

Abstract
Enteropathogenic Escherichia coli (EPEC) primarily infects children in developing countries and causes diarrhea that can be deadly. EPEC pathogenesis occurs through type III secretion system (T3SS)-mediated injection of effectors into intestinal epithelial cells (IECs); these effectors alter actin dynamics, modulate the immune response, and disrupt tight junction (TJ) integrity. The resulting compromised barrier function and increased gastrointestinal (GI) permeability may be responsible for the clinical symptoms of infection. Type I interferon (IFN) mediates anti-inflammatory activities and serves essential functions in intestinal immunity and homeostasis; however, its role in the immune response to enteric pathogens, such as EPEC, and its impact on IEC barrier function have not been examined. Here, we report that IFN-β is induced following EPEC infection and regulates IEC TJ proteins to maintain barrier function. The EPEC T3SS effector NleD counteracts this protective activity by inhibiting IFN-β induction and enhancing tumor necrosis factor alpha to promote barrier disruption. The endoribonuclease RNase L is a key mediator of IFN induction and action that promotes TJ protein expression and IEC barrier integrity. EPEC infection inhibits RNase L in a T3SS-dependent manner, providing a mechanism by which EPEC evades IFN-induced antibacterial activities. This work identifies novel roles for IFN-β and RNase L in IEC barrier functions that are targeted by EPEC effectors to escape host defense mechanisms and promote virulence. The IFN-RNase L axis thus represents a potential therapeutic target for enteric infections and GI diseases involving compromised barrier function.
INTRODUCTION
Enteropathogenic Escherichia coli (EPEC) is an important and deadly cause of diarrhea among infants in developing countries (1). EPEC infects host intestinal epithelial cells (IECs) through a multistep attaching and effacing process, which results in loss of microvilli, disrupted gastrointestinal (GI) barrier function, and increased water and ion permeability (2–4). Intestinal barrier function is regulated by tight junctions (TJs), apical protein complexes that seal the intercellular space between IECs and allow selective permeability. Disruption of TJ organization or function is a critical feature of EPEC infection, as well as chronic GI disorders, including inflammatory bowel disease (IBD) and celiac disease (5, 6). Furthermore, some studies have reported an increased incidence of mucosally adherent E. coli, including EPEC, in IBD patients (7). TJs are comprised of the transmembrane proteins occludin (OCLN) and claudins and the cytoplasmic adaptor proteins zonula occludins 1, -2, and -3 (ZO-1/2/3) (8), which link transmembrane proteins with the actin cytoskeleton (9). In the first step of infection, adhesive type IV bundle-forming pili allow EPEC to bind IECs and form aggregates. Intimate attachment of EPEC to the IEC membrane initiates the formation of actin-rich pedestals and disrupts TJ function (10). Specifically, ZO-1 is dislocated, causing endocytosis and dephosphorylation of occludin (11) and contraction of the perijunctional actin-myosin ring (12). Retraction of the pili facilitates injection of type III secretion system (T3SS) effector proteins directly into the cytosol (10). T3SS effectors mediate sustained loss of barrier function by targeting TJs and subvert the host immune response via multiple mechanisms (13). For example, several EPEC effectors converge on nuclear factor kappa light chain enhancer of activated B cells (NF-κB), a central signaling molecule and transcription factor in the host response to diverse pathogens. Non-locus of enterocyte effacement effector C (NleC) is a metalloprotease, targeting the NF-κB subunits p65, p50, and c-Rel (14). NleB1 inhibits NF-κB expression in response to tumor necrosis factor alpha (TNF-α) (15). NleE also functions to inhibit NF-κB signaling, as it blocks translocation of the p65 subunit to the nucleus (15). NleD is a metalloprotease that specifically cleaves Jun-N-terminal-kinase (JNK) and p38 mitogen-activated protein kinases (MAPKs) that mediate signaling in response to inflammatory stimuli (14). Thus, EPEC effectors mediate pleiotropic activities to perturb host defense mechanisms and promote infection. Consistent with this critical role for effectors in pathogenesis, a mutant EPEC strain that is unable to deliver T3SS effectors is far less virulent in volunteers than wild-type (WT) EPEC (16).
The increased GI permeability following EPEC infection exposes host pattern recognition receptors (PRRs) located on the basolateral surfaces of IECs to EPEC-derived pathogen-associated molecular patterns (PAMPs). Stimulation of PRRs by EPEC PAMPS activates downstream signaling through NF-κB and MAPK pathways. Major EPEC PAMPs include flagellin, which is recognized by Toll-like receptor 5 (TLR5) during attachment to IECs (17), and lipopolysaccharide (LPS), which stimulates TLR4 in lamina propria immune cells (18). Studies of the host immune response to attaching and effacing bacteria have been performed in vivo using the mouse pathogen Citrobacter rodentium, which is an attaching and effacing bacterium similar to EPEC. Infection of mice with C. rodentium has been shown to activate Nod-like receptors (NLRs) and induce inflammasome assembly as part of the innate immune response (19). TLR and NLR activation leads to the induction of proinflammatory cytokines, including TNF-α, gamma interferon (IFN-γ), and interleukin 1β (IL-1β), that modulate intestinal permeability and thus play an important role in EPEC pathogenesis (13, 20). Importantly, TNF-α induces increased permeability in both cultured IECs and mouse intestine (8, 21), and inflammatory cytokines can induce TJ remodeling, during which claudins are selectively endocytosed (22). Although EPEC-derived PAMPS can initiate inflammatory immune responses, several T3SS effectors are capable of inhibiting these responses (23). For example, NleB, NleC, NleD, and NleE directly or indirectly block activation of the NF-κB pathway that mediates induction of proinflammatory cytokines (14, 24–26). These opposing effects of EPEC on inflammatory mediators result in limited inflammation that is thought to enhance virulence by reducing barrier function without inducing a robust host immune response.
The modulation of proinflammatory mediators by EPEC is well studied; however, less is known about the effect of EPEC infection on host anti-inflammatory mechanisms and how this effect may impact pathogenesis. In this regard, type I interferons (primarily IFN-α and IFN-β) mediate anti-inflammatory activity through multiple mechanisms that include the enhanced induction of IL-10 and maintenance of regulatory T cells (Tregs) in a T cell adoptive-transfer model of colitis (27), inhibition of inflammasome activity and IL-1β production in human primary monocytes (28), and suppression of IL-17A in mice (29). Furthermore, several lines of evidence have established IFNs as critical components of host-microbiome interactions in the GI tract (30, 31). Tonic IFN signaling in the GI tract was recently shown to mediate an optimal immune response to systemic pathogen infection (32, 33). In addition, IFN-β has been shown by several groups to be protective against experimental colitis (34–37) and has been used as a therapeutic agent in IBD patients with some efficacy (38, 39). Consistent with its important roles in GI functions, dysregulation of upstream regulators and downstream effectors of IFN is associated with human GI disorders (e.g., nucleotide-binding oligomerization domain-containing protein 2 [NOD2]) (40, 41) and mouse models of GI diseases (42, 43). IFN mediates its biologic activities through the induction of IFN-stimulated genes (ISGs) (44); accordingly, ISGs that function in GI immune and homeostatic activities have been identified (36, 37, 45, 46). Among the gene products, the endoribonuclease RNase L mediates the biologic activities of the IFN-regulated 2-5A pathway and functions as an upstream regulator of IFN induction (47) and a downstream effector of IFN action (48–51). Therefore, RNase L may contribute to IFN-mediated GI functions by multiple mechanisms. In support of this view, we previously identified a role for RNase L in antibacterial immunity (52) and recently reported a protective role for RNase L in the immune response to commensal bacteria following GI injury in a mouse colitis model (45). This protective effect corresponded to an RNase L-dependent increase in IFN induction, suggesting that RNase L is an important mediator of IFN functions in the GI tract.
The roles of IFN in GI immunity and homeostasis and the essential involvement of barrier function in these activities suggested that IFN may regulate IEC barrier function as a host defense mechanism against enteric pathogens that disrupt barrier integrity. Indeed, IFNs promote barrier function in brain endothelial cells (53), and IFN enhanced barrier function in lung epithelium to confer protection from pneumococcal infection (54). However, the regulation and role of IFN in the host response to enteric pathogens and its impact on IEC barrier function have not been examined. Here, we report that IFN-β is induced following EPEC infection and regulates TJ proteins to maintain barrier function. We further show that the EPEC T3SS effector NleD counteracts this protective activity by inhibiting IFN-β induction to promote barrier disruption and evade IFN-mediated antibacterial activities. We determined that RNase L contributes to the IFN-mediated host response to EPEC by stimulating IFN-β induction, downregulating TNF-α, and regulating TJ proteins and barrier integrity. Furthermore, EPEC infection inhibited RNase L activity in a T3SS-dependent manner, providing the first example of RNase L-targeted immune evasion by a bacterial pathogen. Consistent with an important role for RNase L in protection from EPEC pathogenesis, RNase L knockdown dramatically increased EPEC translocation across IEC monolayers, demonstrating its functional impact on permeability. Together, our study identifies IFN-β and RNase L as novel mediators of IEC barrier function that are targeted by EPEC effectors to escape host defense mechanisms and promote virulence. In light of these novel roles, the IFN-RNase L axis represents a potential therapeutic target for enteric infections and GI diseases in which compromised barrier function contributes to pathogenesis.
MATERIALS AND METHODS
Cell culture, treatment, and TEER.
Caco-2 cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA) and cultured in Eagle's minimum essential medium (EMEM) (Quality Biological, Inc., Gaithersburg, MD) with 20% heat-inactivated fetal bovine serum (FBS) (SAFC, St. Louis, MO) and penicillin-streptomycin diluted 1:100 (Gibco, Life Technologies, Carlsbad, CA) used at passages 10 to 25. Stable Caco-2 transfectants expressing nonspecific (shNS) or RNase L-targeted (shRNase L) short hairpin RNA (shRNA) were created by transducing cells with pGIPZ-shRNA lentiviral particles (Thermoscientific, Rockville, MD) following the manufacturer's protocol. Stable transfectants were selected by growth in 9 μg/ml puromycin prior to evaluation of knockdown efficiency and specificity by Western blotting. HT-29 and T84 cells were cultured in media and under conditions recommended by the ATCC. For transepithelial electrical resistance (TEER) experiments, Caco-2 cells were seeded at a density of 0.1 × 106 cells/well onto collagen-coated, 12-mm polycarbonate permeable support cell culture inserts that have a pore size of 0.4 μm in Transwell plates (Costar Corning, Inc., Corning, NY) for 25 to 28 days to form a differentiated monolayer. The medium was changed every 1 to 2 days. TEER was measured using an Evom ohmmeter with the Endohm 12 and STX2 electrodes (World Precision Instruments, Inc., Sarasota, FL). For cytokine treatment, Caco-2 cell monolayers were treated apically with IFN-β (PBL Assay Science, Piscataway, NJ) or IFN-γ (Genscript, Piscataway, NJ) and TNF-α (InvivoGen, San Diego, CA) at the concentrations and times indicated prior to analysis of gene expression, TEER, or EPEC infection.
Construction of strains and plasmids.
The referenced strains, including E2348/69 Nalr and the EPEC deletion mutants listed in Table 1, were described previously (2, 55–57). The ΔnleC and ΔnleD EPEC strains were created using a previously described protocol (58). Briefly, the nleC and nleD genes of EPEC strain E2348/69 were replaced with a 75-bp scar using the phage lambda red recombination method with the following primers: NleC_F1 (5′-ATAAATGATTTGCAGGGTATTAGATATAAACATGAAAATTCCCTCAGTGAAGGCTGGAGCTGCTTC-3′ and NleC_R1 (5′-AATAGTAACCTTATGTCACTGCAAAGACGAATCATCGCTGATTGTGATCCTCCTTAGTTCCTATTCC-3′), and NleD_F1 (5′-CTGCTAATAAGTAGCATTCTCAGGAGTCCTGATGCGCCCTACGTCCGTGAAGGCTGGAGCTGCTTC-3′) and NleD_R1 (5′-ACCAATATATATTCAGCACAAGAAACACAGCTAAAGCAATGGATGATCCTCCTTAGTTCCTATTCC-3′), respectively. The NleD complementation plasmid was constructed by amplifying the nleD gene from the genomic DNA of E2348/69 using primers NleD_F3 (5′-CTAGGAATTCATGCGCCCTACGTCC-3′) and NleD_R3 (5′-CTAGTCTAGACTAAAGCAATGGATGCAGTCTTAC-3′) and cloned into the pTrc99a plasmid. The NleD complementation plasmid was then used to transform ΔnleD EPEC by electroporation, and the transformed bacteria were selected by growth in Luria-Bertani medium (LB) with 200 μg/ml ampicillin. The EscF complementation plasmid was constructed by amplifying the escF gene from the genomic DNA of E2348/69 using primers EscF_F1 (5′-GGGGGGTACCTTTAAGAAGGAGATATACATATGAATTTATCTGAAATTACTCAACAAATGGGTGAAGTAG-3′) and EscF_R3 (5′-GGGGTCTAGATTAAAAACTACGGTTAGAAATGGTTGAGACCAG-3′); it was cloned into the pBad24 vector and was then used to transform ΔescF EPEC by electroporation, and the transformed bacteria were selected using LB with 200 μg/ml ampicillin.
TABLE 1.
Strains used in this studyb
| E. coli strain | Description | Reference |
|---|---|---|
| E2348/69 Nalr | E2348/69 Strr containing plasmid pE2348-2 gyrA ftsK hflD-purB; resistant to nalidixic acid | 55a |
| UMD731 | ΔescF; lacks functioning T3SS | 55a |
| UMD762 | ΔnleD; lacks zinc metalloprotease that degrades JNK and p38 | This study,a 14 |
| UMD761 | ΔnleC; lacks zinc metalloprotease that cleaves NF-κB subunit RelA | This study,a 14 |
| UMD874 | ΔespF; EspF inhibits internalization of nonopsinized EPEC | 2a |
| E2348C_3232a | ΔnleE1; lacks methyltransferase that inhibits NF-κB activation | 56a |
| ICC-250 | ΔnleB1; lacks glycosyltransferase that inhibits NF-κB activation | 57a |
| ICC-251 | ΔnleB2; lacks a homolog of NleB1 | 57a |
| ΔescF escF; T3SS restored | This studya | |
| ΔnleD nleD; zinc metalloprotease that degrades JNK and p38 restored | This studya |
Initial study using the strain.
Nalr, nalidixic acid resistant; Strr, streptomycin resistant.
Bacterial infections.
Wild-type EPEC and deletion mutants were cultured on LB agar plates. Single colonies were selected and grown in LB broth at 37°C overnight. The cultures were diluted 1:50 in Dulbecco's modified Eagle's medium (DMEM)/Ham's f12 and grown to an optical density at 600 nm (OD600) of 0.3 to 0.6. The OD600 of the medium was used to determine the number of CFU per ml broth. EPEC was added to the apical side of the transwell inserts at a multiplicity of infection (MOI) of 50 or 100. For the NleD-complemented strain, single colonies were selected and grown in LB with 200 μg/ml ampicillin and induced with 1 mM IPTG 3 h prior to infection. For the EscF-complemented strain, single colonies were selected and grown in LB with 200 μg/ml ampicillin and induced with 0.2% l-arabinose 3 h prior to infection.
Apoptosis assay.
Apoptosis of Caco-2 cells following EPEC infection was determined using the Vybrant FAM Caspase −3 and −7 Assay kit (Invitrogen, Carlsbad, CA). Briefly, Caco-2 cells were harvested from Transwell plates 2 h postinfection. The cells were then resuspended, incubated with fluorescence-conjugated antibodies, rinsed, and analyzed by flow cytometry following the manufacturer's instructions. Analysis was performed by the University of Maryland Greenebaum Cancer Center Flow Cytometry Core Facility.
ELISA.
Single enzyme-linked immunosorbent assay (ELISA) for human IFN-β was performed using the Pestka Biomedical Laboratories, Inc. (PBL) (Piscataway, NJ), Verikine-HS Human IFN-β Serum ELISA kit following the manufacturer's suggested protocol. Briefly, 50 μl of supernatant was incubated with diluted antibody in precoated microtiter plates; then, the supernatant was aspirated, and horseradish peroxidase (HRP) solution was added. The signal was then developed using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. Finally, the OD450 was measured, and the concentration was determined using a standard curve.
Immunofluorescence.
Caco-2 cells were grown on chamber slides with covers (Lab-Tek, Naperville, IL) to confluence. The cells were fixed and permeabilized using 4% formaldehyde followed by 0.1% Triton X-100 (Sigma, St. Louis, MO). To detect occludin, cells were incubated with 1:500 rabbit anti-OCLN antibody (Zymed, Carlsbad, CA), followed by incubation with 1:500 fluorescence-labeled goat anti-rabbit secondary antibody (Life Technologies, Carlsbad, CA). Cell nuclei were stained using Hoescht dye (Axxora, Farmingdale, NY). The cells were mounted using medium for fluorescence microscopy from KPL (Gaithersburg, MD) and Corning (Corning, NY) coverslips.
Western blot analysis.
Whole-cell lysates were prepared using RIPA lysis buffer supplemented with 10 μg/ml protease inhibitor cocktail (Sigma, St. Louis, MO) and 10 μg/ml leupeptin (Sigma, St. Louis, MO). Protein concentrations were measured by the Bradford method using Bio-Rad Protein Assay Dye Reagent (Bio-Rad, Hercules, CA). Samples were resolved by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) transfer membranes (ThermoScientific, Rockville, MD) using a Bio-Rad transfer apparatus. The blots were blocked as previously described (52), and specific proteins were detected with the following primary antibodies: anti-RNase L clone 2e9 (1:2,000) (Enzo, Farmingdale, NY), anti-claudin 1 (1:500) (Cell Signaling, Danvers, MA), anti-ZO-1 (1:1,000) (Cell Signaling, Danvers, MA), anti-occludin (1:1,000) (BD Transduction Laboratories, San Jose, CA), actin (1:10,000) (Abcam, Cambridge, United Kingdom), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5,000) (Abcam; Cambridge, United Kingdom). The membranes were then washed in Tris-buffered saline with Tween, reacted with HRP-conjugated secondary antibody, and developed with SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific, Rockville, MD). Signals from immunoreactive complexes were visualized by autoradiography (HyBlot CL, Metuchen, NJ) and quantified by densitometry using ImageJ software (NIH).
RNA isolation and qRT-PCR.
Total RNA was isolated from cells using TRIzol Reagent (Ambion, Life Technologies, Carlsbad, CA), and 50 ng was used for each reverse transcription (RT) reaction. For analysis of 16S rRNA in the permeability assay (see Fig. 7), 2 μl of medium from the basolateral chamber was used for quantitative RT-PCR (qRT-PCR). The sequences of the primers used for real-time PCR are as follows: IFN-β F, 5′-TGGGAGGCTTGAATACTGCCTCAA-3′, and IFN-β R, 5′-TCTCATAGATGGTCAATGCGGCGT-3′; TNF-α F, 5′-CCCAGGGACCTCTCTCTAATCA-3′, and TNF-α R, 5′-GCTTGAGGGTTTGCTACAACATG-3′; OCLN F, 5′-GCGAGCGGATTGGTTTATCT-3′, and OCLN R, 5′-TGGACTTTCAAGAGGCCTGG-3′; CLDN1 F, 5′-TTTACTCCTATGCCGGCGAC-3′, and CLDN1 R, 5′-GTTGCTTGCAATGTGCTGCT-3′; GAPDH F, 5′-TTCTTTTGCGTCGCCAGCCG-3′, and GAPDH R, 5′-GCGCCCAATACGACCAAATCCGT-3′; 16S F, 5′-ACTCCTACGGGAGGCAGCAG-3′, and 16S R, 5′-ATTACCGCGGCTGCTGG-3′. The primers were synthesized by IDT Technologies (Coralville, IA). Samples were run on 96-well plates in triplicate using the iScript One-Step RT-PCR kit with SYBR green (Bio-Rad, Hercules, CA), following the manufacturer's protocol. Samples were processed using the CFX96 Real-Time System (Bio-Rad, Hercules, CA).
FIG 7.

RNase L deficiency increases Caco-2 monolayer permeability and bacterial translocation. (A and B) Differentiated shNS and shRNase L monolayers were infected with WT or ΔescF EPEC at an MOI of 50 for 3 h. The supernatants were collected from the basolateral chambers of transwell plates and analyzed by qRT-PCR to detect EPEC 16S rRNA (A) and by culturing overnight to determine the presence of viable bacteria (B). The data points shown are the means of 3 separate experiments. The error bars indicate standard deviations. P values were calculated using the Student t test. **, P < 0.01. (C) Model depicting the roles of IFN-β and RNase L in the regulation of IEC barrier function following EPEC infection. EPEC infection disrupts barrier integrity, leading to activation of PRRs and induction of cytokines, including IFN-β and TNF-α. Injection of T3SS effectors, including nleD, inhibits RNase L, leading to diminished induction of IFN-β and enhanced TNF-α. The opposing effects of these cytokines on IEC barrier function occur through cytokine-mediated regulation of the TJ proteins OCLN and CLDN1. Additional mechanisms that contribute to the regulation of cytokine induction and barrier function are discussed in the text.
rRNA analysis.
rRNA and cleavage products were measured from 500 ng of total RNA using an Agilent Bioanalyzer in the Biopolymer-Genomics Core Facility at the University of Maryland, Baltimore, MD.
RESULTS
IFN-β is induced by EPEC infection and inhibited by the T3SS effector NleD.
Type I IFNs are induced as a primary response to diverse microbial infections (31, 44, 59). In the GI tract, IFNs play integral roles in monitoring commensal bacteria and in regulating the immune response to luminal microbes when intestinal barrier function is compromised (30, 31, 45). EPEC is an important enteric pathogen that disrupts IEC barrier function as a key component of its pathogenesis. A central role for proinflammatory cytokines in EPEC-induced loss of barrier integrity is well established; however, less is known about the impact of anti-inflammatory mediators, such as type I IFNs, on IEC function. Therefore, we investigated the regulation and potential protective role of IFN-β in the host response to EPEC. Caco-2 cells are colon cancer cells that differentiate to form a polarized monolayer with functional TJs and are a well-established model for studies of IEC immune and barrier functions. Accordingly, we chose the Caco-2 cell system to assess the regulation and role of IFN in response to challenge with WT EPEC (strain E2348/69). EPEC infection of Caco-2 cells resulted in a modest 2.5-fold induction of IFN-β mRNA at 3 h (Fig. 1A). To determine whether EPEC effectors impacted IFN-β induction, Caco-2 cells were infected with a T3SS-deficient (ΔescF) EPEC strain (Table 1) that is competent for adherence and viability but cannot inject effectors that mediate pathogenesis (16). Remarkably, nonpathogenic ΔescF EPEC induced dramatically higher levels of IFN-β mRNA than the WT bacteria (Fig. 1A). Increased induction of IFN-β by ΔescF compared to WT EPEC was also observed in HT29 colon cancer cells (see Fig. S1 in the supplemental material), suggesting that T3SS effectors function to inhibit IFN-β induction as a conserved mechanism by which EPEC evades host defenses. Consistent with this T3SS-dependent inhibition of IFN-β induction, infection with an EscF-complemented (ΔescF escF) strain reduced IFN-β induction to a level comparable to that observed with WT EPEC (Fig. 2B and C). In contrast to the T3SS-mediated reduction in the anti-inflammatory cytokine IFN-β, a functional T3SS was required for optimal induction of the proinflammatory cytokine TNF-α (Fig. 1B). Together, the diminished induction of IFN-β and enhanced induction of TNF-α may serve to promote barrier disruption as a component of EPEC pathogenesis.
FIG 1.
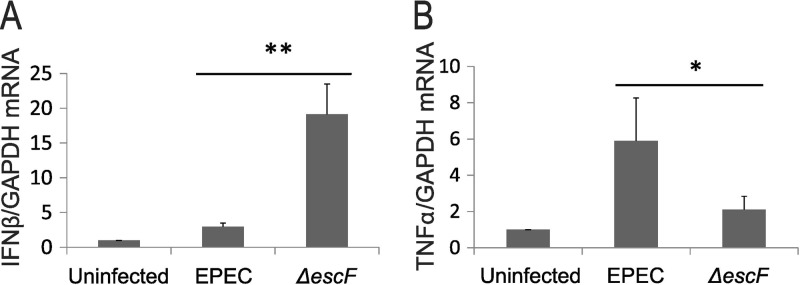
WT and ΔescF EPEC infections have opposing effects on cytokine expression. Caco-2 cells were grown on transwell plates for 28 days to form differentiated monolayers. The cells were apically infected with either WT or ΔescF EPEC at an MOI of 50 for 3 h. Total RNA was analyzed by qRT-PCR for IFN-β (A) and TNF-α (B) expression using GAPDH as a control. The data are the means of three separate experiments. The error bars indicate standard deviations. P values were calculated using the Student t test. *, P < 0.05; **, P < 0.01.
FIG 2.

The T3SS-injected EPEC effector nleD inhibits IFN-β expression. Differentiated Caco-2 cell monolayers were infected with deletion mutant EPEC strains that are deficient in the indicated effectors (A) and that had been complemented with escF and nleD (B and C) at an MOI of 50 for 3 h. Total RNA was analyzed by qRT-PCR for IFN-β with GAPDH as a control. The data points shown are the means of three separate experiments. (C) Secreted IFN-β was measured by ELISA analysis of the supernatant. The error bars indicate standard deviations. P values were calculated using the Student t test. *, P < 0.05.
To identify specific T3SS effectors that are responsible for inhibiting IFN-β induction, we chose EPEC deletion mutants that are deficient in effector proteins known to target components of the innate immune response that may impact IFN-β induction (Table 1) for infection of Caco-2 monolayers. Comparison of IFN-β mRNA induction by WT and ΔescF EPEC with that observed following infection with effector deletion strains revealed that nleD deletion mimicked the enhanced IFN-β induction seen with ΔescF EPEC (Fig. 2A). Consistent with this finding indicating a critical role for NleD in inhibiting IFN-β induction, infection with an NleD-complemented (ΔnleD nleD) strain reduced IFN-β induction to nearly the level observed with WT EPEC (Fig. 2B). An increase in secreted IFN-β, as detected by ELISA, paralleled the increased IFN-β mRNA observed following infection with ΔescF and ΔnleD EPEC compared to WT EPEC, and complementation of EscF or NleD inhibited IFN-β production (Fig. 2C). These data identify NleD as a key effector that inhibits IFN-β induction in the course of EPEC infection. Deletion of other T3SS effector genes (e.g., nleE1) resulted in partial restoration of IFN-β induction, suggesting additional mechanisms may contribute to the inhibition of IFN-β induction by EPEC.
IFN-β protects from EPEC-induced barrier disruption and regulates TJ proteins.
EPEC infection results in loss of IEC barrier function (13, 23, 60, 61). The subsequent stimulation of IEC TLRs by PAMPs, demonstrated by both EPEC infection of human IECs and C. rodentium infection of mice, induces proinflammatory cytokines to maintain GI tract permeability and promote pathogenesis (17, 18). Type I IFNs promote barrier integrity in endothelial cells (53, 62) and following pneumococcal infection of lung epithelium (54) and may thus serve a protective role to maintain IEC barrier function in the host response to EPEC. Therefore, we investigated the functional consequences of T3SS-mediated inhibition of IFN-β induction for IEC barrier function. Caco-2 monolayers were infected with WT and ΔescF EPEC, and barrier function was measured by TEER. An early decrease in TEER was observed in response to both WT and ΔescF EPEC; however, ΔescF EPEC-infected cells subsequently recovered to uninfected levels, whereas WT EPEC-infected cells continued to decline through later times postinfection (Fig. 3A). These data confirmed previous reports of EscF-dependent loss of barrier function (2). The diminished induction of IFN-β thus corresponded to decreased TEER, suggesting that IFN-β functions to protect IEC from EPEC-induced loss of barrier integrity. Consistent with this interpretation, and a critical role for NleD in regulating IFN-β induction (Fig. 2), Caco-2 TEER values were increased following infection with ΔnleD EPEC that induces high levels of IFN-β. Infection with the NleD-complemented ΔnleD EPEC strain in which IFN-β induction is inhibited resulted in decreased TEER values, thus supporting an important role for NleD in mediating reduced TEER that is associated with decreased IFN-β induction (Fig. 3B). These data further suggest that other T3SS effectors that are known to function in EPEC-induced loss of barrier function but did not impact IFN-β induction (e.g., espF) (2) function via IFN-independent mechanisms.
FIG 3.

IFN-β protects from EPEC-induced loss of TEER. (A and B) Differentiated Caco-2 monolayers were apically infected with EPEC strains as indicated at an MOI of 50 for 1, 2, and 3 h (A) or 3 h (B), and permeability was measure by TEER. (C) Caco-2 monolayers were treated with IFN-β for 16 h prior to infection with WT or ΔescF EPEC at an MOI of 50; TEER was measured at 3 h postinfection. The data points shown are the means of three separate experiments. The error bars indicate standard deviations. P values were calculated using the Student t test. *, P < 0.05; **, P < 0.01.
To determine if IFN-β, as opposed to other T3SS-regulated host components, mediated protection from EPEC-induced barrier disruption, TEER was measured in Caco-2 monolayers that had been pretreated with IFN-β and then infected with WT or ΔescF EPEC. Exogenous IFN treatment resulted in a significant increase in TEER values following EPEC infection (Fig. 3C). Specifically, treatment with 100 U/ml IFN-β followed by WT EPEC infection increased TEER values to the same level observed following infection with ΔescF EPEC, in which endogenous IFN is induced. This finding suggested that this dose of exogenous IFN-β was comparable to the endogenous IFN-β secreted by ΔescF-infected cells and is physiologically relevant. Infection with ΔescF induced IFN-β and increased TEER to values that ranged from 80 to 100% of those of uninfected cells (Fig. 2 and 3); this variation may reflect small passage-associated differences in the cells used in different experiments. Exogenous IFN enhanced TEER values above the already elevated levels in ΔescF EPEC-infected cells (Fig. 3C). This protective activity of exogenous IFN was independent of an effect on EPEC-induced apoptosis (see Fig. S2 in the supplemental material). Together, these data are consistent with our model, in which EPEC infection results in the T3SS-mediated inhibition of IFN-β induction that, in turn, contributes to decreased barrier function, as indicated by TEER. An increase in endogenous IFN-β induction following infection with ΔescF or ΔnleD EPEC, or treatment with exogenous IFN-β, protects from EPEC-induced loss of barrier function.
Epithelial barrier function is mediated through tight junction proteins that are dysregulated following EPEC infection to promote GI permeability and pathogenesis (13, 23, 60, 63, 64). This altered TJ function occurs through direct effects of EPEC infection on the IEC cytoskeleton (10, 65) and via the indirect action of EPEC-induced proinflammatory cytokines (17, 66). In light of our data indicating that induction of endogenous IFN-β or treatment with exogenous IFN resulted in protection from EPEC-induced barrier disruption (Fig. 3), we determined if IFN-β induced TJ proteins to promote IEC barrier integrity. Analysis of TJ proteins revealed a marked increase in occludin and claudin 1 following infection with ΔescF compared to WT EPEC. EPEC infection is known to alter the subcellular distribution of ZO-1; accordingly, ZO-1 expression was unchanged (Fig. 4A). The increase in TJ proteins in response to ΔescF corresponded to enhanced induction of IFN-β (Fig. 1A) and suggested that endogenous IFN-β induced TJ proteins in IECs. Consistent with this interpretation, treatment with exogenous IFN dramatically upregulated occludin and claudin 1 proteins (Fig. 4B) and increased occludin accumulation at the cell periphery (see Fig. S3 in the supplemental material). IFN treatment also induced occludin (Fig. 4C) and claudin 1 (Fig. 4D) mRNAs, albeit to a lesser extent than was observed for their corresponding proteins. Our findings thus identify the TJ proteins occludin and claudin 1 as targets of IFN regulation in IECs and provide a potential mechanism by which IFN protects from EPEC-induced barrier disruption.
FIG 4.

Infection with ΔescF EPEC or IFN-β treatment induces the expression of TJ proteins occludin and claudin 1. (A and B) Caco-2 monolayers were infected with WT or ΔescF EPEC at an MOI of 50 for 3 h (A) or treated with 100 U/ml or 500 U/ml IFN-β for 3 or 16 h (B). The cell lysates were then collected and analyzed for ZO-1, occludin (OCLN), and claudin 1 (CLDN1) expression by Western blotting. β-Actin served as a loading control and was used for normalization of densitometric analysis of the Western blot signals (bottom). The Western blot data are representative of 3 separate experiments. (C and D) RNA was isolated from Caco-2 monolayers treated as for panel B and analyzed for occludin (C) and claudin 1 (D) mRNAs by qRT-PCR. GAPDH mRNA served as a control. The data points shown are the means of three separate experiments. The error bars indicate standard deviations. P values were calculated using the Student t test. *, P < 0.05; **, P < 0.01.
RNase L-mediated cytokine regulation is inhibited by EPEC.
Our data demonstrate that EPEC infection inhibited IFN-β induction and decreased IEC barrier function in a T3SS-dependent manner (Fig. 1A and 3A and B), whereas enhanced induction of endogenous IFN-β by ΔescF EPEC, or treatment with exogenous IFN, increased TJ protein expression and promoted barrier integrity (Fig. 1A and 3). These findings suggested that both upstream regulators of IFN induction and downstream mediators of IFN action are impacted by EPEC infection. The 2-5A pathway is among the best-characterized IFN-regulated effector mechanisms (67, 68). RNase L is the terminal component of this pathway that functions in IFN induction in response to microbial infections (47, 69) and is a key downstream mediator of IFN-induced activities (48–50, 70). Furthermore, RNase L functions in the innate immune response to bacteria in the GI tract (45) and thus may be a mechanism by which IFN-β mediates host defense from EPEC infection. To examine a role for RNase L in response to EPEC infection, Caco-2 cells were stably transfected with control (nonspecific sequence; shNS) or RNase L-targeted (shRNase L) shRNA. Analysis of RNase L protein demonstrated efficient knockdown in the shRNase L cells, and no off-target effect on RNase L expression was detected in the shNS cells (see Fig. 6B). Infection of shRNase L and shNS cells with WT EPEC revealed that RNase L knockdown did not significantly alter IFN-β induction, which is efficiently reduced by the presence of NleD (Fig. 2). However, IFN-β induction was markedly compromised in response to ΔescF EPEC infection of shRNase L compared to shNS cells, indicating that this induction is mediated, in part, by RNase L (Fig. 5A). In contrast to the diminished induction of IFN-β by ΔescF EPEC in shRNase L cells, RNase L knockdown significantly enhanced induction of TNF-α by WT EPEC (Fig. 5B). RNase L thus acts as a positive regulator of IFN-β induction and a negative regulator of TNF-α, thereby opposing the T3SS-mediated effects on these cytokines. Accordingly, RNase L knockdown mimicked the T3SS-dependent regulation of IFN-β and TNF-α. For example, the enhanced induction of IFN-β by ΔescF EPEC was reversed by either the presence of T3SS in WT EPEC infection or the absence of RNase L in ΔescF EPEC-infected cells (Fig. 5A). Similarly, the diminished induction of TNF-α by ΔescF EPEC was reversed by the presence of T3SS in WT EPEC-infected shNS cells, with even more TNF-α induced following WT EPEC infection of shRNase L cells (Fig. 5B). Together these data suggested that T3SS effectors inhibit RNase L activity to modulate cytokine expression and promote EPEC pathogenesis. To test this hypothesis, cellular RNA was analyzed for the presence of discrete RNase L-generated rRNA cleavage products as a measure of enzyme activity (71). RNA from control uninfected Caco-2 cells displayed intact 28S and 18S rRNA bands, and this pattern did not change following WT EPEC infection (Fig. 5C). In contrast, infection with ΔescF EPEC produced readily detectable rRNA cleavage products identical to those generated following transfection with poly(I · C) to induce the production of 2-5A and activation of RNase L as a positive control for rRNA cleavage. These data provide evidence that EPEC infection inhibits RNase L in a T3SS-dependent manner. To our knowledge, this is the first example of RNase L inhibition mediated by a bacterial pathogen and supports a model in which evasion of RNase L-mediated cytokine regulation promotes EPEC pathogenesis.
FIG 6.

RNase L deficiency promotes barrier dysfunction following EPEC infection. Caco-2 cells stably transfected with shNS or shRNase L were differentiated for 28 days on transwell plates, and monolayers were infected with WT or ΔescF EPEC at an MOI of 50 for 3 h. (A) TEER was measured at 3 h postinfection, and the data shown are representative of three independent experiments. The data points are the means of three replicate samples. TEER values indicate the percent uninfected control for each cell line independently to measure the impact of RNase L on EPEC-induced changes in TEER independent of basal differences in the cell types (see Fig. S5 in the supplemental material). The error bars indicate standard deviations. P values were calculated using the Student t test. *, P < 0.05. (B) Cell lysates were analyzed for RNase L, OCLN, and CLDN1 protein expression by Western blotting. β-Actin served as a loading control and was used to normalize signals analyzed by densitometry (bottom). The Western blot results are representative of 3 separate experiments.
FIG 5.

RNase L regulates cytokine induction by EPEC. (A and B) Caco-2 cells stably transfected with nonspecific (shNS) or RNase L-targeted (shRNase L) shRNA were differentiated for 28 days on transwell plates, and monolayers were infected with WT or ΔescF EPEC at an MOI of 50 for 3 h. RNA was isolated and analyzed by qRT-PCR for IFN-β (A) and TNF-α (B), and GAPDH was utilized as a control. The data points shown are the means of 3 separate experiments. The error bars indicate standard deviations. P values were calculated using the Student t test. *, P < 0.05. (C) rRNA cleavage was analyzed in the indicated samples using an Agilent Bioanalyzer; RNA from cells transfected with poly(I · C) to produce endogenous 2-5A served as a positive control (+con). Arrows indicate bands which represent rRNA cleavage products. Shown is a representative image from 3 separate experiments.
RNase L modulates barrier function, TJ proteins, and IEC permeability.
To assess the functional consequences of RNase L-dependent cytokine regulation and its inhibition by EPEC, we measured barrier function in the presence and absence of RNase L knockdown. Infection of shRNase L cells with either WT or ΔescF EPEC resulted in decreased TEER compared with that observed following infection of shNS cells (Fig. 6A). The most potent reduction in TEER was observed in shRNase L cells infected with WT EPEC, which likely reflected the knockdown of RNase L expression and inhibition of residual RNase L activity by T3SS effectors. In contrast, sustained RNase L expression in the absence of T3SS-mediated inhibition of its activity in ΔescF EPEC-infected shNS cells resulted in the highest TEER values, which were similar to those in uninfected cells. Inhibition of RNase L activity alone (WT EPEC-infected shNS cells) or RNase L knockdown alone (ΔescF EPEC-infected shRNase L cells) gave intermediate, but nonidentical, TEER values, suggesting that T3SS effectors may modulate barrier function via RNase L-independent mechanisms and that RNase L may contribute to barrier function through activities that are not affected by T3SS effectors.
The effect of RNase L on TEER in EPEC-infected cells (Fig. 6A) correlated with its regulation of cytokine induction (Fig. 5A and B) and represented a potential mechanism by which RNase L impacts IEC barrier function. RNase L also protected from IEC barrier disruption by exogenous treatment with the proinflammatory cytokines TNF-α and IFN-γ (see Fig. S4 in the supplemental material), indicating that RNase L may modulate barrier function through mechanisms that are both upstream and downstream of cytokine induction. Furthermore, RNase L protein was induced during the differentiation of Caco-2 and T84 cell lines to form polarized monolayers with functional TJs (see Fig. S5A in the supplemental material), and basal TEER was reduced by 25% in shRNase L compared to shNS cells (see Fig. S5B in the supplemental material). These findings suggested that RNase L may serve a broader function in IEC barrier integrity. Therefore, we examined a role for RNase L in the regulation of TJ components in EPEC-infected and uninfected cells. Basal levels of occludin protein were comparable in shNS and shRNase L cells; however, RNase L knockdown dramatically reduced occludin induction following infection with WT or ΔescF EPEC (Fig. 6B). Though the induction of claudin 1 following WT or ΔescF EPEC infection was less dramatic, RNase L knockdown also reduced the protein levels following infection. These findings identify the RNase L-dependent regulation of TJ components as a potential mechanism by which it impacts IEC barrier function following EPEC infection.
TEER is a measure of barrier function that correlates with the permeability of IEC monolayers to luminal components and pathogens; accordingly, TEER provides a model of this key feature of GI physiology that is disrupted by enteric pathogens and in GI diseases. Since RNase L knockdown reduced TEER following EPEC infection (Fig. 6A), we directly assessed its impact on IEC permeability. Specifically, we first used qPCR to measure 16S bacterial rRNA that had translocated through Caco-2 monolayers and was present in the basolateral chamber of transwell culture plates. This analysis revealed a dramatic increase in 16S rRNA translocation in EPEC-infected shRNase L compared to shNS cells (Fig. 7A). The increase in permeability of shRNase L monolayers to 16S rRNA reflected a similarly enhanced translocation of viable EPEC to the basolateral chamber (Fig. 7B). ΔescF EPEC did not exhibit significant basolateral translocation consistent with the impaired capacity of that strain to disrupt barrier function. Although WT EPEC is known to disrupt barrier function and increase permeability, 16S rRNA and bacteria were not detected in the basolateral chamber following infection of control shNS samples with WT EPEC. This finding suggested that RNase L knockdown potently enhanced permeability under the infection conditions used and that EPEC-induced permeability in control cells may require an increased MOI.
Together, these data identify roles for RNase L in regulating cytokine induction, barrier function, and TJ proteins in IECs (Fig. 7C). Inhibition of RNase L following EPEC infection counteracts these protective activities as a novel mechanism by which it evades the host immune response.
DISCUSSION
The GI tract is the site of critical interactions between intestinal microbes and the host that are key determinants of physiological and pathological activities (72, 73). IECs that line the intestine play important roles in maintaining a barrier to the potentially pathogenic contents of the GI lumen and in regulating the immune response to microbes that have breached this barrier. Type I IFNs serve essential functions in GI immunity and homeostasis (30, 31) and promote barrier integrity in endothelial cells (53, 62) and following bacterial infection in the lung (54). Therefore, we investigated the regulation of IFN-β and its role in host defense from EPEC, an important human pathogen that disrupts barrier function as a key component of pathogenesis. Our study revealed that IFN-β induction was inhibited and TNF-α was increased in a T3SS-dependent manner following EPEC infection. The regulation of IFN-β and TNF-α by WT EPEC corresponded to reduced barrier function that was not observed following infection with T3SS-deficient EPEC. These findings suggest that the T3SS-dependent inhibition of the anti-inflammatory cytokine IFN-β and induction of TNF-α, a potent proinflammatory cytokine, represent important mechanisms by which EPEC subverts the host immune response to promote pathogenesis. Consistent with this model, exogenous IFN conferred protection from EPEC-induced barrier disruption, providing the first evidence of this role for IFN-β in IECs. Importantly, treatment with 100 U/ml exogenous IFN-β, followed by infection with WT EPEC, which inhibits the induction of endogenous IFN, resulted in TEER values comparable to those observed following infection with T3SS-defective ΔescF EPEC that induce higher levels of endogenous IFN-β. This finding suggested that physiologically attainable amounts of IFN-β mediate protection from EPEC-induced barrier disruption. Indeed, the concentrations of secreted IFN-β detected by ELISA following ΔescF EPEC infection (100 pg/ml) are sufficient to mediate diverse IFN-induced activities. For example, recent studies have revealed the importance of low, basal IFN expression and tonic IFN signaling in GI homeostasis and host defense (32, 33).
The inhibition of IFN-β and enhanced induction of TNF-α by EPEC required a functional T3SS, suggesting that EPEC effectors that are injected into IECs by the T3SS are responsible for this regulation. T3SS effectors modulate the host response to EPEC via multiple mechanisms. Several effectors (e.g., NleC, E1, and B1) inhibit NF-κB (14, 15, 23–26, 74, 75), a key transcription factor in the induction of pro- and anti-inflammatory cytokines, including IFN-β and TNF-α. We identified NleD, a zinc metalloprotease that degrades the MAP kinases JNK and p38, as the major T3SS effector responsible for the diminished induction of IFN-β (Fig. 2); this finding provided the first example of an EPEC effector mechanism that targets IFN-β. Like NF-κB, JNK and p38 contribute to the induction of both IFN-β and TNF-α in response to bacterial pathogens (76–78). The NleD-mediated downregulation of JNK/p38 and inhibition of NF-κB by other T3SS effectors are predicted to diminish induction of both IFN-β and TNF-α and may account, in part, for the reduced induction of IFN-β by EPEC. However, the contrasting effect on TNF-α, in which a functional T3SS is required for optimal induction, indicated that additional targets of T3SS regulation may be involved.
RNase L, an endoribonuclease that functions to regulate IFN induction and as a downstream mediator of IFN action (47, 51, 69, 79, 80), is implicated in IEC activities (45). Therefore, RNase L represented a candidate target of EPEC effectors that may account for the observed T3SS-dependent effects on cytokine induction and barrier function. Consistent with this prediction, RNase L activity was inhibited in a T3SS-dependent manner following EPEC infection (Fig. 5C). Reports from our laboratory and others have demonstrated that RNase L regulates the induction of IFN-β and TNF-α in response to microbial stimuli (45, 47, 52, 69, 81) and suggested a potential mechanism in which the T3SS-dependent regulation of these cytokines is mediated, in part, through RNase L. In support of this model, inhibition of RNase L activity by WT EPEC corresponded to diminished IFN-β induction and increased TNF-α, whereas infection with T3SS-deficient EPEC resulted in an RNase L-dependent increase in IFN-β induction and decrease in TNF-α (Fig. 5A and B). RNase L-mediated changes in IFN-β and TNF-α were correlated with sustained TEER values (Fig. 6A) and reduced IEC permeability (Fig. 6A and B) following EPEC infection, demonstrating the functional significance of this regulation. These findings indicate that RNase L functions to increase IFN-β induction and to reduce TNF-α; thus, the inhibition of RNase L by WT EPEC is an important mechanism by which it modulates host cytokine expression. Furthermore, while viruses have evolved multiple strategies to evade the antiviral activities of RNase L (67, 82), to our knowledge, this is the first example of RNase L inhibition mediated by a bacterial pathogen and supports an important role for RNase L in antibacterial immunity (45, 51, 52).
Regulation of IFN-β by RNase L is thought to occur through an indirect mechanism in which RNase L cleaves host and viral RNAs to produce PAMPs that activate RIG-I-like receptors, cytosolic RNA sensors that, in turn, activate a signaling pathway leading to IFN-β transcription (47, 69). The activation of RNase L-mediated rRNA cleavage and enhanced induction of IFN-β suggested that this pathway is operative following T3SS-deficient EPEC infection. However, the mechanism of RNase L activation and the identities of its RNA substrates in the context of a bacterial infection are not known. In this regard, the interaction of RNase L with the scaffold protein IQGAP1 provides a potential link to EPEC infection (83). IQGAP1 mediates diverse functions, including actin polymerization and is recruited to sites of EPEC attachment to stimulate pedestal formation (84). IQGAP may thus recruit RNase L to sites of EPEC attachment to sense infection and initiate immune signaling. Further studies are required to assess the role of this interaction in the context of EPEC infection. As EPEC infection inhibits RNase L in a T3SS-dependent manner, the established activities of specific EPEC effectors provide another potential source of insight into the pathways involved in RNase L activity. Specifically, JNK is a target of the metaloprotease NleD and is activated downstream of RNase L-mediated rRNA cleavage in the response to picornavirus infection (85, 86). Therefore, RNase L-mediated activities following infection with T3SS-deficient EPEC may similarly involve JNK signaling, and JNK cleavage by NleD may contribute to the T3SS-dependent inhibition of RNase L. Infection with a proteolysis-deficient mutant of NleD may help assess the role of JNK regulation in RNase L-mediated activities (14). However, additional studies are required to dissect how JNK proteolysis by NleD and other EPEC effector mechanisms impacts RNase L activity.
In contrast to IFN-β, RNase L negatively regulated TNF-α expression, suggesting that TNF-α mRNA may be a direct RNase L substrate. The posttranscriptional regulation of TNF-α mRNA stability is known to be a critical mechanism by which its expression is regulated and is mediated by tristetraprolin (TTP) (87). TTP is an RNA binding protein that binds A-U-rich elements in the 3′ untranslated regions of mRNAs encoding TNF-α and other proinflammatory mediators to stimulate their degradation (88). Consistent with a role for RNase L in this regulation, the RNase L protein interacts with TTP and downregulates a subset of TTP targets, suggesting that TTP directs RNase L to cleave specific mRNAs (89). The EPEC-induced inhibition of RNase L and enhanced expression of TNF-α support this model; however, RNase L negatively regulates TTP mRNA as part of an autoregulatory loop (90), and TTP activity is posttranslationally regulated by p38 in response to inflammatory stimuli (91, 92), indicating that a complex regulatory network functions to tightly regulate TNF-α. In addition, the T3SS effector NleD cleaves p38 protein and thus may affect TTP activity as a strategy by which EPEC modulates TNF-α expression. In light of this multifaceted regulation; the potential inhibition by T3SS effectors; and the dynamic nature of RNase L, TTP, and TNF-α expression and activity in the course of an inflammatory response (93), additional studies are required to dissect the roles and mechanisms by which RNase L and TTP regulate TNF-α in the context of EPEC infection. Alternatively, RNase L may regulate TNF-α as a secondary effect of its role in regulating IFN-β. Specifically, IFN-β has been reported to downregulate TNF-α (94, 95); therefore, the T3SS-mediated inhibition of RNase L activity and IFN-β induction may contribute to the observed increase in TNF-α expression. RNase L can thus impact gene expression via direct and indirect mechanisms and represents a potent target for EPEC to reprogram the IEC gene expression profile and modulate the host immune response.
The disruption of IEC barrier integrity by WT EPEC infection occurs through the downregulation and altered organization of TJ proteins that mediate barrier function (13, 60, 64, 96, 97). In contrast, infection with T3SS-deficient EPEC resulted in enhanced IFN-β induction (Fig. 1A) and RNase L activity (Fig. 5C), which corresponded to sustained barrier function (Fig. 3A and 6A) and increased expression of the TJ components claudin 1 and occludin (Fig. 4A and 6B), providing a potential mechanism by which IFN-β and RNase L exert their protective effects. Both the claudin 1 and occludin proteins were dramatically induced following treatment of IECs with exogenous IFN that also resulted in an accumulation of occludin at the cell periphery (see Fig. S3 in the supplemental material). The marked induction of claudin 1 and occludin by IFN corresponded to modest increases in claudin 1 and occludin mRNAs, suggesting that posttranslational mechanisms may be involved (Fig. 4B to D). Consistent with this interpretation, IFN-stimulated response elements have not been identified in the promoters of claudin 1 and occludin genes, and their transcripts are not included in a comprehensive database of known ISGs (98). A potential mechanism by which type I IFNs upregulate TJ proteins was revealed in a recent study describing the interaction of TJ proteins, including occludin and claudin 1, with IFN-induced transmembrane protein 1 (IFITM-1) (99) in hepatocytes. Specifically, IFN induction of IFITM-1 is thought to stabilize components of TJ complexes to inhibit hepatitis C virus entry. IFN induction of TJ proteins was also reported in lung epithelium, indicating that this regulation may be a critical feature in the control of host-microbe barrier function in multiple tissues (54). Thus, like other stimuli that target TJ complexes, IFN may impact TJ function via modulation of protein expression or through the indirect regulation of TJ protein stability and organization. RNase L deficiency markedly reduced expression of claudin 1 and occludin (Fig. 6B and C). Interestingly, this RNase L-dependent regulation did not directly correspond to its regulation of IFN-β (compare Fig. 5A and 6B and C), suggesting that RNase L contributes to expression of these TJ proteins via mechanisms in addition to its regulation of IFN-β.
Together, these findings significantly increase our understanding of the host response to EPEC infection and the mechanisms by which it subverts these protective activities. Specifically, we identified IFN-β and RNase L as novel mediators of IEC barrier function that are targeted by EPEC effectors to evade host defense mechanisms and promote pathogenesis. Future in vivo studies will reveal the full impact of these innate immune effectors in EPEC infection. Consistent with their important functions in the GI tract, defects in IFN-β, RNase L, and their upstream regulators and downstream effectors are associated with GI pathologies in mouse models and human diseases (27, 34–37, 45, 59, 100–104). In light of these findings, the IFN-RNase L axis represents a potential therapeutic target and susceptibility biomarker for enteric pathogens and diseases that disrupt GI barrier function, such as IBD and associated cancers.
Supplementary Material
ACKNOWLEDGMENTS
This investigation was supported by NIAID grant AI077556 to B.A.H. and NIAID Enteric Research Investigational Network (ERIN) grant U19AI090873 to M.S.D. T.M.L. was supported by T32 NIDDK grant 5T32DK067872.
We thank Karen Scanlon (Department of Microbiology and Immunology, UMSOM) and Rao Jaladanki (Department of Surgery, Baltimore Veteran's Administration Medical Center) for valuable discussion, Heather Ezelle (Department of Microbiology and Immunology, UMSOM) for critical reading of the manuscript, and Terez Shea-Donohue (Mucosal Biology Research Center, UMSOM) for assistance with TEER assays.
B.A.H. is an inventor of patents relating to RNase L licensed to AliosBiopharma. There are no other conflicting interests to disclose.
Footnotes
Published ahead of print 14 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00105-14.
REFERENCES
- 1.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2 [DOI] [PubMed] [Google Scholar]
- 2.McNamara BP, Koutsouris A, O'Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. 2001. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J. Clin. Invest. 107:621–629. 10.1172/JCI11138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nougayrede JP, Fernandes PJ, Donnenberg MS. 2003. Adhesion of enteropathogenic Escherichia coli to host cells. Cell. Microbiol. 5:359–372. 10.1046/j.1462-5822.2003.00281.x [DOI] [PubMed] [Google Scholar]
- 4.Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. 2006. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell. Microbiol. 8:634–645. 10.1111/j.1462-5822.2005.00656.x [DOI] [PubMed] [Google Scholar]
- 5.Schumann M, Kamel S, Pahlitzsch ML, Lebenheim L, May C, Krauss M, Hummel M, Daum S, Fromm M, Schulzke JD. 2012. Defective tight junctions in refractory celiac disease. Ann. N. Y. Acad. Sci. 1258:43–51. 10.1111/j.1749-6632.2012.06565.x [DOI] [PubMed] [Google Scholar]
- 6.Hering NA, Schulzke JD. 2009. Therapeutic options to modulate barrier defects in inflammatory bowel disease. Dig. Dis. 27:450–454. 10.1159/000233283 [DOI] [PubMed] [Google Scholar]
- 7.Choi HJ, Kim J, Do KH, Park SH, Moon Y. 2013. Enteropathogenic Escherichia coli-induced macrophage inhibitory cytokine 1 mediates cancer cell survival: an in vitro implication of infection-linked tumor dissemination. Oncogene 32:4960–4969. 10.1038/onc.2012.508 [DOI] [PubMed] [Google Scholar]
- 8.Shen L. 2012. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann. N. Y. Acad. Sci. 1258:9–18. 10.1111/j.1749-6632.2012.06613.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiba H, Osanai M, Murata M, Kojima T, Sawada N. 2008. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 1778:588–600. 10.1016/j.bbamem.2007.08.017 [DOI] [PubMed] [Google Scholar]
- 10.Aroeti B, Friedman G, Zlotkin-Rivkin E, Donnenberg MS. 2012. Retraction of enteropathogenic E. coli type IV pili promotes efficient host cell colonization, effector translocation and tight junction disruption. Gut Microbes 3:267–271. 10.4161/gmic.19814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanajima-Ozawa M, Matsuzawa T, Fukui A, Kamitani S, Ohnishi H, Abe A, Horiguchi Y, Miyake M. 2007. Enteropathogenic Escherichia coli, Shigella flexneri, and Listeria monocytogenes recruit a junctional protein, zonula occludens-1, to actin tails and pedestals. Infect. Immun. 75:565–573. 10.1128/IAI.01479-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. 2002. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 123:163–172. 10.1053/gast.2002.34235 [DOI] [PubMed] [Google Scholar]
- 13.Zhang Q, Li Q, Wang C, Li N, Li J. 2012. Redistribution of tight junction proteins during EPEC infection in vivo. Inflammation 35:23–32. 10.1007/s10753-010-9285-1 [DOI] [PubMed] [Google Scholar]
- 14.Baruch K, Gur-Arie L, Nadler C, Koby S, Yerushalmi G, Ben-Neriah Y, Yogev O, Shaulian E, Guttman C, Zarivach R, Rosenshine I. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30:221–231. 10.1038/emboj.2010.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, Wagstaff KM, Dunstone MA, Sloan J, Whisstock JC, Kaper JB, Robins-Browne RM, Jans DA, Frankel G, Phillips AD, Coulson BS, Hartland EL. 2010. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 6:e1000898. 10.1371/journal.ppat.1000898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacket CO, Sztein MB, Losonsky G, Abe A, Finlay BB, McNamara BP, Fantry GT, James SP, Nataro JP, Levine MM, Donnenberg MS. 2000. Role of EspB in experimental human enteropathogenic Escherichia coli infection. Infect. Immun. 68:3689–3695. 10.1128/IAI.68.6.3689-3695.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salazar-Gonzalez H, Navarro-Garcia F. 2011. Intimate adherence by enteropathogenic Escherichia coli modulates TLR5 localization and proinflammatory host response in intestinal epithelial cells. Scand. J. Immunol. 73:268–283. 10.1111/j.1365-3083.2011.02507.x [DOI] [PubMed] [Google Scholar]
- 18.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect. Immun. 74:2522–2536. 10.1128/IAI.74.5.2522-2536.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W, Lamkanfi M, Kanneganti TD. 2012. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 287:16955–16964. 10.1074/jbc.M112.358705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR. 2006. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology 131:1153–1163. 10.1053/j.gastro.2006.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. 2005. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 166:409–419. 10.1016/S0002-9440(10)62264-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda M, Kubo A, Furuse M, Tsukita S. 2004. A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J. Cell Sci. 117:1247–1257. 10.1242/jcs.00972 [DOI] [PubMed] [Google Scholar]
- 23.Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. 2011. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol. Microbiol. 80:1420–1438. 10.1111/j.1365-2958.2011.07661.x [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Wang X, Pham TH, Feuerbacher LA, Lubos ML, Huang M, Olsen R, Mushegian A, Slawson C, Hardwidge PR. 2013. NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-kappaB activation. Cell Host Microbe 13:87–99. 10.1016/j.chom.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. 2010. NleC, a type III secretion protease, compromises NF-kappaB activation by targeting p65/RelA. PLoS Pathog. 6:e1001231. 10.1371/journal.ppat.1001231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadler C, Baruch K, Kobi S, Mills E, Haviv G, Farago M, Alkalay I, Bartfeld S, Meyer TF, Ben-Neriah Y, Rosenshine I. 2010. The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 6:e1000743. 10.1371/journal.ppat.1000743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kole A, He J, Rivollier A, Silveira DD, Kitamura K, Maloy KJ, Kelsall BL. 2013. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J. Immunol. 191:2771–2779. 10.4049/jimmunol.1301093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. 2011. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223. 10.1016/j.immuni.2011.02.006 [DOI] [PubMed] [Google Scholar]
- 29.Guo B, Chang EY, Cheng G. 2008. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest. 118:1680–1690. 10.1172/JCI33342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McAleer JP, Kolls JK. 2012. Maintaining poise: commensal microbiota calibrate interferon responses. Immunity 37:10–12. 10.1016/j.immuni.2012.07.001 [DOI] [PubMed] [Google Scholar]
- 31.Monroe KM, McWhirter SM, Vance RE. 2010. Induction of type I interferons by bacteria. Cell. Microbiol. 12:881–890. 10.1111/j.1462-5822.2010.01478.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, Artis D. 2012. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37:158–170. 10.1016/j.immuni.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, Diefenbach A. 2012. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37:171–186. 10.1016/j.immuni.2012.05.020 [DOI] [PubMed] [Google Scholar]
- 34.Sainathan SK, Bishnupuri KS, Aden K, Luo Q, Houchen CW, Anant S, Dieckgraefe BK. 2012. Toll-like receptor-7 ligand imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis. Inflamm. Bowel Dis. 18:955–967. 10.1002/ibd.21867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li XD, Chiu YH, Ismail AS, Behrendt CL, Wight-Carter M, Hooper LV, Chen ZJ. 2011. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc. Natl. Acad. Sci. U. S. A. 108:17390–17395. 10.1073/pnas.1107114108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Zhang HX, Sun YP, Liu ZX, Liu XS, Wang L, Lu SY, Kong H, Liu QL, Li XH, Lu ZY, Chen SJ, Chen Z, Bao SS, Dai W, Wang ZG. 2007. Rig-I-/- mice develop colitis associated with downregulation of G alpha i2. Cell Res. 17:858–868. 10.1038/cr.2007.81 [DOI] [PubMed] [Google Scholar]
- 37.Cao SS, Song B, Kaufman RJ. 2012. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm. Bowel Dis. 18:1735–1742. 10.1002/ibd.22878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sumer N, Palabiyikoglu M. 1995. Induction of remission by interferon-alpha in patients with chronic active ulcerative colitis. Eur. J. Gastroenterol. Hepatol. 7:597–602 [PubMed] [Google Scholar]
- 39.Baert F, Rutgeerts P. 1995. Interferon-alpha: an efficacious immune-modulating therapy for ulcerative colitis? Eur. J. Gastroenterol. Hepatol. 7:593–594 [PubMed] [Google Scholar]
- 40.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411:603–606. 10.1038/35079114 [DOI] [PubMed] [Google Scholar]
- 41.Strober W, Kitani A, Fuss I, Asano N, Watanabe T. 2008. The molecular basis of NOD2 susceptibility mutations in Crohn's disease. Mucosal Immunol. 1(Suppl. 1):S5–S9. 10.1038/mi.2008.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pott J, Hornef M. 2012. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 13:684–698. 10.1038/embor.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xavier RJ, Podolsky DK. 2007. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434. 10.1038/nature06005 [DOI] [PubMed] [Google Scholar]
- 44.MacMicking JD. 2012. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12:367–382. 10.1038/nri3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long TM, Chakrabarti A, Ezelle HJ, Brennan-Laun SE, Raufman JP, Polyakova I, Silverman RH, Hassel BA. 2013. RNase-L deficiency exacerbates experimental colitis and colitis-associated cancer. Inflamm. Bowel Dis. 19:1295–1305. 10.1097/MIB.0b013e318281f2fd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solis M, Goubau D, Hiscott J. 2007. RIG-I has guts: identification of a role for RIG-I in colitis development. Cell Res. 17:974–975. 10.1038/cr.2007.107 [DOI] [PubMed] [Google Scholar]
- 47.Malathi K, Dong B, Gale M, Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819. 10.1038/nature06042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou A, Hassel BA, Silverman RH. 1993. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72:753–765. 10.1016/0092-8674(93)90403-D [DOI] [PubMed] [Google Scholar]
- 49.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. 1997. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 16:6355–6363. 10.1093/emboj/16.21.6355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ezelle HJ, Hassel BA. 2012. Pathologic effects of RNase-L dysregulation in immunity and proliferative control. Front. Biosci. (Schol. Ed.) 4:767–786. 10.2741/298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chakrabarti A, Jha BK, Silverman RH. 2011. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 31:49–57. 10.1089/jir.2010.0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li XL, Ezelle HJ, Kang TJ, Zhang L, Shirey KA, Harro J, Hasday JD, Mohapatra SK, Crasta OR, Vogel SN, Cross AS, Hassel BA. 2008. An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc. Natl. Acad. Sci. U. S. A. 105:20816–20821. 10.1073/pnas.0807265105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraus J, Ling AK, Hamm S, Voigt K, Oschmann P, Engelhardt B. 2004. Interferon-beta stabilizes barrier characteristics of brain endothelial cells in vitro. Ann. Neurol. 56:192–205. 10.1002/ana.20161 [DOI] [PubMed] [Google Scholar]
- 54.LeMessurier KS, Hacker H, Chi L, Tuomanen E, Redecke V. 2013. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog. 9:e1003727. 10.1371/journal.ppat.1003727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nisa S, Hazen TH, Assatourian L, Nougayrede JP, Rasko DA, Donnenberg MS. 2013. In vitro evolution of an archetypal enteropathogenic Escherichia coli strain. J. Bacteriol. 195:4476–4483. 10.1128/JB.00704-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eswarappa SM, Janice J, Balasundaram SV, Chakravortty D. 2013. Non-neutral evolution in non-LEE-encoded type III effectors of attaching and effacing Escherichia coli. Microbes Infect. 15:147–151. 10.1016/j.micinf.2012.10.015 [DOI] [PubMed] [Google Scholar]
- 57.Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, Lung TW, Mansell A, Riedmaier P, Oates CV, Zaid A, Muhlen S, Crepin VF, Marches O, Ang CS, Williamson NA, O'Reilly LA, Bankovacki A, Nachbur U, Infusini G, Webb AI, Silke J, Strasser A, Frankel G, Hartland EL. 2013. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501:247–251. 10.1038/nature12524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12:125–135. 10.1038/nri3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Q, Li Q, Wang C, Liu X, Li N, Li J. 2010. Enteropathogenic Escherichia coli changes distribution of occludin and ZO-1 in tight junction membrane microdomains in vivo. Microb. Pathog. 48:28–34. 10.1016/j.micpath.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 61.Canil C, Rosenshine I, Ruschkowski S, Donnenberg MS, Kaper JB, Finlay BB. 1993. Enteropathogenic Escherichia coli decreases the transepithelial electrical resistance of polarized epithelial monolayers. Infect. Immun. 61:2755–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Minagar A, Long A, Ma T, Jackson TH, Kelley RE, Ostanin DV, Sasaki M, Warren AC, Jawahar A, Cappell B, Alexander JS. 2003. Interferon (IFN)-beta 1a and IFN-beta 1b block IFN-gamma-induced disintegration of endothelial junction integrity and barrier. Endothelium 10:299–307. 10.1080/714007544 [DOI] [PubMed] [Google Scholar]
- 63.Li Q, Zhang Q, Wang C, Li N, Li J. 2008. Invasion of enteropathogenic Escherichia coli into host cells through epithelial tight junctions. FEBS J. 275:6022–6032. 10.1111/j.1742-4658.2008.06731.x [DOI] [PubMed] [Google Scholar]
- 64.Glotfelty LG, Hecht GA. 2012. Enteropathogenic E. coli effectors EspG1/G2 disrupt tight junctions: new roles and mechanisms. Ann. N. Y. Acad. Sci. 1258:149–158. 10.1111/j.1749-6632.2012.06563.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zahavi EE, Lieberman JA, Donnenberg MS, Nitzan M, Baruch K, Rosenshine I, Turner JR, Melamed-Book N, Feinstein N, Zlotkin-Rivkin E, Aroeti B. 2011. Bundle-forming pilus retraction enhances enteropathogenic Escherichia coli infectivity. Mol. Biol. Cell 22:2436–2447. 10.1091/mbc.E11-01-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruchaud-Sparagano MH, Maresca M, Kenny B. 2007. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell. Microbiol. 9:1909–1921. 10.1111/j.1462-5822.2007.00923.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silverman RH. 2007. A scientific journey through the 2-5A/RNase L system. Cytokine Growth Factor Rev. 18:381–388. 10.1016/j.cytogfr.2007.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li XL, Ezelle HJ, Hsi TY, Hassel BA. 2011. A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip Rev. RNA 2:58–78. 10.1002/wrna.32 [DOI] [PubMed] [Google Scholar]
- 69.Malathi K, Saito T, Crochet N, Barton DJ, Gale M, Jr, Silverman RH. 2010. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 16:2108–2119. 10.1261/rna.2244210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81:12720–12729. 10.1128/JVI.01471-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silverman RH, Skehel JJ, James TC, Wreschner DH, Kerr IM. 1983. rRNA cleavage as an index of ppp(A2′p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J. Virol. 46:1051–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cerf-Bensussan N, Gaboriau-Routhiau V. 2010. The immune system and the gut microbiota: friends or foes? Nat. Rev. Immunol. 10:735–744. 10.1038/nri2850 [DOI] [PubMed] [Google Scholar]
- 73.Brown EM, Sadarangani M, Finlay BB. 2013. The role of the immune system in governing host-microbe interactions in the intestine. Nat. Immunol. 14:660–667. 10.1038/ni.2611 [DOI] [PubMed] [Google Scholar]
- 74.Sham HP, Shames SR, Croxen MA, Ma C, Chan JM, Khan MA, Wickham ME, Deng W, Finlay BB, Vallance BA. 2011. Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-kappaB and p38 mitogen-activated protein kinase activation. Infect. Immun. 79:3552–3562. 10.1128/IAI.05033-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vossenkamper A, Marches O, Fairclough PD, Warnes G, Stagg AJ, Lindsay JO, Evans PC, Luong LE, Croft ANM, Naik S, Frankel G, MacDonald TT. 2010. Inhibition of NF-kappaB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. J. Immunol. 185:4118–4127. 10.4049/jimmunol.1000500 [DOI] [PubMed] [Google Scholar]
- 76.Symons A, Beinke S, Ley SC. 2006. MAP kinase kinase kinases and innate immunity. Trends Immunol. 27:40–48. 10.1016/j.it.2005.11.007 [DOI] [PubMed] [Google Scholar]
- 77.Hennessy EJ, Parker AE, O'Neill LA. 2010. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9:293–307. 10.1038/nrd3203 [DOI] [PubMed] [Google Scholar]
- 78.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. 1999. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1:94–97. 10.1038/10061 [DOI] [PubMed] [Google Scholar]
- 79.Li XL, Blackford JA, Hassel BA. 1998. RNase L mediates the antiviral effect of interferon through a selective reduction in viral RNA during encephalomyocarditis virus infection. J. Virol. 72:2752–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andersen JB, Mazan-Mamczarz K, Zhan M, Gorospe M, Hassel BA. 2009. Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol. 6:305–315. 10.4161/rna.6.3.8526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yi X, Zeng C, Liu H, Chen X, Zhang P, Yun BS, Jin G, Zhou A. 2013. Lack of RNase L attenuates macrophage functions. PLoS One 8:e81269. 10.1371/journal.pone.0081269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Townsend HL, Jha BK, Han JQ, Maluf NK, Silverman RH, Barton DJ. 2008. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 14:1026–1036. 10.1261/rna.958908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sato A, Naito T, Hiramoto A, Goda K, Omi T, Kitade Y, Sasaki T, Matsuda A, Fukushima M, Wataya Y, Kim HS. 2010. Association of RNase L with a Ras GTPase-activating-like protein IQGAP1 in mediating the apoptosis of a human cancer cell-line. FEBS J. 277:4464–4473. 10.1111/j.1742-4658.2010.07833.x [DOI] [PubMed] [Google Scholar]
- 84.Brown MD, Bry L, Li Z, Sacks DB. 2008. Actin pedestal formation by enteropathogenic Escherichia coli is regulated by IQGAP1, calcium, and calmodulin. J. Biol. Chem. 283:35212–35222. 10.1074/jbc.M803477200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li G, Xiang Y, Sabapathy K, Silverman RH. 2004. An apoptotic signaling pathway in the interferon antiviral response mediated by RNase L and c-Jun NH2-terminal kinase. J. Biol. Chem. 279:1123–1131. 10.1074/jbc.M305893200 [DOI] [PubMed] [Google Scholar]
- 86.Iordanov MS, Wong J, Newton DL, Rybak SM, Bright RK, Flavell RA, Davis RJ, Magun BE. 2000. Differential requirement for the stress-activated protein kinase/c-Jun NH(2)-terminal kinase in RNA damage-induced apoptosis in primary and in immortalized fibroblasts. Mol. Cell Biol. Res. Commun. 4:122–128. 10.1006/mcbr.2000.0266 [DOI] [PubMed] [Google Scholar]
- 87.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brooks SA, Blackshear PJ. 2013. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta 1829:666–679. 10.1016/j.bbagrm.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brennan-Laun SE, Ezelle HJ, Li XL, Hassel BA. 2014. RNase-L control of cellular mRNAs: roles in biologic functions and mechanisms of substrate targeting. J. Interferon Cytokine Res. 34:275–288. 10.1089/jir.2013.0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Al-Haj L, Blackshear PJ, Khabar KS. 2012. Regulation of p21/CIP1/WAF-1 mediated cell-cycle arrest by RNase L and tristetraprolin, and involvement of AU-rich elements. Nucleic Acids Res. 40:7739–7752. 10.1093/nar/gks545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brook M, Tchen CR, Santalucia T, McIlrath J, Arthur JS, Saklatvala J, Clark AR. 2006. Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol. Cell. Biol. 26:2408–2418. 10.1128/MCB.26.6.2408-2418.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tchen CR, Brook M, Saklatvala J, Clark AR. 2004. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem. 279:32393–32400. 10.1074/jbc.M402059200 [DOI] [PubMed] [Google Scholar]
- 93.Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. 2006. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol. Cell. Biol. 26:2399–2407. 10.1128/MCB.26.6.2399-2407.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sauer I, Schaljo B, Vogl C, Gattermeier I, Kolbe T, Muller M, Blackshear PJ, Kovarik P. 2006. Interferons limit inflammatory responses by induction of tristetraprolin. Blood 107:4790–4797. 10.1182/blood-2005-07-3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kovarik P, Sauer I, Schaljo B. 2007. Molecular mechanisms of the anti-inflammatory functions of interferons. Immunobiology 212:895–901. 10.1016/j.imbio.2007.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Simonovic I, Rosenberg J, Koutsouris A, Hecht G. 2000. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell. Microbiol. 2:305–315. 10.1046/j.1462-5822.2000.00055.x [DOI] [PubMed] [Google Scholar]
- 97.Weflen AW, Alto NM, Hecht GA. 2009. Tight junctions and enteropathogenic E. coli. Ann. N. Y. Acad. Sci. 1165:169–174. 10.1111/j.1749-6632.2009.04060.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41:D1040–D1046. 10.1093/nar/gks1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, Tyrrell DL, Gale M., Jr 2013. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 57:461–469. 10.1002/hep.26066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stalnikowicz R, Goder K, Karmeli F, Fiocchi C, Rachmilewitz D. 1985. (2′-5′)oligo adenylate synthetase activity in leucocytes of patients with inflammatory bowel disease. Gut 26:556–561. 10.1136/gut.26.6.556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ting JP, Duncan JA, Lei Y. 2010. How the noninflammasome NLRs function in the innate immune system. Science 327:286–290. 10.1126/science.1184004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dugan JW, Albor A, David L, Fowlkes J, Blackledge MT, Martin TM, Planck SR, Rosenzweig HL, Rosenbaum JT, Davey MP. 2009. Nucleotide oligomerization domain-2 interacts with 2′-5′-oligoadenylate synthetase type 2 and enhances RNase-L function in THP-1 cells. Mol. Immunol. 47:560–566. 10.1016/j.molimm.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Siddiqui MA, Malathi K. 2012. RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 287:43651–43664. 10.1074/jbc.M112.399964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chakrabarti A, Ghosh PK, Banerjee S, Gaughan C, Silverman RH. 2012. RNase L triggers autophagy in response to viral infections. J. Virol. 86:11311–11321. 10.1128/JVI.00270-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.