

Abstract
HU, a widely conserved bacterial histone-like protein, regulates many genes, including those involved in stress response and virulence. Whereas ample data are available on HU-DNA communication, the knowledge on how HU perceives a signal and transmit it to DNA remains limited. In this study, we identify HupB, the HU homolog of the human pathogen Mycobacterium tuberculosis, as a component of serine/threonine protein kinase (STPK) signaling. HupB is extracted in its native state from the exponentially growing cells of M. tuberculosis H37Ra and is shown to be phosphorylated on both serine and threonine residues. The STPKs capable of modifying HupB are determined in vitro and the residues modified by the STPKs are identified for both in vivo and the in vitro proteins through mass spectrometry. Of the identified phosphosites, Thr65 and Thr74 in the DNA-embracing β-strand of the N-terminal domain of HupB (N-HupB) are shown to be crucial for its interaction with DNA. In addition, Arg55 is also identified as an important residue for N-HupB–DNA interaction. N-HupB is shown to have a diminished interaction with DNA after phosphorylation. Furthermore, hupB is shown to be maximally expressed during the stationary phase in M. tuberculosis H37Ra, while HupB kinases were found to be constitutively expressed (PknE and PknF) or most abundant during the exponential phase (PknB). In conclusion, HupB, a DNA-binding protein, with an ability to modulate chromatin structure is proposed to work in a growth-phase-dependent manner through its phosphorylation carried out by the mycobacterial STPKs.
INTRODUCTION
Histone-like proteins (Hlps) are small and basic bacterial proteins involved in maintaining DNA architecture and regulating DNA transactions (1–3). Various Hlps (HU, H-NS, IHF, Dps, Fis, etc.) carry out wrapping, bridging, and bending of the bacterial DNA, resulting in its compaction or topological rearrangement (4–7). Initial studies on HU, an archetypical Hlp, showed that it displayed similarities with the eukaryotic histones in its physiochemical properties and also in its ability to induce topological changes such as supercoiling (8, 9). HU and other Hlps, however, share marginal similarities with the eukaryotic histones at the sequence or the structural level (10). These proteins therefore are now more appropriately termed nucleoid-associated proteins (NAPs), a designation primarily reflecting their localization (10).
Most NAPs display broad specificity toward DNA and can thus have a global effect on gene transcription (10, 11). This is achieved by their ability to maneuver the level of DNA compaction and topology. However, all NAPs do not share a mode of action. Although some act as DNA-bridging molecules (H-NS, MukB, and Lrp), others produce DNA bends (IHF, HU, and Fis) (5, 6). The expression of some of the NAPs also varies with the bacterial growth phase. This in turn guarantees that the nucleoid is modulated as a function of the bacterial growth phase (12, 13).
Apart from working as global regulators, NAPs such as HU also work locally by interacting with other elements of the transcription machinery in the formation of higher order nucleoprotein structures (11, 14). The extent of influence HU exerts on gene expression has been determined for various pathogenic and nonpathogenic bacteria (15–17). In Escherichia coli, HU regulates as many as 8% of the genes with many involved in bacterial adaptation to the environmental shifts and stress response (16). The success of many pathogens is also attributed to the ability of HU to manipulate the expression of genes involved in metabolism and virulence (15, 17). In Mycobacterium sp., the homolog of HU (M-hlp) is implicated in bacterial adaptation to stress conditions, possibly by the inhibition of cellular metabolism and reduction of bacterial growth rate through nucleoid reorganization (18–21). Various studies have reported the accumulation of M-hlp in growth-retarded phases, which is suggestive of a crucial role played by M-hlp in mycobacterial survival during dormancy (19, 22). The accumulation of M-hlp is achieved, in part, by the posttranslational modifications such as methylation, conferring resistance to proteolysis (23).
While HU-DNA communication has been extensively reported, studies on the signaling pathways that lead to a posttranslational modification on the protein remain limited in number (24–26). Even in eukaryotes, the identification of the cellular role of histone phosphorylation has been a subject of active investigation in recent years (27).
In this report we identify the NAP of Mycobacterium tuberculosis, HupB, as a bona fide substrate of serine/threonine protein kinases (STPKs). The identification of HupB as the target of the STPKs indicates that the NAP acts in alliance with the signal transduction machinery to perceive the exogenous cues and carry out necessary physiological changes essential for cellular survival.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
E. coli strain DH5α (Novagen) was used for cloning, and E. coli strain BL21 (Stratagene) was used for the expression of recombinant proteins. E. coli cells were grown and maintained with constant shaking (220 rpm) at 37°C in Luria-Bertani (LB) medium supplemented with 100 μg of ampicillin/ml, when needed. M. tuberculosis H37Ra, cells were maintained in Middlebrook 7H9 broth containing 10% albumin-dextrose-catalase (Difco Middlebrook) and 0.05% Tween 80.
Cloning, expression, and purification of HupB, HupB mutants, and STPKs.
The hupB gene (Rv2986c) and its fragment coding for N-HupB (N-terminal domain from residues 1 to 108) were PCR amplified using M. tuberculosis genomic DNA as a template and forward and reverse primers (Table 1), containing BamHI and EcoRI restriction sites, respectively. The amplified products were digested with restriction enzymes BamHI and EcoRI and ligated into vector pGEX-5X-3 (GE Healthcare Bio-Sciences), previously digested with the same enzymes to yield plasmids pGEX-hupB and pGEX-N-hupB. The pGEX-hupB construct was utilized for generating S163A mutation and pGEX-N-hupB for generating T43A, T45A, T65A, T68A, T74A, R55E, and R55Q mutations. The PknEK45M mutant was generated using pGEX-pknE. pGEX-hupB, pGEX-N-hupB, and pGEX-pknE plasmids were subjected to site-directed mutagenesis using a QuikChange XL site-directed mutagenesis kit (Stratagene) and primer pairs carrying the desired mutations (Table 1). The plasmids thus generated are listed in (Table 2). The integrity of the constructs was confirmed by DNA sequencing (TCGA, New Delhi, India). E. coli BL21 cells were transformed with pGEX-5X-3 vector derivatives expressing HupB, N-HupB, HupB, S163A, N-HupB T43A, N-HupB T45A, N-HupB T65A, N-HupB T68A, N-HupB T74A, N-HupB R55E, and N-HupB R55Q. For overexpression, recombinant E. coli strains harboring the pGEX-5X-3 derivatives were used to inoculate 1,000 ml of LB medium supplemented with ampicillin, followed by incubation at 37°C with shaking (220 rpm) until the optical density at 600 nm (OD600) reached 0.4. IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 1 mM, and growth was continued for an additional 1 h at 37°C. Growth for an extended period resulted in the degradation of the recombinant proteins. Cells were harvested and stored at −80°C. Pellets were thawed on ice and resuspended in cell lysis buffer A (50 mM Tris-Cl [pH 8.0], 300 mM NaCl, 1 mM dithiothreitol [DTT], 1 mM EDTA, 1× protease inhibitor mixture [Roche Applied Science]) and lysed by sonication. The cell lysates were centrifuged at 23,430 × g at 4°C for 20 min. The supernatant containing recombinant proteins was collected and incubated with glutathione-Sepharose 4B affinity resin (GE Healthcare) preequilibrated with buffer A. After extensive washings with buffers A and B (50 mM Tris-Cl [pH 8.0], 1 M NaCl, 1 mM DTT, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10% glycerol), elution was carried out in elution buffer E (50 mM Tris-Cl [pH 8.5], 10% glycerol, 150 mM NaCl, 15 mM glutathione). Fractions were run on a SDS–10% PAGE gel and analyzed by Coomassie blue staining. Pure fractions were dialyzed (20 mM Tris-Cl [pH 8.0], 10% glycerol, 150 mM NaCl), divided into aliquots, and stored at −80°C. Glutathione S-transferase (GST)-tagged PknA (28), PknE, PknF, PknG, PknH, PknI, PknE carrying the mutation K45M (PknEK45M), and His6-tagged PknB were purified according to the protocols described previously (29).
TABLE 1.
Primers used in this study
| Primera | Sequence (5′–3′)b |
|---|---|
| HupB-F | CACTGGATCCGGAGGGTTGGGATGAACAAAGCA (BamHI) |
| HupB-R | GCAAAGGGAATTCGTGTGTCTTGACCTATTTGC (EcoRI) |
| N-HupB-F | CACTGGATCCGGAGGGTTGGGATGAACAAAGCA (BamHI) |
| N-HupB-R | TTCTTGAATTCTCAGGCCCCCACACCACGCTT (EcoRI) |
| N-HupB-R55E-F | GTTCGAACAGCGTCGCGAGGCGGCTCGAGTGGCCC |
| N-HupB-R55E-R | GGGCCACTCGAGCCGCCTCGCGACGCTGTTCGAAC |
| N-HupB-R55Q-F | GTTCGAACAGCGTCGCCAGGCGGCTCGAGTGGCCC |
| N-HupB-R55Q-R | GGGCCACTCGAGCCGCCTGGCGACGCTGTTCGAAC |
| N-HupB-T43A-F | CAAAGGCGACAGCGTCGCCATTACCGGGTTCGGTG |
| N-HupB-T43A-R | CACCGAACCCGGTAATGGCGACGCTGTCGCCTTTG |
| N-HupB-T45A-F | CGACAGCGTCACCATTGCCGGGTTCGGTGTGTTCG |
| N-HupB-T45A-R | CGAACACACCGAACCCGGCAATGGTGACGCTGTCG |
| N-HupB-T65A-F | GCCCGCAATCCGCGTGCCGGCGAGACAGTAAAG |
| N-HupB-T65A-R | CTTTACTGTCTCGCCGGCACGCGGATTGCGGGC |
| N-HupB-T68A-F | CCGCGTACCGGCGAGGCAGTAAAGGTGAAGCCG |
| N-HupB-T68A-R | CGGCTTCACCTTTACTGCCTCGCCGGTACGCGG |
| N-HupB-T74A-F | GTAAAGGTGAAGCCGGCGTCGGTGCCGGCGTTC |
| N-HupB-T74A-R | GAACGCCGGCACCGACGCCGGCTTCACCTTTAC |
| C-HupB-S163A-F | GCTGTCAAGGCCACGAAGGCACCCGCCAAGAAGGTG |
| C-HupB-S163A-R | CACCTTCTTGGCGGGTGCCTTCGTGGCCTTGACAGC |
| PknEK45-M-F | GCGGATCGTGGCACTAATGCTGATGTCGGAGACGC |
| PknEK45-M-R | GCGTCTCCGACATCAGCATTAGTGCCACGATCCGC |
| PknIcat-F | GCGGGCCGTGCCGAATTCGGTTACTATCGGCCA (EcoRI) |
| PknIcat-R | TAGCAACACCTCGAGCGCACCGACCTAGATCCGGCG (XhoI) |
| Rv0884cPromoter region-F | TCGAAGCCTCCCCACCGAGGTGTG |
| Rv0884cPromoter region-R | GCCATCAGGGTAGTGAGGGGTACC |
| Rv1230cPromoter region-F | TTACCGAGTCGGCCATTGCGCCAC |
| Rv1230cPromoter region-R | TCGACCGTCCTCAGTGTGAGCC |
| PknE-F-RT | AACATTCTGGTTAGCGCGGA |
| PknE-R-RT | GCCATGTAGTAGAGGGTGCC |
| PknF-F-RT | TGTGGCATCCACACATCGTC |
| PknF-R-RT | GAAGGGATACGGTGTCGGTG |
| PknB-F-RT | GGTGATGGATTTCGGCATCG |
| PknB-R-RT | CTGTTCGGGTGACAGGTACT |
| HupB-F-RT | GCGCAATTCAAAGCGGTTGT |
| HupB-R-RT | GGTGCCTTCTTCGCTACCTT |
F, forward; R, reverse.
Mutagenized codons are indicated in boldface. Restriction sites are underlined, and the corresponding restriction enzymes are indicated in parentheses.
TABLE 2.
Plasmids used in this study
| Plasmid | Descriptiona | Source or reference |
|---|---|---|
| pGEX-5X-3 | E. coli vector utilized to generate GST fusion proteins and to purify GST protein (GST) | GE Healthcare |
| pGEX-5X-3_hupB | Full-length HupB | This study |
| pGEX-5X-3_N-HupB | pGEX-5X-3 derivative used to purify GST-tagged N-terminal domain (1 to 108 aa) of HupB (N-HupB) | This study |
| pGEX-5X-3_N-HupB_T43A | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the T43A mutation (T43A) | This study |
| pGEX-5X-3_N-HupB_T45A | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the T45A mutation (T45A) | This study |
| pGEX-5X-3_N-HupB_T65A | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the T65A mutation (T65A) | This study |
| pGEX-5X-3_N-HupB_T68A | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the T68A mutation (T68A) | This study |
| pGEX-5X-3_N-HupB_R55E | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the R55E mutation (R55E) | This study |
| pGEX-5X-3_N-HupB_R55Q | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the R55Q mutation (R55Q) | This study |
| pGEX-5X-3_N-HupB_T74A | pGEX-5X-3 derivative used to purify GST-tagged N-HupB carrying the T74A mutation (T74A) | This study |
| pGEX-5X-3_HupB_S163A | pGEX-5X-3 derivative used to purify GST-tagged HupB carrying the S163A mutation (S163A) | This study |
| pGEX-5X-3_pknI(1-1758) | pGEX-5X-3 derivative used to purify GST-tagged PknI (PknI) | This study |
| pGEX-5X-3_pknF(1-1431) | pGEX-5X-3 derivative used to purify GST-tagged PknF (PknF) | 47 |
| pGEX-5X-3_pknG(1-2253) | pGEX-5X-3 derivative used to purify GST-tagged PknG (PknG) | 50 |
| pGEX-5X-3_pknE(1-1701) | pGEX-5X-3 derivative used to purify GST-tagged PknE (PknE) | 28 |
| pGEX-5X-3_pknH(1-1881) | pGEX-5X-3 derivative used to purify GST-tagged PknH catalytic domain1-403 (PknHcat) | 28 |
| ProExHTc_pknE_K45M(1-1701) | pGEX-5X-3 derivative used to purify GST-tagged PknE carrying the K45M mutation (PknEK45M) | This study |
| pProExHTc_pknB(1-1881) | pProExHTc derivative used to purify His6-tagged PknB (PknB) | 29 |
| pProExHTc_pknAcat(1-337) | pProExHTc derivative used to purify His6-tagged PknA catalytic domain1-337 (PknAcat) | 28 |
The name of purified proteins used in the study are indicated in parentheses after the vector description.
In vitro kinase assay.
In vitro kinase assays were performed with 1 to 2 μg of kinase or the kinase-inactive mutant. The kinases and kinase-inactive mutant were added, separately, in 15 μl of kinase buffer (20 mM PIPES [pH 7.2], 5 mM MgCl2, 5 mM MnCl2) with 2 to 4 μg of the desired GST-tagged recombinant protein. Reactions were started with the addition of 2 μCi of [γ-32P]ATP (BRIT, Hyderabad, India) and were incubated at 25°C for 20 min. The reaction with PknE and PknEK45M as the kinase was performed in the reaction buffer (25 mM Tris [pH 7.0], 1 mM DTT, 5 mM MgCl2, 1 mM EDTA) at 25°C for 20 min. The reactions were terminated by the addition of 5× SDS sample buffer, followed by boiling for 5 min. Samples were resolved by using SDS–12% PAGE. Gels were dried and visualized using a FLA-2000 PhosphorImager (Fuji).
Phosphoamino acid analysis.
For phosphoamino acid analysis, N-HupB was phosphorylated by PknE, PknF, and PknB, and the recombinant tag was removed by the addition of factor Xa protease (Novagen) after the kinase reaction, followed by an additional incubation for various time points at 20°C. The reactions were stopped by using 5× SDS buffer, and the reaction mixture with the cleaved protein was separated by SDS-PAGE and electroblotted onto an Immobilon polyvinylidene difluoride (PVDF) membrane (Millipore). The phosphorylated and cleaved N-HupB was detected by autoradiography, and the bands corresponding to the 32P-labeled cleaved N-HupB were excised and hydrolyzed in 6 M HCl for 1 h at 110°C. The hydrolyzed samples containing liberated phosphoamino acids were lyophilized and redissolved for phosphoamino acid analysis as described earlier (30).
Phosphorylation site identification.
For phosphorylation site identification, 2 μg of N-HupB was incubated with PknE in the presence of 50 μM cold ATP at 25°C for 20 min in the reaction buffer containing 25 mM Tris [pH 7.0], 1 mM DTT, 5 mM MgCl2, and 1 mM EDTA. Further, sample preparation was performed as described earlier (31). Briefly, the protein samples were processed by either in-gel or in-solution digestion with trypsin or ArgC. The resulting peptides were loaded directly or after phosphopeptide enrichment on reversed-phase C18 StageTips. The (phospho)peptide mixtures were analyzed using an Easy nLC Nanoflow HPLC system (Proxeon Biosystems, Odense, Denmark [now Thermo Fisher Scientific]) coupled via a nanoelectrospray ion source (Proxeon Biosystems) to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). The raw mass spectrometric data were processed and analyzed using MaxQuant software (v1.2.0.17).
Electrophoretic mobility shift assay (EMSA).
For the protein-DNA binding assay, Rv0884c promoter region (207bp) was end labeled with [γ-32P]ATP using T4 polynucleotide kinase (10 U/20-μl reaction) in accordance with the manufacturer's instructions (Roche). The labeled probe was incubated with various concentrations of N-HupB (10 to 100 μM), and the mutants of N-HupB at room temperature for 30 min in the buffer containing 10 mM Tris-HCl (pH 8.0), 50 mM NaCl, 50 μg of bovine serum albumin (BSA)/ml, 1 mM DTT, 1 mM EDTA, and 5% glycerol in a total volume of 10 μl. The formed complex and free DNA were resolved using 5% nondenaturing polyacrylamide gels in running buffer (0.5× Tris-borate-EDTA). Gels were dried and subjected to autoradiography. To determine the effect of phosphorylation on the DNA-binding ability of N-HupB, the domain was incubated with PknE for 20 min in the presence of cold ATP and assayed with labeled DNA probe as mentioned above. The primers used for amplification of the DNA fragments used in EMSAs are listed in Table 1.
Immunoaffinity purification of endogenous HupB protein from M. tuberculosis H37Ra.
For immunoaffinity purification of endogenous HupB from M. tuberculosis H37Ra, the cells were grown till mid-log phase in Middlebrook 7H9 broth containing 10% ADC and 0.05% Tween 80. The cells were harvested and resuspended in phosphate-buffered saline (PBS) containing 5% glycerol, followed by lysis using a French press. The lysate was centrifuged at 13,000 rpm for 20 min, and the supernatant thus obtained was filtered and loaded onto an anti-HupB immunoaffinity column. The immunoaffinity column was prepared as described previously (32) with minor modifications. Briefly, polyclonal anti-HupB antibody, raised in mice, was affinity purified using HupB-coupled cyanogen bromide-activated Sepharose. Purified anti-HupB antibody was covalently linked to protein A-Sepharose (1 ml) using 20 mM dimethyl pimelimidate. The coupled matrix was regenerated with 10 mM glycine–HCl (pH 2.8) and equilibrated with PBS. Binding of the endogenous HupB in the filtered supernatant to antibody-coupled beads was carried out in PBSN (PBS containing 1% Nonidet P-40). The column was washed with 10 column volumes of PBSN, followed by 10 column volumes of PBS. Bound HupB was eluted with buffer containing 0.1 M glycine-HCl (pH 2.8) and 500 mM NaCl. The protein containing fractions were immediately neutralized with 50 mM Tris-HCl (pH 8.0) and dialyzed against storage buffer (10 mM Tris-HCl, 50 mM KCl, 5% glycerol). For Western blot analysis, 1.5 μg of the purified endogenous HupB was resolved using SDS–15% PAGE with positive and negative controls when required and probed against anti-HupB or anti-phosphothreonine/serine antibodies according to the protocol described below.
Immunoblotting.
To determine the phosphorylation status of the purified endogenous HupB by Western blot analysis, HupB was resolved by SDS-PAGE, along with positive control (RAW cell lysate) and negative control (KpnI), and transferred onto nitrocellulose membrane (Bio-Rad). After overnight blocking of the membrane with 3% BSA in PBST (PBS [pH 7.2], 0.1% Tween 20), the blots were incubated with antibodies directed against phosphoserine or phosphothreonine (Invitrogen) dissolved in PBST at a 1:10,000 dilution for 1 h at room temperature. After five washes with PSBT, the blots were incubated for 1 h at room temperature with anti-rabbit horseradish peroxidase-conjugated polyclonal antibody dissolved in PBST at a 1:10,000 dilution. The blots were developed after five washes with PBST using an ECL Plus kit (GE Healthcare Bio-Sciences) according to the manufacturer's instructions. A similar protocol was followed for the determination of HupB levels in lysates of M. tuberculosis H37Ra obtained from different growth stages. Anti-HupB antibody was used as the primary antibody at a dilution of 1:10,000, and anti-mouse antibody was used as the secondary antibody at a dilution of 1:10,000.
Quantitative RT-PCR.
For quantitative reverse transcription-PCR (RT-PCR), an M. tuberculosis H37Ra culture was grown to different growth stages: early log phase (OD600 = 0.4), mid-log phase (OD600 = 0.8), late log phase (OD600 = 1.5), and stationary phase (OD600 = 2.5). RNA was extracted using TRIzol reagent (Invitrogen Corp., Carlsbad, CA), followed by DNase I treatment using an Ambion DNA-free kit (Invitrogen). First-strand cDNA synthesis was carried out using random primers and Superscript III reverse transcriptase (Invitrogen). The cDNA thus synthesized was used in an RT-PCR supplemented with specific primer pairs (hupB, pknE, pknF, or pknB as listed in Table 1). The data obtained were analyzed using the ΔΔCT method, and the transcript levels were normalized to that of the housekeeping gene sigA. The mRNA levels are an average of two biological replicates.
RESULTS
HupB domain organization and genomic conservation.
The histone-like protein, HU, is a small DNA-binding protein of ca. 9 to 10 kDa. HU exists as a dimer composed of a largely α-helical “body” with two protruding β-ribbon “arms” (33). The β-ribbon arms intercalate with DNA to induce a bend resulting in DNA compaction (33). The HU homologs in Mycobacterium sp. and some other members of actinomycetes show unusual two-domain architecture. The N-terminal domain displays an atypical HU fold, and the C-terminal extension shows the presence of eukaryotic histone H1-like tetrapeptide repeats (AKKA) (34, 35). The presence of histone H1-like repeats is the reason these proteins are described as “histone-like” (10). Figure 1A shows the residues critical for the interaction of Hlps with DNA and their conservation in HupB (34, 36–38). The tetrapeptide repeats (AKKA) similar to eukaryotic histone H1 are also marked (Fig. 1B).
FIG 1.

Sequence alignment of N-terminal and C-terminal domains of HupB. (A) Sequence alignment of the HU fold (1-90 aa) of N-terminal domain of M. tuberculosis H37Rv HupB was performed with the representative members of the bacterial HU-like proteins using the CLUSTAL W program. Residues implicated in HU interaction with DNA and largely conserved in HU homologs are boxed. Among the conserved residues, the residue chosen for mutagenesis in N-HupB is indicated by a star. HUβ, E. coli HU beta subunit; HUα, E. coli HU alpha subunit; HUBst, HU homolog of B. stearothermophilus; N-HupB, 1 to 90 residues representing the HU fold of the N-terminal domain of M. tuberculosis HupB. (B) Sequence alignment of C-HupB (108 to 214 aa) with the C-terminal region of human histone H1.5 (103 to 226 aa). The AKKA tetrapeptide repeats are boxed.
Endogenous HupB from the avirulent strain of M. tuberculosis, H37Ra, is phosphorylated on threonine and serine residues.
In eukaryotes, histones have long been identified as targets of the serine/threonine/tyrosine protein kinases (27). Hence, we set out to determine whether prokaryotic STPKs also identify HupB as their target. To determine this, endogenous HupB from the exponentially growing cells of M. tuberculosis H37Ra strain was immunoaffinity purified using polyclonal anti-HupB antibody as described in Materials and Methods. Notably, HupB of H37Ra, an attenuated strain of H37Rv, displays 100% similarity with its ortholog in H37Rv (see Fig. S1 in the supplemental material). That the protein purified from H37Ra corresponded to HupB was ascertained by Western blot analysis with anti-HupB antibody (Fig. 2A). Of note, HupB and its orthologs purified from various mycobacterial species have been reported to migrate around ∼30 kDa owing to the highly basic nature of the protein, resulting in aberrant migration during electrophoresis (34, 35, 39).
FIG 2.

Endogenous HupB of M. tuberculosis H37Ra is phosphorylated on threonine and serine residues. (A) Endogenous HupB was purified using polyclonal anti-HupB antibody raised in mice. The purified protein was probed against anti-HupB antibody to ensure that the protein thus purified represented the endogenous HupB. The purified protein was resolved using SDS–15% PAGE, electroblotted onto PVDF membrane, and subjected to Western blot analysis. The left panel shows the Ponceau blue-stained PVDF membrane, and the right panel is the corresponding blot. (B and C) To determine the phosphorylation status of the purified protein, endogenous HupB was probed against anti-phosphothreonine and anti-phosphoserine antibodies. RAW cell lysate was included as the positive control (lanes 1) and KpnI was used as the negative control (lanes 3). The left panels show the Ponceau-stained PVDF membrane, and the right panels show the corresponding blot in the respective panels.
The purified protein was analyzed for its phosphorylation status with anti-phosphothreonine/serine antibody. As shown in Fig. 2B and C, endogenous HupB was found to be phosphorylated on both threonine and serine residues. These results indicated that HupB was either autophosphorylated or was a target of the STPKs in vivo.
HupB is phosphorylated by STPKs in vitro.
To determine whether HupB was modified on serine and threonine residues as a result of autophosphorylation or phosphotransfer by the STPKs, HupB was purified as a recombinant GST-tagged full-length protein and incubated with [γ-32P]ATP with or without STPKs. As shown in Fig. 3A, panel I, GST-HupB failed to label itself when incubated with [γ-32P]ATP. However, when incubated in the presence of the STPKs (PknE, PknF, and PknB), GST-HupB was efficiently labeled, indicating that the mycobacterial histone-like protein was indeed targeted by the STPKs (Fig. 3A, panels II, III, and IV). However, the levels of phosphotransfer by the kinases, added in equimolar concentrations, varied (PknE > PknF > PknB). Faint phosphotransfer was observed on HupB when PknA and PknH were used as the kinases, whereas PknG and PknI completely failed to phosphorylate HupB (data not shown). To ascertain that the observed phosphotransfer on HupB was carried out by the purified mycobacterial kinase and not by a contaminating E. coli protein, a kinase-inactive mutant of PknE, PknEK45M, was generated and used as a control. As seen in Fig. 3B, when HupB was incubated with PknEK45M, no phosphotransfer was observed. This result showed that the modification on HupB was PknE specific.
FIG 3.
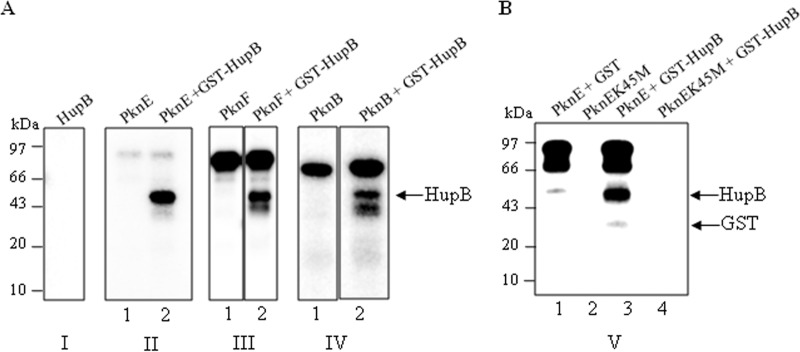
HupB is phosphorylated by serine/threonine protein kinases in vitro (A) Recombinant GST-HupB was incubated in the presence of [γ-32P]ATP for 20 min at 25°C. The reaction was run on an SDS-PAGE gel, and the gel was autoradiographed after drying. As shown in panel I, HupB was incapable of undergoing autophosphorylation. In panels II, III, and IV, 1 to 2 μg of purified STPKs PknE, PknF, or PknB were incubated alone (lane 1) or with 2 μg of recombinant GST HupB (lane 2) in the presence of [γ-32P]ATP for 20 min at 25°C. As shown, transphosphorylation on HupB was visualized in the presence of the STPKs, with PknE displaying most efficient phosphotransfer (panel II, lane 2). (B) PknE and PknEK45M were incubated with GST or GST-HupB. No phosphorylation was observed on GST alone when incubated with PknE (lane 1). PknEK45M, the kinase-inactive mutant, displayed a lack of autophosphorylation ability (lane 2). No phosphotransfer was observed on GST-HupB when incubated with PknEK45M (lane 4).
Further, since HupB protein was purified with a recombinant GST tag, GST alone was used as control to ensure that the observed phosphorylation was on HupB protein and not on the recombinant tag (Fig. 3B, lane 1). Taken together, these results showed that the observed phosphorylation on HupB was a result of phosphotransfer by the STPKs.
Identification of the phosphorylation site(s) in HupB.
To identify the sites targeted by the STPKs, immunoaffinity-purified endogenous HupB from M. tuberculosis H37Ra was subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Since HupB is composed of a large number of lysine and arginine residues, it was prone to degradation and also showed high susceptibility to the proteases used for digestion during MS analysis. After multiple attempts, only a singly phosphorylated peptide was obtained from the endogenous phosphorylated protein. The peptide comprised two phosphorylatable residues (Thr43 and Thr45), one of which represented the potential phosphosite. However, the exact residue on which phosphorylation occurred could not be defined based on fragmentation spectra. No phosphosites could be mapped on the C-terminal domain due to excessive cleavage with the proteases used and hence a limited peptide coverage. Since the endogenous protein was shown to be phosphorylated on serine residues also (Fig. 4), the presence of other residues modified by the STPK cannot be ruled out.
FIG 4.

Identification of the phosphorylation site(s) in HupB and N-HupB using high-accuracy measurements at both the precursor mass and the fragment levels (a “high-high” strategy) on the LTQ Orbitrap Velos. (A) Heated capillary dissociation-based MS/MS spectrum of a tryptic peptide (GDSVTITGFGVFEQR) of endogenous HupB phosphorylated on either Thr43 or Thr45. (B) In vitro phosphorylation by PknE allows identification and localization of phosphorylation on Thr43 (left spectra) and Thr45 (right spectra).
Identification of phosphorylation site(s) in the N-terminal domain of HupB.
Since phosphosites could not be mapped on the C-terminal domain and the presence of the C-terminal repeats resulted in the degradation of full-length protein during purification (40, 41), further studies were conducted on the prokaryotic HU-like N-terminal domain (N-HupB, amino acids 1 to 108). N-HupB was cloned, expressed, and purified as a GST-tagged protein. Purified recombinant GST-N-HupB was less susceptible to degradation than was the full-length GST-HupB (data not shown). As for full-length HupB, N-HupB was assayed with various kinases and was shown to be phosphorylated by PknE, PknB, and PknF, with PknE displaying the most efficient phosphotransfer (Fig. 5A). The PknE phosphorylated GST-N-HupB was subjected to LC-MS/MS for identification of the phosphorylation sites. However, in N-HupB multiple sites were found to be phosphorylated (Thr11, Thr22, Thr31, Thr43, Thr45, Thr65, Thr68, Thr74, Ser75, and Ser90). The presence of multiple phosphorylation sites on the N-terminal domain can partially be attributed to the availability of the residues in the absence of C-terminal domain. Also, going by the analogy with the eukaryotic histones known to be phosphorylated at multiple residues (27), the presence of multiple phosphorylation sites in prokaryotic histone-like protein HupB cannot be ruled out.
FIG 5.

Identification of phosphorylation site(s) in the prokaryotic HU-like N-terminal domain of HupB. (A) In vitro phosphorylation of HupB N-terminal domain by STPKs (left panel, autoradiogram; right panel, corresponding Coomassie blue-stained gel). Although PknE-phosphorylated GST-N-HupB appeared as a single phosphorylated species (panel I, lane 2), PknF and PknB phosphorylated an additional slower-migrating species, possibly an isoform of N-HupB, indicated by arrows. (B) PknE mediated comparative phosphorylation of the wild-type GST-N-HupB (lane 1) with threonine mutants GST-N-HupB T43A (lane 2) and T45A (lane 3). (C) Analysis of the phosphoamino acid content of cleaved N-HupB phosphorylated by PknE (panel I), PknF (panel II), and PknB (panel III). Amino acid standards, phosphoserine (pSer), phosphothreonine (pThr), and phosphotyrosine (pTyr) were added in the radiolabeled sample and visualized by ninhydrin staining (right panel) prior to autoradiography (left panel).
Since one of the residues, i.e., Thr43 or Thr45, was found to be phosphorylated on the native protein and also on the in vitro-phosphorylated N-HupB, these residues were individually mutagenized to alanine (GST-N-HupB-T43A and GST-N-HupB-T45A). However, no major loss of phosphorylation was observed on any of the mutants (Fig. 5B). This was apparently due to the presence of other phosphorylatable serine and threonine residues present in N-HupB, as shown previously (data not shown).
Further, among the identified phosphosites the phospholabel was shown to be localized primarily on the threonine residues as shown by two-dimensional thin-layer electrophoresis experiments. Moreover, as with PknE, PknF and PknB preferred threonine over serine for phosphorylation (Fig. 5C). In conclusion, the N-HupB kinases followed a similar phosphorylation pattern on N-HupB, with threonine as the preferred phosphorylated amino acid.
Residues critical for N-HupB–DNA interaction.
To determine the residues critical for N-HupB interaction, the activity of the purified GST-N-HupB was first subjected to an EMSA. The EMSA was carried out with increasing concentrations of N-HupB (10 to 100 nM) and the promoter region of the Rv0884c gene. Notably, M. tuberculosis HupB and a shorter version of its N-terminal domain (1 to 95 amino acids [aa]) have been previously reported to interact with various forms of DNA in a non-sequence-specific manner (42, 43). Hence, the DNA probe used in the present study was selected randomly and was the promoter region previously shown to be targeted by cyclic AMP receptor protein (CRP) (44). As shown in Fig. 6A. GST-N-HupB showed efficient interaction with double-stranded DNA (dsDNA) and with a higher affinity than previously reported (42). The promoter region of the Rv1230c gene, another member of CRP regulon, also displayed a similar binding affinity with HupB (data not shown).
FIG 6.

DNA-binding activity of GST-N-HupB and identification of Thr65, Thr74, and Arg55 as important residues for N-HupB–DNA interaction. (A) 32P-labeled DNA was incubated with increasing concentrations of GST-N-HupB (10 to 100 nM). As shown, multiple complexes were formed (C1, C2, and C3), primarily due to the nonspecific interaction of N-HupB at multiple sites on the labeled probe. (B) Thr65 and Thr74, the well-conserved residues on the “return” strand of the β-arm, were mutagenized to alanine to generate GST-N-HupB T65A and GST-N-HupB T74A, respectively. Both mutants displayed defective DNA-binding abilities. Lane 1, control; lane 2, GST-N-HupB; lane 3, GST-N-HupB T65A; lane 4, GST-N-HupB T74A. Mutation of T68 residue to alanine had no effect on the DNA-binding efficiency of GST-N-HupB (lane 5). (C) GST-N-HupB T65A and GST-N-HupB T74A mutants were assayed with PknE in the presence of [γ-32P]ATP. (Upper panel) Autoradiogram; (lower panel) corresponding Coomassie blue-stained gel. (D) The surface-exposed Arg55 on the “outgoing” strand of β-arm was mutagenized to Gln (R55Q) and Glu (R55E). The proteins (20 nM) were incubated with 32P-labeled DNA probe. A decrease in the DNA-binding ability of the mutants was observed, clearly indicating at the role of a positively charged residue for the interaction of N-HupB with DNA. (E) The mutants GST-N-HupB R55E and GST-N-HupB R55Q were incubated with PknE in the presence of [γ-32P]ATP. After separation by SDS-PAGE, the gels were dried, and signals were detected using a PhosphorImager. An apparent decrease in phosphorylation on GST-N-HupB R55Q (lane 2) and on GST-N-HupB R55E (lane 3) are visible. (Upper panel) autoradiogram; (lower panel) corresponding Coomassie blue-stained gel.
Next, of the identified phosphosites, Thr65, Thr68, and Thr74 positioned in the DNA interchalating β-arm (45) were mutagenized to alanine to elucidate their role in the interaction with DNA, if any. The residue corresponding to Thr65 of HupB has been shown to be located in the bend of the β-ribbon arm in Bacillus stearothermophilus (46) and the residues corresponding to Thr68 and Thr74 in the return arm of the β-ribbon. Of the three residues, Thr65 and Thr74 showed conservation or conservative substitution to serine in HupB homologs in actinomycetes (see Fig. S2 in the supplemental material). Thr65 was found to be conserved not only in HupB homologs in actinomycetes but also in HU homologs across various species in the bacterial kingdom (data not shown). Interestingly, the mutation of Thr65 and Thr74 compromised the DNA-binding ability of N-HupB, whereas the mutation of Thr68 to alanine showed no effect (Fig. 6B). These results showed that the conserved Thr65 and Thr74 situated at the strategic location are crucial for the DNA-binding ability of N-HupB. These residues were, however, not the primary phosphosites since their mutation did not result in an apparent loss of phospholabeling by PknE (Fig. 6C).
In addition, Arg55 was also identified as a residue critical for N-HupB–DNA interaction. Arg55 of B. stearothermophilus histone-like protein (HUBst) has been reported to mediate the interaction of its β-arm with DNA by forming electrostatic contacts with the negatively charged DNA (38). In multiple sequence alignment of various prokaryotic Hlps, Arg55 was indeed found to be one of the highly conserved residues (see Fig. S3 in the supplemental material). Arg55 was thus mutagenized to negatively charged amino acid Glu (R55E) or neutral amino acid Gln (R55Q) and, using EMSA, we demonstrated that the complex was lost when Arg55 was replaced with either Glu (R55E) or Gln (R55Q) (Fig. 6D). Hence, Arg55 seemingly plays a common role in HU-DNA interaction across various species. Interestingly, when the N-HupB Arg55 mutants Glu(R55E) or Gln(R55Q) were assayed with HupB kinases, a loss in phosphorylation on the mutants was observed (Fig. 6E). This result raises the possibility that the N-HupB β arm is involved in the interaction of the domain with the kinases as well.
Phosphorylation negatively regulates the DNA-binding ability of N-HupB.
Since N-HupB was found to be phosphorylated by the STPKs and has been shown to interact with DNA, it was pertinent to believe that the posttranslational modification would act as an important determinant for the regulation of its DNA-binding ability.
The effect of phosphorylation on the DNA-binding ability of N-HupB was determined. EMSA was performed at lower HupB concentrations (20 nM) to avoid the formation of aggregates. Then, 20 nM N-HupB was incubated with PknE in the presence of cold ATP, prior to its interaction with DNA. The phosphorylated protein was then incubated with 32P-labeled DNA probe. Reactions carried out in the absence of PknE or cold ATP was included as the controls. As shown in Fig. 7, compared to the unphosphorylated N-HupB, the phosphorylated N-HupB showed a significant decrease in its ability to form a complex with the labeled DNA. That the added N-HupB was phosphorylated by PknE was ascertained by Western blotting with anti-phosphothreonine antibody (data not shown). Therefore, the phosphorylation of the N-terminal domain by PknE resulted in the negative regulation of its DNA-binding ability.
FIG 7.

Phosphorylation negatively regulates the DNA-binding ability of N-HupB (A) GST-N-HupB was incubated with PknE and 32P-labeled promoter region in the presence (lane 1) or absence (lane 2) of cold ATP. The visible decrease in complex formation shows that the phosphorylated GST-N-HupB had diminished interaction with DNA. GST-N-HupB incubated with ATP was included as a control to ensure that the observed effect was not because of added ATP (lane 3). (B) Bar diagram showing a quantitative analysis of the complex formed, represented as phosphorimager units. Lane numbers correspond to those of panel A.
Growth phase-dependent expression of hupB and pknE, pknF, and pknB kinases.
It has been shown that the pknB kinase, one of the identified HupB kinases, is maximally expressed during the exponential phase (47). Also, the HupB homologs from M. bovis and M. smegmatis have been shown to be upregulated and/or accumulated during the stationary phase of bacterial growth at the protein level (19, 21, 22). The data available for different strains indicate that the levels of HupB and at least one of the HupB kinases (the pknB kinase) reach a maximum at different stages of growth.
To validate this and to have a systematic view on the levels of hupB and HupB kinases at different stages of the bacterial growth cycle, quantitative RT-PCR was performed on the RNA samples prepared from in vitro grown M. tuberculosis H37Ra cultures. As shown in Fig. 8A, the expression levels of hupB declined in the exponential phase and then showed a steady increase, peaking toward the stationary phase. Among the kinases, the mRNA levels of pknB steadily declined from the exponential phase to the stationary phase (Fig. 8C). The levels of pknE and pknF mRNA remained relatively constant throughout the bacterial growth cycle (Fig. 8D and E, respectively). In addition, the changes in hupB mRNA levels were also shown to correlate with the levels of HupB protein. The cell lysates prepared from different stages were subjected to Western blotting with anti-HupB antibody. As seen in Fig. 8B, the intracellular concentration of HupB increased steadily peaking at the stationary phase, a result in agreement with data obtained from quantitative RT-PCR analysis.
FIG 8.

Growth-phase-dependent expression of hupB and pknE, pknF, and pknB kinases. (A) Total RNA was harvested from M. tuberculosis H37Ra cells from different phases of growth (early log phase [OD600 = 0.4], mid-log phase [OD600 = 0.8], late log phase [OD600 = 1.5], and stationary phase [OD600 = 2.5]). hupB mRNA levels were measured using quantitative RT-PCR and normalized to sigA expression. The standard deviations of duplicate measurements are shown from three independent experiments (*, P < 0.05). (B) Cell lysates were prepared from the M. tuberculosis H37Ra cells derived from the growth stages described above. Immunoblot analysis was carried out with anti-HupB antibody (upper panel), and GroEL2 was used as a loading control (lower panel). (C to E) The mRNA levels of pknE, pknF, and pknB at various stages of growth are shown in panels C, D, and E, respectively. The standard deviations of duplicate measurements are shown from three independent experiments (*, P < 0.05).
DISCUSSION
In this study, we show that the endogenous M. tuberculosis histone-like protein HupB is phosphorylated on serine and threonine residues in vivo and is a bona fide substrate of the STPKs. This is the first report in which a mycobacterial nucleoid associated protein has been shown to be targeted by the kinases and the first to describe the effect of the phosphorylation determined on its DNA-binding ability. Even in other bacterial species, the concept of kinase-mediated regulation of nucleoid-associated proteins (NAPs) is relatively new, with only a few reports validating the same (24–26). Since NAPs can modulate the expression of a large number of genes (15–17), the phosphorylation-related changes in their activity can serve as a tool for bacterium to transmit the environmental cue to a large number of genes and bring about adaptive changes.
Our results showed that the endogenous HupB extracted from the exponentially growing cells of M. tuberculosis H37Ra was phosphorylated on both serine and threonine residues. These results were in line with the data available on eukaryotic histones known to be phosphorylated on serine and threonine residues. Interestingly, the HupB C-terminal domain showed the presence of a motif (159KATKSPAK166) similar to the one in histone H1.5 (184KATKSPAK191). The motif has been shown to be phosphorylated by cyclin-dependent kinases (48). Mutation of Ser163 to Ala163 in the full-length GST-HupB, however, did not result in a detectable decrease in the phosphorylation signal on the protein (data not shown). Hence, Ser163 may not represent a genuine phosphorylation site in the mycobacterial HupB or, alternatively, the presence of multiple phosphorylation sites may have masked the effect produced by the mutation.
In vitro-expressed and -purified GST-HupB was shown to be phosphorylated by the STPKs PknE, PknF, and PknB. PknE displayed maximal phosphorylation on GST-HupB. The site marked by the kinases on endogenous HupB was identified and was situated on a singly phosphorylated peptide in the N-terminal domain comprising of two phosphorylatable residues Thr43 and Thr45. However, no phosphosites could be mapped on the C-terminal domain of HupB. The C-terminal domain (aa 108 to 214) which has a high content of Lys (32.1%) and Arg (5.7%) residues showed high susceptibility to proteases, resulting in low peptide coverage. Recently, Jang et al. reported the phosphorylation of numerous peptides by an M. tuberculosis STPK, PknJ, identified on the basis of its similarity to the consensus sequence derived from the autophosphorylation sites targeted by the kinase (49). Thr43 and Thr45 residues in the peptide sequence derived from HupB (DSVTITGF) were proposed to be the putative target sites of the STPK. Although the prediction of substrates on the basis of phosphorylation of a peptide is highly speculative in nature, our results are in agreement with the results obtained by Jang et al.
Since we could not locate the phosphorylation sites on the C-terminal domain and the presence of the C-terminal domain rendered the protein highly susceptible to proteolytic degradation (data not shown), further studies were carried out on the N-terminal domain.
HupB is a 214-aa protein, and residues 1 to 90 show homology to the prokaryotic histone-like proteins (34, 40). It has been shown that the residues following the conventional HU fold are required for an efficient interaction of HU and HU-like proteins with DNA (40). These residues have been proposed to form an α-helix which is required for a stable interaction of the interchalating β-arms with DNA. Therefore, the N-terminal domain construct (1 to 108 aa) used in the present study was generated with the region encompassing these residues ending just before the first C-terminal domain repeat. Previously, the N-terminal domain (1 to 95 aa) has been shown to interact with DNA, albeit at a lower efficiency compared to the full-length HupB, leading to speculation that the C-terminal domain acts as a DNA clasp (43). We, however, observed that the N-terminal domain (1 to 108 aa) used in the present study demonstrated a higher affinity to dsDNA compared to a shorter fragment of the N-terminal domain (1 to 95 aa) used previously (42). This could partially be attributed to the length of the N-terminal domain and shows that the residues beyond the HU fold may be critical for N-HupB–DNA interaction.
Furthermore, there have been no reports on the identification of the residues that play an important role in the interaction of this domain with DNA. We showed here that the interaction of the N-terminal domain with DNA was indeed dependent on the conserved residue (Arg55), previously shown to be critical for the DNA-binding activity of HU homolog in B. stearothermophilus. In HUBst, of the four arginine residues in the β arm (Arg53, Arg55, Arg58, and Arg61), Arg55 had previously been identified as the most essential amino acid required for maintaining the contact with DNA once the HU-DNA complex had formed (38). The positively charged surface-exposed residues on the β-arm forms salt bridges with the phosphate backbone of DNA (33, 38). Our results also established that the conserved residues Thr65 and Thr74 are crucial for the N-HupB–DNA interaction.
The phosphorylation of N-HupB resulted in the inhibition of the DNA-binding activity of the domain. We further conducted quantitative RT-PCR analysis to estimate the relative mRNA abundance of the kinases and hupB at different stages of mycobacterial growth. The expression of the kinases either remained similar (pknE and pknF) or decreased significantly (pknB) at the stationary phase. However, the levels of hupB reached a maximum during the stationary phase.
Given the role of HupB and its homologs in nucleoid condensation, these results lead us to suggest that the phosphorylation of HupB during the exponential phase by the HupB kinases would limit its interaction with DNA and, as the bacteria transitions into the stationary phase, the abundant nonphosphorylated form of HupB capable of interacting with the genome would bring about its compaction and condensation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Matthias Mann (Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany) for access to his proteomic facility for mass spectrometric analysis. The M. tuberculosis H37Ra strain was kindly provided by V. K. Nandicoori (National Institute of Immunology, New Delhi, India). We thank H. K. Prasad (All India Institute of Medical Sciences, Delhi, India) for providing the antibodies for HupB.
Financial support for the work was provided by Council of Scientific and Industrial Research (BSC-0123).
We thank Anshika Singhal (CSIR-IGIB, Delhi, India) for helping with statistical analysis.
Footnotes
Published ahead of print 9 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01625-14.
REFERENCES
- 1.Drlica K, Rouviere-Yaniv J. 1987. Histone-like proteins of bacteria. Microbiol. Rev. 51:301–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dorman CJ, Deighan P. 2003. Regulation of gene expression by histone-like proteins in bacteria. Curr. Opin. Genet. Dev. 13:179–184. 10.1016/S0959-437X(03)00025-X [DOI] [PubMed] [Google Scholar]
- 3.Hayat MA, Mancarella DA. 1995. Nucleoid proteins. Micron 26:461–480. 10.1016/0968-4328(95)00022-4 [DOI] [PubMed] [Google Scholar]
- 4.Pettijohn DE. 1988. Histone-like proteins and bacterial chromosome structure. J. Biol. Chem. 263:12793–12796 [PubMed] [Google Scholar]
- 5.Dame RT. 2005. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol. Microbiol. 56:858–870. 10.1111/j.1365-2958.2005.04598.x [DOI] [PubMed] [Google Scholar]
- 6.Luijsterburg MS, Noom MC, Wuite GJ, Dame RT. 2006. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: a molecular perspective. J. Struct. Biol. 156:262–272. 10.1016/j.jsb.2006.05.006 [DOI] [PubMed] [Google Scholar]
- 7.Dorman CJ. 2009. Nucleoid-associated proteins and bacterial physiology. Adv. Appl. Microbiol. 67:47–64. 10.1016/S0065-2164(08)01002-2 [DOI] [PubMed] [Google Scholar]
- 8.Rouviere-Yaniv J, Gros F. 1975. Characterization of a novel, low-molecular weight DNA-binding protein from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 72:3428–3432. 10.1073/pnas.72.9.3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rouviere-Yaniv J, Yaniv M, Germond JE. 1979. Escherichia coli DNA binding protein HU forms nucleosome-like structure with circular double-stranded DNA. Cell 17:265–274. 10.1016/0092-8674(79)90152-1 [DOI] [PubMed] [Google Scholar]
- 10.Dillon SC, Dorman CJ. 2010. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8:185–195. 10.1038/nrmicro2261 [DOI] [PubMed] [Google Scholar]
- 11.Browning DF, Grainger DC, Busby SJ. 2010. Effects of nucleoid-associated proteins on bacterial chromosome structure and gene expression. Curr. Opin. Microbiol. 13:773–780. 10.1016/j.mib.2010.09.013 [DOI] [PubMed] [Google Scholar]
- 12.Rimsky S, Travers A. 2011. Pervasive regulation of nucleoid structure and function by nucleoid-associated proteins. Curr. Opin. Microbiol. 14:136–141. 10.1016/j.mib.2011.01.003 [DOI] [PubMed] [Google Scholar]
- 13.Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. 1999. Growth phase dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 181:6361–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kar S, Adhya S. 2001. Recruitment of HU by piggyback: a special role of GalR in repressosome assembly. Genes Dev. 15:2273–2281. 10.1101/gad.920301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangan MW, Lucchini S, Cróinín T, Fitzgerald S, Hinton JC, Dorman CJ. 2011. Nucleoid-associated protein HU controls three regulons that coordinate virulence, response to stress and general physiology in Salmonella enterica serovar Typhimurium. Microbiology 157:1075–1087. 10.1099/mic.0.046359-0 [DOI] [PubMed] [Google Scholar]
- 16.Oberto J, Nabti S, Jooste V, Mignot H, Rouviere-Yaniv J. 2009. The HU regulon is composed of genes responding to anaerobiosis, acid stress, high osmolarity, and SOS induction. PLoS One 4:e4367. 10.1371/journal.pone.0004367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priyadarshini R, Cugini C, Arndt A, Chen T, Tjokro NO, Goodman SD, Davey ME. 2013. The nucleoid-associated protein HUβ affects global gene expression in Porphyromonas gingivalis. Microbiology 159:219–229. 10.1099/mic.0.061002-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumoto S, Yukitake H, Furugen M, Matsuo T, Mineta T, Yamada T. 1999. Identification of a novel DNA-binding protein from Mycobacterium bovis bacillus Calmette-Guérin. Microbiol. Immunol. 43:1027–1036. 10.1111/j.1348-0421.1999.tb01232.x [DOI] [PubMed] [Google Scholar]
- 19.Lee BH, Murugasu-Oei B, Dick T. 1998. Upregulation of a histone-like protein in dormant Mycobacterium smegmatis. Mol. Gen. Genet. 260:475–479. 10.1007/s004380050919 [DOI] [PubMed] [Google Scholar]
- 20.Shires K, Steyn L. 2001. The cold-shock stress response in Mycobacterium smegmatis induces the expression of a histone-like protein. Mol. Microbiol. 39:994–1009. 10.1046/j.1365-2958.2001.02291.x [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto S, Furugen M, Yukitake H, Yamada T. 2000. The gene encoding mycobacterial DNA-binding protein I (MDPI) transformed rapidly growing bacteria to slowly growing bacteria. FEMS Microbiol. Lett. 182:297–301. 10.1111/j.1574-6968.2000.tb08911.x [DOI] [PubMed] [Google Scholar]
- 22.Katsube T, Matsumoto S, Takatsuka M, Okuyama M, Ozeki Y, Naito M, Nishiuchi Y, Fujiwara N, Yoshimura M, Tsuboi T, Torii M, Oshitani N, Arakawa T, Kobayashi K. 2007. Control of cell wall assembly by a histone-like protein in mycobacteria. J. Bacteriol. 189:8241–8249. 10.1128/JB.00550-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pethe K, Bifani P, Drobecq H, Sergheraert C, Debrie AS, Locht C, Menozzi FD. 2002. Mycobacterial heparin-binding hemagglutinin and laminin-binding protein share antigenic methyl-lysines that confer resistance to proteolysis. Proc. Natl. Acad. Sci. U. S. A. 99:10759–10764. 10.1073/pnas.162246899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Udo H, Lam CK, Mori S, Inouye M, Inouye S. 2000. Identification of a substrate for Pkn2, a protein Ser/Thr kinase from Myxococcus xanthus by a novel method for substrate identification. J. Mol. Microbiol. Biotechnol. 2:557–563 [PubMed] [Google Scholar]
- 25.Jin H, Pancholi V. 2006. Identification and biochemical characterization of a eukaryotic-type serine/threonine kinase and its cognate phosphatase in Streptococcus pyogenes: their biological functions and substrate identification. J. Mol. Biol. 357:1351–1372. 10.1016/j.jmb.2006.01.020 [DOI] [PubMed] [Google Scholar]
- 26.Burnside K, Lembo A, de Los Reyes M, Iliuk A, Binhtran NT, Connelly JE, Lin WJ, Schmidt BZ, Richardson AR, Fang FC, Tao WA, Rajagopal L. 2010. Regulation of hemolysin expression and virulence of Staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One 11:e11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee T, Chakravarti D. 2011. A peek into the complex realm of histone phosphorylation. Mol. Cell. Biol. 31:4858–4873. 10.1128/MCB.05631-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sajid A, Arora G, Gupta M, Singhal A, Chakraborty K, Nandicoori VK, Singh Y. 2011. Interaction of Mycobacterium tuberculosis elongation factor Tu with GTP is regulated by phosphorylation. J. Bacteriol. 193:5347–5358. 10.1128/JB.05469-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta M, Sajid A, Arora G, Tandon V, Singh Y. 2009. Forkhead-associated domain-containing protein Rv0019c and polyketide-associated protein PapA5, from substrates of serine/threonine protein kinase PknB to interacting proteins of Mycobacterium tuberculosis. J. Biol. Chem. 284:34723–34734. 10.1074/jbc.M109.058834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyle WJ, van der Geer P, Hunter T. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201:110–149. 10.1016/0076-6879(91)01013-R [DOI] [PubMed] [Google Scholar]
- 31.Sharma K, Kumar C, Kéri G, Breitkopf SB, Oppermann FS, Daub H. 2010. Quantitative analysis of kinase-proximal signaling in lipopolysaccharide-induced innate immune response. J. Proteome Res. 9:2539–2549. 10.1021/pr901192p [DOI] [PubMed] [Google Scholar]
- 32.Manjunatha UH, Dalal M, Chatterji M, Radha DR, Visweswariah SS, Nagaraja V. 2002. Functional characterisation of mycobacterial DNA gyrase: an efficient decatenase. Nucleic Acids Res. 30:2144–2153. 10.1093/nar/30.10.2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka I, Appelt K, Dijk J, White SW, Wilson KS. 1984. 3-A resolution structure of a protein with histone-like properties in prokaryotes. Nature 310:376–381. 10.1038/310376a0 [DOI] [PubMed] [Google Scholar]
- 34.Prabhakar S, Annapurna PS, Jain NK, Dey AB, Tyagi JS, Prasad HK. 1998. Identification of an immunogenic histone-like protein (HLPMt) of Mycobacterium tuberculosis. Tuberc. Lung Dis. 79:43–53. 10.1054/tuld.1998.0004 [DOI] [PubMed] [Google Scholar]
- 35.Shimoji Y, Ng V, Matsumura K, Fischetti VA, Rambukkana A. 1999. A 21-kDa surface protein of Mycobacterium leprae binds peripheral nerve laminin-2 and mediates Schwann cell invasion. Proc. Natl. Acad. Sci. U. S. A. 96:9857–9862. 10.1073/pnas.96.17.9857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grove A, Saavedra TC. 2002. The role of surface-exposed lysines in wrapping DNA about the bacterial histone-like protein HU. Biochemistry 41:7597–7603. 10.1021/bi016095e [DOI] [PubMed] [Google Scholar]
- 37.Rice PA. 1997. Making DNA do a U-turn: IHF and related proteins. Curr. Opin. Struct. Biol. 7:86–93. 10.1016/S0959-440X(97)80011-5 [DOI] [PubMed] [Google Scholar]
- 38.Saitoh F, Kawamura S, Yamasaki N, Tanaka I, Kimura M. 1999. Arginine-55 in the beta-arm is essential for the activity of DNA-binding protein HU from Bacillus stearothermophilus. Biosci. Biotechnol. Biochem. 63:2232–2235. 10.1271/bbb.63.2232 [DOI] [PubMed] [Google Scholar]
- 39.de Melo Marques MA, Mahapatra S, Nandan D, Dick T, Sarno EN, Brennan PJ, Vidal Pessolani MC. 2000. Bacterial and host-derived cationic proteins bind α2-laminins and enhance Mycobacterium leprae attachment to human Schwann cells. Microbes Infect. 2:1407–1417. 10.1016/S1286-4579(00)01294-6 [DOI] [PubMed] [Google Scholar]
- 40.Mukherjee A, Bhattacharyya G, Grove A. 2008. The C-terminal domain of HU-related histone-like protein Hlp from Mycobacterium smegmatis mediates DNA end-joining. Biochemistry 47:8744–8753. 10.1021/bi800010s [DOI] [PubMed] [Google Scholar]
- 41.Mukherjee A, DiMario PJ, Grove A. 2009. Mycobacterium smegmatis histone-like protein Hlp is nucleoid associated. FEMS Microbiol. Lett. 291:232–240. 10.1111/j.1574-6968.2008.01458.x [DOI] [PubMed] [Google Scholar]
- 42.Sharadamma N, Khan K, Kumar S, Patil KN, Hasnain SE, Muniyappa K. 2011. Synergy between the N-terminal and C-terminal domains of Mycobacterium tuberculosis HupB is essential for high-affinity binding, DNA supercoiling and inhibition of RecA-promoted strand exchange. FEBS J. 278:3447–3462. 10.1111/j.1742-4658.2011.08267.x [DOI] [PubMed] [Google Scholar]
- 43.Kumar S, Sardesai AA, Basu D, Muniyappa K, Hasnain SE. 2010. DNA clasping by mycobacterial HU: the C-terminal region of HupB mediates increased specificity of DNA binding. PLoS One 5:e12551. 10.1371/journal.pone.0012551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai G, McCue LA, McDonough KA. 2005. Characterization of Mycobacterium tuberculosis Rv3676 (CRPMt), a cyclic AMP receptor protein-like DNA binding protein. J. Bacteriol. 187:7795–7804. 10.1128/JB.187.22.7795-7804.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao A, Ram G, Saini AK, Vohra R, Kumar K, Singh Y, Ranganathan A. 2007. Synthesis and selection of de novo proteins that bind and impede cellular functions of an essential mycobacterial protein. Appl. Environ. Microbiol. 73:1320–1331. 10.1128/AEM.02461-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boelens R, Vis H, Vorgias CE, Wilson KS, Kaptein R. 1996. Structure and dynamics of the DNA binding protein HU from Bacillus stearothermophilus by NMR spectroscopy. Biopolymers 40:553–559. 10.1002/(SICI)1097-0282(1996)40:5<553::AID-BIP13>3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- 47.Kang CM, Abbott DW, Park ST, Dascher CC, Cantley LC, Husson RN. 2005. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19:1692–1704. 10.1101/gad.1311105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarg B, Helliger W, Talasz H, Förg B, Lindner HH. 2006. Histone H1 phosphorylation occurs site-specifically during interphase and mitosis: identification of a novel phosphorylation site on histone H1. J. Biol. Chem. 281:6573–6580. 10.1074/jbc.M508957200 [DOI] [PubMed] [Google Scholar]
- 49.Jang J, Stella A, Boudou F, Levillain F, Darthuy E, Vaubourgeix J, Wang C, Bardou F, Puzo G, Gilleron M, Burlet-Schiltz O, Monsarrat B, Brodin P, Gicquel B, Neyrolles O. 2010. Functional characterization of the Mycobacterium tuberculosis serine/threonine kinase PknJ. Microbiology 156:1619–1631. 10.1099/mic.0.038133-0 [DOI] [PubMed] [Google Scholar]
- 50.Koul A, Choidas A, Tyagi AK, Drlica K, Singh Y, Ullrich A. 2001. Serine/threonine protein kinases PknF and PknG of Mycobacterium tuberculosis: characterization and localization. Microbiology 147:2307–2314 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.