

Abstract
In this study, we identify a major spore surface protein, BclA, and provide evidence that this protein is glycosylated. Following extraction of the spore surface, solubilized proteins were separated by one-dimensional PAGE and stained with glycostain to reveal a reactive high-molecular-mass region of approximately 600 kDa. Tandem mass spectrometry analysis of in-gel digests showed this band to contain peptides corresponding to a putative exosporangial glycoprotein (BclA3) and identified a number of glycopeptides modified with multiple N-acetyl hexosamine moieties and, in some cases, capped with novel glycans. In addition, we demonstrate that the glycosyltransferase gene sgtA (gene CD3350 in strain 630 and CDR3194 in strain R20291), which is located immediately upstream of the bclA3 homolog, is involved in the glycosylation of the spore surface, and is cotranscribed with bclA3. The presence of anti-β-O-GlcNAc-reactive material was demonstrated on the surface of spores by immunofluorescence and in surface extracts by Western blotting, although each strain produced a distinct pattern of reactivity. Reactivity of the spore surface with the anti-β-O-GlcNAc antibody was abolished in the 630 and R20291 glycosyltransferase mutant strains, while complementation with a wild-type copy of the gene restored the β-O-GlcNAc reactivity. Phenotypic testing of R20291 glycosyltransferase mutant spores revealed no significant change in sensitivity to ethanol or lysozyme. However, a change in the resistance to heat of R20291 glycosyltransferase mutant spores compared to R20291 spores was observed, as was the ability to adhere to and be internalized by macrophages.
INTRODUCTION
Clostridium difficile is a Gram-positive, spore-forming anaerobe and is the major cause of antibiotic-associated diarrhea (1). The incidence of C. difficile infection has been rapidly increasing in North America and Europe in recent years, and this increase in infections has been associated with higher rates of morbidity and mortality (2). Recent estimates of the incidence of C. difficile-associated diarrhea (CDAD) in the United States indicate as many as 500,000 cases per year with up to 20,000 deaths (1, 3). The primary focus of much of the research has been on two toxins, TcdA and TcdB, that are produced by C. difficile and cause tissue damage and a severe inflammatory response, which can lead in the more serious cases to potentially lethal pseudomembranous colitis. While toxin activity is recognized as the major virulence factor associated with CDAD, other aspects of C. difficile virulence are less well understood.
Spore production in C. difficile is an integral part of the infectious process. This recalcitrant, dormant form of C. difficile can survive indefinitely outside the host and is known to persist in the hospital environment (4). It has been demonstrated in mice that antibiotic treatment suppresses the diversity of the gut microbiome and promotes the production of these highly infectious spores, which are then disseminated into the environment (so-called supershedder state) (5). As such, more recently there has been increased attention on the process of spore formation in C. difficile as well as studies of spore structure and biochemical composition (6–12). To date, the major focus of the studies on spore structure has been to identify spore coat proteins and demonstrate enzymatic activity. Pretreatment of spores either by enzymatic digestion or sonication was utilized in these studies to remove the exosporangial layer prior to analysis.
In contrast to spores of C. difficile, the spores of another important toxin-producing, Gram-positive pathogen, Bacillus anthracis, have been extensively characterized. These spores have been shown to be enclosed by an exosporangial layer which is composed of a number of different proteins (13, 14) and which includes an outermost hair-like nap layer. The filaments of the nap layer are primarily composed of a highly immunogenic collagen-like protein, BclA (15–17), as well as a second exosporangial protein, BclB (18). Both BclA and BclB have been well characterized and shown to be glycosylated with an O-linked pentasaccharide that contains the novel terminal sugar 2-O-methyl-4-(3-hydroxy-3-methylbutamido)-4,6-dideoxy-d-glucopyranose, which is also referred to as anthrose (16). The utility of this structure as a target for sensitive spore detection systems (19) or as a target for therapeutic intervention against anthrax has been explored as well (16, 20, 21).
Although considerable progress has been made recently in the analysis of spore coat proteins from C. difficile, the identification and characterization of both the exosporangial and glycan-containing components is less well advanced. In this study, we demonstrate by mass spectrometry (MS) analysis that the BclA3 homolog from C. difficile is a surface-associated glycoprotein modified with a novel oligosaccharide. In addition, we identify a glycosyltransferase gene which is involved in the biosynthesis of surface-associated glycan components.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. difficile strains used in this study are listed in Table 1. Initial experiments were carried out using strains 630Δerm and R20291. Comparisons to other C. difficile strains from a variety of ribotypes (QCD-32g58, BI-6, CD20, CF5, and M68) revealed R20291 to be the more representative strain. R20291 is also a more clinically relevant strain and a better sporeformer than strain 630. For these reasons, later experiments, particularly the biological assays, were focused on R20291 spores. All strains were routinely grown under anaerobic conditions on brain heart infusion agar medium (BD, Sparks, MD) supplemented with 5 g/liter yeast extract, 1.2 g/liter NaCl, 0.5 g liter cysteine HCl, 5 mg/liter hemin, 1 mg/liter vitamin K, and 1 mg/liter resazurin (BHIS). Erythromycin (2.5 μg/ml) and thiamphenicol (15 μg/ml) were added as required for the growth of mutant and complemented mutant strains.
TABLE 1.
C. difficile strains used in this study
| Strain | Characteristic(s) | Reference and/or source |
|---|---|---|
| 630Δerm | Ribotype O12 | N. Minton, University of Nottingham (44) |
| R20291 | Ribotype O27 | B. Wren, LSHTM (45) |
| 630ΔCD3350 | 630Δerm CD3350::erm | This study |
| R20291ΔCDR3194 | R20291 CDR3194::erm | This study |
| 630ΔCD3350p3350 | 630Δerm ΔCD3350 pRPF185-CD3350 | This study |
| R20291ΔCDR3194p3350 | R20291 ΔCDR3194 pRPF185-CD3350 | This study |
| QCD-32g58 | Ribotype 027 | A. Dascal, Jewish General Hospital, Montreal (46) |
| BI-6 | Ribotype 0176 | B. Wren, LSHTM |
| CD20 | Ribotype 023 | B. Wren, LSHTM |
| CF5 | Ribotype 017 | B. Wren, LSHTM (47) |
| M68 | Ribotype 017 | B. Wren, LSHTM (47) |
MS analysis of spores.
Spores were harvested from BHIS agar plates into phosphate-buffered saline (PBS) following 7 days of incubation under anaerobic conditions, heat treated at 56°C for 15 min, collected by centrifugation (500 × g for 30 min), and washed once in PBS. Spore numbers (CFU/ml) were determined by serial dilution and plating on BHI containing 0.1% sodium taurocholate (BHI-ST; Sigma-Aldrich, Oakville, Canada). Approximately 5 × 109 spores were resuspended in 200 μl of extraction buffer (2.4 ml 1 M Tris, pH 6.8, 0.8 g ASB-14, 4 ml 100% glycerol, 1% dithiothreitol [DTT], 3.8 ml double-distilled H2O) and were left for 30 min at room temperature. Spores were removed by centrifugation, and soluble material was collected for analysis.
Protein-containing endospore surface extractions were separated using 3 to 8% NuPAGE Novex Tris-acetate minigels by following the manufacturer's instructions (Invitrogen, Life Technologies). High-molecular-mass HiMark protein standards (31 to 500 kDa) were used as markers. The gel was stained using Emerald-Q glycostain per the manufacturer's instructions (Invitrogen, Life Technologies) and subsequently with nonfixing silver stain (22). Protein bands were excised, reduced for 1 h with 10 mM DTT at 56°C, and alkylated for 1 h with 55 mM iodoacetamide in the dark (23) prior to digestion with trypsin as described previously (24) or with proteinase K. Proteinase K digests were carried out using 100 μg/ml of enzyme in 50 mM ammonium bicarbonate for 15 to 40 h. The resulting peptides were analyzed by nanoliquid chromatography coupled to tandem MS (nLC-MS/MS) using electrospray ionization (ESI) as the ion source, as recently described (24). Briefly, peptides were analyzed by nanoflow reversed-phase liquid chromatography (RPLC) coupled to MS using ESI (nanoRPLC-ESI-MS) using a nanoAcquity UltraPerformance LC (UPLC) system coupled to a quadrupole-time-of-flight (Q-TOF) Ultima hybrid mass spectrometer (Waters, Milford, MA). The peptides were first loaded onto a 180-μm-inner-diameter (ID), 20-mm by 5-μm symmetry C18 trap column (Waters, Milford, MA) and then eluted into a 100-μm-ID, 10-cm by 1.7-μm BEH130 C18 column (Waters, Milford, MA) using a linear gradient from 1% to 45% solvent B (acetonitrile plus 0.1% formic acid) for 18 min, 45% to 85% solvent B for 3 min, and 85% to 1% solvent B for 1 min. Solvent A was 0.1% formic acid in high-performance liquid chromatography (HPLC)-grade water. The peak list files of MS/MS spectra from tryptic digests were searched against the NCBI database using the MASCOT search engine (version 2.3.0; Matrix Science, London, United Kingdom). A mass tolerance for precursor ions of 0.8 Da was used for precursor and fragment ions. Ion scores of 30 and above indicated identity. In addition, all spectral matches were verified manually. Unmatched MS/MS spectra and all MS/MS spectra from proteinase K digests were examined manually to determine the sequences of peptide y- and b-type ions.
Construction of CD3350 and CDR3194 insertional mutants and complemented mutants.
The target site was identified for the CD3350 gene from C. difficile 630 using the TargeTron gene knockout system (Sigma-Aldrich), and it was used to design a 45-bp retargeting sequence for the gene. A derivative of plasmid pMTL007C-E2 carrying the retargeting sequence was obtained from DNA2.0 (Menlo Park, CA) and used to generate mutants in strains 630Δerm and R20291 according to Heap et al. (25, 26). A minimum of two Erm-resistant transconjugants for each strain were checked by PCR using the ErmRAM primers to verify splicing of the group I intron following integration and also using flanking primers for the CD3350 gene to verify the disruption of CD3350 and CDR3194 genes by the erm cassette.
Each of the CD3350 and CDR3194 glycosyltransferase mutant strains were complemented with a wild-type copy of the C. difficile CD3350 gene using plasmid pRPF185 (27). The entire coding sequence of the gene, including the Shine-Dalgarno sequence, was cloned under the control of the inducible Ptet promoter. Plasmids were transferred to ΔCD3350 and ΔCDR3194 mutant strains via conjugation, and gene expression was induced by plating onto BHIS agar containing anhydrous tetracycline at 500 ng/ml after growth to mid- to late-log phase in BHIS broth.
Western blotting.
Spore samples were harvested at 72 h, resuspended to 1 × 107 spores/100 μl in 1× Laemmli loading buffer, and heated to 95°C for 5 min. Spore extracts were separated on 3 to 8% NuPAGE Novex Tris-acetate minigels and blotted onto polyvinylidene difluoride (PVDF). The membrane was probed with a 1:5,000 dilution of anti-β-O-GlcNAc (Covance, Montreal, Canada) in PBS–0.1% Tween 20 (PBS-T). Reactivity was detected with anti-mouse IgM horseradish peroxidase (HRP) conjugate (Sigma-Aldrich, Oakville, Canada) secondary antibody at a 1:10,000 dilution in PBS-T. Blots were imaged with an ECL Prime Western blotting detection kit (GE Healthcare, Baie D'Urfe, QC, Canada) according to the manufacturer's instructions, followed by exposure to X-ray film.
Transcription of CD3350 and BclA3 genes by RT-PCR.
To determine if the CD3350 and bclA3 genes are cotranscribed, reverse transcriptase PCR (RT-PCR) was performed using primers designed to amplify across the intergenic region between the two genes from C. difficile 630. RNA template was extracted from broth-grown cells (4 h) using a TRIzol extraction procedure (28). All RNA samples were treated with RNase-free DNase (Thermo Scientific) to remove contaminating DNA. RNA was quantified and 30 ng was used for each RT-PCR using a Sensi-Script RT kit (Qiagen) and PCR amplification using TopTaq Master. In addition, PCR amplifications were performed with the same primers using genomic DNA to verify amplicon size and specificity of primer pairs. Control PCRs of RNA without reverse transcriptase confirmed the absence of contaminating DNA in samples.
Spore production for biological testing.
For production of mature spores, plates were incubated for 7 days in an anaerobic incubator (Don Whitely Scientific, United Kingdom) on BHI at 37°C. Spores were harvested from agar and heat treated at 60°C for 20 min. To purify spores, samples were washed 10× in H2O and the number of spores (CFU/ml) determined by serial dilution and plating on BHI-ST.
Immunofluorescence.
Spores at 1 × 108/ml were air dried and heat fixed onto glass coverslips (VWR). The spores were blocked with 5% milk-PBS for 30 min at room temperature and then incubated with a 1:100 dilution in PBS of β-O-GlcNAc monoclonal antibody (Covance) for 45 min at room temperature. Coverslips were washed with PBS-T and then incubated with a 1:100 dilution in PBS of anti-mouse IgG plus IgM fluorescein isothiocyanate (FITC) conjugate (Caltag, Burlingame, CA) for 45 min at room temperature in the dark. Coverslips were washed with PBS-T and then mounted onto slides with Vectashield plus 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Slides were examined with an Axioplan 200M (Zeiss) with multiple fields of view observed. The experiment was performed in duplicate on at least three independent occasions. For quantification of GlcNAc reactivity to spores, slides were prepared as stated above using spores that had been grown for 7 days and washed with H2O, and then they were examined by microscopy. Using an Axioplan 200M microscope (Zeiss), at least 8 fields of view were examined per slide, with three replicate slides per sample. At least 100 spores per slide were counted for anti-β-O-GlcNAc binding to phase-bright spores. This was performed on at least three occasions to enumerate the percentage of fully mature spores that could be bound with anti-β-O-GlcNAc.
Spore heat resistance assay.
Heat resistance of C. difficile spores was determined as previously described (7). Briefly, spores of R20291 and CDR3194 mutant strains were resuspended in 5 ml of PBS at 1 × 106/ml, with starting inoculum numbers confirmed by serial dilution and plating on BHI-ST, and then incubated anaerobically for 24 h at 37°C. One-ml aliquots of spores were heated to 80°C for 20 min in a water bath, plated on BHI-ST, and incubated anaerobically for 24 h at 37°C to determine the number of CFU/ml. Percent survival was determined by comparing pre- and postheat treatment CFU/ml. The experiment was performed in triplicate on at least three independent occasions.
Spore lysozyme resistance assay.
Spores of R20291 and CDR3194 mutant strains were diluted to 1 × 106/ml in 5 ml PBS with starting inoculum numbers confirmed by serial dilution as described above. Lysozyme was added to a final concentration of 250 μg/ml, and 1-ml samples were incubated for 1 h at 37°C. The number of CFU/ml was determined by serial dilution and plating on BHI-ST. Percent survival was determined by comparing pre- and postlysozyme treatment CFU/ml. The experiment was performed in triplicate on three independent occasions.
Spore ethanol resistance assay.
Spores of R20291 and CDR3194 mutant strains were diluted to 1 × 106/ml in 5 ml 70% ethanol. The time point at which the spore count was 0 CFU/ml was confirmed by serial dilution and plating on BHI-ST as described above. One-ml aliquots were incubated at room temperature for 20 min, plated on BHI-ST, and incubated anaerobically for 24 h at 37°C to determine the number of CFU/ml. Percent survival was determined by comparing pre- and postethanol treatment CFU/ml. Experiments were performed in triplicate on three independent occasions.
Macrophage assay.
J774A.1 macrophages were cultured at 5 × 105 cells/well on coverslips in a 24-well plate in 1 ml RPMI media supplemented with 10% fetal bovine serum (FBS) (R10) under 5% CO2 at 37°C for 24 h. Spores were diluted to 5 × 106/ml (multiplicity of infection [MOI], 10:1) in R10, and CFU/ml was calculated by dilution series and plating on BHI-ST. J774A.1 cells were washed with PBS, and then 1-ml spores were added to four replicate wells. The plate was incubated for 30 min at 37°C and 5% CO2, and then wells were washed with PBS before cells were fixed with 250 μl 4% formaldehyde for 15 min at room temperature. Cells were washed with PBS and then incubated with a rabbit anti-C. difficile polyclonal antisera (CD3) at a 1:100 dilution in PBS for 45 min at room temperature. Coverslips were washed with PBS and then incubated with anti-rabbit Alexa fluor 488 (Invitrogen) at a 1:1,000 dilution in PBS for 45 min at room temperature. Coverslips were washed with PBS, and then cells were permeabilized with 0.1% Triton-PBS for 15 min at room temperature. Coverslips were washed with PBS and then incubated for 45 min at room temperature in a 1:100 dilution of CD3 antisera in PBS. Coverslips were washed in PBS and then incubated in a 1:1,000 dilution of anti-rabbit Alexa Fluor 594 (Invitrogen) for 45 min at room temperature. Coverslips were washed with PBS and then mounted onto slides with Vectashield plus DAPI (Vector Laboratories) and sealed with nail polish. Slides were examined with an Axioplan 200M microscope (Zeiss). Three coverslips per strain, with 50 J774A.1 cells per coverslip, were counted utilizing z stack images to gain a three-dimensional (3D) representation of the cell. Adhesion and internalization were quantified by counting adhered (red/green) and internalized (red) spores and calculating percent adhered or internalized per J774A.1 cell based on known MOIs. The assay was performed in triplicate on three independent occasions.
Statistical analysis.
Student's t test with Welch's correction was used for pairwise comparisons.
RESULTS
Bioinformatic identification of BclA and BclB homologs in strains of Clostridium difficile.
The Gram-positive spore-forming bacterium Bacillus anthracis elaborates two glycosylated spore surface proteins, denoted BclA (BAS1130; GenBank accession number YP_027402 in strain Sterne) and BclB (BAS2281; YP_028542 in strain Sterne), for Bacillus collagen-like protein (Fig. 1a). Homologs of BclA have also been found within the genome sequences of Bacillus cereus and Bacillus thuringiensis (29). BLAST searches of the BclA and BclB sequences against genome sequences of three C. difficile strains revealed Bcl protein homologs. C. difficile 630, the first strain to have a completed genome sequence, had three ORFs with homology to BclA and BclB. These were found in different regions of the C. difficile 630 genome, as shown in Fig. 1b. Percent homology and expect (E) values of significance are shown in Table 2. The open reading frame (ORF) CD3349 was annotated BclA3 (YP_001089866) despite showing greater homology to BclB; therefore, we refer to this protein as BclA3 to remain consistent with the genome annotation. Figure 1b also shows C. difficile 630 CD3349 (BclA3; YP_001089866) to be located 66 bp downstream of a putative glycosyltransferase (CD3350; YP_001089867). The glycosyltransferase gene has high homology to a B. anthracis glycosyltransferase (BAS 1131; YP_027403) that is likely responsible for the transfer of carbohydrate components to exosporangial proteins in this species. The proximity of the genes and high homology with genes of known function within B. anthracis provided early suggestions that C. difficile exosporangial proteins are glycosylated.
FIG 1.

Schematic diagram showing the genetic organization of the putative C. difficile exosporium glycoprotein genes and related glycosyltransferase gene. (a) Bacillus anthracis Sterne strain has been shown to possess two exosporium glycoprotein genes, BAS 1130 and 2281 (colored black in the figure) denoted bclA and bclB. A glycosyltransferase has also been identified lying adjacent to bclA (BAS 1131, denoted white). Genetic organization of the bcl homologs (black arrows) and putative glycosyl transferase sgtA (white arrows) in C. difficile 630 (b), R20291 (c), and QCD-32g58 (d).
TABLE 2.
BLAST search of Bacillus anthracis exosporangial glycoprotein (BclA and BclB) translated gene sequences against selected Clostridium difficile translated genomes
| B. anthracis strain Sterne protein | % Sequence identity toa: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
C. difficile strain 630 |
C. difficile strain R20291 |
C. difficile strain QCD-32g58 |
|||||||
| Exosporium glycoprotein BclA1 | Exosporium glycoprotein BclA2 | Exosporium glycoprotein BclA3 | Glycosyl transferase | Exosporium glycoprotein BclA2 | Exosporium glycoprotein BclA3 | Glycosyl transferase | Putative exosporium glycoprotein BclA3 | Glycosyl transferase | |
| BclA | 60 (2e−68) | 57 (4e−49) | 49 (1e−41) | 56 (1e−52) | 56 (7e−32) | 62 (7e−41) | |||
| BclB | 69 (1e−38) | 62 (2e−34) | 50 (1e−35) | 64 (8e−37) | 50 (1e−35) | 50 (3e−35) | |||
| Glycosyl transferase | 47 (7e−114) | 46 (4e−113) | 46 (4e−113) | ||||||
Shown are the percent sequence identities and E values (in parentheses).
We next searched the genomes of C. difficile strains QCD-32g58 and R20291. Within the strain QCD-32g58 genome, only a single ORF had significant homology to the Bcl proteins; CdifQ_040500019311 (WP_009891815) showed 62% homology to BclA and 50% homology to BclB of B. anthracis. A BLAST search of the glycosyltransferase gene BAS 1131 against the QCD-32g58 genome sequence showed a homolog, a putative glycosyltransferase (CdifQ_040500019316; WP_009891817), 78 bp upstream of the putative exosporium glycoprotein gene (CdifQ_040500019311). The genome of strain R20291 contained two bcl gene homologs, with the translated product of CDR20291_3090 (BclA2; YP_003219565) showing 64% homology to B. anthracis BclB and that of CDR20291_3193 (BclA3; YP_003219669) showing 56% homology to B. anthracis BclA. In addition, CDR20291_3194 (YP_003219670), which lies 78 bp downstream of BclA3, showed 46% identity to the B. anthracis glycosyltransferase (Table 2).
In all three strains, the Bcl protein homologs have only short, trypsin-susceptible regions (as predicted by amino acid sequence) at the N and C termini. The central region of the Bcl protein homologs is comprised of approximately 40 kDa of collagen-like repeats with no predicted trypsin cleavage site. The three C. difficile glycosyltransferase homologs have identical sequences apart from a single conserved amino acid substitution at the third amino acid in the sequence.
Characterization of C. difficile exosporangial surface extraction.
Previous studies have demonstrated that C. difficile spores possess an exosporangial layer which surrounds the spore coat, and this layer has been shown to be structurally variable among isolates (6, 30, 31). Various treatments of spores have been utilized to remove this layer, which allowed the characterization of the underlying spore coat (7, 8, 30, 32). The structural components of the exosporangial layer have yet to be characterized; thus, in this study we focused on identifying and characterizing spore surface-associated protein components. Using a detergent-based extraction, we removed surface-associated components from spore preparations which had not been extensively water washed or treated with enzymes/sonication to facilitate retention of surface structures. Endospores of strains 630, QCD-32g58, and R20291 were incubated in detergent solutions to extract the spore surface proteins, and then intact spores were removed by centrifugation. The protein-containing supernatants were resolved by a 3 to 8% NuPAGE gradient gel. The high-molecular-mass region of the gel shows diffuse banding patterns reactive with both silver stain (Fig. 2a) and glycostain (Fig. 2b), suggesting high-molecular-mass complexes containing glycoproteins. Proteinase K digestion of spores had only a marginal effect on the migration of this material, suggesting a more complex composition. A pattern of staining in this region distinct from that of strain 630 was obtained for R20291 and QCD32g58 spore extracts. Glycostaining revealed a reactive high-molecular-mass band in R20291 and QCD32g58 extracts only.
FIG 2.

NuPAGE gel analysis of C. difficile endospore surface protein extracts. (a) Silver-stained 3 to 8% NuPAGE; (b) Pro-Emerald Q glycostain 3 to 8% NuPAGE. Lane 1, HiMark-prestained molecular mass marker; lane 2, 630Δerm spore surface extract; lane 3, R20291 spore surface extract; lane 4, QCD-32g58 spore surface extract. Arrows indicate regions of the gel that were excised, enzymatically digested, and analyzed by nLC-MS/MS.
Initially, extraction of all gel bands of molecular masses of <160 kDa was performed, and each band was digested with either trypsin or proteinase K and analyzed by nLC-MS/MS. No BclA protein identification was made from these analyses. Subsequently, the high-molecular-mass region (>160 kDa) of each lane was excised in bands and digested with trypsin or proteinase K. The trypsin digests for all three strains did not result in any protein identifications by nLC-MS/MS. Analysis of the MS/MS spectra from proteinase K digests, however, yielded several spore surface protein identifications, as indicated by the numbered annotations in Fig. 2a, lane 3, and summarized in Table 3 for R20291 and Fig. S1 in the supplemental material for QCD-32g58.
TABLE 3.
nLC-MS/MS analysis of glycoreactive peptidesa
| Gel band | Accession no. (designation) | Protein (molecular mass, kDa) | Peptide sequence | Glycan mass, Da (monosaccharide neutral losses, Da) |
|---|---|---|---|---|
| 1 | YP_003219669 (CDR20291_3193) | Exosporium glycoprotein BclA3 (59.5) | 308AGLIGPTGATGV319 | 609 (203-203-203) |
| 145TGPTGATGADGITGP159 | 983 (203-203-203-374) | |||
| 344VGPTGATGA352b | 406 (203-203) | |||
| 189GLIGPTGATGTPGA202 | 406 (203-203) | |||
| 278TGATGLIGPTGATGA292 | 1,186 (203-203-203-203-374) | |||
| 278TGATGLIGPTGATGA292 | 1,241 (203-203-203-203-429) | |||
| 212TGIGITGPTGATGA225c | 1,184 (203-203-203-203-372) | |||
| 212TGIGITGPTGA222d | 1,095 (203-203-203-486) | |||
| 309GLIGPTGATGVTGA322 | 1,071 (203-203-203-462) | |||
| 299TGVTGATGAAGLIGP313 | 609 (203-203-203) | |||
| 2 | YP_003219669 (CDR20291_3193) | Exosporium glycoprotein BclA3 (59.5) | 278TGATGLIGPTGATGA202 | 1,187 (203-203-203-203-375) |
| 191IGPTGATGTPGATGPTGA208 | 1,661 (203-203-203-222-203-203-424) | |||
| 424TGPTGATGPTGADGL438 | 609 (203-203-203) | |||
| 3 | YP_003219669 (CDR20291_3193) | Exosporium glycoprotein BclA3 (59.5) | 119GVTGPTGPTGPTGATGA135 | 609 (203-203-203) |
| 169GVTGPTGPTGATGV182 | 609 (203-203-203) | |||
| 344VGPTGATGATGADGL358 | 609 (203-203-203) | |||
| 359VGPTGPTGATGV370 | 609 (203-203-203) | |||
| 191IGPTGATGTPGATGPTGA208 | 609 (203-203-203) | |||
| 311IGPTGATGVTGADGA325 | 609 (203-203-203) | |||
| 439VGPTGATGATGL450 | 609 (203-203-203) | |||
| 391VGPTGATGATGADGV405 | 609 (203-203-203) | |||
| 359VGPTGPTGATGV370 | 203 | |||
| 656ATASGLSLVNTVA668 |
High-molecular-mass gel bands of R20291 spore surface extracts were digested with proteinase K, and the numbering of gel bands refers to that of Fig. 2. MS/MS spectra were de novo sequenced, and the identified peptides and observed glycan moieties are indicated.
This sequence was also found at amino acids 391 to 399, 439 to 447, and 487 to 495.
This sequence was also found at amino acids 259 to 272.
This sequence was also found at amino acids 259 to 269.
De novo sequencing of the peptide MS/MS spectra from the proteinase K digests of gel bands 1 to 3 from surface extract of R20291 spores revealed a number of peptides that corresponded to the putative exosporium glycoprotein BclA3 (CDR20291_3193). Further inspection of the MS/MS spectra showed peptides with ions that did not correspond to peptide y- or b-type ions but were characteristic of carbohydrate-associated fragment ions. For example, from tandem mass spectrometry analyses of band 1 (Fig. 2a), which migrated to a molecular mass of greater than 600 kDa, the MS/MS spectrum of the putative glycoprotein peptide AGLIGPTGATGV modified with three N-acetyl hexosamine (HexNAc) moieties is shown in Fig. 3A. The glycan modification was observed as sequential neutral losses of 203 Da from the glycopeptide precursor ion in the high-m/z region of the MS/MS spectrum. In addition, an intense glycan oxonium ion was observed at m/z 204 which was common to all of the identified glycopeptides. In addition, more complex glycosylation patterns were observed in some cases, with intense ions observed in glycopeptide spectra that did not correspond to HexNAc residues or peptide type y or b fragment ions (for example, oxonium ions corresponding to masses of 486 Da, 372 Da, and 374 Da). Figure 3b shows an MS/MS spectrum of the BclA peptide TGPTGATGADGITGP modified with three HexNAc moieties and an additional mass of 374 Da. The sequential neutral losses suggest that HexNAc is the linking sugar. This glycan neutral mass was also linked to a putative glycan oxonium ion at m/z 375. Other intense ions were also observed in the low-m/z region of this MS/MS spectrum, including putative glycan fragment ions at m/z 300 and 272. The absence of potential N-linked glycosylation sites suggested the glycans are O-linked through threonine residues within each peptide. Observed sequential neutral losses of 203 Da in the high-m/z region of the spectrum and the presence of an intense ion corresponding to the unmodified form of the peptide suggest the glycan is composed of oligosaccharide chains attached to a single-amino-acid residue in each identified glycopeptide.
FIG 3.

Mass spectrometry analysis of peptides from proteinase K digestion of C. difficile R20291 endospore surface extracts. (a) nLC-MS/MS spectrum of the doubly protonated glycopeptide ion at m/z 811.8. Peptide type y and b ions were visible and gave the peptide sequence AGLIGPTGATGV, a peptide from the BclA3 protein. The spectrum was dominated in the high-m/z region by sequential neutral losses of 203 Da, with the unmodified peptide ion observed at m/z 1,013.5. Combined with the observed intense glycan oxonium ion at m/z 204 and neutral losses of water to give glycan-related ions at m/z 186 and 168, this spectrum suggested the peptide was modified with a chain of 3 HexNAc moieties. (b) nLC-MS/MS spectrum of a doubly protonated glycopeptides ion at m/z 1,129. Peptide type y and b ions corresponded to a sequence of TGPTGATGADGITGP, corresponding to the BclA3 protein. The high-m/z region of the spectrum was dominated by sequential neutral losses of 374 Da, 203 Da, 203 Da, and 203 Da. An intense oxonium ion was observed at m/z 375, and a very weak oxonium ion was observed at m/z 204 (not indicated). Glycan-related fragment ions were observed at m/z 300 and 272. (c) Peptide sequence coverage map of BclA3 protein homolog from spore surface protein extraction (band 1). Boldface and underlining indicate peptides modified with glycan moieties. Two of the peptides shown to be modified with glycan are shown in boldface gray text to indicate that the amino acid sequence appears in the BclA3 protein more than once. Dotted underlining indicates a glycopeptide sequence that is common to both BclA3 and BclA2 proteins.
Table 3 shows the complete list of surface protein peptides and glycopeptides identified from each of the annotated bands indicated in Fig. 2a. The unknown glycans varied in observed mass from 281 to 486 Da, and it is possible that these are modified HexNAc moieties. Tandem mass spectrometry analysis of proteinase K digests of bands 2 and 3 showed BclA3 glycopeptides modified with chains of HexNAc moieties, predominantly in trimers. In these cases, only peptides modified with HexNAc moieties were observed at detectable levels.
All of the identified glycopeptides reside within the central collagen-like repeating domain of the BclA3 protein; however, sample limitations prohibited the identification of the precise sites of modification within the respective peptides or further characterization of the glycan moieties. The central collagen-like repeat domains of the putative exosporial proteins contained some nonunique regions, which resulted in the identification of multiple glycopeptides with amino acid sequences that repeat within the protein sequence (for example, TGIGITGPTGA occurs in BclA3 at residues 212 to 222 and 259 to 269). One of the identified peptides, which also happens to be repeated in BclA3 four times, is also common to both BclA3 and BclA2 (VGPTGATGA). The nonspecific cleavage by proteinase K produced a number of glycopeptides with overlapping sequences. Furthermore, multiple glycopeptides were identified that possessed identical peptide sequences but different glycans.
As indicated above, strains R20291 and QCD-32g58 showed similar protein staining patterns for both silver stain and glycostains, and nLC-MS/MS analysis also showed that the Bcl protein of QCD-32g58 is similarly glycosylated predominantly with HexNAc moieties (see Fig. S1 in the supplemental material). nLC-MS/MS analysis of the gel digests from strain 630, which showed significantly different staining patterns in the high-molecular-mass region of the gel compared to the other two strains, did not yield any protein or glycoprotein identifications.
Anti-β-O-GlcNAc reactivity of C. difficile spores.
As the most abundant glycan modification observed in the MS analysis of spore surface-extracted material was shown to have a mass corresponding to an N acetyl-hexosamine moiety, we next examined the ability of spores to bind to an O-linked N-acetylglucosamine (β-O-GlcNAc) antibody. A monoclonal antibody (MAb) which recognizes O-GlcNAc in a β-O-glycosidic linkage to both threonine and serine was utilized in immunofluorescence experiments with intact spores from a number of C. difficile clinical isolates (Fig. 4a). This antibody had been used previously to demonstrate the presence of β-O-GlcNAc attached to serine and threonine residues of Listeria monocytogenes flagellin (33).
FIG 4.

Immunofluorescence of anti-β-O-GlcNAc binding to spores. (a) Wild-type spores of C. difficile strains from a range of ribotypes and geographical locations. For 630Δerm, GlcNAc binding at poles is marked with arrows. (b) sgtA mutant spores of strains R20291 and 630Δerm. First row, merged image of phase contrast, DAPI, and FITC; 2nd row, DAPI channel only; 3rd row, FITC channel only; 4th row, phase-contrast image only. GlcNAc was visualized with mouse anti-β-O-GlcNAc and anti-mouse IgM-FITC conjugate. (c) Percentage of spores reacting with anti-β-O-GlcNAc after 7 days of growth. At least 100 spores were counted in triplicate on three independent occasions for anti-β-O-GlcNAc binding to the surface, as analyzed by immunofluorescence microscopy.
When the β-O GlcNAc antibody was used in immunofluorescence reactions with spores of R20291 and 630Δerm, both spore preparations reacted strongly with the antibody. Interestingly, distinct patterns of reactivity with the spore surface were observed by this immunofluorescence method for each strain (Fig. 4a). With R20291 spores, anti-β-O-GlcNAc was uniformly reactive over the entire spore surface, while with strain 630Δerm, anti-β-O-GlcNAc reactivity was restricted to the poles of the spores with only limited labeling of the central surface (Fig. 4a, arrows). Vegetative cells of both strains showed no reactivity with anti-β-O-GlcNAc (see Fig. S2 in the supplemental material). To confirm the conservation of β-O-GlcNAc on the surface of multiple C. difficile strains, a range of spores from different ribotypes and geographic locations were also tested for anti-β-O-GlcNAc binding. The reactivity pattern observed for R20291 spores was found to be conserved in all strains examined, with the only exception being spores of 630Δerm (Fig. 4a). Any unstained cells in the images were either immature spores or cell debris from the washing process. DAPI binding was observed only with vegetative cells and immature phase-dark spores; phase-bright spores were considered mature.
RT-PCR of CD3350-bclA3 gene locus.
As indicated in Fig. 1, CD3350 (B. anthracis exosporangial glycosyltransferase gene homolog) and bclA3 lie immediately adjacent to each other and are orientated in the same direction on the chromosome in both 630 and R20291 strains. The two genes are separated by only a short intergenic region, suggesting they form a single transcriptional unit. Primers which amplified across this intergenic region were used to determine if the genes were cotranscribed. RNA samples extracted from C. difficile 630 cells were subjected to reverse transcription, and an amplification product of 257 bp linking CD3350 and bclA3 was obtained, confirming cotranscription of these two genes (see Fig. S3 in the supplemental material). PCRs using the same primers and total RNA that had not undergone a reverse transcriptase reaction did not yield any amplification product, demonstrating that the RNA was free of contaminating DNA.
Mutagenesis of CD3350 and CDR3194 and spore characterization.
We next generated insertionally inactivated glycosyltransferase mutants in strains 630Δerm and R20291 (ΔCD3350 and ΔCDR3194) by using ClosTron technology as previously described (26). Insertion of the TargeTron Erm resistance marker was confirmed by PCR using primers flanking the gene of interest and with primers specific to the TargeTron Erm resistance marker (data not shown). Vegetative cell growth of both ΔCD3350 and ΔCDR3194 was unchanged compared to that of their respective parent strains, and motility was unaffected (data not shown). Immunofluorescence of spores with anti-β-O-GlcNAc antibody revealed a complete loss of reactivity for both ΔCD3350 and ΔCDR3194 compared to the respective parent strains (Fig. 4b). The percentage of wild-type spores compared to mutant phase-bright spores reacting with anti-β-O-GlcNAc antibody was quantified microscopically and shown to be 80 to 95% compared to less than 1% for the respective mutants (Fig. 4c).
Both ΔCDR3194 and ΔCD3350 strains were complemented with wild-type copies of CD3350 using pRPF185 (27), as evidenced by both Western blotting and immunofluorescence studies (Fig. 5; also see Fig. S4 in the supplemental material). As can be seen in Fig. 5, lanes 2 and 5, a positive reaction was observed in Western blotting with spore surface extracts from both R20291 and 630Δerm spores, respectively. Spore extracts of R20291 displayed reactivity with the region of gel corresponding to band 4 (approximately 400 kDa) from MS analysis. In addition, a second strongly reactive band migrating at a molecular mass of approximately 170 kDa on 3 to 8% NuPAGE gel was observed, although we were unable to identify peptides from a proteinase K digestion of this region of the gel by MS analysis. For strain 630Δerm, no reactivity was observed in the corresponding higher-molecular-mass region of the gel, and a series of three distinct reactive bands was observed at approximately 170 kDa. All reactivity was lost in CD3350 and CDR3194 mutant strains, while the strain-specific pattern of reactivity was restored upon complementation (Fig. 5; also see Fig. S4). On the basis of these results, we now propose that this gene be renamed sgtA (spore glycosyl transferase).
FIG 5.

Restoration of anti-GlcNAc reactivity through complementation. Western blot of cultures grown on plates for 72 h and run on 3 to 8% Nu-PAGE gel. Complemented strains were induced with 500 ng anhydrotetracycline. Lane 1, HiMark (Invitrogen); lane 2, R20291; lane 3, R20291ΔCDR3194; lane 4, R20291Δ3194p3350; lane 5, 630Δerm; lane 6, 630Δ3350; lane 7, 630Δ3350p3350.
Characterization of ΔsgtA spore surface extract.
In parallel with the spore surface protein extracts of the wild-type strain, spore surface extracts of the R20291ΔsgtA mutant strain were also prepared and analyzed by 3 to 8% NuPAGE Tris-acetate gels to resolve high-molecular-mass material. The protein-stained gel shows a diffuse area of staining at 460 kDa and greater; however, the distinct ∼600-kDa band was not observed. Similarly, glycostaining of the same gel showed no detectable reactivity at ∼600 kDa (see Fig. S5 in the supplemental material). In contrast to MS studies of gel bands of R20291 spore surface extracts, which identified peptides/glycopeptides in proteinase K digests, the equivalent region of the NuPAGE gel of the spore surface protein extraction of ΔsgtA did not yield any peptide or glycopeptide identifications. In addition, to date, our analyses of lower-molecular-mass protein bands from spore surface extracts have shown no evidence of unglycosylated BclA3.
Resistance of R20291ΔsgtA spores.
As the more clinically relevant strain and as shown by immunofluorescence to be a more representative strain of C. difficile spore morphology, phenotypic assays were undertaken on R20291 wild-type spores and ΔsgtA spores. The heat resistance of spores was examined as previously described (7). When incubated at 80°C for 20 min, ΔsgtA spores showed significantly lower survival rates than the parent R20291 spores (Fig. 6). The susceptibility of spores to 70% ethanol and 250 μg/ml lysozyme was also examined, but no significant difference was observed between wild-type and ΔsgtA spores (see Fig. S6 in the supplemental material).
FIG 6.
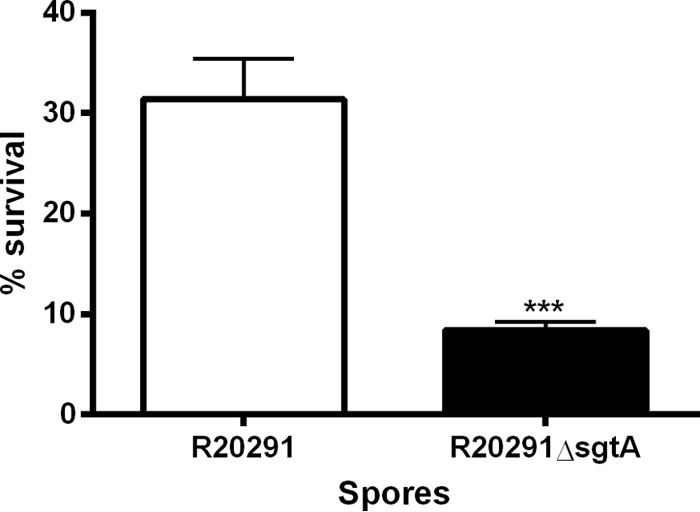
Resistance of R20291 wild-type and ΔsgtA spores to 80°C for 20 min. Spores were incubated for 20 min in a water bath at 80°C, and then the number of CFU/ml was determined. Percent survival was calculated by comparing inocula to postheat treatment samples. P < 0.0001 by t test with Welch's correction.
Role of sgtA in adherence and internalization of macrophage cells.
To gain insight into a possible biological role for the SgtA glycosyltransferase, we next investigated the ability of spores to adhere to and be internalized by the J774A.1 macrophage cell line (ATCC TIB-106). Spores were counted based on association with J774A.1 cells and counted as adhered if green/red and internalized if red. Spores not associated with cells were ignored, as were any remaining vegetative cells based on rod shape. As can be seen in Fig. 7, it is clear that adherence and internalization of J774A.1 macrophage cells by C. difficile R20291 spores was affected following inactivation of the sgtA gene, with significantly greater numbers of ΔsgtA spores being internalized compared to the wild type.
FIG 7.
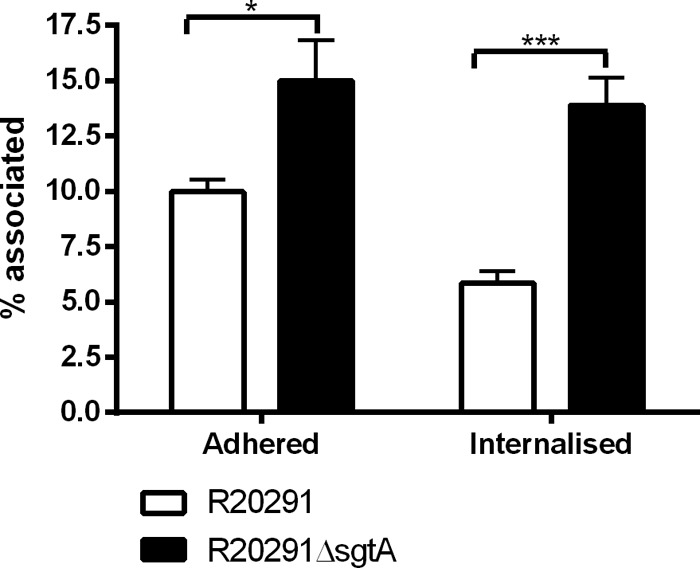
Adherence and invasion of J774A.1 macrophage cells. Shown are the percentages of spores adhering to or internalized into J774A.1 macrophages after 30 min of incubation at 37°C under 5% CO2. Percentages were calculated based on known MOIs and final adhered (green/red) or internalized (red) spores. Fifty J774A.1 cells were counted in triplicate on three independent occasions. Statistical analysis was performed with a t test with Welch's correction (*, P < 0.05; ***, P < 0.0001).
DISCUSSION
This study presents, to our knowledge, the first characterization of glycoproteins from C. difficile spores and provides direct evidence demonstrating that BclA3 is a glycoprotein which is glycosylated with chains of β-O-linked GlcNAc as well as with additional glycans of novel masses. Bioinformatic analysis of clostridial genomes revealed the presence of Bacillus anthracis exosporangial bclA and bclB gene homologs. In addition, immediately upstream of the B. anthracis bclB gene homologs in all C. difficile strains, and shown in this study to be cotranscribed with bclA3, lies a homolog of the neighboring B. anthracis glycosyltransferase gene (BAS1131). While the predicted molecular mass of BclA3 based on the translated amino acid sequence is ∼57 kDa, identification of BclA3 was made only in higher-molecular-mass material found to be migrating in NuPAGE gels at masses corresponding to >460 kDa and which stained in a diffuse pattern by silver staining. In addition, reactivity with glycostain in this region of the gel was observed, suggesting that the higher-molecular-mass material also contained carbohydrate. After repeated attempts to identify the composition of the high-molecular-mass material using trypsin digestion of selected regions of the gel, we were finally successful in obtaining peptide identifications following extensive proteinase K digestion of the gel bands. This nLC-MS/MS analysis identified BclA3 peptides and also provided the first evidence that this protein is a glycoprotein. In a fashion similar to that of B. anthracis, it appears that the BclA3 protein of C. difficile is glycosylated with predominantly novel tri- or pentasaccharide oligosaccharides composed of chains of N-acetyl hexosamine sugars, which may be capped with novel glycan moieties. Due to sample limitations and capping glycan heterogeneity, we were unable to determine the precise molecular structure, although it is clear from glycan component neutral masses that the structural composition of the C. difficile BclA3 glycan is quite distinct from that previously reported for B. anthracis (16). Gel migration characteristics do suggest that C. difficile BclA3 monomers from R20291 and QCD-32g58 form some sort of stable, higher-molecular-mass complex, which is resistant to denaturation by heating and detergents. In contrast, spore surface extracts of C. difficile 630 appear to form distinct, lower-molecular-mass complexes which do not appear to contain any BclA3 protein. In addition, spores of 630 produced a distinct pattern of β-O-linked GlcNAc reactivity by immunofluorescence studies, and the significance of these distinct differences in spore structure between isolates remains to be determined. However, β-O-linked GlcNAc reactivity of spores from a number of clinical isolates demonstrates, for the first time, the conserved nature of this posttranslational modification on C. difficile spores. Insertional inactivation of the glycosyltransferase gene, sgtA (CD3350 and CDR3194), provided direct evidence for a role of the glycosyltransferase enzyme in this spore surface β-O-linked GlcNAc reactivity as well as in the production of glycosylated BclA3. While previous work examining sporulation-related gene expression demonstrated that the bclA3 gene and the adjacent glycosyltransferase gene (sgtA) were activated by the sporulation sigma factor, σk (34, 35), this is the first study to link the sgtA gene to a specific spore glycan-associated function.
A role for surface-associated bacterial glycans in host interactions is well documented for many bacterial species (36–40). In the current study, we show that glycans on the spore surface impart resistance of spores to heat treatment as well as appear to play a role in macrophage interactions. In contrast to a previous study, where removal of the exosporangial layer by sonication did not affect binding and internalization of C. difficile spores by Raw 264.7 peritoneal macrophage cells (31, 41), in the current study the loss of the ability to glycosylate surface-associated proteins does appear to affect uptake by macrophages. It should be noted, however, that distinct strains of C. difficile and different macrophage cell lines were used in each study, which may explain the different results. It is clear, however, that C. difficile spores do interact and are internalized by macrophage cells and that the role of surface-expressed glycans now can be more fully explored. While an ability of C. difficile spores to avoid uptake by macrophages might be advantageous during infection, further studies will be required to define the precise role of glycan components in macrophage interactions. It should be noted that exosporium structures of B. anthracis appear to mask epitopes recognized by macrophages which are involved in induction of cytokines (42).
During the preparation of the manuscript, a detailed characterization of BclA1 from C. difficile 630 spores was reported (43). It appears that the BclA1 protein forms high-molecular-mass complexes in extracts of 630 spores in a similar fashion. It is significant that the genomes of strains R20291 and QCD-32g58, which were examined in the current study, encode a truncated bclA1 gene, which may explain why this protein was never identified in extracts. In our analyses of the high-molecular-mass complexes of C. difficile 630 spores, we did not identify any BclA1 peptides and/or glycopeptides, suggesting it forms a distinct complex. Both the current study and the recent BclA1 study also present evidence of additional glycoreactive bands in spore extracts, which will likely be important in our comprehensive understanding of the glycobiology of C. difficile spores.
While known to be recalcitrant to proteolytic digestion and structural characterization, the spores of Gram-positive bacterial pathogens have gained considerable attention in recent years. The initial demonstration that the exosporangial proteins of Bacillus anthracis were glycoproteins which carried a pentasaccharide glycan containing the novel sugar, anthrose was the first example of glycoproteins as a part of spore biology. This study has demonstrated that spores of a second important Gram-positive pathogen, C. difficile, also carry novel glycoproteins on surface-associated structures. Defining the role of these structures in the infectious process, as well as exploiting their potential in therapeutic and diagnostic applications, now can be further pursued. This work adds another example to the constantly expanding data set of protein glycosylation systems in bacterial pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Siklenka, S. O'Hara, and M. David for preliminary gel and mass spectrometry data analyses. We thank R. Fagan and N. Fairweather for provision of pRPF185 plasmid, A. Dascal for provision of C. difficile QCD-32g58, B. Wren for C. difficile strains R20291, BI-6, CD20, CF5, and M68, and N. Minton for C. difficile 630Δerm and the ClosTron mutagenesis system.
This work was funded by the National Research Council Canada.
Footnotes
Published ahead of print 9 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01469-14.
REFERENCES
- 1.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7:526–536. 10.1038/nrmicro2164 [DOI] [PubMed] [Google Scholar]
- 2.Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, Kuijper EJ, Wilcox MH. 2010. The changing epidemiology of Clostridium difficile infections. Clin. Microbiol. Rev. 23:529–549. 10.1128/CMR.00082-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ananthakrishnan AN. 2011. Clostridium difficile infection: epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 8:17–26. 10.1038/nrgastro.2010.190 [DOI] [PubMed] [Google Scholar]
- 4.Gerding DN, Muto CA, Owens RC., Jr 2008. Measures to control and prevent Clostridium difficile infection. Clin. Infect. Dis. 46(Suppl 1):S43–S49. 10.1086/521861 [DOI] [PubMed] [Google Scholar]
- 5.Lawley TD, Clare S, Walker AW, Goulding D, Stabler RA, Croucher N, Mastroeni P, Scott P, Raisen C, Mottram L, Fairweather NF, Wren BW, Parkhill J, Dougan G. 2009. Antibiotic treatment of clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect. Immun. 77:3661–3669. 10.1128/IAI.00558-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawley TD, Croucher NJ, Yu L, Clare S, Sebaihia M, Goulding D, Pickard DJ, Parkhill J, Choudhary J, Dougan G. 2009. Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J. Bacteriol. 191:5377–5386. 10.1128/JB.00597-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Permpoonpattana P, Phetcharaburanin J, Mikelsone A, Dembek M, Tan S, Brisson MC, La Ragione R, Brisson AR, Fairweather N, Hong HA, Cutting SM. 2013. Functional characterization of Clostridium difficile spore coat proteins. J. Bacteriol. 195:1492–1503. 10.1128/JB.02104-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Permpoonpattana P, Tolls EH, Nadem R, Tan S, Brisson A, Cutting SM. 2011. Surface layers of Clostridium difficile endospores. J. Bacteriol. 193:6461–6470. 10.1128/JB.05182-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paredes-Sabja D, Bond C, Carman RJ, Setlow P, Sarker MR. 2008. Germination of spores of Clostridium difficile strains, including isolates from a hospital outbreak of Clostridium difficile-associated disease (CDAD). Microbiology 154:2241–2250. 10.1099/mic.0.2008/016592-0 [DOI] [PubMed] [Google Scholar]
- 10.Burns DA, Heap JT, Minton NP. 2010. SleC is essential for germination of Clostridium difficile spores in nutrient-rich medium supplemented with the bile salt taurocholate. J. Bacteriol. 192:657–664. 10.1128/JB.01209-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burns DA, Heeg D, Cartman ST, Minton NP. 2011. Reconsidering the sporulation characteristics of hypervirulent Clostridium difficile BI/NAP1/027. PLoS One 6:e24894. 10.1371/journal.pone.0024894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abhyankar W, Hossain AH, Djajasaputra A, Permpoonpattana P, Ter Beek A, Dekker HL, Cutting SM, Brul S, de Koning LJ, de Koster CG. 2013. In pursuit of protein targets: proteomic characterization of bacterial spore outer layers. J. Proteome Res. 12:4507–4521. 10.1021/pr4005629 [DOI] [PubMed] [Google Scholar]
- 13.Charlton S, Moir AJ, Baillie L, Moir A. 1999. Characterization of the exosporium of Bacillus cereus. J. Appl. Microbiol. 87:241–245. 10.1046/j.1365-2672.1999.00878.x [DOI] [PubMed] [Google Scholar]
- 14.Redmond C, Baillie LW, Hibbs S, Moir AJ, Moir A. 2004. Identification of proteins in the exosporium of Bacillus anthracis. Microbiology 150:355–363. 10.1099/mic.0.26681-0 [DOI] [PubMed] [Google Scholar]
- 15.Sylvestre P, Couture-Tosi E, Mock M. 2002. A collagen-like surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol. Microbiol. 45:169–178. 10.1046/j.1365-2958.2000.03000.x [DOI] [PubMed] [Google Scholar]
- 16.Daubenspeck JM, Zeng H, Chen P, Dong S, Steichen CT, Krishna NR, Pritchard DG, Turnbough CL., Jr 2004. Novel oligosaccharide side chains of the collagen-like region of BclA, the major glycoprotein of the Bacillus anthracis exosporium. J. Biol. Chem. 279:30945–30953. 10.1074/jbc.M401613200 [DOI] [PubMed] [Google Scholar]
- 17.Steichen C, Chen P, Kearney JF, Turnbough CL., Jr 2003. Identification of the immunodominant protein and other proteins of the Bacillus anthracis exosporium. J. Bacteriol. 185:1903–1910. 10.1128/JB.185.6.1903-1910.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waller LN, Stump MJ, Fox KF, Harley WM, Fox A, Stewart GC, Shahgholi M. 2005. Identification of a second collagen-like glycoprotein produced by Bacillus anthracis and demonstration of associated spore-specific sugars. J. Bacteriol. 187:4592–4597. 10.1128/JB.187.13.4592-4597.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamborrini M, Holzer M, Seeberger PH, Schurch N, Pluschke G. 2010. Anthrax spore detection by a Luminex assay based on monoclonal antibodies that recognize anthrose-containing oligosaccharides. Clin. Vaccine Immunol. 17:1446–1451. 10.1128/CVI.00205-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong S, McPherson SA, Tan L, Chesnokova ON, Turnbough CL, Jr, Pritchard DG. 2008. Anthrose biosynthetic operon of Bacillus anthracis. J. Bacteriol. 190:2350–2359. 10.1128/JB.01899-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta AS, Saile E, Zhong W, Buskas T, Carlson R, Kannenberg E, Reed Y, Quinn CP, Boons GJ. 2006. Synthesis and antigenic analysis of the BclA glycoprotein oligosaccharide from the Bacillus anthracis exosporium. Chemistry 12:9136–9149. 10.1002/chem.200601245 [DOI] [PubMed] [Google Scholar]
- 22.Blum H, Beier H, Gross HJ. 1987. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8:93–99. 10.1002/elps.1150080203 [DOI] [Google Scholar]
- 23.Gharahdaghi F, Weinberg CR, Meagher DA, Imai BS, Mische SM. 1999. Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: a method for the removal of silver ions to enhance sensitivity. Electrophoresis 20:601–605 [DOI] [PubMed] [Google Scholar]
- 24.Fulton KM, Zhao X, Petit MD, Kilmury SL, Wolfraim LA, House RV, Sjostedt A, Twine SM. 2011. Immunoproteomic analysis of the human antibody response to natural tularemia infection with type A or type B strains or LVS vaccination. Int. J. Med. Microbiol. 301:591–601. 10.1016/j.ijmm.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heap JT, Cartman ST, Kuehne SA, Cooksley C, Minton NP. 2010. ClosTron-targeted mutagenesis. Methods Mol. Biol. 646:165–182. 10.1007/978-1-60327-365-7_11 [DOI] [PubMed] [Google Scholar]
- 26.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 80:49–55. 10.1016/j.mimet.2009.10.018 [DOI] [PubMed] [Google Scholar]
- 27.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J. Biol. Chem. 286:27483–27493. 10.1074/jbc.M111.263889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aubry A, Hussack G, Chen W, KuoLee R, Twine SM, Fulton KM, Foote S, Carrillo CD, Tanha J, Logan SM. 2012. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect. Immun. 80:3521–3532. 10.1128/IAI.00224-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Todd SJ, Moir AJ, Johnson MJ, Moir A. 2003. Genes of Bacillus cereus and Bacillus anthracis encoding proteins of the exosporium. J. Bacteriol. 185:3373–3378. 10.1128/JB.185.11.3373-3378.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Escobar-Cortes K, Barra-Carrasco J, Paredes-Sabja D. 2013. Proteases and sonication specifically remove the exosporium layer of spores of Clostridium difficile strain 630. J. Microbiol. Methods 93:25–31. 10.1016/j.mimet.2013.01.016 [DOI] [PubMed] [Google Scholar]
- 31.Paredes-Sabja D, Cofre-Araneda G, Brito-Silva C, Pizarro-Guajardo M, Sarker MR. 2012. Clostridium difficile spore-macrophage interactions: spore survival. PLoS One 7:e43635. 10.1371/journal.pone.0043635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barra-Carrasco J, Olguin-Araneda V, Plaza-Garrido A, Miranda-Cardenas C, Cofre-Araneda G, Pizarro-Guajardo M, Sarker MR, Paredes-Sabja D. 2013. The Clostridium difficile exosporium cysteine (CdeC)-rich protein is required for exosporium morphogenesis and coat assembly. J. Bacteriol. 195:3863–3875. 10.1128/JB.00369-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schirm M, Kalmokoff M, Aubry A, Thibault P, Sandoz M, Logan SM. 2004. Flagellin from Listeria monocytogenes is glycosylated with beta-O-linked N-acetylglucosamine. J. Bacteriol. 186:6721–6727. 10.1128/JB.186.20.6721-6727.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fimlaid KA, Bond JP, Schutz KC, Putnam EE, Leung JM, Lawley TD, Shen A. 2013. Global analysis of the sporulation pathway of Clostridium difficile. PLoS Genet. 9:e1003660. 10.1371/journal.pgen.1003660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saujet L, Pereira FC, Serrano M, Soutourina O, Monot M, Shelyakin PV, Gelfand MS, Dupuy B, Henriques AO, Martin-Verstraete I. 2013. Genome-wide analysis of cell type-specific gene transcription during spore formation in Clostridium difficile. PLoS Genet. 9:e1003756. 10.1371/journal.pgen.1003756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szymanski CM, Burr DH, Guerry P. 2002. Campylobacter protein glycosylation affects host cell interactions. Infect. Immun. 70:2242–2244. 10.1128/IAI.70.4.2242-2244.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott AE, Twine SM, Fulton KM, Titball RW, Essex-Lopresti AE, Atkins TP, Prior JL. 2011. Flagellar glycosylation in Burkholderia pseudomallei and Burkholderia thailandensis. J. Bacteriol. 193:3577–3587. 10.1128/JB.01385-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fletcher CM, Coyne MJ, Villa OF, Chatzidaki-Livanis M, Comstock LE. 2009. A general O-glycosylation system important to the physiology of a major human intestinal symbiont. Cell 137:321–331. 10.1016/j.cell.2009.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindenthal C, Elsinghorst EA. 1999. Identification of a glycoprotein produced by enterotoxigenic Escherichia coli. Infect. Immun. 67:4084–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benz I, Schmidt MA. 2001. Glycosylation with heptose residues mediated by the aah gene product is essential for adherence of the AIDA-I adhesin. Mol. Microbiol. 40:1403–1413. 10.1046/j.1365-2958.2001.02487.x [DOI] [PubMed] [Google Scholar]
- 41.Paredes-Sabja D, Sarker MR. 2012. Adherence of Clostridium difficile spores to Caco-2 cells in culture. J. Med. Microbiol. 61:1208–1218. 10.1099/jmm.0.043687-0 [DOI] [PubMed] [Google Scholar]
- 42.Basu S, Kang TJ, Chen WH, Fenton MJ, Baillie L, Hibbs S, Cross AS. 2007. Role of Bacillus anthracis spore structures in macrophage cytokine responses. Infect. Immun. 75:2351–2358. 10.1128/IAI.01982-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pizarro-Guajardo M, Olguin-Araneda V, Barra-Carrasco J, Brito-Silva C, Sarker MR, Paredes-Sabja D. 2013. Characterization of the collagen-like exosporium protein, BclA1, of Clostridium difficile spores. Anaerobe 25:18–30. 10.1016/j.anaerobe.2013.11.003 [DOI] [PubMed] [Google Scholar]
- 44.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J. Med. Microbiol. 54:137–141. 10.1099/jmm.0.45790-0 [DOI] [PubMed] [Google Scholar]
- 45.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102. 10.1186/gb-2009-10-9-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forgetta V, Oughton MT, Marquis P, Brukner I, Blanchette R, Haub K, Magrini V, Mardis ER, Gerding DN, Loo VG, Miller MA, Mulvey MR, Rupnik M, Dascal A, Dewar K. 2011. Fourteen-genome comparison identifies DNA markers for severe-disease-associated strains of Clostridium difficile. J. Clin. Microbiol. 49:2230–2238. 10.1128/JCM.00391-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HM, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J. 2010. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. U. S. A. 107:7527–7532. 10.1073/pnas.0914322107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.