

Abstract
Reactivation of chronic infection with Toxoplasma gondii can cause life-threatening toxoplasmic encephalitis in immunocompromised individuals. We examined the role of VCAM-1/α4β1 integrin interaction in T cell recruitment to prevent reactivation of the infection in the brain. SCID mice were infected and treated with sulfadiazine to establish a chronic infection. VCAM-1 and ICAM-1 were the endothelial adhesion molecules detected on cerebral vessels of the infected SCID and wild-type animals. Immune T cells from infected wild-type mice were treated with anti-α4 integrin or control antibodies and transferred into infected SCID or nude mice, and the animals received the same antibody every other day. Three days later, sulfadiazine was discontinued to initiate reactivation of infection. Expression of mRNAs for CD3δ, CD4, CD8β, gamma interferon (IFN-γ), and inducible nitric oxide synthase (NOS2) (an effector molecule to inhibit T. gondii growth) and the numbers of CD4+ and CD8+ T cells in the brain were significantly less in mice treated with anti-α4 integrin antibody than in those treated with control antibody at 3 days after sulfadiazine discontinuation. At 6 days after sulfadiazine discontinuation, cerebral tachyzoite-specific SAG1 mRNA levels and numbers of inflammatory foci associated with tachyzoites were markedly greater in anti-α4 integrin antibody-treated than in control antibody-treated animals, even though IFN-γ and NOS2 mRNA levels were higher in the former than in the latter. These results indicate that VCAM-1/α4β1 integrin interaction is crucial for prompt recruitment of immune T cells and induction of IFN-γ-mediated protective immune responses during the early stage of reactivation of chronic T. gondii infection to control tachyzoite growth.
INTRODUCTION
Toxoplasma gondii, an obligate intracellular protozoan parasite, is an important food-borne pathogen in humans. Acute infection is characterized by proliferation of tachyzoites in a variety of cells throughout the body and can cause various diseases, including lymphadenitis and congenital infection of fetuses (1). Gamma interferon (IFN-γ)-dependent, cell-mediated immune responses (2–4) and, to a lesser degree, humoral immunity (5–7) can suppress proliferation of tachyzoites, but the parasite establishes a chronic infection by forming tissue cysts, primarily in the brain. Chronic infection with T. gondii is one of the most common parasitic infections in humans (8, 9). It is estimated that 500 million to 2 billion people worldwide are chronically infected with the parasite (8, 10). The importance of immune responses in maintaining the latency of the chronic infection is clearly evident in the development of life-threatening toxoplasmic encephalitis (TE), caused by reactivation of the chronic infection in immunocompromised individuals, such as those with AIDS and organ transplants (11, 12). However, the mechanisms by which the immune system maintains the latency of chronic infection with T. gondii in the brain and prevents TE still need to be elucidated.
T. gondii has three predominant genotypes (I, II, and III), and infection with all the genotypes occurs in humans (13–15). However, type II is predominant in the strains isolated from patients with TE in North America and Europe (16, 17). Because TE mostly occurs due to reactivation of chronic infection with the parasite, mouse strains that can establish a latent, chronic infection with type II strains of the parasite appear to be an ideal animal model to analyze the mechanisms by which the immune system maintains the latency of the chronic infection in the brain. In this regard, resistance to chronic infection with type II T. gondii is under genetic control in mice, and strains of inbred mice can be generally divided into two groups. Strains with the H-2b (e.g., C57BL/6) or H-2k (e.g., CBA/Ca) haplotype are susceptible and develop progressive and ultimately fatal TE without immunosuppressive treatment (18, 19). In contrast, strains with the H-2d haplotype (e.g., BALB/c) are resistant and establish a latent, chronic infection (18, 19), as do immunocompetent humans. Therefore, BALB/c mice appear to provide an excellent model to analyze how the immune system functions to maintain the latency of chronic type II T. gondii infection in the brain.
Infecting BALB/c-background SCID or athymic nude mice with a type II (ME49) strain and treating them with sulfadiazine enables them to establish a chronic infection in their brains (20, 21). Discontinuation of sulfadiazine treatment induces reactivation of the chronic infection in the brain in these immunodeficient mice, and adoptive transfer of immune T cells from infected wild-type BALB/c mice into these animals can prevent the reactivation of infection (20, 22, 23). Therefore, this T cell transfer system in BALB/c-background SCID and nude mice provides an excellent model to analyze the mechanisms by which the immune system prevents reactivation of the infection in the brain and development of TE.
The blood-brain barrier prevents most intravascular leukocytes from entering the parenchyma of the normal brain (24). However, leukocytes are able to migrate from blood vessels into the brain when infection, ischemia, or an autoimmune disease, such as multiple sclerosis, occurs. This migration is mediated, in part, by endothelial adhesion and activation molecules that are found in injured brain but not in normal brain (24). In the present study, we utilized the SCID and nude mouse model of reactivation of cerebral T. gondii infection and analyzed vascular endothelial adhesion molecules important for T cell recruitment into the brain and prevention of reactivation of the infection. We found that interactions between VCAM-1 expressed on cerebrovascular endothelial cells and α4β1 integrin expressed on the surfaces of immune T cells are crucial for recruiting the T cells into the brain and inducing IFN-γ-mediated protective immunity during the early stage of reactivation of cerebral infection with T. gondii. Such prompt induction of T cell migration and IFN-γ-mediated immune responses was associated with effective control of tachyzoite proliferation and prevention of TE.
MATERIALS AND METHODS
Mice.
Female BALB/c and BALB/c-background SCID mice were obtained from the Jackson Laboratories (Bar Harbor, ME). Female BALB/c-background athymic nude mice were from the Jackson Laboratories or Taconic (Germantown, NY). Female Swiss-Webster mice were from Taconic. All the mice were housed under specific-pathogen-free conditions. There were 3 to 6 mice in each experimental group.
Infection with T. gondii and reactivation of cerebral infection.
Cysts of the ME49 strain of T. gondii were obtained from brains of chronically infected Swiss-Webster mice (25). The mice were euthanized by asphyxiation with CO2, and their brains were removed and triturated in phosphate-buffered saline (PBS) (pH 7.2). An aliquot of the brain suspension was examined microscopically for the number of cysts, and after appropriate dilution in PBS, BALB/c, SCID, and nude mice were infected with 10 cysts orally by gavage (20). SCID and nude mice received sulfadiazine-supplemented drinking water (400 mg/liter) beginning 7 or 8 days after infection for 3 to 4 weeks to control the proliferation of tachyzoites and establish a chronic infection in their brains (20, 21). Reactivation of cerebral infection with T. gondii was induced in the SCID and nude mice by discontinuing sulfadiazine treatment (20, 22, 23). In one experiment, one group of wild-type BALB/c mice received sulfadiazine for 11 days beginning 14 days after infection to examine whether sulfadiazine treatment could affect the expression of cerebrovascular endothelial adhesion molecules during the infection. Mouse care and experimental procedures were performed in accordance with established institutional guidance and approved protocols from the Institutional Animal Care and Use Committee.
Immunohistochemistry for vascular adhesion molecules.
Brains from BALB/c and SCID mice were removed 25 days after infection, which is the time that the SCID mice were receiving sulfadiazine; bisected along the median sagittal plane; and snap-frozen in OCT compound (Sakura Finetek, Torrance, CA). Acetone-fixed frozen sections were stained as previously described with monoclonal antibodies (MAbs) against ICAM-1 (clone YN1/1.7; ATCC, Manassas, VA, and Abcam, Cambridge, MA), VCAM-1 (MK-2.7; ATCC and Abcam), E-selectin (10E9.6; BD PharMingen, San Diego, CA), P-selectin (RB40; ATCC), MAdCAM-1 (MECA-367; provided by E. Butcher, Stanford, CA), PNAd (MECA-79; provided by E. Butcher), and irrelevant negative-control antigens (26). Briefly, the sections were sequentially incubated with rat primary antibody, biotin-conjugated anti-rat IgG, or anti-rat IgM; peroxidase-streptavidin; diaminobenzidine/hydrogen peroxide; and methylene blue or hematoxylin counterstain. There were two washes with PBS between the steps. Three or more sections, at least 200 μm apart, were stained for each adhesion molecule in each brain. The slides were examined by light microscopy. The numbers of vessels expressing VCAM-1 or ICAM-1 per mm2 of brain area were determined by image analysis, as described previously (27).
Flow cytometry to detect expression of α4β1 integrin on CD4+ and CD8+ immune T cells from BALB/c mice chronically infected with T. gondii.
Spleen cells were obtained from chronically infected BALB/c mice and suspended in Hanks' balanced salt solution (HBSS) (HyClone, Logan, UT) containing 2% fetal bovine serum (FBS) (Sigma, St. Louis, MO). After depletion of red blood cells with 160 mM NH4Cl (pH 7.2), the cells were incubated on ice for 10 min with a predetermined optimal amount of anti-FcγII/III receptor MAb (clone 2.4G2). The cells were then incubated for 30 min with an allophycocyanin (APC)-conjugated MAb to CD4 (clone RM4-5) or an APC-conjugated MAb to CD8β (clone 53-6.7), in combination with phycoerythrin (PE)-conjugated MAb to α4 integrin (clone 9C10), fluorescein isothiocyanate (FITC)-conjugated MAb to β1 integrin (clone Ha2/5), and PE-Cy5-conjugated MAb to CD44 (clone IM7). Isotype control antibodies were used as negative controls. All but FITC-hamster IgM(κ) (BioLegend, San Diego, CA) were purchased from BD Biosciences (Mountain View, CA). Cells were analyzed on a FACSCalibur using CellQuest software (BD Biosciences). At least 20,000 cells in the gated area for lymphocytes were analyzed.
Adoptive transfer of T cells and blocking of T cell homing by MAb against α4 integrin.
Spleen cells were obtained from chronically infected BALB/c mice and suspended in HBSS containing 2% FBS. Immune T cells were purified by treating the immune spleen cells with magnetic-bead-conjugated anti-CD4 and anti-CD8α MAb (Miltenyi Biotech, Sunnyvale, CA) for magnetic cell sorting (MACS), as previously described (20–23). The purity of the MACS-purified T cells was >95%. Immune T cells were treated with 10 μg/ml of anti-α4 integrin MAb (PS/2, grown from a hybridoma obtained from ATCC [28]) or a control MAb (9B5 [28] [anti-human CD44] or RTK4530 [anti-trinitrophenol-KLH] [BioLegend]) on ice for 10 min (28). The cells (0.9 × 107 to 1.5 × 107) were transferred into infected and sulfadiazine-treated SCID or nude mice at 4 weeks after infection. Thereafter, the animals received the MAb (200 or 250 μg in 0.2 ml PBS) intraperitoneally every other day. Sulfadiazine treatment was discontinued 3 days after the cell transfer to initiate reactivation of T. gondii infection.
Immunohistochemistry for T cells that infiltrated into the brain after T cell transfer.
SCID mice were infected, treated with sulfadiazine, and received immune T cells in combination with treatment with anti-α4 integrin MAb or isotype control MAbs as described above. At 3 days after discontinuation of sulfadiazine treatment, their brains were snap-frozen in OCT compound in the same manner as described in “Immunohistochemistry for vascular adhesion molecules” above. Sections were stained with anti-CD3 (17A2; BioLegend) or isotype control (RTK4530) diluted in PBS containing 2.5% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) in the same manner as described for staining for vascular adhesion molecules with hematoxylin counterstain.
Immunohistochemical staining of T. gondii.
The brains of infected nude mice that had received immune T cells in combination with treatment with anti-α4 integrin MAb or isotype control MAbs were obtained 6 days after discontinuation of sulfadiazine treatment and fixed with 10% formalin, 70% ethanol, and 5% acetic acid. Staining for T. gondii on the histological sections of the brains was performed as described previously (29). Briefly, the sections were sequentially incubated with rabbit anti-T. gondii primary antibody, goat anti-rabbit IgG antibody, peroxidase-rabbit anti-peroxidase complex, diaminobenzidine/hydrogen peroxide, and hematoxylin counterstain. Three sections, at least 50 μm apart, were stained in each brain. In counting parasitophorous vacuoles (PV) containing tachyzoites in inflammatory foci, a photograph of each of the foci containing large numbers of PV was taken at a high magnification, and the PV in each focus were counted on a large image or print of the photograph.
Flow cytometry to detect T cells that migrated into the brains of infected nude mice after T cell transfer.
Sulfadiazine treatment of infected nude mice was discontinued 3 days after receiving immune T cells to initiate reactivation of cerebral infection with T. gondii. Three days after discontinuation of sulfadiazine treatment, anesthetized animals were perfused intracardially with PBS to remove intravascular leukocytes. Mononuclear cells were isolated from brains from each group as described previously (30). After treating the cells with anti-FcγII/III receptor MAb, the cells were incubated for 30 min with a PE-conjugated MAb to CD4 (clone RM4-5) or a FITC-conjugated MAb to CD8β (clone 53-6.7) in combination with peridinin chlorophyll protein (PerCP)-conjugated MAb to CD3ε (clone 145-2C11). All MAbs were from BD Biosciences. Fluorescence-activated cell sorter (FACS) analysis was performed in the same manner as described for detection of α4β1 integrin on immune T cells.
Quantification of mRNAs for CD3δ, CD4, CD8β, IFN-γ, NOS2, VCAM-1, MAdCAM-1, tachyzoite-specific SAG1, and bradyzoite-specific BAG1 in the brain.
Total RNAs were purified from brains of SCID and nude mice using RNA Stat-60 (Tel-Test B, Inc., Friendswood, TX), and 1 μg of RNA was treated with DNase I (Invitrogen, Carlsbad, CA) and reverse transcribed into cDNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) according to the manufacturer's instructions (20). To quantify tachyzoites in the mouse brains, real-time PCR was conducted in triplicate to measure the amounts of mRNA for tachyzoite-specific SAG1 using TaqMan reagents and the StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) as described previously (31, 32). mRNA levels of CD3δ, CD4, CD8β, IFN-γ, and BAG1 were also measured with the following primers and probes: CD3δ, AAGCCCCAATTGCAACAAAACT (forward), GGCAGGTTTGATCTCCGTTCT (reverse), and TCAGTGTAGCAATGATTTC (probe); CD4, CATCTCTCTTAGGCGCTTGCT (forward), GCGTCTTCCCTTGAGTGACA (reverse), and CAGCTGTCACAACTCC (probe); CD8β, CCTCGGAGTGGCCGTCTA (forward), GCGCTGATCATTTGTGAAACTGTTT (reverse), and CCGCACACAGTAAAAG (probe); IFN-γ, CATTGAAAGCCTAGAAAGTCTGAATAA (forward), TGGCTCTGCAGGATTTTCATG (reverse), and TCACCATCCTTTTGCCAGTTCCTCCAG (probe); BAG1, TCACGTGGAGACCCAGAGT (forward), CTGGCAAGTCAGCCAAAATAATCAT (reverse), and TTTGCTGTCGAACTCC (probe). The IFN-γ probe was labeled with 6-carboxyfluorescein (FAM) at the 5′ end and TAMRA at the 3′ end. All the other probes were labeled with FAM at the 5′ ends and minor groove binder (MGB)-nonfluorescent quencher (NFQ) at the 3′ ends. Mouse β-actin was used as an endogenous control and, the comparative threshold cycle (Ct) method was applied to analyze the data. Mouse β-actin (mouse ACTB; 4352933E), NOS2 (Mm00440489_g1), VCAM-1 (Mm01320970_m1), and MAdCAM-1 (Mm00522088_m1) gene expression kits and all the primers and probes were from Applied Biosystems.
Statistical analysis.
Numerical data are presented as means and standard errors. Differences between groups were analyzed with one-way analysis of variance (ANOVA) with a Newman-Keuls posttest or Mann-Whitney test. A P value of <0.05 was considered to be significant.
RESULTS
Expression of endothelial adhesion molecules on cerebral vessels of SCID and wild-type BALB/c mice infected with T. gondii.
We previously reported that VCAM-1 and ICAM-1 are the vascular endothelial adhesion molecules whose expression is upregulated in the brains of BALB/c mice during chronic infection with T. gondii (33). To examine whether BALB/c-background SCID mice with chronic infection express the same vascular adhesion molecules in their brains as infected wild-type mice, the two strains of mice were infected and the SCID mice received treatment with sulfadiazine from 7 days after infection to control the proliferation of tachyzoites and establish a chronic infection (20, 21). We stained frozen sections of brains from SCID and wild-type mice with antibodies against ICAM-1, VCAM-1, E-selectin, P-selectin, MAdCAM-1, and PNAd 25 days after infection. A few vessels in the brains of uninfected SCID and wild-type mice expressed VCAM-1 (1.71 ± 0.41 and 4.51 ± 1.47 vessels/mm2 of tissue, respectively) (Fig. 1A). Infection with T. gondii caused an increase in vascular VCAM-1 expression in both strains (P < 0.05) (Fig. 1A). The mean number of VCAM-1-expressing vessels in the brains of infected wild-type mice tended to be slightly greater than in infected SCID mice, but the difference did not reach statistical significance (Fig. 1A). Very few vessels in the brains of uninfected SCID and wild-type mice had detectable levels of ICAM-1 (Fig. 1B). After T. gondii infection, both strains had 7- to 30-fold increases in the number of ICAM-1+ vessels (P < 0.05) (Fig. 1B).
FIG 1.

VCAM-1 and ICAM-1 are the endothelial adhesion molecules expressed on cerebral vessels in SCID and wild-type BALB/c mice with chronic infection with T. gondii. SCID and wild-type BALB/c mice were infected with 10 cysts of the ME49 strain orally. The SCID mice were treated with sulfadiazine beginning 7 days after infection. One group of BALB/c mice also received sulfadiazine beginning 14 days after infection. All the mice were sacrificed 25 days after infection. (A and B) The number of cerebral vessels that expressed VCAM-1 or ICAM-1 was determined by frozen-section immunohistochemistry staining and quantitative image analysis (see Materials and Methods). (C) Examples of VCAM-1 and ICAM-1 staining in the brains of uninfected and chronically infected BALB/c mice. The arrows indicate representative VCAM-1+ and ICAM-1+ vessels. (D) Expression of mRNA for VCAM-1 and MAdCAM-1 in the brains of uninfected and infected SCID mice. The mRNA levels were determined by real-time RT-PCR (see Materials and Methods). *, P < 0.05; **, P < 0.01. The error bars indicate standard errors.
We did not detect expression of E-selectin, P-selectin, MAdCAM-1, or PNAd on cerebral vessels in infected or uninfected mice of either strain. Treatment of infected wild-type mice with sulfadiazine did not alter the expression of vascular adhesion molecules in their brains (Fig. 1A and B). Examples of staining for VCAM-1 and ICAM-1 in uninfected and chronically infected BALB/c mice are shown in Fig. 1C. These results indicate that ICAM-1 and VCAM-1 are the cerebrovascular endothelial adhesion molecules whose expression is upregulated during chronic infection with T. gondii in both SCID and wild-type BALB/c mice. These results strongly suggest that a model using infected, sulfadiazine-treated SCID mice and a transfer of immune T cells is suitable to examine the roles of the cerebrovascular endothelial adhesion molecules in T cell recruitment into the brain to prevent reactivation of chronic infection with T. gondii. Our results in T. gondii infection are different from those observed in mice with experimental allergic encephalitis and a model of Alzheimer's disease, in which P-selectin expression is enhanced (34–36), suggesting that expression of cerebrovascular endothelial adhesion molecules differs depending on the stimuli present under the specific conditions.
Importance of α4 integrin for recruiting CD4+ and CD8+ immune T cells into the brain during the early stage of reactivation of T. gondii infection.
We previously showed that IFN-γ plays an important role in recruiting immune T cells into the brains of T. gondii-infected BALB/c mice and that expression of endothelial VCAM-1, but not ICAM-1, in their brains is largely dependent on IFN-γ (33). Our previous study also showed that endothelial VCAM-1 is important for migration of carboxyfluorescein succinimidyl ester (CFSE)-labeled immune CD8+ T cells into the brains of infected BALB/c mice (33). Therefore, we utilized a reactivation model of T. gondii infection in SCID and nude mice and examined the role of VCAM-1/α4β1 integrin interaction in recruitment of immune T cells to prevent reactivation of the infection. In this model, SCID and nude mice were infected and treated with sulfadiazine to establish a chronic infection in their brains (20, 22). Discontinuation of sulfadiazine treatment initiates reactivation of the cerebral infection and results in mortality of the animals within 2 weeks after the initiation of reactivation of the infection (20, 22). Adoptive transfer of immune T cells from chronically infected BALB/c mice can prevent the reactivation of infection and the mortality (20, 22).
Infected, sulfadiazine-treated SCID mice received a systemic transfer of immune T cells purified from the spleens of chronically infected BALB/c mice in combination with treatment with anti-α4 integrin or control MAb at 3 weeks after infection. The anti-α4 integrin MAb is known to block α4β1 integrin/VCAM-1 and α4β7 integrin/MAdCAM-1 binding (37, 38). However, because MAdCAM-1 was not detected on intracerebral vessels in the infected mice in the immunohistochemistry, any inhibitory effect of the MAb in this mouse model would be on α4β1/VCAM-1 binding. In order to further confirm that MAdCAM-1 is not expressed in the brains of infected SCID mice, we performed real-time reverse transcription (RT)-PCR to measure amounts of mRNA for VCAM-1 and MAdCAM-1 expressed in the brains of uninfected and infected SCID mice. The amounts of VCAM-1 mRNA were significantly greater in the brains of infected than uninfected SCID mice (P < 0.05) (Fig. 1D). This is in agreement with the detection of larger numbers of cerebral vessels expressing this vascular adhesion molecule in the brains of infected than in those of uninfected animals in the immunohistochemistry shown in Fig. 1A. In contrast, MAdCAM-1 mRNA levels did not differ between the brains of uninfected and infected SCID mice (Fig. 1D), and mRNA levels for MAdCAM-1 in the brains of infected SCID mice were approximately 280 times lower than those for VCAM-1 in these animals (Fig. 1D). Therefore, the infection did not stimulate expression of MAdCAM-1 mRNA in their brains, and the mRNA expression remained at a very low level. These results are consistent with our observation in immunohistochemistry, in which cerebral vessels expressing MAdCAM-1 were not detected in these animals. These results indicate that the inhibitory effect of anti-α4 integrin MAb on T cell recruitment into the brain in this mouse model is on α4β1/VCAM-1 binding.
Three days after the transfer of immune T cells into infected SCID mice in combination with treatment with anti-α4 integrin or control MAb, sulfadiazine treatment was discontinued to initiate reactivation of the infection in the brain (20, 22, 23). Three days after discontinuation of sulfadiazine treatment, we examined the amounts of mRNA for T cell markers (CD3δ, CD4, and CD8β) in the brains of the SCID mice. The amounts of mRNAs for CD3δ, CD4, and CD8β were 27.9, 10.5, and 34.1 times less, respectively, in the brains of mice treated with anti-α4 integrin MAb than in the brains of animals treated with control MAb (P < 0.001) (Fig. 2A). In a control group that did not receive a transfer of T cells, the amounts of mRNA for these T cell markers in their brains were close to detectable limits or very low even when detected (Fig. 2A). Immunohistochemistry detecting CD3+ T cells demonstrated the presence of large numbers of T cells in the parenchyma of the brains of SCID mice treated with control MAb (Fig. 2B, middle), but not in the brains of the animals treated with anti-α4 integrin MAb (Fig. 2B, right). CD3+ T cells were also detectable in association with some cerebral vessels, in addition to the parenchyma, of the SCID mice treated with control MAb (Fig. 2B, left). We also measured the amounts of mRNA for VCAM-1 expressed in the brains of infected SCID mice on the last day of sulfadiazine treatment and 3 days after discontinuation of the treatment. Comparable levels of cerebral VCAM-1 mRNA were detected before and after initiation of reactivation of cerebral T. gondii infection (data not shown). These results strongly suggested that VCAM-1/α4β1 integrin interaction is crucial for mediating infiltration of both CD4+ and CD8+ immune T cells into the brain parenchyma during the early stage of reactivation of cerebral infection with T. gondii.
FIG 2.

Cerebral expression of mRNA for T cell markers (CD3δ, CD4, and CD8β) (A) and frequency of CD3+ T cells in the brain tissue (B) are markedly less during the early stage of reactivation of T. gondii infection in the brains of SCID mice that received immune T cells with anti-α4 integrin MAb treatment than in those with the cell transfer with control MAb. SCID mice were infected with 10 cysts of the ME49 strain orally and treated with sulfadiazine for 3 weeks beginning 7 days after infection. Groups of animals received immune T cells (1.2 × 107 cells) pretreated with either anti-α4 integrin or control MAb intravenously 4 weeks after infection. Thereafter, the mice received the MAb (250 μg) intraperitoneally every other day. Sulfadiazine treatment was discontinued 3 days after the cell transfer, and the brains were collected 3 days after discontinuation of sulfadiazine treatment. (A) mRNA levels for the T cell markers were measured by real-time RT-PCR. **, P < 0.01; ***, P < 0.001. (B) Frozen sections of the brains were stained with anti-CD3 (top row) or isotype control MAbs (bottom row) (see Materials and Methods). The arrows indicate representatives of CD3+ T cells. The error bars indicate standard errors.
To further confirm the importance of α4 integrin in recruitment of immune T cells into the brain during reactivation of infection, we also examined cerebral expression of mRNA for T cell markers 3 days after discontinuation of sulfadiazine treatment in infected nude mice that had received immune T cells in combination with anti-α4 integrin or control MAb treatment. Whereas SCID mice lack both T and B cells, nude mice have B cells. Therefore, by using nude mice, we were able to examine the mechanisms of T cell recruitment into the brain to prevent reactivation of chronic T. gondii infection in the presence of B cells. Consistent with those observed in the brains of infected SCID mice, the amounts of mRNA for CD3δ, CD4, and CD8β were 3.0, 2.6, and 3.9 times less, respectively, in the brains of the nude mice treated with anti-α4 integrin MAb than in the brains of animals treated with control MAb (P < 0.05) (Fig. 3A).
FIG 3.

mRNA levels for T cell markers (CD3δ, CD4, and CD8β) (A) and numbers of CD4+ and CD8+ T cells (B) are markedly lower during the early stage of reactivation of T. gondii infection in the brains of nude mice that received immune T cells with anti-α4 integrin MAb treatment than in those with the cell transfer with control MAb. Nude mice were infected with 10 cysts of the ME49 strain orally and treated with sulfadiazine for 3 weeks beginning 7 days after infection. Groups of animals received immune T cells (0.9 × 107 cells) pretreated with either anti-α4 integrin or a control MAb intravenously 4 weeks after infection. Thereafter, the mice received the MAb (200 or 250 μg) intraperitoneally every other day. Sulfadiazine treatment was discontinued 3 days after the cell transfer, and mRNA levels for the T cell markers and numbers of CD4+ and CD8+ T cells in the brains were determined by real-time RT-PCR and flow cytometry 3 days after discontinuation of sulfadiazine treatment (see Materials and Methods). *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard errors.
We also performed flow cytometric analyses to examine the numbers of CD4+ and CD8+ T cells recruited into the brains of the infected nude mice after they received immune T cells in combination with treatment with anti-α4 integrin or control MAb. The numbers of mononuclear cells purified from the brains of the former and the latter groups of mice were 0.93 × 106 cells/mouse and 1.24 × 106 cells/mouse, respectively. The percentages of CD4+ and CD8+ T cells in the mononuclear cell preparations were 14.0 ± 1.15 and 8.83 ± 1.33, respectively, in the mice treated with anti-α4 integrin MAb, and they were14.3 ± 0.81 and 15.5 ± 2.14, respectively, in those treated with control MAb. Figure 3B shows the numbers of CD4+ and CD8+ T cells that had migrated into the brains of the two groups of mice, calculated from these numbers. In agreement with the mRNA expression data, CD4+ and CD8+ T cells detected in the brain were significantly fewer in nude mice treated with anti-α4 MAb than in the brains of animals treated with control MAb (P < 0.001) (Fig. 3B). These results from SCID and nude mice indicate that the binding of lymphocyte α4β1 integrin to endothelial VCAM-1 on cerebral vessels plays a crucial role in recruitment of CD4+ and CD8+ immune T cells into the brains of mice during the early stage of reactivation of infection with T. gondii.
Expression of α4β1 integrin on the CD44high population of both CD4+ and CD8+ immune T cells in the spleens of BALB/c mice chronically infected with T. gondii.
We previously demonstrated that both CD4+ and CD8+ T cells that infiltrated into the brains of chronically infected BALB/c mice are a CD44high LFA-1high CD62Lneg effector/memory T cell population (33). To further confirm the importance of VCAM-1/α4β1 integrin interactions in recruiting immune T cells into the brains of chronically infected SCID and nude mice from the experiments described above, we examined the expression of α4β1 integrin on the surfaces of the CD44high population of splenic immune T cells of chronically infected BALB/c mice, which were transferred into infected recipients. A majority (71 to 79%) of the CD44high populations of both CD4+ and CD8+ immune T cells were positive for α4β1 integrin (Fig. 4). Interestingly, large portions (42 to 59%) of these CD44high T cell populations expressed high levels of this adhesion molecule, indicated as α4β1 integrinhigh in Fig. 4. As a comparison, we also examined the expression of α4β1 integrin on a CD44low population of splenic immune T cells, which do not enter the brains of infected mice (33). Many T cells in the CD44low population were also detected as α4β1 integrin+ (Fig. 4), but only low percentages (4.6 to 12%) of these T cells expressed high levels of the adhesion molecule (Fig. 4).
FIG 4.

Expression of high levels of α4β1 integrin on the CD44high population of CD4+ and CD8+ splenic immune T cells in BALB/c mice chronically infected with T. gondii. BALB/c mice were infected with 10 cysts of the ME49 strain orally, and their spleen cells were obtained during the chronic stage of infection for analysis of expression of CD44, α4 integrin, and β1 integrin on CD4+ and CD8+ T cells by flow cytometry (see Materials and Methods). Arrows in the top row indicate the upper right quadrant of the plots for α4β1 integrin+ and α4β1 integrinhigh populations.
Importance of α4 integrin for inducing IFN-γ-mediated protective immune responses in the brain during the early stage of reactivation of T. gondii infection.
Because IFN-γ production by T cells is required for the prevention of reactivation of T. gondii infection (29, 33) and because IFN-γ-mediated expression of NOS2 is important for preventing tachyzoite growth in the brain (39, 40), we next examined mRNA expression of IFN-γ and NOS2 in the brains of SCID mice that received immune T cells with anti-α4 integrin or control MAb treatment. Three days after discontinuation of sulfadiazine, the amounts of mRNAs for IFN-γ and NOS2 detected in the brains of SCID mice treated with anti-α4 integrin MAb were 5.7 and 9.2 times less than those in the brains of animals treated with control MAb (P < 0.05 and P < 0.01) (Fig. 5A). mRNA levels for both IFN-γ and NOS2 in control mice that had not received T cells were very low or close to detectable limits (Fig. 5A), indicating that the majority of IFN-γ and NOS2 expressed in the brains of animals with the T cell transfer were from or induced by T cells that had migrated into their brains. The amounts of mRNA for tachyzoite-specific SAG1 in the brains of the animals were low and close to detectable limits in most of the mice at this early stage of reactivation of infection, and there were no significant differences in their amounts between the experimental groups (data not shown).
FIG 5.

IFN-γ and NOS2 expression is markedly suppressed during the early stage of reactivation of T. gondii infection in the brains of SCID (A) and nude (B) mice that received immune T cells with anti-α4 integrin MAb treatment compared to animals with the cell transfer with control MAb. Mice were infected, treated with sulfadiazine, and received T cells with treatment with anti-α4 integrin or a control MAb as described in the legends to Fig. 2 and 3. Sulfadiazine treatment was discontinued 3 days after the cell transfer, and mRNA levels for IFN-γ and NOS2 were determined by real-time RT-PCR 3 days after discontinuation of sulfadiazine treatment (see Materials and Methods). *, P < 0.05; **, P < 0.01. The error bars indicate standard errors.
We also compared expression of IFN-γ and NOS2 in the brains of nude mice that had received immune T cells in combination with anti-α4 integrin or control MAb treatment. Three days after discontinuation of sulfadiazine treatment, the amounts of mRNAs for IFN-γ and NOS2 were 2.8 and 3.8 times less, respectively, in the brains of anti-α4 integrin MAb-treated mice than those in control MAb-treated animals (P < 0.05) (Fig. 5B). The levels of IFN-γ and NOS2 mRNAs in the brains of the former were as low as those detected in the brains of control nude mice that had not received T cells (Fig. 5B). The expression levels of IFN-γ and NOS2 in the control animals without T cell transfer (Fig. 5B) were higher than those in the control SCID mice without T cell transfer (Fig. 5A). Such differences between the control nude and SCID mice could be due to the presence of low numbers of thymus-independent T cells (Fig. 3B) only in the former animals. These results from both SCID and nude mice strongly suggest that recruitment of immune T cells mediated by VCAM-1/α4β1 integrin interaction is critical for inducing IFN-γ-mediated protective immune responses against T. gondii in the brain during the early stage of reactivation of infection with T. gondii.
Importance of α4 integrin for prevention of proliferation of tachyzoites in the brain during reactivation of T. gondii infection.
We next examined whether the inhibition of induction of T cell-dependent, IFN-γ-mediated protective immune responses in the brain by anti-α4 integrin MAb assists in proliferation of tachyzoites during reactivation of cerebral infection with T. gondii. For this purpose, we examined the amounts of mRNA for tachyzoite-specific SAG1 in the brains of infected nude mice 6 days after discontinuation of sulfadiazine treatment. Nude rather than SCID mice were used in this experiment because the former have B cells and we are able to examine the mechanisms of T cell recruitment into the brain to prevent tachyzoite growth in the presence of B cells. The amounts of SAG1 mRNA detected in the brains of nude mice that had received immune T cells along with treatment with anti-α4 integrin MAb were 17 times greater than those detected in the brains of animals that had received the T cells with control MAb (P < 0.01) (Fig. 6A). The amounts of SAG1 mRNA in the former were 15 times less than those detected in the brains of animals that did not receive T cells (P < 0.001) (Fig. 6A). These results indicate that T cell recruitment mediated by binding of lymphocyte α4β1 integrin to endothelial VCAM-1 is critical for preventing tachyzoite growth during reactivation of chronic infection with T. gondii in the brain, although T cell migration not mediated by the VCAM-1/α4β1 integrin interaction is also able to partially inhibit tachyzoite proliferation.
FIG 6.

(A to C) Greater levels of tachyzoite-specific SAG1 (A) and bradyzoite-specific BAG1 (C) mRNAs and higher ratios of the SAG1 mRNA level to the BAG1 mRNA level (B) are detected during the later stage of reactivation of T. gondii infection in the brains of nude mice that had received immune T cells with anti-α4 integrin MAb treatment than in animals that had received the T cells with a control MAb. (D and E) Greater numbers of foci associated with tachyzoites (D) and PV containing tachyzoites (E) are also detected in the brains of the former than of the latter. Mice were infected, treated with sulfadiazine, and received T cells (1.5 × 107 cells) with treatment with anti-α4 integrin or a control MAb as described in the legend to Fig. 3. Sulfadiazine treatment was discontinued 3 days after the cell transfer, and mRNA levels for SAG1 and BAG1 and numbers of tachyzoite-associated foci and PV were determined 6 days after discontinuation of sulfadiazine treatment (see Materials and Methods). *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard errors. (F and G) Representative inflammatory foci associated with tachyzoites in the brains of mice treated with control MAb (F) or anti-α4 integrin MAb (G). The sections were conterstained with hematoxylin to visualize the nuclei of the host cells. The arrows indicate representative PV containing tachyzoites.
In reactivation of chronic T. gondii infection, tachyzoite proliferation occurs after the rupture of cysts, followed by invasion of host cells by released bradyzoites and conversion of bradyzoites to tachyzoites. Therefore, taking the ratios of mRNA levels for tachyzoite-specific SAG1 to those for bradyzoite (cyst)-specific BAG1 in the brain can be a useful indicator of the occurrence of reactivation of the infection. In the brains of control mice that did not receive T cells, almost equal amounts of mRNAs for tachyzoite-specific SAG1 and bradyzoite-specific BAG1 were detected (Fig. 6B), indicating the occurrence of reactivation of chronic T. gondii infection. The SAG1/BAG1 mRNA ratios in the brains of nude mice that had received immune T cells with treatment with control MAb were markedly (7.5 times) less than those of the control animals without the T cell transfer (Fig. 6B) (P < 0.05), demonstrating that the reactivation of cerebral T. gondii infection was efficiently inhibited in the former animals. In contrast, the SAG1/BAG1 mRNA ratios in the brains of mice that had received immune T cells with treatment with anti-α4 integrin MAb were equivalent to those of the control animals without the T cell transfer (Fig. 6B). These results support the importance of T cell recruitment mediated by binding of lymphocyte α4β1 integrin to endothelial VCAM-1 for preventing reactivation of chronic T. gondii infection and cerebral tachyzoite growth.
When BAG1 mRNA levels in the brain were compared between the groups of nude mice, animals treated with anti-α4 integrin MAb had 53% greater amounts of the mRNA than those treated with control MAb (Fig. 6C). Because the former had 17 times greater levels of SAG1 mRNA than the latter, a small portion of tachyzoites proliferating in the brains of the former appear to have converted to bradyzoites to form new cysts.
We performed immunohistochemistry detecting T. gondii to further confirm the importance of VCAM-1/α4β1 integrin interactions for T cell recruitment into the brain to prevent reactivation of T. gondii infection. The numbers of foci associated with tachyzoites were 11 to 16 times less in the brains of nude mice that had received immune T cells with treatment with control MAb than in those of the animals that had received the T cells with treatment with anti-α4 integrin MAb and animals without the cell transfer (Fig. 6D) (P < 0.01 and P < 0.001, respectively). In addition, most of the foci associated with tachyzoites in the animals treated with control MAb contained only small numbers of tachyzoites, and such tachyzoites were associated with large numbers of inflammatory cells infiltrated into the areas (Fig. 6F). In contrast, those foci in the brains of animals treated with anti-α4 integrin MAb contained large numbers of tachyzoites, but only small numbers of inflammatory cells were detected in most of those areas (Fig. 6G). Such inflammatory foci in these animals were often associated with necrosis of brain tissue, as seen in Fig. 6G. In addition, the numbers of parasitophorous vacuoles containing tachyzoites detected in the brain sections were 20 times fewer in the brains of mice treated with control MAb than in those of the animals treated with anti-α4 integrin MAb (Fig. 6E) (P < 0.05). These observations strongly suggest that the VCAM-1/α4β1 integrin interaction is important for recruiting immune T cells that can infiltrate the areas where tachyzoites started proliferating to prevent reactivation of chronic cerebral infection with T. gondii.
The amounts of mRNA for T cell markers (CD3δ, CD4, and CD8β) were 46%, 47%, and 30% less in the nude mice with T cell transfer in combination with anti-α4 integrin MAb than in those treated with control MAb (P < 0.01, P < 0.001, and P < 0.05) (Fig. 7) 6 days after discontinuation of sulfadiazine. The former had markedly larger numbers of inflammatory foci associated with tachyzoites than the latter, as shown in Fig. 6D. Therefore, these results suggest that fewer T cells are associated with each of these inflammatory foci to prevent tachyzoite growth in the brains of mice treated with anti-α4 integrin MAb than in those treated with control MAb. Immunohistochemistry detecting T. gondii eventually demonstrated the presence of fewer inflammatory cells in the areas of foci associated with tachyzoites in the former than the latter (Fig. 6F and G), as described above. The differences in mRNAs for the T cell markers between these two groups were less than those observed at 3 days after discontinuation of sulfadiazine treatment (Fig. 3A). These results support the possibility that T cells can infiltrate into the brain through a mechanism(s) not mediated by α4 integrin during the later stage of reactivation of the infection. Local proliferation of T cells in the brain may also have contributed, in part, to the smaller differences in the amounts of mRNAs for these T cell markers between the two groups of mice.
FIG 7.

mRNA levels for T cell markers (CD3δ, CD4, and CD8β) are lower during the later stage of reactivation of T. gondii infection in the brains of nude mice that received immune T cells with anti-α4 integrin MAb treatment than in those with the cell transfer with control MAb. Mice were infected, treated with sulfadiazine, and received T cells with treatment with anti-α4 integrin or a control MAb as described in the legend to Fig. 5. mRNA levels for the T cell markers were measured by real-time RT-PCR 6 days after discontinuation of sulfadiazine treatment (see Materials and Methods). *, P < 0.05; **, P < 0.01; ***, P < 0.001. The error bars indicate standard errors.
In contrast to 3 days after discontinuation of sulfadiazine, the amounts of mRNA for IFN-γ and NOS2 were 40% and 56% greater in the brains of nude mice treated with anti-α4 integrin MAb than in those of animals treated with control MAb at the later stage of reactivation of T. gondii infection (P < 0.05 only for NOS2) (Fig. 8). Such upregulated expression of these molecules in anti-α4 integrin MAb-treated animals could be due to the presence of greater stimulation of both innate immune cells and T cells by markedly increased numbers of tachyzoites in the brains of these animals, as shown in Fig. 6A. The capability of innate immune cells to express upregulated IFN-γ and NOS2 in response to increased numbers of tachyzoites is obvious from the evidence of the presence of large amounts of mRNA for these molecules in the brains of control animals that received no T cells (Fig. 8), and it is consistent with our previous observations on production of IFN-γ by NK cells, microglia, and macrophages during reactivation of the infection (41). In addition, the animals that had received immune T cells with anti-α4 integrin MAb treatment had suppressed but still significant T cell infiltration into their brains at the later stage of reactivation of the infection, as described above and shown in Fig. 7. Those T cells in the presence of large numbers of tachyzoites could certainly contribute to the upregulated IFN-γ responses and induction of NOS2, most likely by microglia (42), in these animals. However, it is apparent that the upregulated expression of IFN-γ and NOS2 in the anti-α4 integrin MAb-treated mice in this later stage of reactivation of infection is not efficient in controlling the parasite, because large numbers of foci associated with tachyzoites and large amounts of tachyzoite-specific SAG1 mRNA were present in their brains (Fig. 6A and D). In addition, brain tissue necrosis had already developed in many of the areas of inflammatory foci associated with tachyzoites in these mice by this later stage of reactivation of infection, as shown in Fig. 6G. It appears that induction of increased expression of IFN-γ and NOS2 after large numbers of tachyzoites had already grown is not effective in controlling the parasite in the brain. These results strongly suggest that prompt recruitment of immune T cells mediated by VCAM-1/α4β1 integrin interaction to induce IFN-γ-mediated protective immune responses during the early stage of reactivation of infection is crucial for preventing proliferation of tachyzoites in the brain and TE, although T cells can infiltrate into the brain in an α4 integrin-independent manner during the later stage of infection.
FIG 8.
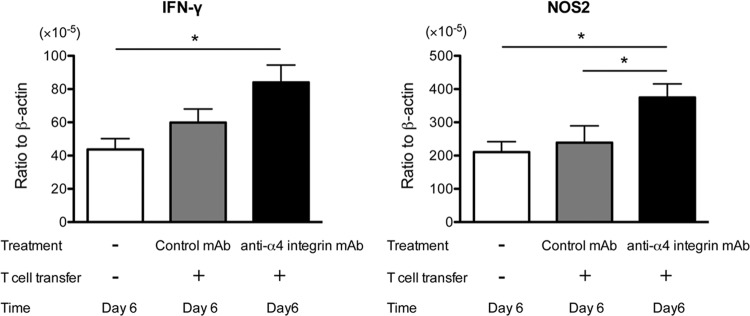
IFN-γ and NOS2 expression is increased during the later stage of reactivation of T. gondii infection in the brains of nude mice that received immune T cells with anti-α4 integrin MAb compared to animals with the cell transfer with control MAb. Mice were infected, treated with sulfadiazine, and received T cells with treatment with anti-α4 integrin or a control MAb as described in the legend to Fig. 5. mRNA levels for IFN-γ and NOS2 were measured by real-time RT-PCR 6 days after discontinuation of sulfadiazine treatment (see Materials and Methods). *, P < 0.05. The error bars indicate standard errors.
DISCUSSION
T cells and IFN-γ-mediated immune responses are essential for prevention of reactivation of cerebral infection with T. gondii (20, 22, 23). In the present study, we utilized a murine model of reactivation of chronic infection with the parasite and examined the adhesion molecules important for recruiting immune T cells into the brain to prevent reactivation of the cerebral infection. In this animal model, infected, sulfadiazine-treated SCID and nude mice received a systemic transfer of immune T cells, and reactivation of cerebral T. gondii infection was initiated by discontinuation of sulfadiazine treatment (20, 22, 23). Immunohistochemical study revealed that VCAM-1 and ICAM-1 are the two cerebrovascular endothelial adhesion molecules expressed in both wild-type and SCID mice during the chronic stage of infection. Although the cerebrovascular VCAM-1 expression following the infection is largely dependent on IFN-γ (33), microglia in SCID and nude mice and NK cells are able to produce this cytokine in response to T. gondii (41, 43, 44). IFN-γ production by these non-T cells appears to be sufficient to upregulate VCAM-1 expression on the cerebral vessels in infected SCID and nude mice. Therefore, the infected SCID and nude mice provided a suitable model to analyze the adhesion molecules important for recruiting the T cells into the brain to maintain the latency of chronic infection with T. gondii in the brain.
In the present study, we utilized inhibition of binding of lymphocyte α4β1 integrin to endothelial VCAM-1 on cerebral vessels by anti-α4 integrin MAb to analyze the role of interactions between these adhesion molecules in T cell recruitment and prevention of reactivation of chronic T. gondii infection in the brains of infected SCID and nude mice that received a systemic transfer of immune T cells. Flow cytometric analyses demonstrated that the numbers of CD4+ and CD8+ immune T cells that had migrated into the brain within 3 days after the initiation of reactivation of the cerebral infection were significantly lower in nude mice treated with anti-α4 integrin MAb than in animals treated with control MAb. These observations were consistent with the detection of markedly smaller amounts of mRNA for the T cell markers CD3δ, CD4, and CD8β in the brains of SCID and nude mice treated with anti-α4 integrin MAb than in those treated with control MAb at this early stage of reactivation of infection. Immunohistochemical studies also demonstrated the presence of large numbers of CD3+ T cells in the brain parenchyma of the SCID mice treated with control MAb, but not in the animals treated with anti-α4 integrin MAb. These results indicate that VCAM-1/α4β1 integrin interaction is crucial for recruitment of both CD4+ and CD8+ immune T cells into the brain during the early stage of reactivation of infection with T. gondii.
Our previous study showed that treatment of mice with the anti-α4 integrin MAb does not affect homing of systemically transferred lymphocytes labeled with tetramethyl rhodamine isocyanate (TRITC) to the spleen or peripheral lymph nodes but reduces their homing to bronchus- and gut-associated lymphoid tissues in short-term lymphocyte-homing assays (28). The blocking effects of the MAb resulted in increased numbers of the labeled lymphocytes present in the peripheral blood. Therefore, anti-α4 integrin would not have reduced the numbers of T cells in the peripheral blood available to infiltrate into the brain in the present study. Therefore, the effect of anti-α4 integrin MAb observed in the present study is the inhibition of recruitment of immune T cells from the periphery into the brain during reactivation of infection with T. gondii.
IFN-γ is essential to maintain the latency of chronic infection with T. gondii in the brain and to prevent TE (4, 20, 45). In vitro studies showed that IFN-γ activates microglia to prevent tachyzoite growth (46, 47), and the antimicrobial activity of the murine microglia is mediated by production of nitric oxide by NOS2 (42). Murine models also previously demonstrated the importance of NOS2 in controlling the parasite in the brain during the later stage of infection (39, 40). The present study demonstrated that 3 days after discontinuation of sulfadiazine treatment, IFN-γ and NOS2 mRNA levels were 3 to 9 times lower in the brains of SCID and nude mice that had received immune T cells with anti-α4 integrin MAb than in those that had received the T cells with control MAb. Since the expression levels of these two molecules in the brains of control mice that had received no T cells were equivalent to or lower than those in animals with T cell transfer in combination with anti-α4 integrin MAb, the impaired production of IFN-γ by T cells is most likely the major cause of the suppressed NOS2 expression in animals treated with anti-α4 integrin MAb. Therefore, VCAM-1/α4β1 integrin interaction is critical for promptly recruiting immune T cells capable of producing IFN-γ into the brain and inducing expression of NOS2 during the early stage of reactivation of cerebral T. gondii infection. To our knowledge, the present study provides the first evidence indicating the importance of VCAM-1/α4β1 integrin interaction for inducing IFN-γ-dependent protective immune responses against T. gondii in the brain.
At a later stage of reactivation of cerebral T. gondii infection (6 days after discontinuation of sulfadiazine treatment), 17 times greater amounts of mRNA for tachyzoite-specific SAG1 were detected in the brains of nude mice that had received immune T cells with anti-α4 integrin MAb than in animals that had received the cells with control MAb. The presence of large amounts of SAG1 mRNA in the former was associated with high SAG1/BAG1 mRNA ratios, indicating the occurrence of reactivation of cerebral T. gondii infection in these animals. In contrast, SAG1/BAG1 mRNA ratios remained low in the animals treated with control MAb. This is consistent with the presence of markedly greater numbers of foci associated with tachyzoites and greater numbers of parasitophorous vacuoles containing tachyzoites in the sections of the brains of mice treated with anti-α4 integrin MAb than in animals that had received the cells with control MAb. Thus, T cell recruitment into the brain through VCAM-1/α4β1 integrin interaction is crucial to effectively inhibit reactivation of chronic T. gondii infection and prevent cerebral tachyzoite growth.
Interestingly, there was more cerebral mRNA for IFN-γ and NOS2 in the animals treated with anti-α4 integrin MAb than in animals treated with control MAb at the later stage of reactivation of infection, probably due to greater stimulation of innate immune cells and T cells by large numbers of tachyzoites in the brains of the former animals. However, such upregulated expression of IFN-γ and NOS2 in the anti-α4 integrin MAb-treated mice does not seem to prevent pathological effects of tachyzoite growth, because inflammatory foci associated with large numbers of tachyzoites and tissue necrosis in the brain have already developed by this stage of reactivation of infection. Thus, it is most likely that quick recruitment of immune T cells mediated by VCAM-1/α4β1 integrin interaction into the brain and prompt induction of cerebral expression of IFN-γ and NOS2 during the early stage of reactivation of T. gondii infection are crucial for successful control of the parasite and prevention of TE. To our knowledge, the importance of T cell recruitment mediated by VCAM-1/α4β1 integrin interaction for prevention of reactivation of chronic T. gondii infection by induction of IFN-γ-dependent protective immunity has not been reported before.
The present study used BALB/c-background SCID and nude mice infected with a type II strain of T. gondii as a model of reactivation of chronic T. gondii infection. In contrast to BALB/c mice, which are genetically resistant to the infection and maintain the latency of chronic infection, C57BL/6 mice are genetically susceptible to the infection and develop progressive and ultimately fatal TE during the later stage of the infection. A previous study using C57BL/6 mice infected with a T. gondii strain genetically modified to secrete ovalbumin (OVA) and a transfer of OVA-specific T cell receptor transgenic mouse (OT-1) CD8+ T cells activated with OVA in vitro showed that α4β1 integrin is important for recruitment of the OVA-specific OT-1 CD8+ T cells into the brains of the animals and for suppressing parasite DNA levels during progressive TE (48). Another study using a susceptible strain of mice showed that neonatal inactivation of the VCAM-1 gene did not impair leukocyte recruitment into the brain after T. gondii infection (49). The reasons for the differences in the observations in these two studies in TE-susceptible strains of mice are unclear. It is possible that neonatal inactivation of VCAM-1 had resulted in expression of a molecule that is not usually expressed in wild-type mice to compensate for the absence of VCAM-1. It is also possible that T. gondii infection induces the expression of multiple adhesion molecules, including α4β1 integrin, in wild-type C57BL/6 mice and that these adhesion molecules have redundant roles in recruiting the T cells into the brains of infected animals, whereas adhesion molecules expressed on the OT-1 CD8+ T cells activated with OVA in vitro are limited to α4β1 integrin.
The present study demonstrated the importance of VCAM-1/α4β1 integrin interaction for recruitment of immune T cells into the brain to prevent reactivation of chronic cerebral infection with T. gondii. The importance of VCAM-1/α4β1 integrin interactions in recruitment of T cells has also been reported in cerebral infections with coronavirus and Listeria monocytogenes (50, 51), whereas the involvement of ICAM-1 was shown in T cell recruitment into the brain during infections with Sinbis and Theiler's viruses (52, 53). Therefore, it is important to elucidate the pathogen-specific mechanisms of T cell recruitment into the brain for controlling different infections. In the present study, we showed that treatment of T. gondii-infected mice with anti-α4 integrin MAb markedly inhibited recruitment of CD4+ and CD8+ immune T cells into their brains and induction of IFN-γ-dependent protective immune responses to prevent reactivation of cerebral infection with the parasite. Anti-α4 integrin humanized MAb (natalizumab) has been used for treatment of two diseases, multiple sclerosis and Crohn's disease (54, 55). Since 500 million to 2 billion people are estimated to be chronically infected with T. gondii worldwide, it is possible that individuals with chronic infection with the parasite will receive the anti-α4 integrin MAb for treatment of multiple sclerosis or Crohn's disease. It is possible that the anti-α4 integrin MAb intended to treat these diseases causes reactivation of T. gondii infection in the brains of the patients who have been chronically infected with the parasite. Although the occurrence of ocular toxoplasmosis during treatment of multiple sclerosis with natalizumab has recently been reported (56), occurrence of cerebral toxoplasmosis associated with the treatment has not been reported yet. It may be important to consider the possible occurrence of reactivation of cerebral T. gondii infection as a side effect of treatment with anti-α4 integrin MAb in patients with multiple sclerosis or Crohn's disease.
ACKNOWLEDGMENTS
We thank Jennifer Strange and Greg Bauman in the Flow Cytometry Core Facility for their technical assistance.
This work was supported in part by grants from the National Institutes of Health (AI078756, AI095032, and AI077887 to Y.S.) and a grant (08R-2047 to Y.S.) from the Stanley Medical Research Institute.
Footnotes
Published ahead of print 21 April 2014
REFERENCES
- 1.McCabe RE, Remington JS. 1990. Toxoplasma gondii, p 2090 In Mandell GL, Douglas RG, Bennett JE. (ed), Principles and practice of infectious diseases. Churchill Livingstone Inc., New York, NY [Google Scholar]
- 2.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240:516–518. 10.1126/science.3128869 [DOI] [PubMed] [Google Scholar]
- 3.Suzuki Y, Remington JS. 1990. The effect of anti-IFN-gamma antibody on the protective effect of Lyt-2+ immune T cells against toxoplasmosis in mice. J. Immunol. 144:1954–1956 [PubMed] [Google Scholar]
- 4.Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A. 1991. Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-gamma production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J. Immunol. 146:286–292 [PubMed] [Google Scholar]
- 5.Kang H, Remington JS, Suzuki Y. 2000. Decreased resistance of B cell-deficient mice to infection with Toxoplasma gondii despite unimpaired expression of IFN-gamma, TNF-alpha, and inducible nitric oxide synthase. J. Immunol. 164:2629–2634. 10.4049/jimmunol.164.5.2629 [DOI] [PubMed] [Google Scholar]
- 6.Frenkel JK, Taylor DW. 1982. Toxoplasmosis in immunoglobulin M-suppressed mice. Infect. Immun. 38:360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson LL, Sayles PC. 2002. Deficient humoral responses underlie susceptibility to Toxoplasma gondii in CD4-deficient mice. Infect. Immun. 70:185–191. 10.1128/IAI.70.1.185-191.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denkers EY, Gazzinelli RT. 1998. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 11:569–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill D, Dubey JP. 2002. Toxoplasma gondii: transmission, diagnosis and prevention. Clin. Microbiol. Infect. 8:634–640. 10.1046/j.1469-0691.2002.00485.x [DOI] [PubMed] [Google Scholar]
- 10.Boyer K, Marcinak J, McLeod R. 2007. Toxoplasma gondii (toxoplasmosis), p 1267–1288 In Long S, Pickering LK, Prober CG. (ed), Principles and practice of pediatric infectious diseases, 3rd ed. Churchill Livingstone, New York, NY [Google Scholar]
- 11.Israelski DM, Remington JS. 1993. Toxoplasmosis in the non-AIDS immunocompromised host. Curr. Clin. Top. Infect. Dis. 13:322–356 [PubMed] [Google Scholar]
- 12.Wong SY, Remington JS. 1994. Toxoplasmosis in the setting of AIDS, p 223–257 In Broder S, Mergan TC, Jr, Bolognesi D. (ed), Textbook of AIDS medicine. Williams & Wikins, Baltimore, MD [Google Scholar]
- 13.Howe DK, Sibley LD. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis. 172:1561–1566. 10.1093/infdis/172.6.1561 [DOI] [PubMed] [Google Scholar]
- 14.Grigg ME, Bonnefoy S, Hehl AB, Suzuki Y, Boothroyd JC. 2001. Success and virulence in Toxoplasma as the result of sexual recombination between two distinct ancestries. Science 294:161–165. 10.1126/science.1061888 [DOI] [PubMed] [Google Scholar]
- 15.Darde ML, Bouteille B, Pestre-Alexandre M. 1992. Isoenzyme analysis of 35 Toxoplasma gondii isolates and the biological and epidemiological implications. J. Parasitol. 78:786–794. 10.2307/3283305 [DOI] [PubMed] [Google Scholar]
- 16.Ajzenberg D, Cogne N, Paris L, Bessieres MH, Thulliez P, Filisetti D, Pelloux H, Marty P, Darde ML. 2002. Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis, and correlation with clinical findings. J. Infect. Dis. 186:684–689. 10.1086/342663 [DOI] [PubMed] [Google Scholar]
- 17.Howe DK, Honore S, Derouin F, Sibley LD. 1997. Determination of genotypes of Toxoplasma gondii strains isolated from patients with toxoplasmosis. J. Clin. Microbiol. 35:1411–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki Y, Joh K, Orellana MA, Conley FK, Remington JS. 1991. A gene(s) within the H-2D region determines the development of toxoplasmic encephalitis in mice. Immunology 74:732–739 [PMC free article] [PubMed] [Google Scholar]
- 19.Brown CR, Hunter CA, Estes RG, Beckmann E, Forman J, David C, Remington JS, McLeod R. 1995. Definitive identification of a gene that confers resistance against Toxoplasma cyst burden and encephalitis. Immunology 85:419–428 [PMC free article] [PubMed] [Google Scholar]
- 20.Kang H, Suzuki Y. 2001. Requirement of non-T cells that produce gamma interferon for prevention of reactivation of Toxoplasma gondii infection in the brain. Infect. Immun. 69:2920–2927. 10.1128/IAI.69.5.2920-2927.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki Y, Wang X, Jortner BS, Payne L, Ni Y, Michie SA, Xu B, Kudo T, Perkins S. 2010. Removal of Toxoplasma gondii cysts from the brain by perforin-mediated activity of CD8+ T cells. Am. J. Pathol. 176:1607–1613. 10.2353/ajpath.2010.090825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang H, Liesenfeld O, Remington JS, Claflin J, Wang X, Suzuki Y. 2003. TCR V beta 8+ T cells prevent development of toxoplasmic encephalitis in BALB/c mice genetically resistant to the disease. J. Immunol. 170:4254–4259. 10.4049/jimmunol.170.8.4254 [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Claflin J, Kang H, Suzuki Y. 2005. Importance of CD8(+)Vbeta8(+) T Cells in IFN-gamma-mediated prevention of toxoplasmic encephalitis in genetically resistant BALB/c mice. J. Interferon Cytokine Res. 25:338–344. 10.1089/jir.2005.25.338 [DOI] [PubMed] [Google Scholar]
- 24.Hickey WF. 2001. Basic principles of immunological surveillance of the normal central nervous system. Glia 36:118–124. 10.1002/glia.1101 [DOI] [PubMed] [Google Scholar]
- 25.Suzuki Y, Orellana MA, Wong SY, Conley FK, Remington JS. 1993. Susceptibility to chronic infection with Toxoplasma gondii does not correlate with susceptibility to acute infection in mice. Infect. Immun. 61:2284–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikulowska-Mennis A, Xu B, Berberian JM, Michie SA. 2001. Lymphocyte migration to inflamed lacrimal glands is mediated by vascular cell adhesion molecule-1/alpha(4)beta(1) integrin, peripheral node addressin/l-selectin, and lymphocyte function-associated antigen-1 adhesion pathways. Am. J. Pathol. 159:671–681. 10.1016/S0002-9440(10)61738-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson GG, Mikulowska A, Butcher EC, McEvoy LM, Michie SA. 1999. Anti-CD43 monoclonal antibody L11 blocks migration of T cells to inflamed pancreatic islets and prevents development of diabetes in nonobese diabetic mice. J. Immunol. 163:5678–5685 [PubMed] [Google Scholar]
- 28.Xu B, Wagner N, Pham LN, Magno V, Shan Z, Butcher EC, Michie SA. 2003. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd, alpha4beta1 integrin/VCAM-1, and LFA-1 adhesion pathways. J. Exp. Med. 197:1255–1267. 10.1084/jem.20010685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Kang H, Kikuchi T, Suzuki Y. 2004. Gamma interferon production, but not perforin-mediated cytolytic activity, of T cells is required for prevention of toxoplasmic encephalitis in BALB/c mice genetically resistant to the disease. Infect. Immun. 72:4432–4438. 10.1128/IAI.72.8.4432-4438.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki Y, Rani S, Liesenfeld O, Kojima T, Lim S, Nguyen TA, Dalrymple SA, Murray R, Remington JS. 1997. Impaired resistance to the development of toxoplasmic encephalitis in interleukin-6-deficient mice. Infect. Immun. 65:2339–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh J, Graniello C, Ni Y, Payne L, Sa Q, Hester J, Shelton BJ, Suzuki Y. 2010. Toxoplasma IgG and IgA, but not IgM, antibody titers increase in sera of immunocompetent mice in association with proliferation of tachyzoites in the brain during the chronic stage of infection. Microbes Infect. 12:1252–1257. 10.1016/j.micinf.2010.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hester J, Mullins J, Sa Q, Payne L, Mercier C, Cesbron-Delauw MF, Suzuki Y. 2012. Toxoplasma gondii antigens recognized by IgG antibodies differ between mice with and without active proliferation of tachyzoites in the brain during the chronic stage of infection. Infect. Immun. 80:3611–3620. 10.1128/IAI.00604-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Michie SA, Xu B, Suzuki Y. 2007. Importance of IFN-gamma-mediated expression of endothelial VCAM-1 on recruitment of CD8+ T cells into the brain during chronic infection with Toxoplasma gondii. J. Interferon Cytokine Res. 27:329–338. 10.1089/jir.2006.0154 [DOI] [PubMed] [Google Scholar]
- 34.Askarova S, Sun Z, Sun GY, Meininger GA, Lee JC. 2013. Amyloid-beta peptide on sialyl-Lewis(X)-selectin-mediated membrane tether mechanics at the cerebral endothelial cell surface. PLoS One 8:e60972. 10.1371/journal.pone.0060972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrithers MD, Visintin I, Kang SJ, Janeway CA., Jr 2000. Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain 123:1092–1101. 10.1093/brain/123.6.1092 [DOI] [PubMed] [Google Scholar]
- 36.Kerfoot SM, Kubes P. 2002. Overlapping roles of P-selectin and alpha 4 integrin to recruit leukocytes to the central nervous system in experimental autoimmune encephalomyelitis. J. Immunol. 169:1000–1006. 10.4049/jimmunol.169.2.1000 [DOI] [PubMed] [Google Scholar]
- 37.Gurish MF, Tao H, Abonia JP, Arya A, Friend DS, Parker CM, Austen KF. 2001. Intestinal mast cell progenitors require CD49dbeta7 (alpha4beta7 integrin) for tissue-specific homing. J. Exp. Med. 194:1243–1252. 10.1084/jem.194.9.1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mori Y, Shimizu N, Dallas M, Niewolna M, Story B, Williams PJ, Mundy GR, Yoneda T. 2004. Anti-alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 104:2149–2154. 10.1182/blood-2004-01-0236 [DOI] [PubMed] [Google Scholar]
- 39.Collazo CM, Yap GS, Hieny S, Caspar P, Feng CG, Taylor GA, Sher A. 2002. The function of gamma interferon-inducible GTP-binding protein IGTP in host resistance to Toxoplasma gondii is Stat1 dependent and requires expression in both hematopoietic and nonhematopoietic cellular compartments. Infect. Immun. 70:6933–6939. 10.1128/IAI.70.12.6933-6939.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scharton-Kersten TM, Yap G, Magram J, Sher A. 1997. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185:1261–1273. 10.1084/jem.185.7.1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki Y, Claflin J, Wang X, Lengi A, Kikuchi T. 2005. Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. Int. J. Parasitol. 35:83–90. 10.1016/j.ijpara.2004.10.020 [DOI] [PubMed] [Google Scholar]
- 42.Chao CC, Anderson WR, Hu S, Gekker G, Martella A, Peterson PK. 1993. Activated microglia inhibit multiplication of Toxoplasma gondii via a nitric oxide mechanism. Clin. Immunol. Immunopathol. 67:178–183. 10.1006/clin.1993.1062 [DOI] [PubMed] [Google Scholar]
- 43.Hunter CA, Subauste CS, Van Cleave VH, Remington JS. 1994. Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect. Immun. 62:2818–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sher A, Oswald IP, Hieny S, Gazzinelli RT. 1993. Toxoplasma gondii induces a T-independent IFN-gamma response in natural killer cells that requires both adherent accessory cells and tumor necrosis factor-alpha. J. Immunol. 150:3982–3989 [PubMed] [Google Scholar]
- 45.Suzuki Y, Conley FK, Remington JS. 1989. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J. Immunol. 143:2045–2050 [PubMed] [Google Scholar]
- 46.Chao CC, Gekker G, Hu S, Peterson PK. 1994. Human microglial cell defense against Toxoplasma gondii. The role of cytokines. J. Immunol. 152:1246–1252 [PubMed] [Google Scholar]
- 47.Chao CC, Hu S, Gekker G, Novick WJ, Jr, Remington JS, Peterson PK. 1993. Effects of cytokines on multiplication of Toxoplasma gondii in microglial cells. J. Immunol. 150:3404–3410 [PubMed] [Google Scholar]
- 48.Wilson EH, Harris TH, Mrass P, John B, Tait ED, Wu GF, Pepper M, Wherry EJ, Dzierzinski F, Roos D, Haydon PG, Laufer TM, Weninger W, Hunter CA. 2009. Behavior of parasite-specific effector CD8+ T cells in the brain and visualization of a kinesis-associated system of reticular fibers. Immunity 30:300–311. 10.1016/j.immuni.2008.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deckert M, Lutjen S, Leuker CE, Kwok LY, Strack A, Muller W, Wagner N, Schluter D. 2003. Mice with neonatally induced inactivation of the vascular cell adhesion molecule-1 fail to control the parasite in Toxoplasma encephalitis. Eur. J. Immunol. 33:1418–1428. 10.1002/eji.200322826 [DOI] [PubMed] [Google Scholar]
- 50.Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonniere L, Poirier J, Duquette P, Talbot PJ, Arbour N, Prat A. 2011. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 134:3560–3577. 10.1093/brain/awr268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young KG, Maclean S, Dudani R, Krishnan L, Sad S. 2011. CD8+ T cells primed in the periphery provide time-bound immune-surveillance to the central nervous system. J. Immunol. 187:1192–1200. 10.4049/jimmunol.1100695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Irani DN, Griffin DE. 1996. Regulation of lymphocyte homing into the brain during viral encephalitis at various stages of infection. J. Immunol. 156:3850–3857 [PubMed] [Google Scholar]
- 53.Njenga MK, Marques C, Rodriguez M. 2004. The role of cellular immune response in Theiler's virus-induced central nervous system demyelination. J. Neuroimmunol. 147:73–77. 10.1016/j.jneuroim.2003.10.042 [DOI] [PubMed] [Google Scholar]
- 54.Fernandez O. 2013. Best practice in the use of natalizumab in multiple sclerosis. Ther. Adv. Neurol. Disord. 6:69–79. 10.1177/1756285612470401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLean LP, Shea-Donohue T, Cross RK. 2012. Vedolizumab for the treatment of ulcerative colitis and Crohn's disease. Immunotherapy 4:883–898. 10.2217/imt.12.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zecca C, Nessi F, Bernasconi E, Gobbi C. 2009. Ocular toxoplasmosis during natalizumab treatment. Neurology 73:1418–1419. 10.1212/WNL.0b013e3181bd114f [DOI] [PubMed] [Google Scholar]