

Abstract
The iron binding siderophore pyoverdine constitutes a major adaptive factor contributing to both virulence and survival in fluorescent pseudomonads. For decades, pyoverdine production has allowed the identification and classification of fluorescent and nonfluorescent pseudomonads. Here, we demonstrate that PvdP, a periplasmic enzyme of previously unknown function, is a tyrosinase required for the maturation of the pyoverdine chromophore in Pseudomonas aeruginosa. PvdP converts the nonfluorescent ferribactin, containing two iron binding groups, into a fluorescent pyoverdine, forming a strong hexadentate complex with ferrous iron, by three consecutive oxidation steps. PvdP represents the first characterized member of a small family of tyrosinases present in fluorescent pseudomonads that are required for siderophore maturation and are capable of acting on large peptidic substrates.
INTRODUCTION
Iron is an essential element used as a cofactor in a large number of reactions in all living organisms. However, in the presence of oxygen, iron occurs predominantly in the Fe3+ state, which is characterized by a very low solubility, and thus it is also one of the major growth-limiting factors. To overcome this problem, a large number of microorganisms produce siderophores, iron-chelating molecules that are secreted into the environment where they strongly bind and facilitate the import of Fe3+ inside the cells (1–7). Fluorescent pseudomonads, including the human pathogen Pseudomonas aeruginosa and the plant pathogen Pseudomonas syringae, rely on pyoverdine (PVD) siderophores to perform this task. PVDs are the major siderophores in fluorescent pseudomonads and consist of a variable short peptide chain (containing two iron binding residues) linked to a conserved catecholate chromophore moiety (that provides a third iron binding site) (7). Three types of PVDs (PVDI, PVDII, and PVDIII) have been found to be produced by different strains of P. aeruginosa (8). Hereby, we will refer to PVDI when describing PVD type I, the most studied P. aeruginosa PVD type found in multiple strains, including the well-characterized PAO1 and PA14. The PVDI pathway is complex, comprising approximately 20 different proteins involved in its regulation, synthesis, maturation, transport, and uptake (Fig. 1) (7). While the synthesis and regulation of PVDI have been well documented in the past years, its maturation process is not elucidated. PVDI maturation starts with the transport of a PVDI precursor (PVDIq) from the cytoplasm to the periplasm of P. aeruginosa by the ABC transporter PvdE (9, 10). It was recently demonstrated that this precursor (PVDIq) is a C14-acylated ferribactin that once in the periplasm is cleaved by the acylase PvdQ, leading to the formation of ferribactin (11, 12). Ferribactin is a yellowish precursor of PVDI in which the cyclization of the third ring in the catechol moiety has not been accomplished. This molecule lacks the typical greenish coloration of PVDI and its third stable iron binding group. Little is known about the following steps in the maturation of ferribactin leading to PVDI. The scarce data on additional enzymes such as PvdN, PvdM, PvdO, or PvdP involved in PVDI maturation in the periplasm originate from the quantification of PVDI production in a set of mutants initially identified as iron-responsive (13).
FIG 1.

Production and export of the siderophore PVDI. PVDI biosynthesis starts when the low concentration of iron abrogates the formation of the stable Fe-ferric uptake regulator (FUR) complex which otherwise binds to the pvdS promoter, blocking the production of the extracytoplasmic function (ECF)-sigma factor PvdS. Upon translation, the regulator, PvdS, binds to the promoter IS boxes of PVDI genes, initiating transcription and translation of PVDI proteins. The nonribosomal peptide synthases (NRPS) PvdL (41), PvdI, PvdJ, and PvdF initiate the assembly of the precursor siderophore PVDIq, which starts with a C14 fatty acid (FA) being attached to l-Glu and finishes with the release of PVDIq from PvdD by the thioesterase module (TE) after the last l-Thr has been incorporated. Enzymes PvdH, PvdA, and PvdF are responsible for the synthesis of the nonnatural amino-acids l-diaminobutyrate (l-Dab) and l-formyl-OH-ornithine that are incorporated into PVDIq by the four NRPS proteins. PvdE transports PVDIq to the periplasm, where the myristoleic acid is removed by the acylase PvdQ prior to the maturation of the chromophore, generating the precursor ferribactin (FB). The role of the uncharacterized PvdM, PvdN, and PvdO proteins in the maturation of ferribactin remains unknown; one possibility is that at least one of these proteins transforms the l-glutamate into one of the three residues found at this position in PVDI. Mature PVDI is then secreted to the surrounding environment by PvdRT-OpmQ and by an as-yet-unidentified transporter. Immature precursors, such as ferribactin (42) and PVDIq (this study), have also been found in the secreted fraction of fluorescent pseudomonads, indicating that these precursors can also be secreted into the extracellular medium (dashed lines). Once in the medium, PVDI binds iron, forming the PVDI-Fe complex, which can be taken up by the cell, where further steps are conducted.
Here, we demonstrate that PvdP, a previously uncharacterized periplasmic protein of unknown function in the maturation of PVDI, is capable of converting the precursor, ferribactin, into the green fluorescent PVDI. Mechanistically, PvdP appears to hydroxylate the d-tyrosine moiety of the tetrahydropyrimidine ring, resulting in a catechol functionality, which is followed by the formation of a third ring in the chromophore, leading to the formation of the final PVDI chromophore (Fig. 2). In a final reaction sequence, PvdP restores the catechol functionality and creates the third iron binding site in PVDI, needed for the high affinity of Fe3+ (14). Thus, PvdP catalyzes three steps in the maturation of ferribactin into fluorescent PVDI.
FIG 2.
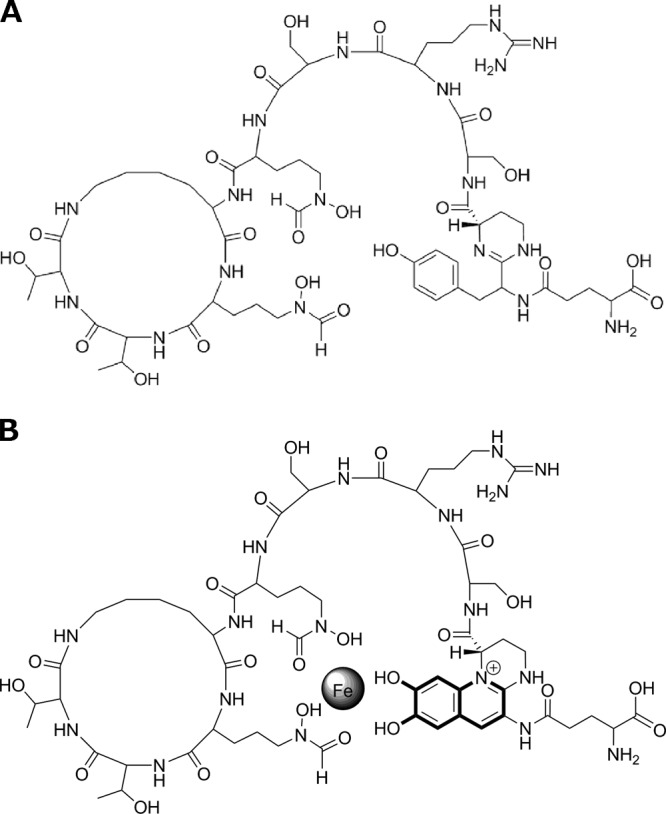
Structure of ferribactin (A) and its product, PVDI(l-Glu) (B), after incubation with PvdP.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
P. aeruginosa PAO1 was used as a source for the pvdP gene, which was PCR amplified and cloned with a C-terminal His tag fusion in pET26b (Novagen) using standard cloning methods. For PvdP production, Escherichia coli BL21(DE3) harboring plasmid pET26B-pvdP_H6 cultures were grown overnight in LB medium and 1 ml was added to 1 liter of 2× TY medium (15) (both media supplemented with 50 mg/liter kanamycin), incubated at 30°C with shaking at 250 rpm, and induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.6, followed by further incubation for approximately 16 h at 30°C. PvdQ was produced in E. coli DH10B (Invitrogen) harboring plasmid pMCT-pvdQ as previously described (16). For protein localization assays, pvdP_H6 was PCR amplified from pET26B-pvdP_H6 and cloned in the shuttle vector pME6032 (17), obtaining the plasmid pME-pvdP_H6. This plasmid was then transformed in the E. coli HB2151 wild type (WT) and the E. coli HB2151 ΔtatC mutant (18).
PvdP purification.
Induced E. coli BL21(DE3)/pET26B-pvdP_H6 cells were resuspended in 3 ml of sonication buffer (50 mM Tris-HCl, 500 mM NaCl, 10% glycerol [vol/vol], 20 mM imidazole, 14 mM β-mercaptoethanol, 1% Tween [pH 8]) per gram of wet cells, lysed by sonication, and centrifuged at high speed to remove cell debris (1 h, 18,000 rpm). The supernatant was filtered using a cellulose acetate filter (0.2-μm pore size) and subsequently loaded on a nickel-charged HiTrap HP (5 ml) column (GE Healthcare). After the column was washed with 3 column volumes of washing buffer (50 mM Tris-HCl, 500 mM NaCl, 10% glycerol [vol/vol], 20 mM imidazole, 14 mM β-mercaptoethanol [pH 8]), elution was performed with a linear gradient from 20 to 500 mM imidazole in elution buffer (50 mM Tris-HCl, 500 mM NaCl, 10% glycerol [vol/vol], 500 mM imidazole, 14 mM β-mercaptoethanol [pH 8]). PvdP was eluted with approximately 100 mM imidazole. The elution buffer was exchanged to 20 mM Tris-HCl buffer, pH 8, using a desalting HiTrap HP (5-ml) column (GE Healthcare) and concentrated to 5 mg/ml using a 20,000 MWCO Vivaspin column (Sartorius Stedim Biotech S.A., France). The protein was aliquoted, snap-frozen in liquid nitrogen, and stored at −80°C till further use.
PvdP cell localization. (i) Cell fractionation.
To determine the cellular localization of PvdP, pvdP_H6 was amplified from pET26b-pvdP_H6 and cloned in the shuttle vector pME6032 (17) to obtain the plasmid pME-pvdP_H6. This plasmid was electroporated into the E. coli HB2151 WT and the E. coli HB2151 ΔtatC mutant (18). The two strains were grown overnight in 2× TY of LB medium at 37°C and 250 rpm. After incubation, cells were harvested and resuspended in phosphate-buffered saline (PBS) to an OD580 of 1. A 1-ml aliquot of cells was pipetted to a new tube and centrifuged, and the pellet was washed 3 times with 1 ml of PBS to remove extracellular proteins. After the last washing step, the cells were resuspended in 1 ml of ice-cold Tris-HCl buffer (100 mM Tris [pH 8]) containing 20% (wt/vol) sucrose. A total of 1 ml of a Tris-HCl buffer (100 mM Tris [pH 8]) containing 5 mM EDTA was added to the tube together with 10 μl of lysozyme (2 mg/ml). The cells were incubated for 30 min at room temperature and centrifuged for 20 min at 10,000 rpm. The supernatant containing the periplasmic fraction was collected and precipitated overnight using 5% (wt/vol) trichloroacetic acid (TCA). Subsequently, the remaining pellet was resuspended in 1 ml of an ice-cold Tris-HCl buffer and the cells were lysed by sonication (2 min, 50%). After sonication, the cells were centrifuged for 1 h at 13,000 × g. The supernatant containing the cytoplasmic proteins was precipitated overnight using 5% (wt/vol) TCA.
(ii) PvdP detection.
After the periplasmic and cytoplasmic fractions were separated, 80 μg of total protein was added to a 10% SDS polyacrylamide gel. The proteins were then transferred to a nitrocellulose membrane, blocked with 5% skim milk, and probed with anti-6×His tag antibody (Sigma) to detect the presence of PvdP_H6. A control with a GroEL antibody (Sigma) was used to assess the proper separation of the periplasmic and the cytoplasmic fractions.
Isolation of PVDI and PVDI precursors. (i) PVDI.
PVDI was isolated from 1-liter cell-free culture supernatants of P. aeruginosa PAO1 WT grown at 30°C, 250 rpm, for 48 h in low-iron CAA medium (16). After centrifugation, the medium was separated with H2O and methanol by using a C18 solid-phase extraction (SPE) column (Varian Megabond Elut C18; Agilent) separating three fractions: unbound fraction, 50% methanol-eluting fraction, and 100% MetOH-eluting fraction. The 50% eluted sample containing PVDI was collected, air dried at room temperature, resuspended in 1 ml H2O, and stored at −20°C till further analysis.
(ii) PVDIq.
PVDIq was isolated from 1-liter cell-free culture supernatants of the P. aeruginosa PAO1 ΔpvdQ mutant (19) grown at 30°C, 250 rpm, for 48 h in low-iron medium (CAA). After centrifugation, the medium was separated with H2O and methanol using a C18-SPE column, as in the case of PVDI, but the 100% methanol elution containing the more hydrophobic PVDIq was collected. The sample was air dried at room temperature, resuspended in 1 ml of 100% methanol, and stored at −20°C till further analyses.
(iii) Ferribactin.
Ferribactin was obtained after mixing the 1 ml methanol solution containing PVDIq with 50 ml of 50 mM Tris-HCl, pH 8.8, adding 1 mg of purified PvdQ (1 mg/ml) protein, and incubating 4 h at 30°C. The mixture was again separated using the same SPE process described above for PVDI, but the 50% methanol fraction was collected, air dried, resuspended in 1 ml H2O, and stored at −20°C till further analysis.
(iv) PVDI(l-Glu).
PVDI(l-Glu) was obtained by mixing the 1 ml ferribactin-containing solution with 50 ml of 50 mM CHES (N-cyclohexyl-2-aminoethanesulfonic acid) buffer and 250 μM CuSO4 at pH 9 and 1 mg of purified PvdP and incubating the mixture for 4 h at 30°C. The sample was again separated using the same SPE process described for ferribactin, air dried, resuspended in 1 ml of H2O, and stored at −20°C till further analysis.
Analysis of PVDI precursor molecules.
To verify the identity of the reaction products ferribactin and PVDI(l-Glu), the samples were analyzed by isoelectric focusing and liquid chromatography-mass spectrometry (LC-MS). The concentrations of the samples were calculated after measuring their dry weight and considering all the samples (with the exception of PVDI) of a high purity as determined by high-performance liquid chromatography (HPLC) and gas chromatography (GC)-MS analyses.
Isoelectric focusing.
PVDI and its precursors [PVDIq, ferribactin, and PVDI(l-Glu)] were separated based on their isoelectric point (pI) using isoelectric focusing (IEF) gels (8, 20, 21) at pH 3.5 to 9.5 (Invitrogen). PVDI, PVDIq, ferribactin, and PVDI(l-Glu) were aliquoted in different tubes containing the same concentration of the starting material (±1 mg/ml) to ensure that the properties displayed in the IEF gels were not biased by concentration differences. After separation, gels were visualized under UV light to detect the formation of the fluorescent chromophore. Here, the IEF gel was overlaid with a fresh 1-mm CAS agarose gel till orange bands, indicative of iron chelation, developed (20).
Fluorescence spectrometry.
A PVDIq sample, obtained as previously described, was divided into 3 aliquots and resuspended in 2 ml of either 100% methanol (in the case of PVDIq due to its low solubility) or 0.1 M acetate buffer (pH 5). The final concentration of PVDIq was 1 mg/ml in all three aliquots. A total of 20 μl of purified PvdQ (1 mg/ml) was added to one of the samples to obtain ferribactin. A total of 20 μl of PvdQ (1 mg/ml) and 10 μl of purified PvdP (2 mg/ml) were added to the second sample. No additional enzymes were added to the third PVDIq sample. Additionally, a fourth sample containing PVDI isolated from the supernatant of P. aeruginosa PAO1 was also tested as a control. CuSO4 was added to all 4 samples to a final concentration of 250 μM, after which the samples were incubated for 24 h at room temperature prior to the analysis. PVDIq, ferribactin, PVDI(l-Glu), and PVDI were analyzed using a Varian Cary Eclipse fluorescence spectrophotometer. Emission was scanned from 400 to 600 nm after excitation at a wavelength of 390 nm. The excitation and emission slit was set at 10 nm, and the scan was performed at a rate of 300 nm/min, with an average time of 0.1 s and a data interval of 0.5 nm. FeCl3 was added to each of the samples at various final concentrations (10 μM, 20 μM, 200 μM, and 400 μM) to determine the ability of each molecule to bind iron and consequently quench fluorescence (see Fig. S1 in the supplemental material).
LC-MS.
Liquid chromatography-electrospray mass spectrometry was performed on an API3000 triple quadrupole mass spectrometer (PE Sciex), equipped with a turbo ion spray source coupled to a Shimadzu LC system equipped with LC-20AD gradient pumps and a SIL-20AC autosampler. Analyst 1.5.1 software was used for data acquisition and evaluation. Each sample containing approximately 1 mg/ml (suspended in 100% methanol) was diluted 20 times in methanol before injection, and 30 μl was injected on a C18 Phenomenex Gemini 5-μm, 110-Å, 250- by 4.6-mm column. The gradient mobile phase composition was a mixture of solvent A (consisting of 99.9% H2O and 0.1% formic acid) and solvent B (consisting of 50% acetonitrile and 50% acetone). The main flow was set at 0.6 ml/min, and an additional postcolumn flow was added, providing a flow of 0.2 ml/min of solvent D (consisting of 99.9% acetonitrile and 0.1% formic acid) to improve spray conditions. Elution was performed using a 25-min linear gradient from 5% to 90% solvent B.
Enzymatic assays.
Functional prediction based on the fold algorithm recognition server FFAS (22–24) suggested that PvdP shares homology with polyphenol oxidases and tyrosinases. The activity of PvdP was monitored at 30°C over a range of substrates (0.5 to 6.5 mM) that included l-Tyr, d-Tyr, and l-Dopa (Sigma-Aldrich) by measuring A475 for the formation of dopachrome (ε = 3,600 M−1 cm−1) and ferribactin by measuring A405 (25) for the formation of the PVDI chromophore (ε = 4,160 M−1 cm−1). A range of pH (3 to 12) was tested using 50 mM CHES and 10 mM phosphate buffers to determine optimal conditions for enzyme activity. All measurements were performed at 30°C. The optimal pH of the reaction mixture was determined to be 9, and all subsequent kinetic experiments were performed in 50 mM CHES buffer at pH 9. All reactions were performed in the presence of 250 μM CuSO4, which was determined to be the optimal concentration for enzyme activity. Mushroom tyrosinase (T3824-25KU; Sigma-Aldrich) was used as a control to evaluate the specificity of PvdP toward ferribactin.
Kinetic studies were performed at 30°C in 50 mM CHES buffer and 250 μM CuSO4 at pH 9. Under these conditions, substrate concentrations ranging from 0.5 to 6.5 mM were incubated with 22.5 μg of PvdP in a 1-ml volume, and the absorbance was monitored every 2 min at either 475 nm for l-Tyr, d-Tyr, and l-Dopa or 405 nm for ferribactin. In the case of l-Tyr, d-Tyr, and l-Dopa, kinetic parameters were obtained by applying the Michaelis-Menten equation (v = Vmax × S/Km + S), with v = rate of reaction, Vmax = maximum velocity, Km = Michaelis-Menten constant, and S = substrate concentration. In the case of ferribactin, kinetic parameters were calculated using the substrate inhibition equation (26, 27), where v = Vmax/[1 + (Km/S) + (S/Ksi)], with Ksi representing the constant describing the substrate inhibition interaction. The reaction rates were corrected for the rate of the spontaneous reaction.
PvdP structural model.
A PvdP model was created using the same FFAS03 server that was used to predict its protein function through a fold recognition algorithm. PvdP was modeled based on the closest homologue determined by the server, which corresponded to the arthropodan hemocyanin from the California spiny lobster Panulirus interruptus (Protein Data Bank [PDB] file 1HC1). Given the low amino acid identity between these two proteins, minimization on the PvdP model was performed using Discovery Studio 3.0 (Accelrys, USA), consisting of 1,000 steps of steepest descent followed by 5,000 iterations of the adopted basis set Newton-Raphson algorithm using an energy tolerance of 0.001 kcal · mol−1 · Å−1 to reduce as much as possible the errors.
Site-directed mutagenesis and analysis of PvdP mutants.
PvdP structure model suggested His216, His220, His271, His375, His379, and His432 as the six histidine residues forming the di-copper active site. Protein sequence alignment of PvdP and protein homologues in other fluorescent pseudomonads was performed using an identity score matrix (Blosum62) in Geneious 7.0.6 (see Fig. S2 in the supplemental material). Using standard methods (28), site-directed mutagenesis was performed to substitute each individual histidine to an aspartate. Enzymatic activity of each mutant enzyme was assayed as described in the previous section. Circular dichroism (CD) was performed using a Jasco J-715 CD spectrophotometer equipped with a PFD350S Peltier temperature-control unit (Jasco). Rectangular quartz 1-mm path length cuvettes were used. Protein samples were dialyzed against PBS (pH 7.3) and adjusted to a final concentration of 1 mg/ml. Wavelength spectra were recorded between 250 and 205 nm using a 0.2-nm step size and 1-nm bandwidth at 25°C for the wild type and each of the mutant proteins to verify secondary structure integrity.
RESULTS
PvdP is a copper-dependent tyrosinase.
During a bioinformatics search, using the “fold and function assignment system” (FFAS) algorithm (22–24) to determine the function of the uncharacterized proteins predicted to be involved in the maturation of the PVDI chromophore of P. aeruginosa (PvdM, PvdN, PvdO, and PvdP), we obtained several hits for PvdP that were all linked to closely related proteins: hemocyanin, phenoloxidase, and tyrosinase. Despite the low amino acid identity between PvdP and these proteins (7 to 12%), the presence of a d-tyrosine in the immature chromophore of the PVDI precursor ferribactin (29) prompted us to investigate whether PvdP might have tyrosinase activity. After production in Escherichia coli, purified PvdP was tested in a standard spectrophotometric assay for tyrosinase activity. In a first assay with l- and d-tyrosine, the presence of PvdP induced a weak brown coloration at A475 typical for the formation of melanin. Since tyrosinases are characterized by a type 3 copper center (30, 31), and given the slow reaction rate in our preliminary assay, we decided to titrate different amounts of copper. Copper titration revealed a 10-fold increase in activity on l- and d-tyrosine in the presence of 250 μM CuSO4 while decreasing at higher concentrations (see Fig. S3 in the supplemental material), demonstrating that PvdP requires copper as a cofactor.
PvdP is a tyrosinase essential for chromophore maturation in PVDI synthesis.
In our search for the substrate of PvdP in the PVDI synthesis pathway, we tested a pvdP-knockout strain, the P. aeruginosa PAO1 pvdP::Kn strain (32), and tried to isolate the substrate for PvdP. Unfortunately, we were unable to isolate and purify this compound from this strain due to low quantities and multiple forms of PVDI precursors. In an attempt to overcome this problem, we used another P. aeruginosa mutant, the P. aeruginosa PAO1 ΔpvdQ mutant, as starting material to obtain the C14-acylated precursor, PVDIq (see Materials and Methods). Subsequently, PVDIq was converted in vitro with purified PvdQ and isolated. This method allowed us to produce semipreparative amounts of product, with both high yield and purity. In accordance with earlier publications (7, 10, 11), the product was verified to be ferribactin using LC-MS analysis (Fig. 3) and other tests (see below).
FIG 3.

Mass spectra of ferribactin (eluting between 3.204 and 3.04 min on a C18 Phenomenex Gemini column [5 μm, 110 Å, 250 by 4.6 mm]) (a) and PVDI(l-Glu) (eluting between 2.937 and 3.137 min) (b). The calculated uncharged masses of ferribactin and PVDI(l-Glu) are 1,351.2 Da and 1,364.5 Da, respectively. Notice that PVDI(l-Glu) carries already one positive charge in the chromophore of PVDI(l-Glu) (Fig. 2).
The biosynthetic route from ferribactin to the chromophore of PVDI has been suggested to involve several oxidation steps (7, 33) but so far no enzyme responsible has been identified. Incubation of ferribactin with PvdP resulted in an increase of A405, indicative of the formation of the mature PVDI chromophore. Incubation of commercially available mushroom tyrosinase together with ferribactin did not result in any changes of A405 even after 24 h of incubation. The maturation reaction as proposed by Dorrestein et al. (33) involves 3 oxidation steps and shows high similarity to the tyrosinase activity on l-Tyr and l-Dopa to dopachrome (Fig. 4). The incubation of ferribactin with purified PvdP resulted in an absorbance increase at 405 nm, appearance of fluorescence, and an increase in iron chelation capability (Fig. 5). We named this product PVDI(l-Glu) to differentiate it from PVDI isolated from P. aeruginosa PAO1 and PA14 that contain different substitutions of the l-Glu chain presumably as a consequence of the enzymatic reaction with other PVD enzymes of as-yet-unknown function. Importantly, whereas ferribactin is a nonfluorescent substrate, the product of the PvdP-catalyzed reaction is a green fluorescent molecule; thus, the reaction proceeds past the nonfluorescent dihydroPVD (34). All products have been analyzed by different assays for identification (see below).
FIG 4.

Comparison of the tyrosinase reaction leading from tyrosine to dopachrome (A) versus the same reaction leading from ferribactin to PVDI (B). The analogy between the two reactions provides an explanation to the formation of the third ring in the PVDI chromophore.
FIG 5.
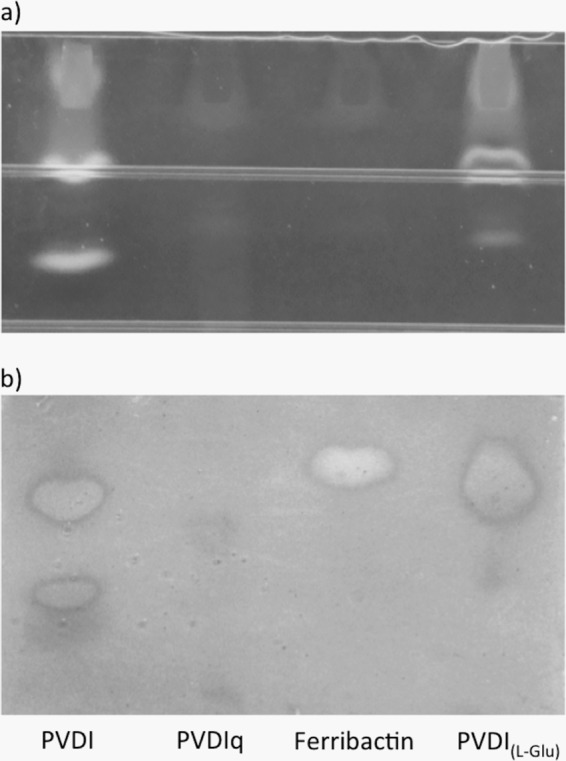
UV and CAS overlay detection of siderophores after migrating through an IEF gel. (a) Fluorescence under UV light of PVDI, PVDIq, PVDIq incubated with PvdQ (ferribactin), and PVDIq digested with PvdQ-PvdP [PVDI(l-Glu)]. (b) After detection of the fluorescent chromophores, the gel was covered with 1 mm of CAS agarose, revealing orange spots that indicate iron-chelating activities of PVDI and its precursors.
PvdP is a periplasmic protein requiring a TAT secretion system.
PvdP has been proposed to be a periplasmic protein due to the presence of a putative TAT signal peptide (35, 36). To verify this, we constructed the plasmid pME-pvdP_H6 and transformed it into the E. coli HB2151 WT and the E. coli HB2151 ΔtatC mutant and analyzed the periplasmic and cytoplasmic fractions of each strain for the presence of PvdP_H6. The absence of PvdP in the periplasmic fraction of the E. coli HB2151 ΔtatC mutant (Fig. 6) clearly demonstrates that PvdP is a periplasmic protein that requires a TAT secretion system for its secretion to the periplasm. A small fraction of PvdP remains in the cytoplasm of the E. coli HB2151 WT, which may be a consequence of the overexpression of this protein and consequent saturation of the TAT system.
FIG 6.
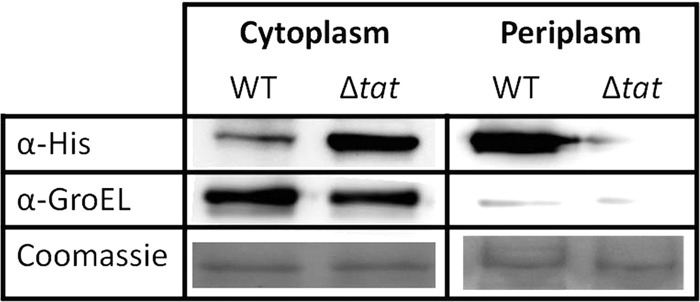
Periplasmic localization and TAT secretion system dependency of PvdP. After separating the cytoplasmic and periplasmic proteins, PvdP_H6 was detected using an anti-6×His antibody. After cell fractionation, PvdP_H6 was detected in an E. coli wild-type strain having a functional TAT system and in a ΔtatC mutant. GroEL detection was used as a control for the cytoplasmic fraction. PvdP_H6 is absent in the periplasmic fraction of the E. coli HB2151 ΔtatC mutant (1st lane, right side), indicating the requirement of the TAT system for the periplasmic localization of PvdP. A large amount of PvdP can be detected in a wild-type E. coli, confirming that PvdP is a periplasmic protein (1st lane, right side). Coomassie staining served as the loading control.
Analysis of products PVDI, PVDIq, ferribactin, and PVDI(l-Glu).
The spectral properties of the four products PVDIq, ferribactin, PVDI(l-Glu), and PVDI were examined by fluorescence spectroscopy. Although PVDIq and ferribactin show some fluorescence at 450 nm, the fluorescence of PVDI(l-Glu) and PVDI is 35-fold higher than that of PVDIq and ferribactin (see Fig. S1 in the supplemental material). The latter two fluorescent products have a nearly identical spectrum, in line with the expectation that they have a very similar structure. The addition of ferrous iron resulted in fluorescence quenching, indicating that all products can bind Fe3+. Due to the very high affinity of PVD for ferrous iron, this assay does not allow to efficiently compare the affinities of the different species.
In parallel, we have examined these same molecules using siderotyping. This technique allows us to visualize siderophores and classify bacterial strains by the study of the migration behavior of their respective siderophores (8, 20, 21). This technique was exploited to analyze the different properties of PVDI as isolated from P. aeruginosa PAO1, in comparison to the three compounds isolated in our experiments: PVDIq, ferribactin, and PVDI(l-Glu). UV illumination demonstrates that PVDIq and ferribactin lack the fluorescence typical of PVDIs (Fig. 5a), in line with the absence of a mature chromophore. Clearly, PVDI and PVDI(l-Glu) do show fluorescence, and one of the bands runs at comparable height in the gel, suggesting these compounds to be very similar. In the CAS overlay, PVDI and PVDI(l-Glu) show a band at the same height in the IEF gel, with very similar staining (Fig. 5b), demonstrating a comparable, strong Fe3+ binding capacity. These results strongly suggest that these two compounds are very similar if not identical. The weaker staining with ferribactin suggests a lower affinity for Fe3+, in line with the assumption that this compound equals the low affinity molecule ferribactin (34). Obviously, iron staining is hardly present in the compound PVDIq (Fig. 5b). We hypothesize that this feature is a consequence of the PVDIq structural conformation where the immature chromophore may be adapting a distant position from the other two iron binding groups as a result of the attached C14-acyl chain (11).
The compounds ferribactin and PVDI(l-Glu) were also analyzed by LC-MS. The mass for ferribactin was calculated based on the previously published structures (7, 29) and considering the acyl chain (often represented as –R due to the different possible substitutions) to be the original glutamate present after NRPS synthesis. Similarly, the mass of PVDI(l-Glu) was calculated from ferribactin using the reaction scheme in Fig. 4B. The results of the LC-MS analysis (Fig. 3) demonstrate that the experimentally determined molecular mass of ferribactin, 1,351.2 Da, corresponds to the calculated mass of 1,351.42 Da. Similarly, the calculated and experimental molecular masses of PVDI(l-Glu) are practically identical: 1,364.4 and 1,364.5 Da. These results strongly underline our interpretation of the catalytic activity of PvdQ and PvdP in the PVDIq and ferribactin maturation processes, respectively.
Kinetic analysis of PvdP.
The kinetic parameters of the PvdP-catalyzed reactions were measured for l-Tyr, d-Tyr, l-Dopa, and ferribactin. The enzyme exhibited typical Michaelis-Menten kinetics on l-Tyr, d-Tyr, and l-Dopa and substrate inhibition on ferribactin with a Ksi of 3.37 ± 0.54. The Km and kcat values do not differ much for the various substrates, with the exception of a significantly lower kcat value for l-Dopa (Table 1).
TABLE 1.
Kinetic values of PvdP
| Substrate | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) |
|---|---|---|---|
| l-Tyr | 1.3 ± 0.05 | 1.38 ± 0.09 | 1.06 |
| d-Tyr | 2.2 ± 1.65 | 0.77 ± 0.33 | 0.35 |
| l-Dopa | 0.75 ± 0.28 | 0.13 ± 0.01 | 0.17 |
| Ferribactin | 1.6 ± 0.23 | 1.30 ± 0.0 | 0.81 |
PvdP structural model.
Modeling the active site of PvdP based on its closest structure-solved homologue (PDB file 1HC1) reveals the presence of the six histidines (His216, His220, His271, His375, His379, and His432) presumably responsible for the formation of the characteristic di-copper active site of tyrosinases (Fig. 7). To verify this, we constructed single amino acid mutants where each of the six histidines was substituted with an aspartic acid and tested the resulting proteins for enzymatic activity toward tyrosine or ferribactin. Mutated proteins behaved identically to WT PvdP during purification. In addition, the presence of secondary structures in each purified mutant protein was checked by circular dichroism and showed results comparable to those of WT PvdP, indicating that the mutations had no effect on the structural integrity of the proteins. None of the mutants demonstrated tyrosinase activity against any of the previously tested substrates. These results are in agreement with the proposed structural model that indicates that these six histidines are essential to form the di-copper active site of PvdP.
FIG 7.
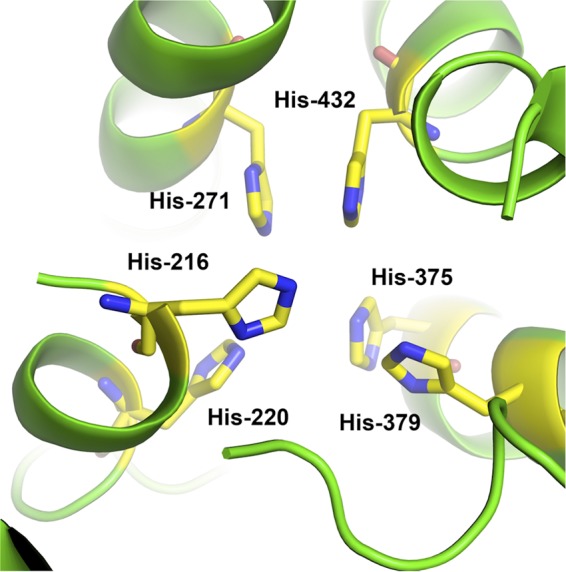
Model of the active site of PvdP obtained using the FFAS03 server after minimization with Discovery Studio 3.0. The 6 histidines involved in copper binding are highlighted in yellow.
PvdP is the first member of a tyrosinase subfamily devoted to PVDI maturation.
The small percent identity between PvdP and other tyrosinases and the exceptionally large size of its substrate suggest that PvdP is the first characterized tyrosinase member of a protein family exclusive to fluorescent pseudomonads. In P. aeruginosa and probably other pseudomonads species, PvdP is dedicated to the maturation of PVD and PVD-like siderophores. To confirm this hypothesis, we performed a phylogenetic analysis using the amino acid sequence of the closest PvdP homologues (as determined by a protein BLAST) and representing one single PvdP sequence of each representative fluorescent pseudomonad and a few examples outside this clade. Our results (Fig. 8) confirm that PvdP (as other PVDI enzymes) is absent in nonfluorescent pseudomonads, with the closest homologue outside the fluorescent pseudomonads group sharing less than 40% identity. The level of identity between PvdP and homologues in other fluorescent pseudomonads is high (51 to 66%) (Fig. 8) but still divergent, which may be related to the structural differences in the PVDs produced in different species (8, 32, 37) that during evolution would have driven PvdP toward a coevolutive process in order to better fit each molecule. Furthermore, the absence of activity of commercial mushroom tyrosinase on ferribactin (data not shown) underlines the hypothesis that this new family of PvdP-like tyrosinases has evolved to accommodate large peptidic substrates and accomplish its function on PVDI biosynthesis. An alignment of the protein sequences used for the phylogenetic analysis reveals that all six histidines involved in the copper binding are strictly conserved in all fluorescent pseudomonads (see Fig. S2 in the supplemental material).
FIG 8.
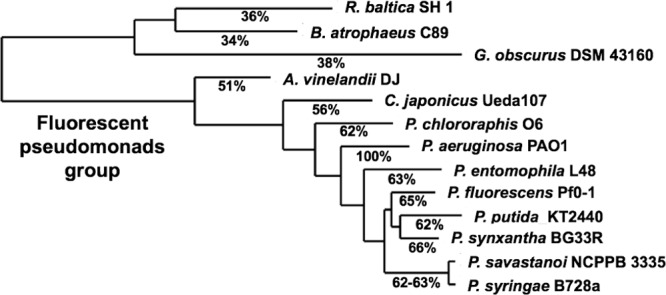
Phylogenetic tree of PvdP homologues from fluorescent pseudomonads (Azotobacter vinelandii, Cellvibrio japonicus, Pseudomonas chlororaphis, Pseudomonas aeruginosa, Pseudomonas entomophila, Pseudomonas fluorescens, Pseudomonas putida, Pseudomonas synxantha, Pseudomonas savastanoi, and Pseudomonas syringae) and the three closest protein homologues outside the Pseudomonadales clade (Rhodopirellula baltica, Bacillus atrophaeus, and Geodermatophilus obscurus). Numbers in red indicate percentages of amino acid identity related to P. aeruginosa PAO1 PvdP.
DISCUSSION
Previous studies have shown the involvement of PvdP in pyoverdine biosynthesis (32); however, the exact function of PvdP has remained elusive. Its low homology to other annotated proteins is evident when using a basic BLAST sequence analysis. In the present study, by using the FFAS tool (22–24) that predicts protein function based on folding, we were able to obtain several hits, suggesting that PvdP could act as a tyrosinase. We pursued this hypothesis encouraged by the presence of a d-tyrosine in the PVDI precursor, ferribactin (29), in the exact location where the PVDI chromophore is located. Looking at the previously described products being formed from tyrosine in the consecutive steps of a tyrosinase reaction, we further hypothesized that the same reaction on ferribactin would lead to the formation of the PVDI chromophore (Fig. 4A and B). Purified PvdP was then tested against both l-Tyr and d-Tyr. As expected, PvdP transformed these two colorless substrates into a dark-brown compound indicative of a melanization reaction typical of tyrosinases, in which after the enzymatic conversion of tyrosine to l-Dopa and subsequently dopaquinone, a series of spontaneous reactions takes place, leading to the formation of melanin. Despite the positive result, the low activity of PvdP on these substrates was surprising. We hypothesized that the two copper atoms present in all tyrosinases and responsible for the activity of these proteins (30) may not be present in all of our purified PvdP. This could either be a consequence of the large amounts of PvdP generated after overexpression in E. coli or of the absence of copper in the PvdP fraction that is still in the cytoplasm, presumably due to a saturation of the TAT pathway after PvdP overexpression. The activity of our batch of purified PvdP was then tested in the presence of various amounts of CuSO4, revealing a maximum activity toward tyrosine around 125 to 250 μM (see Fig. S3 in the supplemental material). It seems that the copper concentrations in these assay conditions are ideally balanced. Concentrations higher than 250 μM seem to disturb the interaction between the substrate and the metal, either by competition for the oxygen site, a phenomenon that is often seen for metalloproteins, or by competitive inhibition where the copper-bound substrate would be unable to fit in the copper-bound active site of PvdP. These results confirmed our hypothesis and also provide an explanation for the previously reported requirement for copper for maximum PVDI production in P. aeruginosa (38).
The next step was to analyze the reaction of PvdP on the PVD precursor in P. aeruginosa from the cellular and extracellular fractions of a P. aeruginosa pvdP::Kn mutant (32). Our attempts to find this molecule proved unsuccessful due to the low concentration and the diversity of other similar molecules obtained in this P. aeruginosa mutant. To overcome this problem, we decided to purify a previous substrate (PVDIq), which had been extensively studied in our lab and reported by others (11, 12). We obtained ferribactin by digesting PVDIq with PvdQ protein, which was used as a template to test the activity of PvdP. A reaction of ferribactin with PvdP transformed the initially faint yellow molecule into a fluorescent green molecule with a strong absorption at A405 nm, confirming our prediction that PvdP was responsible for the formation of the PVDI chromophore. While this approach allowed us to corroborate the function of PvdP in the PVDI pathway, we are unable to propose ferribactin as the natural substrate of PvdP, since other proteins, such as PvdM, PvdN, and PvdO, with an as-yet-unassigned function, may be converting ferribactin into yet another product prior to PvdP. For this reason, we named PVDI(l-Glu) the product of the reaction of ferribactin plus PvdP.
The identity of the molecules was verified using LC-MS: the peaks corresponding to ferribactin and PVDI(l-Glu) were found with masses almost identical to those calculated: 1,351.2 Da compared to the calculated a mass of 1,351.42 Da in the case of ferribactin and 1,364.4 compared to the calculated mass of 1,364.5 Da for PVDI(l-Glu) (Fig. 3a and b).
Fluorescence spectrometry was performed on all four compounds [PVDIq, ferribactin, PVDI(l-Glu), and PVDI] to evaluate the fluorescence properties of each molecule. We compared their fluorescence after PvdP addition and tested their ability to bind iron by testing fluorescence quenching. The results obtained clearly indicate that despite all sharing a similar fluorescent profile, there is a 35-fold increase in fluorescence in PVDI(l-Glu) and PVDI compared to the other two PVD precursors. Unfortunately, this technique did not allow us to establish whether PVDI(l-Glu) and PVDI have a higher affinity for iron as a consequence of the formation of the PVD chromophore due to the high differences in total fluorescence among the molecules. To overcome this, we decided to use siderotyping (8, 20, 39). This technique allows us to compare the four molecules according to their different migration on an IEF gel and examine their fluorescence and iron binding properties (Fig. 5). Both PVDIq and ferribactin revealed undetectable fluorescence under a standard UV light in accordance with the absence of a chromophore in these molecules. Additionally PVDI(l-Glu) displayed a stronger coloration on the same gel than ferribactin, a clear indication of the increase in affinity for Fe3+ of this molecule as a consequence of the formation of the third iron binding group (Fig. 2A and B). Kinetic analysis of PvdP activity toward the different substrates revealed that PvdP is neither specific for our proposed natural substrate (ferribactin) nor its target amino acid d-Tyr. Nevertheless, time of expression (13) and cellular localization (periplasm) may be the reason a higher activity of PvdP toward ferribactin is not required. Tyrosine is synthesized in the cytoplasm, where it is used in protein synthesis, while PvdP as a Cu2+-dependent enzyme will be active only once it has reached the periplasm, where it can convert ferribactin. In comparison to PvdP, commercially available mushroom tyrosinase did not show activity on ferribactin even after 24 h of incubation. This strongly supports the hypothesis that PvdP belongs to a new family of tyrosinases. Phylogenetic analysis suggests that PvdP is a member of a group of tyrosinases, each of which may be involved in the maturation of their canonical PVDI chromophore.
PVD siderophores require a large array of proteins for their synthesis, transport, and maturation. A full elucidation of the complete PVDI pathway is crucial to understand the chemical nature of this complex siderophore. Additionally, since iron is one of the most important limiting factors for bacterial growth, interfering with siderophore production may contribute to the development of novel antimicrobial agents capable of interfering with siderophore synthesis. The addition of PvdP to the list of enzymes with an assigned reaction in the biosynthesis of PVDI leaves the conversion of the attached l-Glu side chain into the various side chains observed in PVDI (40) as the only reactions left for the remaining uncharacterized enzymes, e.g., PvdM, PvdN, and PvdO.
Supplementary Material
ACKNOWLEDGMENTS
This work was partly funded by EU grant Antibiotarget MEST-CT-2005-020278 to P.N.-J., G.K., and W.J.Q.
We are grateful to Isabelle J. Schalk and Herbert Budzikiewicz for useful discussion and analysis of the PVDI maturation and synthesis metabolites, Ian Lamont for providing us with the P. aeruginosa PAO1 PvdP::Kn strain, Wesley R. Browne for assistance with the circular dichroism analysis, Lígia O. Martins and Vânia Brissos for assistance with the fluorescence measurements, and Jessica Thompson and Hjalmar P. Permentier for carefully reading the manuscript.
Footnotes
Published ahead of print 9 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01376-13.
REFERENCES
- 1.Wandersman C, Delepelaire P. 2004. Bacterial iron sources: from siderophores to hemophores. Annu. Rev. Microbiol. 58:611–647. 10.1146/annurev.micro.58.030603.123811 [DOI] [PubMed] [Google Scholar]
- 2.Crosa JH. 1989. Genetics and molecular biology of siderophore-mediated iron transport in bacteria. Microbiol. Rev. 53:517–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faraldo-Gomez JD, Sansom MS. 2003. Acquisition of siderophores in Gram-negative bacteria. Nat. Rev. Mol. Cell Biol. 4:105–116. 10.1038/nrm1015 [DOI] [PubMed] [Google Scholar]
- 4.Budzikiewicz H. 2010. Microbial siderophores. Fortschr. Chem. Org. Naturst. 92:1–75. 10.1007/978-3-211-99661-4_1 [DOI] [PubMed] [Google Scholar]
- 5.Miethke M, Marahiel MA. 2007. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71:413–451. 10.1128/MMBR.00012-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schalk IJ, Guillon L. 2013. Pyoverdine biosynthesis and secretion in Pseudomonas aeruginosa: implications for metal homeostasis. Environ. Microbiol. 15:1661–1673. 10.1111/1462-2920.12013 [DOI] [PubMed] [Google Scholar]
- 7.Visca P, Imperi F, Lamont IL. 2007. Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol. 15:22–30. 10.1016/j.tim.2006.11.004 [DOI] [PubMed] [Google Scholar]
- 8.Meyer JM, Stintzi A, De Vos D, Cornelis P, Tappe R, Taraz K, Budzikiewicz H. 1997. Use of siderophores to type pseudomonads: the three Pseudomonas aeruginosa pyoverdine systems. Microbiology 143(Part 1):35–43 [DOI] [PubMed] [Google Scholar]
- 9.McMorran BJ, Merriman ME, Rombel IT, Lamont IL. 1996. Characterisation of the pvdE gene which is required for pyoverdine synthesis in Pseudomonas aeruginosa. Gene 176:55–59. 10.1016/0378-1119(96)00209-0 [DOI] [PubMed] [Google Scholar]
- 10.Yeterian E, Martin LW, Guillon L, Journet L, Lamont IL, Schalk IJ. 2010. Synthesis of the siderophore pyoverdine in Pseudomonas aeruginosa involves a periplasmic maturation. Amino Acids 38:1447–1459. 10.1007/s00726-009-0358-0 [DOI] [PubMed] [Google Scholar]
- 11.Drake EJ, Gulick AM. 2011. Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6:1277–1286. 10.1021/cb2002973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hannauer M, Schafer M, Hoegy F, Gizzi P, Wehrung P, Mislin GL, Budzikiewicz H, Schalk IJ. 2012. Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586:96–101. 10.1016/j.febslet.2011.12.004 [DOI] [PubMed] [Google Scholar]
- 13.Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML. 2002. GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 45:1277–1287. 10.1046/j.1365-2958.2002.03084.x [DOI] [PubMed] [Google Scholar]
- 14.Budzikiewicz H. 2001. Siderophores of the human pathogenic fluorescent pseudomonads. Curr. Top. Med. Chem. 1:1–6. 10.2174/1568026013395560 [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J, Green M. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 16.Nadal Jimenez P, Koch G, Papaioannou E, Wahjudi M, Krzeslak J, Coenye T, Cool RH, Quax WJ. 2010. Role of PvdQ in Pseudomonas aeruginosa virulence under iron-limiting conditions. Microbiology 156:49–59. 10.1099/mic.0.030973-0 [DOI] [PubMed] [Google Scholar]
- 17.Heeb S, Blumer C, Haas D. 2002. Regulatory RNA as mediator in GacA/RsmA-dependent global control of exoproduct formation in Pseudomonas fluorescens CHA0. J. Bacteriol. 184:1046–1056. 10.1128/jb.184.4.1046-1056.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Droge MJ, Boersma YL, Braun PG, Buining RJ, Julsing MK, Selles KG, van Dijl JM, Quax WJ. 2006. Phage display of an intracellular carboxylesterase of Bacillus subtilis: comparison of Sec and Tat pathway export capabilities. Appl. Environ. Microbiol. 72:4589–4595. 10.1128/AEM.02750-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papaioannou E, Wahjudi M, Nadal-Jimenez P, Koch G, Setroikromo R, Quax WJ. 2009. Quorum-quenching acylase reduces the virulence of Pseudomonas aeruginosa in a Caenorhabditis elegans infection model. Antimicrob. Agents Chemother. 53:4891–4897. 10.1128/AAC.00380-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koedam N, Wittouck E, Gaballa A, Gillis A, Hofte M, Cornelis P. 1994. Detection and differentiation of microbial siderophores by isoelectric focusing and chrome azurol S overlay. Biometals 7:287–291 [DOI] [PubMed] [Google Scholar]
- 21.Meyer JM, Geoffroy VA, Baida N, Gardan L, Izard D, Lemanceau P, Achouak W, Palleroni NJ. 2002. Siderophore typing, a powerful tool for the identification of fluorescent and nonfluorescent pseudomonads. Appl. Environ. Microbiol. 68:2745–2753. 10.1128/AEM.68.6.2745-2753.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rychlewski L, Jaroszewski L, Li W, Godzik A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9:232–241. 10.1110/ps.9.2.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaroszewski L, Rychlewski L, Godzik A. 2000. Improving the quality of twilight-zone alignments. Protein Sci. 9:1487–1496. 10.1110/ps.9.8.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. 2005. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 33:W284–W288. 10.1093/nar/gki418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stintzi A, Evans K, Meyer JM, Poole K. 1998. Quorum-sensing and siderophore biosynthesis in Pseudomonas aeruginosa: lasR/lasI mutants exhibit reduced pyoverdine biosynthesis. FEMS Microbiol. Lett. 166:341–345. 10.1111/j.1574-6968.1998.tb13910.x [DOI] [PubMed] [Google Scholar]
- 26.Cornish-Bowden A. 1995. Analysis of enzyme kinetics data. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 27.Houston JB, Kenworthy KE. 2000. In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab. Dispos. 28:246–254 [PubMed] [Google Scholar]
- 28.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 29.Hohlneicher U, Schafer M, Fuchs R, Budzikiewicz H. 2001. Ferribactins as the biosynthetic precursors of the Pseudomonas siderophores pyoverdins. Z. Naturforsch. C. 56:308–310 [DOI] [PubMed] [Google Scholar]
- 30.Claus H, Decker H. 2006. Bacterial tyrosinases. Syst. Appl. Microbiol. 29:3–14. 10.1016/j.syapm.2005.07.012 [DOI] [PubMed] [Google Scholar]
- 31.Fairhead M, Thony-Meyer L. 2012. Bacterial tyrosinases: old enzymes with new relevance to biotechnology. N. Biotechnol. 29:183–191. 10.1016/j.nbt.2011.05.007 [DOI] [PubMed] [Google Scholar]
- 32.Lamont IL, Martin LW. 2003. Identification and characterization of novel pyoverdine synthesis genes in Pseudomonas aeruginosa. Microbiology 149:833–842. 10.1099/mic.0.26085-0 [DOI] [PubMed] [Google Scholar]
- 33.Dorrestein PC, Poole K, Begley TP. 2003. Formation of the chromophore of the pyoverdine siderophores by an oxidative cascade. Org. Lett. 5:2215–2217. 10.1021/ol034531e [DOI] [PubMed] [Google Scholar]
- 34.Philson SB, Llinas M. 1982. Siderochromes from Pseudomonas fluorescens. I. Isolation and characterization. J. Biol. Chem. 257:8081–8085 [PubMed] [Google Scholar]
- 35.Ochsner UA, Snyder A, Vasil AI, Vasil ML. 2002. Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 99:8312–8317. 10.1073/pnas.082238299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravel J, Cornelis P. 2003. Genomics of pyoverdine-mediated iron uptake in pseudomonads. Trends Microbiol. 11:195–200. 10.1016/S0966-842X(03)00076-3 [DOI] [PubMed] [Google Scholar]
- 37.Palanche T, Blanc S, Hennard C, Abdallah MA, Albrecht-Gary AM. 2004. Bacterial iron transport: coordination properties of azotobactin, the highly fluorescent siderophore of Azotobacter vinelandii. Inorg. Chem. 43:1137–1152. 10.1021/ic034862n [DOI] [PubMed] [Google Scholar]
- 38.Teitzel GM, Geddie A, De Long SK, Kirisits MJ, Whiteley M, Parsek MR. 2006. Survival and growth in the presence of elevated copper: transcriptional profiling of copper-stressed Pseudomonas aeruginosa. J. Bacteriol. 188:7242–7256. 10.1128/JB.00837-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuchs R, Schafer M, Geoffroy V, Meyer JM. 2001. Siderotyping—a powerful tool for the characterization of pyoverdines. Curr. Top. Med. Chem. 1:31–57. 10.2174/1568026013395542 [DOI] [PubMed] [Google Scholar]
- 40.Budzikiewicz H. 2004. Siderophores of the Pseudomonadaceae sensu stricto (fluorescent and non-fluorescent Pseudomonas spp.). Fortschr. Chem. Org. Naturst. 87:81–237. 10.1007/978-3-7091-0581-8_2 [DOI] [PubMed] [Google Scholar]
- 41.Mossialos D, Ochsner U, Baysse C, Chablain P, Pirnay JP, Koedam N, Budzikiewicz H, Fernandez DU, Schafer M, Ravel J, Cornelis P. 2002. Identification of new, conserved, nonribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol. Microbiol. 45:1673–1685. 10.1046/j.1365-2958.2002.03120.x [DOI] [PubMed] [Google Scholar]
- 42.Budzikiewicz H, Schafer M, Fernandez DU, Matthijs S, Cornelis P. 2007. Characterization of the chromophores of pyoverdins and related siderophores by electrospray tandem mass spectrometry. Biometals 20:135–144. 10.1007/s10534-006-9021-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.