

Abstract
Regulated intramembrane proteolysis of membrane-embedded substrates by site-2 proteases (S2Ps) is a widespread mechanism of transmembrane signal transduction in bacteria and bacterial pathogens. We previously demonstrated that the Mycobacterium tuberculosis S2P Rip1 is required for full virulence in the mouse model of infection. Rip1 controls transcription in part through proteolysis of three transmembrane anti-sigma factors, anti-SigK, -L, and -M, but there are also Rip1-dependent, SigKLM-independent pathways. To determine the contribution of the sigma factors K, L, and M to the Δrip1 attenuation phenotype, we constructed an M. tuberculosis ΔsigKΔ sigL ΔsigM mutant and found that this strain fails to recapitulate the marked attenuation of Δrip1 in mice. In a search for additional pathways controlled by Rip1, we demonstrated that the SigD regulon is positively regulated by the Rip1 pathway. Rip1 cleavage of transmembrane anti-SigD is required for expression of SigD target genes. In the absence of Rip1, proteolytic maturation of RsdA is impaired. These findings identify RsdA/SigD as a fourth arm of the branched pathway controlled by Rip1 in M. tuberculosis.
INTRODUCTION
Extracytoplasmic function (ECF) sigma factors are transcriptional regulators that allow bacteria to modulate gene expression in response to external stimuli (1). In the absence of an activating extracytoplasmic stimulus, the ECF sigma factor is often held inactive by a cognate negative regulator known as an anti-sigma factor (2). In many cases, the anti-sigma factor is a single-pass transmembrane protein, which sequesters the sigma factor to the cytoplasmic side of the plasma membrane, thereby preventing its interaction with RNA polymerase. In the presence of the activating signal for the pathway, the anti-sigma factor is degraded by two coupled proteolytic events: cleavage by a site-1 protease (S1P), immediately followed by a second intramembrane cleavage by a site-2 protease (S2P), effectively liberating the anti-sigma/sigma factor complex into the cytoplasm, where it is available to associate with RNA polymerase (RNAP) and mediate a transcriptional change in response to the initial stimulus (3).
The M. tuberculosis S2P Rip1 is required for the full virulence of M. tuberculosis. M. tuberculosis lacking rip1 is defective for initial growth in the lungs of mice and also substantially impaired for persistence during chronic infection (4). Rip1 cleaves not one but three different membrane-embedded anti-sigma factors: anti-sigma factor K (RskA), anti-sigma factor L (RslA), and anti-sigma factor M (RsmA) (5), which negatively regulate ECF sigma factors K (SigK), L (SigL), and M (SigM), respectively. This multiplicity of substrates has also been noted for other prokaryotic S2Ps (6, 7). Through Rip1-mediated proteolysis of RskA, RslA, and RsmA, anti-sigma factor inhibition of SigK, SigL, and SigM is relieved and these transcription factors are subsequently free to associate with RNAP and transcribe their respective regulons. Thus, activation of the SigK, SigL, and SigM regulons depends upon Rip1 activity. Taken together, these observations suggest that the Δrip1 mutant could be attenuated through the collective loss of the SigK, SigL, and SigM regulons. Potentially consistent with this idea, M. tuberculosis strains lacking sigL or sigM have been tested in the mouse model of infection and have mild virulence defects (8–11), suggesting that inactivation of more than one of these pathways may be required for full attenuation of virulence.
Genetic dissection of the relationships between Rip1 and these three sigma factor regulons has indicated the presence of both Rip1- and SigKLM-dependent pathways and Rip1-dependent, SigKLM-independent pathways. Analysis of the transcriptomes of Δrip1, ΔsigK, ΔsigL, and ΔsigM mutants revealed that induction of the gene encoding the catalase-peroxidase KatG and the upstream gene encoding the iron-dependent repressor FurA is dependent on Rip1, SigK, and SigL (5). In contrast, several other genes whose wild-type (WT) expression pattern required Rip1 were not affected by the loss of SigK, SigL, or SigM individually. The gene encoding the resuscitation-promoting factor C (rpfC), believed to play a role in dormancy (12, 13), and the mycobacterial β-ketoacyl acyl carrier protein (ACP) synthase gene kasA were both underexpressed in the Δrip1 mutant but unaffected by any individual sigma factor deletion (5).
Given the prominent role of prokaryotic S2Ps in ECF anti-sigma factor degradation, and our prior findings that Rip1 is a multi-ECF anti-sigma factor protease, we hypothesized that Rip1 may degrade additional ECF anti-sigma factors in M. tuberculosis. Indeed, M. tuberculosis encodes seven other ECF sigma factors, most with a putative cognate anti-sigma factor (14). We explored the collective impact of the sigma factors K, L, and M on M. tuberculosis virulence and the possibility that Rip1 degrades additional membrane-embedded anti-sigma factors. Here we identify anti-sigma factor D (RsdA)—the negative regulator of sigma factor D—as an additional Rip1 substrate. We propose a model in which Rip1 is required for the activation of four ECF sigma factor regulons.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains for this study are listed in Table S1, plasmids in Table S2, and oligonucleotides in Table S3 in the supplemental material. Escherichia coli DH5α was used for all recombinant DNA manipulations and grown in Luria-Bertani broth at 37°C. M. tuberculosis strains (all based on the wild-type strain Erdman EG1, which is animal passaged) were grown aerobically at 37°C in 7H9 (broth) or 7H10 (agar) (Difco) medium with oleic acid-albumin-dextrose-catalase (OADC) enrichment, 0.5% glycerol, and 0.05% Tween 80 (broth medium only). Mycobacterium smegmatis strains were cultured at 37°C on Luria-Bertani (LB) medium containing 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 (broth). When appropriate, the following were added into growth medium for E. coli and mycobacteria, respectively: hygromycin B (Boehringer Mannheim) at 150 and 50 μg ml−1, kanamycin (Sigma) at 40 and 20 μg ml−1, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal; Fisher Scientific) at 50 μg ml−1, and 5-bromo-4-chloro-3-indolylphosphate (BCIP; Sigma) at 60 μg ml−1.
Transmembrane prediction and testing.
Transmembrane predictions were performed on the TMpred server accessed at http://www.ch.embnet.org/software/TMPRED_form.html.
PhoA/LacZ contruction and testing.
Fusions of the C termini of RsdA and RsgA to alkaline phosphatase (PhoA) or β-galactosidase (β-Gal) were constructed by ablating the termination codon of RsdA or RsgA such that the terminal amino acid (H299 for RsdA or T244 for RsgA) was fused to the coding sequence of either lacZ or phoA. Alkaline phosphatase and β-galactosidase assays were performed as previously described (15).
Deletion of rsdA and sigD from M. tuberculosis.
M. tuberculosis ΔsigD and ΔrsdA mutants were constructed via specialized transduction using the temperature-sensitive phage phAE87 as previously described and were verified by Southern blotting (16). The ΔsigD allele replaces sigD (leaving a remnant of the first two and the last four sigD codons, including the stop codon) with a hygromycin cassette flanked by loxP sites. The 3′ flanking region of sigD was used as a probe of chromosomal DNA digested with SmaI to distinguish between wild-type sigD and ΔsigD alleles. The predicted size for wild-type sigD is 941 bp, while the ΔsigD allele is 2,972 bp. The ΔrsdA allele was verified using a 5′ flanking region of rsdA as a probe of chromosomal DNA digested with BamHI. The predicted size for wild-type rsdA is 1,148 bp, and the ΔrsdA allele is 7,288 bp.
Quantitative RT-PCR.
mRNA levels were measured with quantitative reverse transcriptase PCR (RT-qPCR). Cells were grown into mid-log phase in 7H9 with appropriate antibiotics, collected by centrifugation at 3,000 × g, and resuspended in TRIzol reagent (Life Technologies). Cells were disrupted mechanically with zirconia beads in a FastPrep instrument (QBiogene), and nucleic acid was extracted with chloroform, precipitated with isopropyl alcohol, washed with 70% ethanol, and dried. RNA was treated with a TURBO DNA-free kit (Ambion) and then cleaned with RNeasy minikits (Qiagen). A total of 500 ng of RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen) using random hexamer primers (Invitrogen). Real-time PCR was performed using SYBR green and an Opticon2 real-time fluorescence detector (MJ Research) on RNA samples from biologic triplicates. Single amplification products were confirmed for each PCR by the presence of melting curves with a single peak. The absence of amplification signal that could arise from contaminating chromosomal DNA was confirmed by control PCRs without reverse transcriptase. The cycle threshold value (CT) observed for each sample was normalized to the housekeeping gene sigA from the same cDNA sample by the formula ΔCT = CT, gene − CT, sigA. Relative levels of mRNA were calculated using the formula 2(−ΔCT) to generate an expression level for a gene.
Immunoblotting procedures.
M. smegmatis and M. tuberculosis strains were grown to mid-log phase in LB medium or 7H9, respectively, with appropriate antibiotics. Cells were then collected by centrifugation, frozen on dry ice, lysed by incubation at 37°C in Tris-EDTA (TE) with 10 mg ml−1 of lysozyme for 45 min, and then incubated for 10 min at 100°C in SDS-PAGE loading buffer (20% glycerol, 125 mM Tris-HCl [pH 6.8], 4% SDS, 0.2% bromophenol blue, 100 mM dithiothreitol [DTT]). Proteins were resolved by electrophoresis on NuPAGE 4 to 12% bis-tris polyacrylamide gels (Invitrogen). Separated proteins were transferred to a nitrocellulose membrane and probed with mouse monoclonal anti-maltose-binding protein (anti-MBP, clone B48; NEB) for detection of MBP-RsdA species and monoclonal anti-RNAPβ (clone 8RB13; NeoClone) for detection of RNA polymerase beta-subunit as a loading control. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Invitrogen) and the ECL kit (GE Healthcare) were used to visualize proteins in autoradiography film.
Murine infection.
Murine experiments were performed in accordance with National Institutes of Health guidelines for housing and care of laboratory animals and were approved by the Memorial Sloan-Kettering Institutional Animal Care and Use Committee (IACUC). M. tuberculosis was grown into log phase (optical density at 600 nm [OD600] = 0.5), washed twice with phosphate-buffered saline (PBS) containing 0.05% Tween 80, and briefly sonicated to disperse clumps. C57BL/6 mice were exposed to 8 × 107 CFU of the appropriate strain in a Middlebrook aerosol exposure system (Glas-Col). Bacterial burdens were determined by plating serial dilutions of lung and spleen homogenates on 7H10 agar media. Each time point represents the average bacterial counts from 4 or 5 mice enumerated after incubation at 37°C in 5% CO2 for 3 to 5 weeks.
Construction of the M. tuberculosis ΔsigKLM mutant.
All M. tuberculosis mutants were constructed via specialized transduction using the temperature-sensitive phage phAE87. To remove the Hygr cassette from the first sigL::hyg intermediate during triple sigma factor mutant construction, cells were transformed with pMSG381-1, a plasmid expressing HSP60-Cre, which contains an unstable MF1 origin of replication. After 3 weeks of growth on 7H10 plates containing kanamycin, transformants were picked and grown in 7H9 medium without antibiotics for 1 week. After reaching confluence, 10-μl aliquots were subcultured into 10 ml of 7H9 medium without antibiotics. After 1 week of growth, cells from these cultures were struck onto nonselective 7H10 agar plates. Single colonies from these plates were scored for kanamycin and hygromycin sensitivity, with loss of hygromycin resistance indicating successful loxP recombination and loss of kanamycin resistance indicating curing of the Cre-expressing plasmid. Loss of Hygr through LoxP recombination was verified using PCR. The sigL::loxP strain is MGM3254.
RESULTS
Deletion of sigma factors K, L, and M in M. tuberculosis.
Based on our model of the Rip1 pathway in M. tuberculosis, we hypothesized that the severe attenuation of the Δrip1 strain results from the inability to activate three ECF sigma factor regulons (4, 5). To test this model, we constructed a triple sigma factor mutant (ΔsigKLM) and monitored its virulence in a mouse model of aerosol infection. The triple mutant was constructed in three steps. First, we removed the Hygr cassette from a sigL::loxP-hyg-loxP strain by expression of the Cre recombinase (5), resulting in an unmarked, in-frame sigL deletion (MGM3254). This strain served as a recipient for a specialized transducing phage (5, 17) containing a sigM::hyg null allele. Hygromycin-resistant transductants were confirmed by Southern hybridization to contain two sigma factor deletions (sigL::loxP sigM::hyg; MGM3255). In the final step, the sigL::loxP sigM::hyg strain was transduced with a phage carrying sigK::zeo. Zeocin-resistant transductants were screened by Southern blotting for the sigK::zeo allele (Fig. 1A). The final strain is a triple sigma factor mutant (sigL::loxP sigM::hyg sigK::zeo; MGM3256).
FIG 1.

ΔsigKLM deletion does not phenocopy Δrip1 in mouse infection. (A) Confirmation of the M. tuberculosis ΔsigKLM strain. Southern blot analysis of M. tuberculosis chromosomal DNA was used to confirm the genotype of a ΔsigKLM triple mutant. When probed with the 3′ flanking region of sigM, EcoRI-digested genomic DNA produces a 12.2-kb band in ΔsigL (ΔL) cells, whereas the ΔsigLM (ΔLM) strain has a 5.2-kb band representing the sigM deletion. When probed with the 5′ flanking region of sigK, PstI-digested genomic DNA produces a 4.8-kb band in the ΔLM strain, indicating a wild-type sigK allele, whereas the ΔsigKLM (ΔKLM) triple mutant strain has a 551-bp band, representing the ΔsigK allele. (B) Phenotype of the Δrip1 strain in mice. Recovered CFU from lungs of mice infected via aerosol with wild-type M. tuberculosis and the Δrip1 strain are depicted. Error bars represent standard deviations and when not visible are within the symbol. (C) Recovered CFU from lungs of mice infected via aerosol with wild-type M. tuberculosis and the ΔsigKLM mutant. (D) Recovered CFU from spleens of mice infected via aerosol with wild-type M. tuberculosis and the ΔsigKLM mutant.
M. tuberculosis ΔsigKLM does not phenocopy Δrip1 in murine infection.
The Δrip1 strain of M. tuberculosis has a growth defect in the first 3 weeks of infection in mice and a persistence defect during the chronic phase of infection (4). To more fully characterize the previously reported virulence defect of the Δrip1 strain, we infected mice by aerosol with wild-type and Δrip1 M. tuberculosis and determined bacterial loads in lungs over the course of a 300-day infection. We observed a severe initial growth defect of the Δrip1 strain compared to the WT over the first 3 weeks of infection (Fig. 1B), similar to findings from our previous study (4). We also observed a severe defect in persistence reflected in the steady decline in Δrip1 strain titers over the course of the 300-day infection. Our prior report of the Δrip1 persistence defect included data up to 22 weeks, at which point the Δrip1 strain titers were steadily declining. To examine whether the Δrip1 mutant would be completely cleared from the lungs, we followed this infection out to 300 days. At the 300-day time point, 3 of 4 mice had no detectable CFU in the lungs (limit of detection 5 CFU). One mouse had 128 CFU remaining (Fig. 1B). These data indicate that the Rip1 mutant is cleared spontaneously from the lungs and is completely defective for maintenance of chronic infection.
To test whether the M. tuberculosis ΔsigKLM strain phenocopies the Δrip1 strain in murine infection, we infected mice via aerosol and examined bacterial titers in the lungs and spleen. Despite having been grown for over 6 months in vitro in order to genetically ablate the three sigma factors, the ΔsigKLM strain was not impaired for bacterial growth during the acute phase of infection or during the chronic phase of infection in either the lungs (Fig. 1C) or spleen (Fig. 1D). These data demonstrated that the three sigma factors are not required for virulence of M. tuberculosis and strongly suggest that the virulence defect of the Rip1 mutant is not caused by the combined inactivation of the SigKLM regulon. This result also strongly suggests that RskA, RslA, and RsmA are unlikely to represent the full complement of Rip1 substrates.
Membrane-embedded anti-sigma factors in M. tuberculosis.
Though ECF sigma factors are regulated by extracytoplasmic stimuli, only some are held inactive by a cognate membrane-embedded anti-sigma factor which would be a candidate substrate for Rip1. We previously demonstrated the topology of anti-sigma factors K, L, and M as transmembrane proteins with extracytoplasmic C termini (15). To search for additional anti-sigma factors that might be candidate Rip1 substrates, we analyzed additional anti-sigma factors annotated in the M. tuberculosis genome for putative transmembrane domains and determined their topology in the mycobacterial membrane. Transmembrane prediction using primary sequence hydrophobicity identified the anti-sigma factors for sigma factors K, L, M, and D as likely single-pass transmembrane proteins (5, 18). Of the remaining annotated sigma factors with candidate anti-sigma factors adjacently encoded, we identified anti-sigma factor G (rsgA, rv0181c), a 244-residue protein. RsgA is encoded immediately downstream of sigma factor G (sigG) and contains a predicted transmembrane domain falling on the borderline of probability by transmembrane prediction (Fig. 2A).
FIG 2.

Topology of anti-sigma factors D and G in the mycobacterial membrane. (A) The amino acid sequence of anti-sigma factor G was analyzed for potential transmembrane domains using the TMpred server. The red line indicates the threshold for significance. (B) β-Gal or PhoA was fused to the C terminus of anti-sigma factor G (G), anti-sigma factor D (D), or anti-sigma factor L (L), and the plasmids encoding these fusions were transformed into M. smegmatis along with a vector control (vect) or a positive control (+) for lacZ (consisting of unfused lacZ) or phoA (an antigen85-phoA fusion [26]). The left side shows three replicates of each strain with β-galactosidase fusions cultured on medium containing X-Gal, and the right side shows three replicates of M. smegmatis with each PhoA fusion cultured on medium containing BCIP.
To further assess whether RsgA and RsdA are transmembrane proteins, we determined the topology of these proteins in the mycobacterial membrane by constructing enzymatic fusions to alkaline phosphatase (encoded by phoA) and β-galactosidase (encoded by lacZ), as previously performed for RskA, RslA, and RsmA (5). Alkaline phosphatase is active in the periplasm and produces a blue colony on agar media containing BCIP, whereas β-galactosidase is active in the cytoplasm and produces a blue colony on agar medium containing X-Gal. Mycobacterium smegmatis containing a PhoA fusion to the C terminus of RsdA was blue on agar containing BCIP, but the corresponding β-Gal fusion was white on X-Gal (Fig. 2B). In contrast, M. smegmatis expressing a C-terminal fusion of PhoA to RsgA was white on BCIP, but a C-terminal fusion to β-Gal was blue on X-Gal, indicating that the C terminus of RsgA is cytoplasmic (Fig. 2B). Thus, RsdA is topologically similar to other Rip1 substrates with a periplasmic C terminus.
Deletion of anti-sigma factor D restores sigma factor D activity in the Δrip1 mutant.
In addition to regulating the SigK, SigL, and SigM regulons, the Δrip1 mutant exhibits sigKLM-independent phenotypes. One of these phenotypes is the significant underexpression of resuscitating promoter factor C (rpfC) mRNA, a gene hypothesized to function in mycobacterial dormancy (12, 13, 19, 20). rpfC is among the most strongly regulated genetic targets of SigD, as determined by two independent research groups (21, 22). The genetic links between rip1 and rpfC expression and sigD and rpfC expression render RsdA an attractive candidate Rip1 substrate. Our previous attempt to identify RsdA as a Rip1 substrate failed to show accumulation of a Rip1-uncleaved RsdA intermediate in the Δrip1 mutant, in contrast to RskA, RslA, and RsmA, in which a Rip1-uncleaved intermediate was visible by Western blotting (5). However, this cleavage assay relies on the accumulation of the site-1 protease-cleaved anti-sigma factor intermediate in the rip1 mutant, which may not be present if this intermediate is short-lived in the membrane.
We sought to alternatively address the question of whether Rip1 controls the SigD regulon by cleavage of RsdA through the genetic criteria applied previously to the SigKLM regulons (5). Specifically, if rpfC transcription is activated by Rip1 cleavage of anti-SigD with consequent liberation of SigD, then the following criteria should be met: (i) rpfC transcription should be abolished in the ΔsigD and Δrip1 strains, and (ii) rpfC transcription should be restored in the Δrip1 strain by deletion of rsdA, as the protease should not be required for SigD activation in the absence of the anti-sigma factor. To explore this hypothesis, we deleted sigD and rsdA in both the wild-type and Δrip1 M. tuberculosis backgrounds (Fig. 3). Through Southern blot analysis, we confirmed deletion of sigD and rsdA, indicating that they are not essential in either background (Fig. 3).
FIG 3.

Allelic exchange of rsdA and sigD in M. tuberculosis. (A and B) Targeted allelic exchange strategy for replacement of sigD (A) or rsdA (B) with a hygromycin resistance cassette flanked by loxP sites. (C) Southern blot analysis of M. tuberculosis chromosomal DNA was used to confirm the genotype of ΔsigD and ΔsigD Δrip1 strains. Hybridization of the 3′ flanking region of SigD to SmaI-fragmented genomic DNA produces a 941-bp hybridization product in the wild type, whereas the ΔsigD mutant produces a 2,972-bp hybridization product. (D) When probed with the 5′ flanking region of rsdA, BamHI-digested chromosomal DNA produces a 1,148-bp hybridization product, whereas a ΔrsdA strain produces a 7,288-bp hybridization product.
We then interrogated the effect of rsdA deletion on SigD-dependent gene expression in the Δrip1 strain by quantitating the mRNAs encoding RpfC and Rv1815c, both reported SigD target genes (21, 22). As previously reported, rpfC expression is undetectable in both the Δrip1 and ΔsigD mutants (Fig. 4A). We observed that deletion of rsdA in the Δrip1 background partially restores rpfC mRNA levels, though the deletion has no impact in a wild-type background (Fig. 4A). Deletion of rsdA restores expression of rv1815c, also a SigD target, in the Δrip1 strain (Fig. 4B). Taken together, these data provide genetic evidence that Rip1 controls the SigD regulon through cleavage of anti-SigD.
FIG 4.
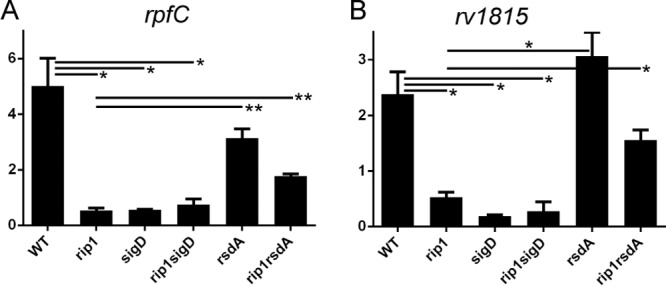
Activation of SigD-dependent transcription requires Rip1 cleavage of RsdA. Quantitative real-time PCR was used to measure the mRNA levels of rpfC (A) or rv1815c (B) in the strains as indicated. Strains were grown in 7H9 medium to log phase for RNA collection. Relative gene expression was normalized to sigA (housekeeping) gene expression. Significance is indicated by horizontal bars connecting specific pairs of measurements, with single asterisks indicating a P value of <0.01 and double asterisks indicating a P value of <0.001.
Rip1 is required for anti-sigma factor D degradation.
Because our genetic analysis strongly suggests that Rip1 controls SigD via RsdA, we reattempted to demonstrate Rip1-mediated RsdA proteolysis. To test whether anti-sigma factors are Rip1 substrates, we previously constructed recombinant anti-sigma factors with N-terminal hemagglutinin (HA) tags and examined their degradation pattern in wild-type and Δrip1 M. tuberculosis. Full-length HA-tagged RskA, RslA, RsmA, and RsdA all accumulate in wild-type cells, but an additional smaller product accumulates in the Δrip1 mutant for all anti-sigma factors except RsdA (5). These intermediates indicate that in the absence of rip1, RskA, RslA, and RsmA undergo incomplete proteolysis, consistent with S1P but not S2P (i.e., Rip1) degradation. Coupled with the genetic analysis mentioned above, this supported our conclusion that RskA, RslA, and RsmA are bona fide Rip1 substrates in vivo.
Because our prior failure to observe the accumulation of S1P-cleaved RsdA in the rip1 mutant could be explained by a short-lived RsdA intermediate that does not accumulate, we constructed an RsdA fusion to maltose-binding protein (MBP) at the RsdA N terminus. We reasoned that MBP might enhance the stability of an RsdA S1P-cleaved intermediate (Fig. 5A). To confirm that anti-sigma factor MBP fusions recapitulated our prior findings, we also constructed an MBP-RsmA fusion and confirmed that an S1P-cleaved intermediate accumulates in the M. smegmatis Δrip1 strain (Fig. 5B). We next expressed MBP-RsdA in wild-type M. smegmatis and ΔMSMEG_2579 (the M. smegmatis rip1 ortholog) M. smegmatis strains and detected the MBP-RsdA fusion protein by immunoblotting with anti-MBP antibodies. The ΔMSMEG_2579 mutant recapitulates the anti-sigma factor degradation patterns of the M. tuberculosis Δrip1 strain (5). Full-length RsdA accumulated at its predicted unprocessed size in both wild-type M. smegmatis and the ΔMSMEG_2579 mutant (Fig. 5C). However, in contrast to our prior experiments using HA-tagged RskA, RslA, and RsmA, we observed less full-length RsdA in the rip1 mutant. In the rip1 mutant the majority of the MBP-RsdA accumulated at the size of the predicted S1P-cleaved intermediate (Fig. 5C). The accumulation of this truncated intermediate in Δrip1 suggests that this species of the anti-sigma factor is processed by Rip1 in wild-type cells and is similar to the unprocessed fragments of RskA, RslA, and RsmA that accumulate in the M. tuberculosis Δrip1 mutant. We performed the same experiment examining MBP-RsgA in M. smegmatis but could not detect any differences in degradation between the wild type and the ΔMSMEG_2579 mutant (Fig. 5D), indicating that this anti-sigma factor is not a Rip1 substrate. We next examined the degradation pattern of MBP-RsdA in M. tuberculosis. The fusion protein was unstable, and multiple degradation products were present. However, a clear truncation product of MBP-RsdA was present in the Δrip1 strain but not the wild type, suggesting impaired maturation of RsdA in the absence of Rip1 (Fig. 5E). Taken together, these results indicate that Rip1 proteolytically degrades RsdA.
FIG 5.

Rip1 is required for the proteolytic processing of RsdA. (A) Experimental schematic for degradation of RsdA with an N-terminal MBP tag in the mycobacterial membrane. Shown are the possible species of MBP-RsdA after proteolysis by site-1 (S1P) and site-2 (i.e., Rip1) proteases. Lysates from the M. smegmatis wild type (WT) and ΔMSMEG_2579 mutant (ΔMsrip1) with plasmids encoding MBP-RsmA (B), MBP-RsdA(C), and MBP-RsgA (D) were analyzed by immunoblotting with antibodies recognizing MBP or RNAPβ as a loading control. (E) Immunoblots for MBP (top) or RNAPβ (bottom) of MBP-RsdA expressed in biologic triplicates of WT or Δrip1 M. tuberculosis.
DISCUSSION
We have investigated the downstream pathways responsible for the marked attenuation of the M. tuberculosis Δrip1 mutant in murine aerosol infection by testing the hypothesis that the combined loss of the SigKLM pathways, all of which require Rip1 for activation, would phenocopy the Δrip1 phenotype in mice. Surprisingly, we found that ΔsigKLM strain is not attenuated in mice, strongly indicating that Rip1-dependent, SigKLM-independent pathways are the critical virulence mediators controlled by Rip1.
Based on these results, we searched for additional pathways controlled by Rip1. We have previously demonstrated Rip1-dependent, SigKLM-independent pathways, including the target genes rpfC and the mycolic acid biosynthetic gene kasA. Here we have demonstrated that rip1 is required for the proteolytic degradation of at least one additional anti-sigma factor substrate, RsdA, which negatively regulates sigma factor D. As a consequence, Rip1 controls the SigD regulon, including the SigD targets rpfC and rv1815c. Thus, our present model is that Rip1 controls four sigma factor regulons, SigK, SigL, SigM, and SigD. The SigD axis of the Rip1 pathway adds another potential downstream pathway that may contribute to the virulence functions controlled by Rip1.
Genetic ablation of sigma factor L, M, or D causes mild effects on M. tuberculosis virulence (8–11, 21, 22), and the data presented here indicate that combined deletion of SigKLM also does not attenuate M. tuberculosis. However, given that sigma factors K and L exert combinatorial control on at least one gene, katG (5), it is plausible that there is extensive redundancy between the SigK, -L, -M, and -D regulons, akin to the sigma factor redundancy that exists in other bacteria (23). The natural hypothesis that arises out of this work is that an M. tuberculosis ΔsigKLMD strain may approach the attenuation of the Δrip1 strain, a hypothesis that will be tested in future work by construction of this quadruple mutant and testing its phenotype in mouse infection.
It is also possible that there are additional pathways controlled by Rip1 that remain to be identified and that these pathways contribute to virulence. Rip1 has been shown to cleave at least one additional non-anti-sigma factor substrate, PBP 3, under specific conditions of mutation of its binding partner, Wag31, and oxidative stress (24). Furthermore, a recent study with E. coli suggests that RseP, the Rip1 S2P homolog, has an essential function outside the realm of ECF anti-sigma factor proteolysis as a signal peptide peptidase (25). The severe attenuation phenotype of the Δrip1 strain, which is among the most severe reported for single-gene deletion mutations in M. tuberculosis and which is accompanied by spontaneous clearance of the infection, indicates that understanding the pathways controlled by Rip1 will provide further insight into the strategies employed by this successful pathogen.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AI080628, P30 CA008748, and T32 AI007621.
Footnotes
Published ahead of print 9 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01537-14.
REFERENCES
- 1.Helmann JD. 2002. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 46:47–110. 10.1016/S0065-2911(02)46002-X [DOI] [PubMed] [Google Scholar]
- 2.Hughes KT, Mathee K. 1998. The anti-sigma factors. Annu. Rev. Microbiol. 52:231–286. 10.1146/annurev.micro.52.1.231 [DOI] [PubMed] [Google Scholar]
- 3.Urban S. 2009. Making the cut: central roles of intramembrane proteolysis in pathogenic microorganisms. Nat. Rev. Microbiol. 7:411–423. 10.1038/nrmicro2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makinoshima H, Glickman MS. 2005. Regulation of Mycobacterium tuberculosis cell envelope composition and virulence by intramembrane proteolysis. Nature 436:406–409. 10.1038/nature03713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sklar JG, Makinoshima H, Schneider JS, Glickman MS. 2010. M. tuberculosis intramembrane protease Rip1 controls transcription through three anti-sigma factor substrates. Mol. Microbiol. 77:605–617. 10.1111/j.1365-2958.2010.07232.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hastie JL, Williams KB, Ellermeier CD. 2013. The activity of sigmaV, an extracytoplasmic function sigma factor of Bacillus subtilis, is controlled by regulated proteolysis of the anti-sigma factor RsiV. J. Bacteriol. 195:3135–3144. 10.1128/JB.00292-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schöbel S, Zellmeier S, Schumann W, Wiegert T. 2004. The Bacillus subtilis sigmaW anti-sigma factor RsiW is degraded by intramembrane proteolysis through YluC. Mol. Microbiol. 52:1091–1105. 10.1111/j.1365-2958.2004.04031.x [DOI] [PubMed] [Google Scholar]
- 8.Agarwal N, Woolwine SC, Tyagi S, Bishai WR. 2007. Characterization of the Mycobacterium tuberculosis sigma factor SigM by assessment of virulence and identification of SigM-dependent genes. Infect. Immun. 75:452–461. 10.1128/IAI.01395-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dainese E, Rodrigue S, Delogu G, Provvedi R, Laflamme L, Brzezinski R, Fadda G, Smith I, Gaudreau L, Palu G, Manganelli R. 2006. Posttranslational regulation of Mycobacterium tuberculosis extracytoplasmic-function sigma factor sigma L and roles in virulence and in global regulation of gene expression. Infect. Immun. 74:2457–2461. 10.1128/IAI.74.4.2457-2461.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hahn MY, Raman S, Anaya M, Husson RN. 2005. The Mycobacterium tuberculosis extracytoplasmic-function sigma factor SigL regulates polyketide synthases and secreted or membrane proteins and is required for virulence. J. Bacteriol. 187:7062–7071. 10.1128/JB.187.20.7062-7071.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karls RK, Guarner J, McMurray DN, Birkness KA, Quinn FD. 2006. Examination of Mycobacterium tuberculosis sigma factor mutants using low-dose aerosol infection of guinea pigs suggests a role for SigC in pathogenesis. Microbiology 152:1591–1600. 10.1099/mic.0.28591-0 [DOI] [PubMed] [Google Scholar]
- 12.Gupta RK, Srivastava BS, Srivastava R. 2010. Comparative expression analysis of rpf-like genes of Mycobacterium tuberculosis H37Rv under different physiological stress and growth conditions. Microbiology 156:2714–2722. 10.1099/mic.0.037622-0 [DOI] [PubMed] [Google Scholar]
- 13.Mukamolova GV, Turapov OA, Young DI, Kaprelyants AS, Kell DB, Young M. 2002. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol. Microbiol. 46:623–635. 10.1046/j.1365-2958.2002.03184.x [DOI] [PubMed] [Google Scholar]
- 14.Rodrigue S, Provvedi R, Jacques PE, Gaudreau L, Manganelli R. 2006. The sigma factors of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 30:926–941. 10.1111/j.1574-6976.2006.00040.x [DOI] [PubMed] [Google Scholar]
- 15.Schneider JS, Reddy SP, E HY, Evans HW, Glickman MS. 2013. Site-2 protease substrate specificity and coupling in trans by a PDZ-substrate adapter protein. Proc. Natl. Acad. Sci. U. S. A. 110:19543–19548. 10.1073/pnas.1305934110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barkan D, Liu Z, Sacchettini JC, Glickman MS. 2009. Mycolic acid cyclopropanation is essential for viability, drug resistance, and cell wall integrity of Mycobacterium tuberculosis. Chem. Biol. 16:499–509. 10.1016/j.chembiol.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017 [DOI] [PubMed] [Google Scholar]
- 18.Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132. 10.1016/0022-2836(82)90515-0 [DOI] [PubMed] [Google Scholar]
- 19.Downing KJ, Mischenko VV, Shleeva MO, Young DI, Young M, Kaprelyants AS, Apt AS, Mizrahi V. 2005. Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect. Immun. 73:3038–3043. 10.1128/IAI.73.5.3038-3043.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kana BD, Gordhan BG, Downing KJ, Sung N, Vostroktunova G, Machowski EE, Tsenova L, Young M, Kaprelyants A, Kaplan G, Mizrahi V. 2008. The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol. Microbiol. 67:672–684. 10.1111/j.1365-2958.2007.06078.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calamita H, Ko C, Tyagi S, Yoshimatsu T, Morrison NE, Bishai WR. 2005. The Mycobacterium tuberculosis SigD sigma factor controls the expression of ribosome-associated gene products in stationary phase and is required for full virulence. Cell. Microbiol. 7:233–244. 10.1111/j.1462-5822.2004.00454.x [DOI] [PubMed] [Google Scholar]
- 22.Raman S, Hazra R, Dascher CC, Husson RN. 2004. Transcription regulation by the Mycobacterium tuberculosis alternative sigma factor SigD and its role in virulence. J. Bacteriol. 186:6605–6616. 10.1128/JB.186.19.6605-6616.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo Y, Asai K, Sadaie Y, Helmann JD. 2010. Transcriptomic and phenotypic characterization of a Bacillus subtilis strain without extracytoplasmic function sigma factors. J. Bacteriol. 192:5736–5745. 10.1128/JB.00826-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukherjee P, Sureka K, Datta P, Hossain T, Barik S, Das KP, Kundu M, Basu J. 2009. Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol. Microbiol. 73:103–119. 10.1111/j.1365-2958.2009.06750.x [DOI] [PubMed] [Google Scholar]
- 25.Saito A, Hizukuri Y, Matsuo E, Chiba S, Mori H, Nishimura O, Ito K, Akiyama Y. 2011. Post-liberation cleavage of signal peptides is catalyzed by the site-2 protease (S2P) in bacteria. Proc. Natl. Acad. Sci. U. S. A. 108:13740–13745. 10.1073/pnas.1108376108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braunstein M, Griffin TI, Kriakov JI, Friedman ST, Grindley ND, Jacobs WR., Jr 2000. Identification of genes encoding exported Mycobacterium tuberculosis proteins using a Tn552′phoA in vitro transposition system. J. Bacteriol. 182:2732–2740. 10.1128/JB.182.10.2732-2740.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.