPs in clinical atherosclerosis are carotid plaque instability, coronary plaque rupture and development of aneurysms22,23.
A very few number of genetic studies revealed MMP genes associated with atherosclerosis, but no available evidence suggests a common and functionally evaluated polymorphism in humans.
MMP-2 expression seems to be stimulated by thrombin, in human macrophage and SMC located in normal carotid artery, in serum levels in acute coronary syndrome and in abdominal aortic aneurysm (AAA). In rabbits, pigs and rats, experimental lesions of macrophages in carotid artery are stimulated by reactive oxygen species, oxidative stress (ROS), high-flow state, hypercholesterolemia, balloon injury and low-flow state5,24-29.
MMP-9 expression is found to be stimulated by ox-LDL in human macrophage and SMC, located in coronary atherectomy specimens in the setting of unstable angina, in plasma levels in acute coronary syndrome (ACS) and AAA3,5,7. In pigs, rabbits and rats, experimental lesions of macrophage and SMC in saphenous vein grafted into carotid position, carotid artery and aorta were stimulated by ROS, high-flow state, hypercholesterolemia, balloon injury, flow cessation and hyperhomocysteinemia25-27,29-33.
MMP expression and activity are associated with development of neointimal arterial lesions and SMC migration.
Recent studies suggest that MMP-mediated vascular remodeling could reduce MMP-2 and -9 expression and impair their migration by in vitro endothelial nitric oxide synthase (ecNOS) gene transfer to SMCs. Simultaneous production of nitric oxide (NO) and superoxide and other reactive species such as hydrogen peroxide, can also modulate the activity of MMPs. The current concept of atherosclerosis emphasizes the remodeling of the arterial wall, provides a framework for weakening and destabilization and reveals that angiographically undetectable moderate lesions could become culprits for ACS.
The action of MMPs has been studied in relation to formation of intimal lesion and overall geometrical remodeling, as these both require sustained changes in the structure and dimensions of the arterial wall5.
Expression of MMPs is increased in atherosclerotic lesions. Inflammatory cell stimulation of cell-cell and cell-matrix interactions can diversify the MMP spectrum. Degradation of matrix by activated MMPs, detectable in vessels undergoing remodeling may facilitate cell migration and reorganization of vascular tissue. Finally, MMPs may weaken the arterial wall, contributing to destabilization and rupture of atherosclerotic plaques3,4.
Arterial remodeling may progress by (1) expansive remodeling of the wall, tending to preserve the lumen in the face of increased lesion burden, and (2) plaque rupture that also occurs in arteries have undergone outward remodeling, suggesting the role of MMPs in matrix degradation that leads to weakening and destabilization. Aneurysmal dilation is an extreme case of expansive remodeling. MMP expression and activation have been associated with neointimal growth after balloon angioplasty of experimental arteries. Changes that decrease the lumen of a remodeling artery include intimal thickening and constrictive geometric remodeling of the wall3-5.
MMPs and Proteinuria
Several studies in humans suggested that abnormal persistence of α1(IV), α2(IV) chains of col-IV instead of α3(IV), α4(IV) and α5(IV) isoforms is associated with an increased susceptibility to proteases. Proteolysis in humans, as well as in mouseAlport BM, was significantly enhanced by MMP-2, -3 and -9.
Recently an animal model of Alport’s syndrome (AS) deficient in the α3 chain of col-IV (α3[IV]) made significant advances in understanding the role of MMPs, particularly gelatinases, in the evolution of proteinuria and progression of renal disease34.
At one month before the onset of proteinuria in α3(IV)-/-mice, MMP-2, -3 and -9 levels were significantly upregulated in glomeruli, whereas after two months, when glomerular kidney disease was established and progressed toward tubulointerstitial fibrosis, MMP expression spread from the glomeruli into the tubulointerstitial compartment.
Combined pharmacological inhibitors of MMP-2, -3, and -9 were administered orally to the α3 (ΙV) -/- mice before the onset of proteinuria and GBM structural defects led to significant attenuation in disease progression and markedly prolonged survival. In contrast, inhibition of MMPs after induction of proteinuria led to acceleration of disease, associated with extensive interstitial fibrosis and early death.
These findings suggest that, at early stages, MMPs help to breakdown the GBM, whereas at later stages, they may help to remove ECM, associated with scarring and fibrosis. This model reflects the relation between MMPs and proteinuria in many pathological conditions34,35.
As very recently reported, serum levels of gelatinase A in CKD is an independent correlate of proteinuria36.
Glomerulonephritis (GN) progresses to renal failure with an accumulation of the mesangial matrix (MM) and a thickening of the GBM. The accumulation and thickening of these components are controlled by the stimulation of TGF-β, VEGF, thrombin and ILs on the mesangial, epithelial and endothelial cells of the glomerulus, while the degradation is conducted by MMPs and TIMPs. Additional experimental data suggests that MMP-2 reflects the degradation of the GBM, while MMP-9 represents the degradation of the MM23,34-38.
ΜΜPs in kidney disease
MMPs and the kidney has been the subject of a limited number of investigations relating to their role in nephrogenesis, tumorogenesis, renal damage and renal remodeling. Specifically, very few reports on glomerular MMP activities and their relation to glomerular damage and remodeling have been reported. MMPs play a prominent role in glomerular inflammatory diseases, including pauci-immune, rapidly progressive GN and IgA nephritis (N). Most of the reports dealt with MMP-2 and -9, because of their action on col-IV, the main ECM protein in GBM and mesangium7. The exact localization of both MMPs and TIMPs has not yet been completely elucidated. Gelatinases are expressed in human glomerulus but seem confined, while expression in the proximal and distal tubule of the monkey and in the collecting duct of the rabbit is reported1. Gelatinases, as recently reported, are expressed in glomeruri (predominantly in human mesangial and epithelial cells) and in tubuli (mainly in proximal tubular cells)37.
MMP in GN
Advanced glomerular diseases are characterized by the accumulation of ECM components, mainly col-IV, in the mesangial matrix (MM) and glomerular BM (GBM) various glomerular diseases progress to end-stage renal disease (ESRD) due to an accumulation of the MM and a thickening of the GBM. Both the MM and GBM are consistently metabolized through the synthesis and destruction of the matrix. Synthesis, accumulation, and thickening seem to be influenced by TGF-b and other factors (IL-1, TNFa, thrombin) on the mesangial, epithelial, and endothelial cells of the glomerulus, while degradation is mediated by MMPs and TIMPs. The main constituent of MM and GBM is col-IV, which is degraded by MMP-2 and MMP-9, whereas its degradation is mainly inhibited by TIMP-1.
MMP-2 synthesis has been shown to be produced by fibroblasts, epithelial and mesangial cells (MCs), and MMP-9 synthesis is produced by glomerular epithelial cells and MCs. These MMP-2, MMP-9 and TIMP-1 are reported to be restored in the MM and GBM, becoming active in local tissues, and thereby playing important roles in the progression and resolution of experimental Thy 1.1 nephritis and Heymann nephritis, as well as in human GN.
Additional evidence, regarding the MMP secretion from the glomerulus, imply that serum MMP-2 reflects the degradation of the GBM, while MMP-9 represents the degradation of the MM. TGF-b1 was found in the tissues of both thickened GBM, which was associated with the increased level of MMP-2, as well as in the expanded MM area, which was not necessarily associated with an increased MMP-9 level. These observations suggest that TGF-b1 stimulates the glomerular epithelial and mesangial cells to generate MMP-2, while other factors also play a role in the generation of MMP-9 by these cells1,7,37,38.
In inflammatory glomerular diseases, increased levels of MMP are generally associated with disease activity and the influx of inflammatory cells. Both the level and the duration of MMP elevation may determine the extent of glomerular damage1,7,13,37,38 (Figure 1).
Figure 1. Figure 1: Schematic overview of the inflammatory mechanisms concerning the primary glomerulonephritis process to the progression to glomerulosclerosis. (modified from reference 13 with permission).
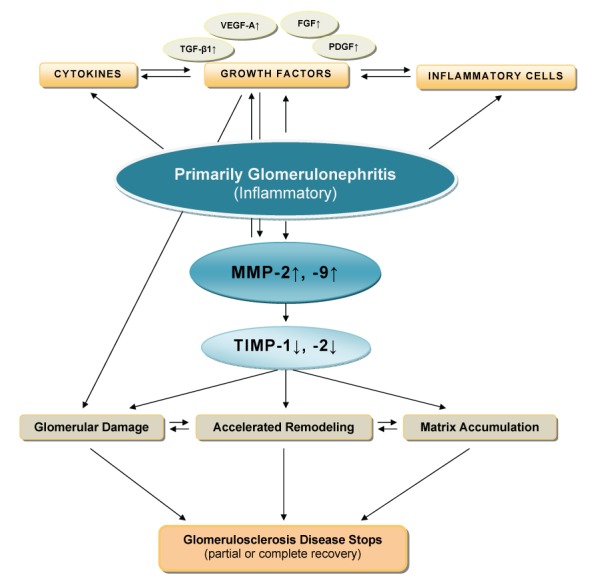
The first best characterized model of glomerular inflammatory disease showed that MMP-2 induced directly on MCs an inflammation and regulated matrix remodeling by a protease activity and a proinflammatory action, and functioned as a growth factor in rat kidneys. This model described the anti-inflammatory effects of the MMP inhibitor BB-1101 on this experimental acute nephritis (anti-Thy 1.1) in the rat as an animal model of immune complex - mediated, mesangial proliferative GN. In humans, mesangial proliferative GN such as IgA N is an important cause of ESRD. The treatment of this disease is still controversial because no current (treatment) modality has shown a convincing benefit. BB-1101 inhibited all inflammatory factors studied, decreased MC proliferation, ECM accumulation, glomerular diameter and proteinuria. Zymography demonstrated strong MC MMP-2 inhibition by BB-1101, when the MMP inhibitor was administered to nephrotic animals. Zymography represents a very sensitive and specific method for the detection of MMP activity. The effect of BB-1101 on other MMPs was confirmed by zymography demonstrating inhibition of purified human MMP-2, -3 and -9 by this compound (data not shown, personal communication with HP Marti)38.
Other treatment modalities were investigated in experimental nephritis with different effective targets. The proteoglycan decorin, a natural inhibitor of TGF-β activity, by intravenous application or by overexpression, decreased ECM accumulation and proteinuria in nephrotic rats, by administering neutralizing antibodies raised against TGF-β1 or antisense oligonucleotides. Antisense RNA with specific anti-MMP-2 ribozymes decreased the synthesis of MMP-2. The mechanism of these beneficial effects remains to be elucidated2,35,38.
MMPs in acute kidney injury
MMPs are involved in acute kidney injury and changes in the vascular endothelium, glomeruli, and tubular epithelial cells (TECs). Increased expression of MMP-2,-9, TIMP-2 and decreased TIMP-1 in glomeruli rats, tubules and interstitium were observed after a few days of reperfusion following one hour of ischemia. Increased MMP-9 activity was associated with degradation of occludin in ECs and increased vascular permability.
MMPs in chronic kidney disease (CKD)
An early increase in MMP-2, TIMP-1 and -3 expression and activity, and a decrease MMP-1 and -9 activity, are associated with the development of tubulointerstitial fibrosis and glomerulosclerosis in rats and humans. These observations suggest increased ECM turnover following injury, but decreased degradation, promoting the development of tubolointerstitial fibrosis at later periods1,7,37.
According to the current concept, the glomerular and tubulointerstitial scarring processes are thought to involve, initially, interactions between infiltrating inflammatory cells and resident renal cells, resulting in loss of renal cells and their replacement by collagenous ECM.
Vascular endothelial growth factor (VEGF) is thought to mediate reactive endothelial proliferation and the regeneration of the glomerular capillary endothelium in glomerular damage. In an experimental model of GN, the administration of VEGF 165 enhanced glomerular capillary repair and accelerated the healing of the extensive endothelial damage39-42.
TGF-β1 controls mesangial activation, proliferation, and ECM synthesis. It activates mesangial a-SMA expression, facilitating the inhibition of mesangial proliferation and glomerular proliferative healing. On the other hand, TGF-β1 stimulates mesangial synthesis of col I and III, contributing to irreversible glomerular sclerosis. This effect may be mediated by the activation of kinases called Smads. Smads are also involved in the stimulation of the essential expression of connective tissue growth factor (CTGF), a key factor in TGF-β induced fibrogenesis. This is why the role of TGF-β1 is so controversial in the relevant literature38-41.
In the animal renovascular hypertensive model, after inhibiting NO synthesis, the following are observed: a deterioration of renal function, a TGF-β expression, col I and IV in renal vessels, excessive glomerular accumulation of ECM and increase activity of MMP-2 and -9 after 1 month of induced hypertension. When the rats were administered an Ang II receptor antagonist rapidly decreased col I and IV, TGF-β gene and protein, without altering MMP-2 and -9 activity. After one month, glomerulosclerosis regressed, and renal function and structure were restored. Inhibited collagen synthesis by AT1 blocker and activated MMPs may reflect the degree of fibrosis43. In experimental models and human diseases, fibrosis usually sustains increased MMP expression. It is likely that integrins and other ECM receptors, as previously reported in this review, give the message of ECM accumulation in the cellular environment that lead to sustained MMP synthesis and activation. The onset of Ang II – induced hypertension is accompanied by increased MMP-9 activity in conducted vessels and the absence of MMP-9 activity results in vessel stiffness and increased pulse or blood pressure. MMP activation seems to exert a beneficial effect by counteracting the prohypertensive stimulus. Earlier, MMP-2 may alter structurally the tubular BM, which leads to tubular EMT resulting in tubular atrophy, fibrosis and renal disease43,44
Challenge approaches and future perspectives
The role of MMPs in atherosclerosis and proteinuria in kidney disease progression, through most forms of GN, appears accumulated evident, but not yet completely elucidated.
The relative contribution of the kidney to the altered MMP expression patterns in both the plasma and urine is still unclear. More studies are required to determine the plasma levels of MMP-2, -9, TGF-β and TIMP-1 of patients with different types of primary GN to evaluate the possible relationship of these parameters with the degree of interstitial and glomerular fibrosis.
The contribution of SMC migration, and thus the need for degradation of the internal elastic lamina, remains to be demonstrated in pathogenesis of human lesions. The clinical interest in investigations targeted at human endothelium will elucidate MMPs’ pathophysiological implications in these specific type cells. Venous dilatation and wall thickening are part of the maturation of an arteriovenous fistula (AVF) and MMPs have also been implicated, but their role in venous remodeling has not yet been reported.
A simplistic, but also realistic, approach to the MMPs could consider them as good relative factors of angiogenesis before the onset of proteinuria and bad ones after the early stages of atherosclerosis, nephritis and GBM structural defects, accelerating the progression of diseases.
The definition of the role of MMPs in kidney inflammatory diseases, proteinuria and atherosclerosis may lead to a new target therapeutic, offering challenge approaches and future perspectives.
References
- 1.Catania JM, Chen G, Parrish AR. Role of matrix metalloproteinases in renal pathophysiologies. Am J Physiol Renal Physiol. 2007;292:F905–F911. doi: 10.1152/ajprenal.00421.2006. [DOI] [PubMed] [Google Scholar]
- 2.Ronco P, Lelongt B, Piedagnel R, Chatziantoniou C. Matrix metalloproteinases in kidney disease progression and repair : a case of flipping the coin. Semin Nephrol. 2007;27:352–362. doi: 10.1016/j.semnephrol.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 4.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 5.Donnelly R, Donnelly J, Manning G. Hypertension, matrix metalloproteinases and target organ damage. J Hypertens. 2003;21:1627–1630. doi: 10.1097/00004872-200309000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Xue M, Thompson PJ, Clifton-Bligh R, Fulcher G, Gallery ED, Jackson C. Leucocyte matrix metalloproteinase-9 is evelated and contributes to lemphocyte activation of type I diabetes. Int J Biochem Cell Biol. 2005;37:2406–2416. doi: 10.1016/j.biocel.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Keeling J, Herrera GA. Human matrix metalloproteinases: characteristics and pathologic role in altering mesangial homeostasis. Microsc Res Tech. 2008;71:371–379. doi: 10.1002/jemt.20565. [DOI] [PubMed] [Google Scholar]
- 8.Ghilardi G, Biondi ML, DeMonti M, Turri O, Guagnellini E, Scorza R. Matrix metalloproteinase-1 and matrix metalloproteinase-3 gene promoter polymorphisms are associated with carotid artery stenosis. Stroke. 2002;33:2408–2412. doi: 10.1161/01.str.0000031929.05665.da. [DOI] [PubMed] [Google Scholar]
- 9.Derosa G, Cicero AF, Scalise F, Avanzini MA, Tinelli C, Piccinni MN, et al. Metalloproteinase-2 and -9 in diabetic and nondiabetic subjects during acute coronary syndromes. Endothelium. 2007;14:45–51. doi: 10.1080/10623320601177064. [DOI] [PubMed] [Google Scholar]
- 10.Pastercamp G, Schoneveld AH, Hijnen DJ, de Kleijn DP, Teepen H, van der Wal AC, et al. Atherosclerotic arterial remodeling and the localization of macrophages and metalloproteases 1, 2 and 9 in the human coronary artery. Atherosclerosis. 2000;150:245–253. doi: 10.1016/s0021-9150(99)00371-8. [DOI] [PubMed] [Google Scholar]
- 11.Zeng B, Prasan A, Fung KC, Solanki V, Bruce D, Freedman SB, et al. Elevated circulating levels of metalloproteinase-9 and -2 in patients with symptomatic coronary artery disease. J Intern Med. 2005;35:331–335. doi: 10.1111/j.1445-5994.2005.00822.x. [DOI] [PubMed] [Google Scholar]
- 12.Newby AC, Johnson JL. Genetic strategies to elucidate the roles of matrix metalloproteinases in atherosclerotic plaque growth and stability. Circ Res. 2005;97:958–960. doi: 10.1161/01.RES.0000193565.23357.c0. [DOI] [PubMed] [Google Scholar]
- 13.Lenz O, Elliot SJ, Stetler-Stevenson WG. Matrix metalloproteinases in renal development and disease. J Am Soc Nephrol. 2000;11:574–581. doi: 10.1681/ASN.V113574. [DOI] [PubMed] [Google Scholar]
- 14.Schäfer B, Marg B, Gschwind A, Ullrich A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem. 2004;279:47929–47938. doi: 10.1074/jbc.M400129200. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Abair TD, Ibanez MR, Su Y, Frey MR, Dise RS, et al. Integrin alpha1beta1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol Cell Biol. 2007;27:3313–3326. doi: 10.1128/MCB.01476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Dai Y, Wu C, Liu Y. PINCH-1 promotes tubular epithelial-to-mesenchymal transition by interacting with integrin-linked kinase. J Am Soc Nephrol. 2007;18:2534–2543. doi: 10.1681/ASN.2007030315. [DOI] [PubMed] [Google Scholar]
- 17.Hu K, Wu C, Mars WM, Liu Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J Clin Invest. 2007;117:3821–3832. doi: 10.1172/JCI32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson TS, Fisher M, Haylor JL, Hau Z, Skill NJ, Jones R, et al. Transglutaminase inhibition reduces fibrosis and preserves function in experimental chronic kidney disease. J Am Soc Nephrol. 2007;18:3078–3088. doi: 10.1681/ASN.2006070690. [DOI] [PubMed] [Google Scholar]
- 19.Flamant M, Placier S, Rodenas A, Curat CA, Vogel WF, Chatziantoniou C, et al. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J Am Soc Nephrol. 2006;17:3374–3381. doi: 10.1681/ASN.2006060677. [DOI] [PubMed] [Google Scholar]
- 20.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada M, Yamabe H, Osawa H, Nakamura N, Kumasaka R, Murakami R, et al. Extracellular matrix metalloproteinase inducer is expressed in the proximal tubular epithelial cells of the human kidney. Nephrology (Carlton) 2009;14:171–178. doi: 10.1111/j.1440-1797.2008.01033.x. [DOI] [PubMed] [Google Scholar]
- 22.Abilleira S, Bevan S, Marcus HS. The role of genetic variants of matrix metalloproteinases in coronary and carotid atherosclerosis. J Med Genet. 2006;43:897–901. doi: 10.1136/jmg.2006.040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimas GG, Iliadis FS, Tegos TJ, Makedou KJ, Didaggelos TP, Pitsalidis CG, et al. Evaluation of serum circulating MMPs-2 and -9 as biomarkers of atherosclerosis in patients with early stages of chronic kidney disease. Nephrol Dial Transplant-Plus Suppl. 2011;6 (ERA-EDTA) [Google Scholar]
- 24.Huang Y, Mironova M, Lopes-Virella MF. Oxidized LDL stimulates matrix metalloproteinase-1 expression in human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19:2640–2647. doi: 10.1161/01.atv.19.11.2640. [DOI] [PubMed] [Google Scholar]
- 25.Rekhter MD, Hicks GW, Brammer DW, Hallak H, Kindt E, Chen J, et al. Hypercholesterolemia causes mechanical weakening of rabbit atheroma: local collagen loss as a prerequisite of plaque rupture. Circ Res. 2000;86:101–108. doi: 10.1161/01.res.86.1.101. [DOI] [PubMed] [Google Scholar]
- 26.Southgate KM, Mehta D, Izzat MB, Newby AC, Angelini GD. Increased secretion of basement membrane-degrading metalloproteinases in pig saphenous vein into carotid artery interposition grafts. Arterioscler Thromb Vasc Biol. 1999;19:1640–1649. doi: 10.1161/01.atv.19.7.1640. [DOI] [PubMed] [Google Scholar]
- 27.Chesler NC, Ku DN, Galis ZS. Transmural pressure induces matrix-degrading activity in porcine arteries ex vivo. Am J Physiol. 1999;277:H2002–H2009. doi: 10.1152/ajpheart.1999.277.5.H2002. [DOI] [PubMed] [Google Scholar]
- 28.Bassiouny HS, Song RH, Hong XF, Singh A, Kocharyan H, Glagov S. Flow regulation of 72-kD collagenase IV (MMP-2) after experimental artery injury. Circulation. 1998;98:157–163. doi: 10.1161/01.cir.98.2.157. [DOI] [PubMed] [Google Scholar]
- 29.Kai H, Ikeda H, Yasukawa H, Kai M, Seki Y, Kuwahara F, et al. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Cardiol. 1998;32:368–372. doi: 10.1016/s0735-1097(98)00250-2. [DOI] [PubMed] [Google Scholar]
- 30.Xu XP, Meisel SR, Ong JM, Kaul S, Cercek B, Rajavashisth TB, et al. Oxidized low-density lipoprotein regulates matrix metalloproteinase-9 and its tissue inhibitor in human monocyte-derived macrophages. Circulation. 1999;99:993–998. doi: 10.1161/01.cir.99.8.993. [DOI] [PubMed] [Google Scholar]
- 31.Ganné F, Vasse M, Beaudeux JL, Peynet J, François A, Mishal Z, et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits urokinase/urokinase-receptor expression and MMP-9 secretion by peripheral blood monocytes--a possible protective mechanism against atherothrombosis. Thromb Haemost. 2000;84:680–688. [PubMed] [Google Scholar]
- 32.Godin D, Ivan E, Johnson C, Magid R, Galis ZS. Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation. 2000;102:2861–2866. doi: 10.1161/01.cir.102.23.2861. [DOI] [PubMed] [Google Scholar]
- 33.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, et al. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ronco P, Chatziantoniou C. Matrix metalloproteinases and matrix receptors in progression and reversal of kidney disease: therapeutic perspectives. Kidney Int. 2008;74:873–878. doi: 10.1038/ki.2008.349. [DOI] [PubMed] [Google Scholar]
- 36.Nagano M, Fukami K, Yamagishi S, Ueda S, Kaida Y, Matsumoto T, et al. Circulating matrix metalloproteinase-2 is an independent correlate of proteinuria in patients with chronic kidney disease. Am J Nephrol. 2009;29:109–115. doi: 10.1159/000151439. [DOI] [PubMed] [Google Scholar]
- 37.Bauvois B, Mothu N, Nguyen J, Nguyen-Khoa T, Nöel LH, Jungers P. Specific changes in plasma concentrations of matrix metalloproteinase-2 and -9, TIMP-1 and TGF-beta1 in patients with distinct types of primary glomerulonephritis. Nephrol Dial Transplant. 2007;22:1115–1122. doi: 10.1093/ndt/gfl743. [DOI] [PubMed] [Google Scholar]
- 38.Steinmann-Niggli K, Ziswiler R, Küng M, Marti HP. Inhibition of matrix metalloproteinases attenuates Anti-Thy1.1 nephritis. J Am Soc Nephrol. 1998;9:397–407. doi: 10.1681/ASN.V93397. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai H, Nigam SK. Transforming growth factor-beta selectively inhibits branching morphogenesis but not tubulogenesis. Am J Physiol. 1997;272:F139–F146. doi: 10.1152/ajprenal.1997.272.1.F139. [DOI] [PubMed] [Google Scholar]
- 40.Campbell RC, Ruggenenti P, Remuzzi G. Halting the progression of chronic nephropathy. J Am Soc Nephrol. 2002;13 Suppl 3:S190–S195. doi: 10.1097/01.asn.0000032522.29672.0a. [DOI] [PubMed] [Google Scholar]
- 41.Kang DH, Johnson RJ. Vascular endothelial growth factor: a new player in the pathogenesis of renal fibrosis. Curr Opin Nephrol Hypertens. 2003;12:43–49. doi: 10.1097/00041552-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 42.Dimas GC, Iliadis FS, Tegos TJ, Makedou KJ, Didaggelos TP, Pitsalidis CG, et al. Increased circulating levels of VEGF-A as an independent correlate of proteinuria in early stages of chronic kidney disease. BANTAO Journal. 2011;Vol 9: 2 Suppl.1 [Google Scholar]
- 43.Flamant M, Placier S, Dubroca C, Esposito B, Lopes I, Chatziantoniou C, et al. Role of metalloproteinases in early hypertensive vascular remodeling. Hypertention. 2007;50:212–218. doi: 10.1161/HYPERTENSIONAHA.107.089631. [DOI] [PubMed] [Google Scholar]
- 44.Cheng S, Pollock AS, Mahimkar R, Olson JL, Lovett DH. Matrix metalloproteinase 2 and basement membrane integrity: a unifying mechanism for progressive renal injury. FASEB J. 2006;20:1898–1900. doi: 10.1096/fj.06-5898fje. [DOI] [PubMed] [Google Scholar]