

Abstract
ALOX12 is a gene encoding arachidonate 12-lipoxygenase (12-LOX), a member of a nonheme lipoxygenase family of dioxygenases. ALOX12 catalyzes the addition of oxygen to arachidonic acid, producing 12-hydroperoxyeicosatetraenoic acid (12-HPETE), which can be reduced to the eicosanoid 12-HETE (12-hydroxyeicosatetraenoic acid). 12-HETE acts in diverse cellular processes, including catecholamine synthesis, vasoconstriction, neuronal function, and inflammation. Consistent with effects on these fundamental mechanisms, allelic variants of ALOX12 are associated with diseases including schizophrenia, atherosclerosis, and cancers, but the mechanisms have not been defined. Toxoplasma gondii is an apicomplexan parasite that causes morbidity and mortality and stimulates an innate and adaptive immune inflammatory reaction. Recently, it has been shown that a gene region known as Toxo1 is critical for susceptibility or resistance to T. gondii infection in rats. An orthologous gene region with ALOX12 centromeric is also present in humans. Here we report that the human ALOX12 gene has susceptibility alleles for human congenital toxoplasmosis (rs6502997 [P, <0.000309], rs312462 [P, <0.028499], rs6502998 [P, <0.029794], and rs434473 [P, <0.038516]). A human monocytic cell line was genetically engineered using lentivirus RNA interference to knock down ALOX12. In ALOX12 knockdown cells, ALOX12 RNA expression decreased and levels of the ALOX12 substrate, arachidonic acid, increased. ALOX12 knockdown attenuated the progression of T. gondii infection and resulted in greater parasite burdens but decreased consequent late cell death of the human monocytic cell line. These findings suggest that ALOX12 influences host responses to T. gondii infection in human cells. ALOX12 has been shown in other studies to be important in numerous diseases. Here we demonstrate the critical role ALOX12 plays in T. gondii infection in humans.
INTRODUCTION
Toxoplasma gondii is an obligately intracellular apicomplexan parasite that is capable of infecting a wide range of vertebrate hosts, including humans. Infection typically occurs after ingestion of T. gondii cysts from the tissues of infected animals or exposure to oocysts excreted in the feces of cats. Approximately one-third of the world's population is seropositive, indicating that they are infected with T. gondii (1). Healthy individuals who are infected with T. gondii have a chronic, lifelong infection that is generally asymptomatic and is characterized by the formation of dormant bradyzoites in cysts in the tissues. However, in immunocompromised or congenitally infected individuals, toxoplasmosis can develop into an extremely severe disease. In these individuals, T. gondii infection can cause encephalitis, myocarditis, and severe eye damage (2).
Recently, a region of the rat genome, named Toxo1, has been shown to be associated with resistance to T. gondii (3, 4). Compared to rats of the susceptible Brown Norway (BN) strain, LEW (Lewis) strain rats are extremely resistant to T. gondii infection: few to no parasites are found postinoculation, very few encysted bradyzoites are found, no antibody is produced, and there is no transmission to their pups. Reciprocal LEW × BN lines congenic for Toxo1 have allowed for the mapping of the genes responsible for the robust resistance phenotype to a region on rat chromosome 10 (4). These studies further demonstrated the central role played by macrophages in the immune defense against T. gondii, known for humans (5) and clearly seen in rat macrophages (3, 4). Rats with the LEW Toxo1 region were able to control the proliferation of T. gondii within parasitophorous vacuoles and significantly inhibited the parasite growth rate in peritoneal macrophages (3).
Following this discovery, we noted that an orthologous TOXO1 region is present in humans, on human chromosome 17. Many of the genes within the TOXO1 gene region are cell death genes, including the gene at the top of the region, ALOX12. ALOX12 encodes the enzyme arachidonate 12-lipoxygenase (12-LOX). 12-LOX (ALOX12) is a member of a family of dioxygenases that are involved in the metabolism of fatty acids into hydroperoxides (6). ALOX12 specifically adds molecular oxygen to arachidonic acid, leading to the production of the eicosanoid 12-HETE (12-hydroxyeicosatetraenoic acid). 12-HETE is an important signaling molecule that has been shown to play a role in everything from vasoconstriction to catecholamine synthesis, inflammation, and immune cell recruitment (6–8).
We hypothesized that in the pathogenesis of T. gondii infection in humans, the ALOX12 gene might play a significant role that could be demonstrated by finding susceptibility alleles of ALOX12 for human congenital toxoplasmosis. Our cohort in the National Collaborative Chicago-Based Congenital Toxoplasmosis Study (NCCCTS) has been a powerful tool for identifying such susceptibility alleles (9). Thus, in the present study, we utilized the same methodology. We conducted transmission disequilibrium testing (TDT) to determine whether certain alleles within the human ALOX12 gene are associated with congenital toxoplasmosis in families in this cohort. In addition to genetic analysis, we also attempted to understand the role ALOX12 plays during T. gondii infection in human monocytic cells by studying the effects of silencing ALOX12 gene expression in a human monocytic cell line by RNA interference (RNAi). We found that ALOX12 has susceptibility alleles and that silencing of ALOX12 increases arachidonic acid levels in human monocytic cells and, concomitantly, leads to the progression of T. gondii infection in those cells.
MATERIALS AND METHODS
Patient cohort and genotyping of ALOX12.
We used samples from patient-parent trios (a congenitally infected individual and his/her biological parents) from the National Collaborative Chicago-Based Congenital Toxoplasmosis Study (NCCCTS). We extracted DNA from peripheral blood mononuclear cells (PBMC) obtained from 149 patient-parent groups that were genotyped at 12 single nucleotide polymorphism tags (tag-SNPs) throughout the ALOX12 gene. As in earlier studies (10), tag-SNPs were then selected from the International HapMap Project, release 21 (http://www.hapmap.org). This was done using a 10-kb flanking sequence on either side of the ALOX12 gene. A minor allele frequency (MAF) cutoff of 5% in Utah residents with Northern and Western European ancestry (CEU) and an r2 threshold of 0.8 were used. Tag-SNPs were selected using the Tagger tool in the Haploview program. UNPHASED (https://sites.google.com/site/fdudbridge/software/unphased-2-404) was used for the allelic association analysis of 124 infected children in the NCCCTS cohort who had a confirmed presentation of clinical disease involving the eyes and/or brain.
Plasmid constructs.
Short hairpin RNA (shRNA) sequences were designed for the human ALOX12 gene coding sequence (GenBank accession number NM_000697) as well as for the Salmonella enterica tetracycline repressor (TetR) gene coding sequence (GenBank accession number NC_006856). The sense and antisense shRNA sequences for the ALOX12 coding sequence were 5′-caccAAAGCTGTGCTAAACCAATTCCGAACAGAcgaaTCTGTTCGGAATTGGTTTAGCACAGC-3′ and 5′-aaaaGCTGTGCTAAACCAATTCCGAACAGAttcgTCTGTTCGGAATTGGTTTAGCACAGCTTT-3′. Similarly, the sense and antisense shRNA sequences for the TetR coding sequence were 5′-caccAACGGCCGACGCGCAGCTTCGCTTCCTCTGcgaaCAGAGGAAGCGAAGCTGCGCGTCGGCCGTA-3′ and 5′-aaaaTACGGCCGACGCGCAGCTTCGCTTCCTCTGttcgCAGAGGAAGCGAAGCTGCGCGTCGGCCGTT-3′. In these sequences, the first four nucleotides (shown in lowercase) allow for directional cloning, the target sequence is shown in boldface, the lowercase sequence in the middle creates the loop formation, and underlining indicates the antisense sequence of the target sequence. The target sequence for ALOX12 was bp 1870 to 1889, while for TetR, the target sequence was bp 3360 to 3389 of the coding sequence. Once the ALOX12 and TetR shRNAs were designed, the double-stranded shRNAs were cloned into a Gateway-adapted entry vector, pENTR.H1/TO (Invitrogen), by annealing and directionally ligating the sense and antisense shRNA sequences into the entry vector, according to the instructions in the manufacturer's user manual (11). The recombinant plasmids were sequenced after sufficient propagation to ensure that the shRNA insert was successful. Once confirmed, the ALOX12 and TetR shRNAs were transferred into expression vector pLenti4/Block-iT-DEST through an LR recombination reaction (12). The newly designed recombinant plasmids were propagated in Escherichia coli and were then extracted and sequenced to ensure that the inserted shRNAs were accurate.
Generation of lentiviruses expressing ALOX12 and TetR.
Stocks of lentiviruses expressing ALOX12 or TetR shRNA were produced by transfecting the pLenti4/Block-iT-DEST expression vector carrying ALOX12 or TetR shRNA into 293FT cells, along with the optimized ViraPower packaging mix, which contains plasmids pLP1, pLO2, and pLO/VSVG to help facilitate viral packaging, according to the manufacturer's instructions (Invitrogen) (11). The transfected cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM), containing 10% fetal calf serum, 2 mM l-glutamine, 0.1 mM MEM nonessential amino acids, 1 mM sodium pyruvate, and 1% penicillin-streptomycin, for 72 h. The cell culture was then centrifuged, and the supernatant, containing the lentivirus, was harvested and stored in aliquots at −70°C. Lastly, titrations to determine the viral yield were performed according to the instructions in the manual.
Establishment of human monocytic cell lines with stable expression of ALOX12 or TetR shRNA.
In vitro culture-adapted human monocytes (MonoMac6 [MM6] cells) were used to create human monocytic cell lines stably expressing ALOX12 or TetR shRNA. MonoMac6 cells were seeded at a density of 4 × 105/well in a 24-well plate with RPMI medium supplemented with 10% fetal calf serum, 2.05 mM l-glutamine, 1× nonessential amino acids (Sigma), OPI medium supplement (Hybri-Max; Sigma), and 1% penicillin-streptomycin. To each cell culture, 250 μl of a lentiviral stock expressing either ALOX12 or TetR shRNA (3 × 106 transducing units [TU]/ml) was added, followed by gentle mixing and maintenance at 37°C under 5% CO2. In addition, untransduced, wild-type MonoMac6 cells were cultured under the same conditions as a control. To select for transduced cells, 100 μg/ml of Zeocin (Invitrogen) was added to the cultures after 24 h of incubation. The cell culture medium with Zeocin was changed every 2 days for approximately 3 weeks, after which the only cells that propagated were those resistant to Zeocin (13).
Analysis of RNAi-based knockdown of ALOX12 gene transcription.
To determine the efficiency of lentivirus-expressed ALOX12 shRNA at silencing ALOX12 gene expression in human monocytic cells, we used quantitative real-time PCR (qRT-PCR) to measure and compare ALOX12 gene expression in ALOX12 knockdown and wild-type MonoMac6 cells. First, wild-type MonoMac6 cells and MonoMac6 cells engineered to express either ALOX12 shRNA or TetR shRNA were seeded in 12-well plates at 1 × 105 cells/well. After 72 h, the cells were collected by centrifugation. Total RNA was extracted from each cell culture using TRIzol reagent, and to ensure that no residual genomic DNA lingered in the RNA, each sample was treated with DNase I. Exactly 2 μg of each DNase I-treated RNA sample was subsequently reverse transcribed into first-strand cDNA using the Invitrogen SuperScript III First-Strand Synthesis SuperMix kit. To generate standards for real-time PCR quantification of ALOX12 and human actin gene transcripts, we performed conventional PCR on the cDNA using primer sets specific for amplifying ∼300-bp fragments of ALOX12 or the actin gene (forward and reverse primers were ALOX12-F/ALOX12-R and ACTIN-F/ACTIN-R, respectively) (Table 1). After electrophoretic separation, both the ALOX12 and actin fragments were extracted and were used to create serial 10-fold dilutions so as to generate a standard curve for the qRT-PCR analysis. The real-time PCR mixture was made up of 1 μl of the primer mixture (500 nM each), 10 μl of SsoFast EvaGreen supermix (Bio-Rad), and 1 μl of the cDNA template, and the final volume was made up to 20 μl with RNase/DNase-free water. Cycling was performed using the CFX96 real-time system (Bio-Rad). The cycling conditions consisted of initial denaturation for 30 s at 95°C; 40 cycles of 95°C for 5 s, 57°C for 5 s, and 60°C for 10 s; and finally cooling to 40°C. The software for the CFX96 real-time system generated the transcript quantities by using the standard curves generated from the serial dilutions.
TABLE 1.
Primers used
| Primer | Sequence |
|---|---|
| ALOX12-F | 5′-AGAAAAGTTGACTAGTCCAGTGTGGTGAA-3′ |
| ALOX12-R | 5′-AAAAGCTGTGCTAAACCAATTCCGAACAGATTCTCA-3′ |
| ACTIN-F | 5′-CTCTTCCAGCCCTCCTTCTT-3′ |
| ACTIN-R | 5′-GACGTTCCGTCAGATCCT-3′ |
| IL-1β-F | 5′-TGGACAAGCTGAGGAAGATGCTGGT-3′ |
| IL-1β-R | 5′-AGGACATGGAGAACACCACTTGTTGCT-3′ |
| IL-6-F | 5′-AGAAGATTCCAAAGATGTA-3′ |
| IL-6-R | 5′-TCACTACTCTCAAATCTGTT-3′ |
| TNFα-F | 5′-TCTTCTCCTTCCTGATCGTGGCA-3′ |
| TNFα-R | 5′-ACCACCAGCTGGTTATCTCTCA-3′ |
| Casp-F | 5′-AGCTATGCCCACATCCTCAGGCTCAGA-3′ |
| Casp-R | 5′-AAATGCCTCCAGCTCTGTAGTCATGT-3′ |
Cell viability assay.
To determine if ALOX12 gene knockdown affects the viability of human monocytic cells when they are infected with the virulent type I T. gondii strain RH in culture, wild-type MonoMac6 cells and MonoMac6 cells stably expressing ALOX12 shRNA or TetR gene shRNA were seeded at a density of 104 cells/well in 96-well plates and were maintained in 200 μl of supplemented RPMI medium. Type I T. gondii strain RH parasites were added to the cell cultures at a multiplicity of infection (MOI) of 1:2 (parasites to monocytes), while uninfected cell cultures were maintained simultaneously as a control. The cells were cultured for 5 days, and at each day (days 0, 1, 2, 3, and 4), the viability of cells in each culture was quantified using the Cell Proliferation Reagent WST-1 colorimetric assay (Roche). For the assay, 10 μl of the WST-1 reagent was added to each well; the contents of each well were mixed; the 96-well plates were wrapped in aluminum foil; and the cultures were incubated at 37°C under 5% CO2 for 1 h. Metabolically active or viable cells produce a formazan dye, for which the absorbance was read using a scanning multiwell spectrophotometer at a wavelength of 420 nm (SpectraMax 250; Molecular Devices).
Preparation and microscopic analysis of Giemsa-stained MonoMac6 cytocentrifuge preparations.
To visualize the effect of silencing of ALOX12 gene expression through RNA interference on the viability of monocytic cells cultured with or without parasites, MonoMac6 cells genetically engineered to express ALOX12 shRNA or TetR shRNA, along with wild-type Monomac6 cells, were seeded at 105/well in 24-well plates. T. gondii strain RH parasites were added to half of the wells at an MOI of 1:4 (parasites to monocytes), while the remaining cells were cultured without parasites. At days 0, 2, 4, and 6 postinfection, the cultures were gently mixed by pipetting up and down, and 50 μl of the suspension was applied to a glass slide. The slides were air dried, fixed in amino acridine, and stained with Giemsa stain. The cells were examined by light microscopy using a 20× objective. To replenish the medium, after every 2 days of continuous culture, half of the culture medium in each well was carefully aspirated out (without disturbing the cells at the bottom of the culture) and was replaced with an equal volume of fresh medium.
Preparation and fluorescence analysis of T. gondii replication in MonoMac6 cells.
In order to measure the replication of T. gondii in monocytic cells, wild-type MonoMac6 cells and MonoMac6 cells genetically modified for the stable knockdown of the ALOX12 or TetR gene were seeded in 24-well plates at a density of 105/well and were cultured with or without T. gondii parasites engineered to express yellow fluorescent protein (YFP) in the cytoplasm at an MOI of 1:2 (parasites to monocytes). At different time intervals (days 0, 1, 2, 3, and 4), the cultures were observed by light microscopy and were photographed. Simultaneously, the three types of cells (wild-type, ALOX12 knockdown, and TetR knockdown MonoMac6 cells) were seeded in triplicate in 96-well plates at a density of 105/well and were cultured with or without YFP-expressing parasites at an MOI of 1:2 (parasites to monocytes). At days 0, 1, 2, 3, and 4 postinfection, fluorescence was measured using a scanning multiwell spectrophotometer (SpectraMax 250; Molecular Devices) at wavelengths of 514 and 540 nm.
Quantitation of arachidonic acid.
Arachidonic acid levels in wild-type and ALOX12 or TetR knockdown MonoMac6 cells were measured by mass spectrometry (Kansas Lipidomics Research Center). Wild-type MonoMac6 cells, ALOX12 knockdown cells, and TetR knockdown cells were seeded in 1-in-diameter glass dishes at a density of 106 cells with or without T. gondii at an MOI of 4:1 (parasites to cells) for 24 h, and samples were collected and were frozen until analysis. Samples were analyzed using an API 4000 mass spectrometer (Applied Biosystems, Foster City, CA). Samples in chloroform-methanol-300 mM ammonium acetate in water (66.5:30:3.5) were introduced by direct infusion to an electrospray ionization source at 30 μl per min. Samples were analyzed by mass spectrometry in negative mode. Arachidonic acid was quantified by comparison of the intensity of the mass spectral peak for arachidonic acid (20:4) ([M − H]−) with the intensity of the spectral peak for the internal standard, pentadecanoic acid (15:0) (1.0 nmol added) ([M − H]−).
Quantitation of expression of cytokines and caspase-1.
To assess the effect of ALOX12 knockdown in MonoMac6 cells on the expression of the cytokines interleukin 6 (IL-6), IL-1β, tumor necrosis factor alpha (TNF-α), and caspase-1, triplicate sets of wild-type MonoMac6 cells and MonoMac6 cells engineered to express either ALOX12 shRNA or TetR shRNA were cultured with or without parasites at an MOI of 1:4 for 36 h. The cells were harvested, and total RNA was extracted for transcript analysis. Total RNA was extracted using the TRIzol reagent, followed by first-strand cDNA synthesis. Quantitative real-time PCR was performed on each sample of cDNA to determine the transcript levels. The primers used were designed to amplify a fragment of approximately 300 bp for the coding sequences of IL-6, IL-1β, TNF-α, and caspase-1 (GenBank accession numbers M54894, NM_000576, NM_000594, and NM_033294, respectively). The primer sets used are shown in Table 1. The respective gene fragments were amplified by conventional PCR from the cDNA and were sequenced to confirm their identity. The real-time PCR mixture and cycling conditions were essentially those described above. The transcript levels of the human actin gene (GenBank accession number NM_001100) were determined for each sample and were used for normalization.
Statistical analysis.
In the genetics study, allelic association was analyzed using a conventional transmission disequilibrium test (TDT), and P values were calculated using Haploview (http://www.broadinstitute.org/haploview), where P values less than or equal to 0.05 were considered significant for association with disease. Statistical analyses for all in vitro assays were performed using a 2-tailed Student t test. Due to evidence of nonnormality in arachidonic acid levels, nonparametric tests were used. Since the levels in the MM6 (wild-type) and TetR knockdown groups were similar, they were first combined and then compared to the levels in the ALOX12 knockdown group. To account for infection status, a stratified Wilcoxon rank-sum test was performed with infection status as the stratification factor.
Modeling of human 12-lipoxygenase.
A model for the structure of human 12-lipoxygenase was generated using the PHYRE2 Web-based modeling tool (http://www.sbg.bio.ic.ac.uk/phyre2/).
RESULTS
Congenital toxoplasmosis is associated with ALOX12 allelic variants in humans.
The association of ALOX12 allelic variants with congenital toxoplasmosis was shown by genotyping 12 tag-SNPs that were selected from the human ALOX12 genes of patient-parent trios from the NCCCTS. Transmission disequilibrium testing (TDT) was carried out to show overtransmission of ALOX12 alleles in patient-parent trios (Fig. 1). A total of 124 congenitally infected children, many with clinical presentation of ocular and/or brain damage caused by T. gondii, were tested at an r2 threshold of 0.8. Ultimately, SNPs that had a call rate of >90% and were in Hardy-Weinberg equilibrium in the parents were further analyzed.
FIG 1.

Analysis of ALOX12 SNPs. (Upper diagram) Positions of genotyped SNPs within the gene. (Lower diagram) LD plots generated in Haploview using ALOX12 gene data from the NCCCTS cohort. The LD (D′ × 100) between any 2 markers is indicated at the intercept of the markers on the matrix. Where no value is given, linkage disequilibrium (D′) is 1 (LD is 100). Red diamonds indicate the greatest linkage disequilibrium and white diamonds the least. Black lines outline SNPs in haplotype blocks, and SNPs in perfect linkage disequilibrium with each other are represented by red blocks.
Of the 12 SNPs that were tested, ALOX12 rs6502997 (P, <0.000309), rs312462 (P, <0.028499), rs6502998 (P, <0.029794), and rs434473 (P, <0.038516) were found to be associated with congenital toxoplasmosis.
An etiological variant of SNPs in strong linkage disequilibrium (LD) with these particular markers, rather than SNPs within the ALOX12 gene itself, could potentially account for the association with susceptibility to congenital toxoplasmosis.
Generation of human monocytic cell lines for stable expression of ALOX12 or TetR shRNA.
To establish human monocytic cell lines stably expressing ALOX12 or TetR gene shRNA so as to facilitate RNAi silencing of the respective target gene, lentiviral stocks expressing these shRNAs were engineered and were transduced into human monocytic cells; the ALOX12 shRNA-expressing lentivirus was used for targeted silencing of the endogenous ALOX12 gene in monocytic cells, and the TetR shRNA was used as an off-target control. The promoter region of the lentivirus was designed to allow regulation of the target gene shRNA in the presence or absence of a tetracycline repressor protein. Therefore, in the absence of a tetracycline repressor protein, the transduced cell line would constitutively express the target gene shRNA (ALOX12 or TetR). However, we found that even without the tetracycline repressor expression vector, there was no difference in appearance or viability between cells expressing ALOX12 shRNA or TetR shRNA and wild-type MonoMac6 cells (data not shown). Given these data, we then generated stable monocytic cell lines without the tetracycline repressor expression vector, which were capable of constitutively expressing either the ALOX12 or the TetR shRNA. After transducing MonoMac6 cells with the lentivirus, we were able to further select for successfully transduced cells by treating the cells with the antibiotic Zeocin. The lentivirus shRNA expression plasmid contains the Sh ble gene, which confers resistance to Zeocin. Therefore, after approximately 4 weeks of Zeocin treatment, only transduced cells containing the ALOX12 or TetR lentivirus propagated. Importantly, these transduced cells did not require the use of tetracycline for expression of shRNAs.
Expression of ALOX12 shRNA silences endogenous ALOX12 gene expression in human monocytic cells.
We were able to demonstrate the effectiveness of the lentivirus expressing ALOX12 shRNA in silencing endogenous ALOX12 expression in human monocytic cells by performing quantitative real-time PCR (qRT-PCR) analysis to measure relative ALOX12 gene expression in knockdown and wild-type MonoMac6 cells. Wild-type MonoMac6 cells and MonoMac6 cells genetically engineered to express ALOX12 shRNA or off-target TetR shRNA were cultured for 72 h before they were collected and used to extract and reverse transcribe total RNA. When the cells were cultured, there was no difference in physical appearance or cell viability between the two transduced cell lines and wild-type, nontransduced MonoMac6 cells.
After running quantitative real-time PCR and normalizing the ALOX12 gene transcript levels to human actin transcript levels, we observed a significant (P, <0.001) ∼10-fold decrease in the level of ALOX12 gene transcription in ALOX12 shRNA-expressing MonoMac6 cells from that in wild-type or TetR gene shRNA-expressing MonoMac6 cells.
RNAi silencing of ALOX12 augments arachidonic acid levels in human monocytic cells.
To determine whether decreased ALOX12 gene transcription correlated with an increase in the level of the 12-lipoxygenase substrate, arachidonic acid, in infected and uninfected cells, mass spectrometry-based lipidomics analysis of wild-type MonoMac6 cells and ALOX12 and TetR knockdown MonoMac6 cells was performed. Significantly higher levels of arachidonic acid were observed in MonoMac6 cells expressing ALOX12 shRNA than in TetR knockdown and wild-type MonoMac6 cells (Fig. 2). Due to evidence of nonnormality, nonparametric tests were used. Since the MM6 and TetR groups were similar, they were combined and compared to the ALOX12 group. In an overall analysis, stratified by infection status, the ALOX12 group had significantly higher values than the MM6-plus-TetR group (P, 0.05 by a stratified Wilcoxon rank-sum test). These results indicated a correlation of the increase in arachidonic acid levels to the decrease in ALOX12 gene transcript levels quantified by real-time PCR analysis. Arachidonic acid levels were also measured in the three cell lines (wild-type, ALOX12 knockdown, and TetR knockdown) when they were infected with T. gondii in order to determine whether infection with the parasite affected arachidonic acid levels. If T. gondii infection does, in fact, trigger ALOX12 activity, arachidonic acid levels should be lower in cells infected with T. gondii. Levels of arachidonic acid were lower in all of the monocytic cell lines when they were infected with T. gondii (Fig. 2).
FIG 2.

Quantitation of arachidonic acid levels in wild-type MonoMac6 cells (MM6) and in MonoMac6 cells engineered to express either a lentivirus shRNA that knocks down ALOX12 or a control, off-target lentivirus shRNA that knocks down TetR. The data are from a minimum of five independent experiments. Error bars indicate standard errors. The mean arachidonic acid levels (standard errors of the means) are 1.78 (±1.38) nmol/mg for uninfected cells and 1.27 (±0.37) nmol/mg for infected cells in the ALOX12 knockdown group, 0.97 (±0.43) nmol/mg in uninfected cells and 0.44 (±0.10) nmol/mg in infected cells in the wild-type group, and 0.72 (±0.13) nmol/mg in uninfected cells and 0.41 (±0.09) nmol/mg in infected cells in the TetR siRNA group. When the MM6 and TetR siRNA groups are combined as the ALOX12 wild-type group, the mean arachidonic acid levels (standard errors of the means) are 0.85 (±0.22) nmol/mg in uninfected cells and 0.43 (±0.07) nmol/mg in infected cells. Uninfected cells with ALOX12 shRNA, no shRNA, or TetR shRNA are shown. Cells infected with T. gondii for 24 h that show the same pattern of shRNA for ALOX12 have higher levels of the substrate, arachidonic acid, than wild-type MonoMac6 cells and off-target shRNA controls. The results listed here in the figure legend are with the raw, untransformed data. The graph, on the other hand, is plotted using log-transformed data. For each cell type, the number of uninfected- and infected-cell samples combined was 32, 34, or 32, respectively. Due to evidence of nonnormality, nonparametric tests were used. Since the MM6 and TetR groups were similar, they were combined and compared to the ALOX12 group. In an overall analysis, stratified by infection status, the ALOX12 group had significantly higher values than the MM6-plus-TetR group (P, 0.05 by a stratified Wilcoxon rank-sum test).
ALOX12 knockdown reduces the viability of human monocytic cells infected with T. gondii in vitro.
In order to determine the effect of knocking down ALOX12 gene expression in human monocytic cells during T. gondii infection, we measured the viability of ALOX12 shRNA-expressing MonoMac6 cells infected with T. gondii strain RH relative to those of wild-type and TetR shRNA-expressing MonoMac6 cells cultured under the same conditions. The colorimetric assay we used, with the WST-1 reagent, utilizes the ability of cellular mitochondrial dehydrogenase to cleave tetrazolium salts and produce formazan dye, the intensity of which can be read as an absorbance. Viable and metabolically active cells possess high mitochondrial dehydrogenase activity that results in the production of the formazan dye from the WST-1 reagent.
Absorbance measurements were taken at the following time points: days 0, 1, 2, 3, and 4 postinfection. The measurement for each T. gondii-infected cell culture was normalized to that for uninfected MonoMac6 cells at each time point (A420 for infected cells/A420 for uninfected cells). This ratio was taken as the relative cell viability of the sample.
When absorbance was measured immediately after the establishment of the cell and cell-parasite cultures (day 0), there was no significant difference in cell viability between infected wild-type, ALOX12 knockdown, and TetR shRNA-expressing cells. However, by day 3 postinfection, while the ratio of the absorbance of infected ALOX12 knockdown cells to that of uninfected ALOX12 knockdown cells stayed relatively constant, the absorbance ratios of wild-type MonoMac6 cells and off-target tetracycline repressor knockdown cells showed marked decreases. By day 4 postinfection, the difference was more marked: wild-type MonoMac6 cells and off-target TetR knockdown cells displayed steady declines in cell viability, while infected ALOX12 knockdown cells maintained relatively the same cell viability as that observed in the preceding 2 days postinfection (P, <0.040 for ALOX12 knockdown versus MM6 cells) (Fig. 3A). This indicated that there were more viable cells in ALOX12 knockdown cultures than in wild-type MonoMac6 and TetR knockdown cell cultures at 4 days postinfection. These data are corroborated by Giemsa staining of samples of MonoMac6, ALOX12 knockdown, and TetR knockdown cells (Fig. 3B). In the images of the wild-type and transduced cell cultures, it was notable that while ALOX12 knockdown MonoMac6 cells infected with T. gondii were still relatively intact, the wild-type MonoMac6 cells and the off-target TetR knockdown cells had become extremely sparse and had largely disintegrated.
FIG 3.
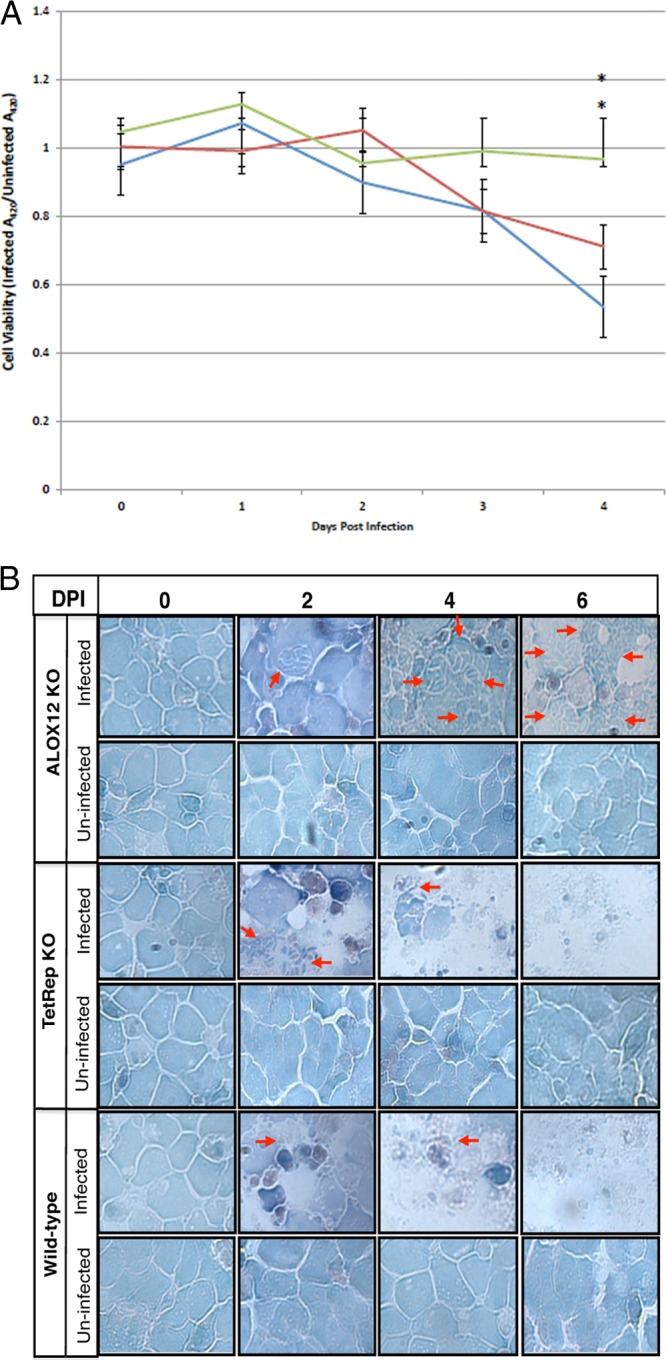
(A) Relative viabilities of wild-type MonoMac6 cells and MonoMac6 cells engineered to express ALOX12 or TetR shRNA after infection with T. gondii. Values were obtained by dividing the absorbance of infected cells by the absorbance of uninfected cells. Blue line, MonoMac6 cells; red line, TetR knockdown cells; green line, ALOX12 knockdown cells. Asterisks indicate a significant difference between ALOX12 knockdown cells and wild-type MM6 cells (P, <0.040). (B) Giemsa-stained cytospin preparations of monocytic cells examined by light microscopy to visualize the effect of ALOX12 gene knockdown on the viability of monocytic cells cultured with or without parasites. Wild-type monocytic cells and monocytic cells modified for the stable knockdown of ALOX12 (ALOX12KO) or off-target tetracycline repressor (TetRep KO) gene transcription were cultured with or without parasites. Cytospin smears were prepared and stained with Giemsa stain for each culture at different days postinfection. Representative images for three independent experiments are shown. Arrows point to infected cells.
ALOX12 knockdown increases T. gondii proliferation in human monocytic cells during in vitro infection.
To measure the effect of ALOX12 knockdown on the relative growth rate of T. gondii parasites in monocytic cells, wild-type, ALOX12 knockdown, and TetR knockdown MonoMac6 cells were cultured with or without T. gondii parasites engineered to express YFP, and fluorescence was measured at different time points postinfection. In order to visualize any effects of ALOX12 knockdown on the proliferation of T. gondii, the same cells were simultaneously cultured with and without YFP-expressing parasites in 24-well plates and were photographed at each time point postinfection. At 3 days postinfection, ALOX12 knockdown MonoMac6 cells exhibited a dramatic increase in fluorescence, correlating with a dramatic increase in parasite proliferation (Fig. 4). As shown for day 4 postinfection, the parasites had ceased to grow in wild-type and TetR knockdown MonoMac6 cells, while parasite numbers continued to increase in MonoMac6 cells expressing ALOX12 shRNA.
FIG 4.

Measurement of parasite growth based on fluorescence counts of YFP-expressing parasites. Exactly 104 wild-type MonoMac6, TetR knockdown, or ALOX12 knockdown cells were infected with 5 × 103 YFP-expressing parasites in 96-well plates. Blue line, MonoMac6 cells; red line, TetR knockdown cells; green line, ALOX12 knockdown cells. Asterisks indicate a significant difference between ALOX12 knockdown cells and wild-type MM6 cells (P, <0.0482).
ALOX12 knockdown modulates the expression of IL-6, IL-1β, TNF-α, and caspase-1 during T. gondii infection.
To determine the effects of ALOX12 knockdown on the expression of IL-6, IL-1β, TNF-α, and caspase-1 in human monocytic cells during infection with T. gondii, quantitative real-time PCR analysis of the gene transcripts was conducted on cDNA synthesized from total RNA extracted from wild-type MonoMac6 cells and MonoMac6 cells engineered to express either ALOX12 gene shRNA or tetracycline repressor gene shRNA. The transcript values obtained were normalized to human actin transcript levels. IL-6 transcript levels were found to be significantly (P, <0.05) upregulated in infected cells with ALOX12 knockdown (Fig. 5B). On the other hand, the upregulation of IL-1β, TNF-α, and caspase-1 was found to be significantly inhibited in infected cells with ALOX12 knockdown (Fig. 5A, C, and D).
FIG 5.

Analysis of the effects of ALOX12 gene knockdown on the expression of cytokines and caspase-1 in a human monocytic cell line (MonoMac6). Quantitative real-time PCR was performed on cDNA synthesized using equal amounts of total RNA from wild-type MonoMac6 cells (MM) and MonoMac6 cells engineered to express either ALOX12 gene shRNA (ALOX12-KD) or tetracycline repressor gene shRNA (TetR) that had been cultured with (shaded bars) or without (open bars) parasites for 36 h. The IL-1β (A), IL-6 (B), TNF-α (C), and caspase-1 (D) transcript levels were divided by the actin transcript level for each respective sample to yield the relative gene transcript levels. Asterisks indicate significant augmentation of gene expression attributable to infection (P, <0.05). Data are means of results from three independent experiments; error bars, standard errors.
Model of 12-LOX structure.
The PHYRE2 program was used to generate a model of the structure of 12-LOX (ALOX12). Although no structure exists for type 12 lipoxygenase, there are structures for the closely related type 5 and 15 isoforms, which share 42 and 62% sequence identity, respectively. This close similarity led to the production of a model covering 99% of the sequence with a 100% confidence value. In humans, some functional allelic variants of ALOX12 (associated with different cell death and hypertension phenotypes) have an Arg substituted for Gln at position 261 and an Asn substituted for Ser at position 322 in LD (Fig. 6). The model shows that these changes affect the surface charge of 12-LOX (ALOX12). This may change the dimerization of 12-LOX (ALOX12) or its association with other molecules. It is possible that the promoter region polymorphisms also may contribute to differing outcomes. Further work will be required to fully characterize these changes.
FIG 6.

Model of human leukocyte 12-LOX (ALOX12). Amino acids shown in red are encoded by functional SNPs. SNPs in ALOX12 (e.g., rs6502997) are associated with susceptibility to congenital toxoplasmosis (P, <0.0005). In humans, some functional allelic variants of ALOX12 (associated with different cell death or hypertension phenotypes) have an Arg substituted for Gln at position 261 and an Asn substitution for Ser at position 322 in LD. This likely would change the surface charge of 12-LOX (ALOX12), which might change the dimerization of 12-LOX (ALOX12) or its association with other molecules. Promoter region polymorphisms also may contribute to differing outcomes.
DISCUSSION
In this study, we investigated the role of ALOX12 in T. gondii infection in humans. T. gondii is an obligately intracellular parasite that can infect any nucleated cell (1, 5). Once inside its host cell, T. gondii resides and proliferates in specialized parasitophorous vacuoles during active infection, effectively evading host cell killing until the parasites egress from the cell (∼48 h) and can then go on to infect new cells.
ALOX12 is a gene located centromerically in the TOXO1 gene region on human chromosome 17 that produces the lipoxygenase that oxidizes arachidonic acid into 12-HETE. In order to elucidate the role of ALOX12 during T. gondii infection, we successfully genetically engineered human monocytic cells (MonoMac6 cells) for ALOX12 gene knockdown by RNAi. After confirming that the gene knockdown did not affect the viability of the cell when it was not infected with the parasite, we cultured MonoMac6 cells expressing ALOX12 shRNA with T. gondii and compared the results with those for wild-type MonoMac6 cells infected with the same number of parasites. We found that parasites were able to proliferate more aggressively in ALOX12 knockdown monocytic cells than in the wild type, and although there were more parasites in the ALOX12 knockdown cells, there were also more surviving, intact cells than in the wild type, where a majority of the cells started dying by day 4 postinfection. These results suggest that ALOX12 prevents T. gondii tachyzoites from proliferating by increasing host cell death.
Despite suggestions that genetic factors are important in determining the outcomes of T. gondii infection, few studies have investigated the influence of immunogenetics in human toxoplasmosis. In this study, we have shown that ALOX12 possesses alleles associated with susceptibility to congenital toxoplasmosis. In particular, four SNPs that we genotyped were shown to be significantly associated with congenital toxoplasmosis. One of the SNPs, which is strongly associated with congenital toxoplasmosis, is located in the promoter region of the ALOX12 gene and could potentially affect the control of ALOX12 activation and transcription. The sequence at the rs6502997 SNP locus substitutes adenosine (A) for cytosine (C), and this difference may result in lower levels of ALOX12 expression in some individuals. Lower levels of ALOX12 expression would lead to a less robust protective response and, therefore, increased susceptibility when an individual becomes infected with T. gondii. The other three SNPs are found closer to the center of the gene sequence and either substitute an adenosine (A) for a guanine (G) or vice versa. These SNPs could potentially alter the translation and folding of the 12-lipoxygenase protein and result in decreased activity of the 12-lipoxygenase protein. A model of the structure of 12-LOX (ALOX12) shows that these changes are distant from the active site and so are unlikely to directly affect function; however, their position on the surface may influence protein-protein or protein-ligand interactions. Another possibility is that the SNPs themselves are not functional in coding but rather are simply in linkage with another part of the ALOX12 gene that is important in congenital toxoplasmosis. Further investigation into the protein structure and binding partners of this 12-lipoxygenase could help clarify the reasons for the association of ALOX12 variants with susceptibility to congenital toxoplasmosis.
rs434473 and rs1126667 (in perfect linkage with rs6502998) are the only two common missense SNPs in ALOX12. Thus, we typed the two missense SNPs in ALOX12 (one directly and one indirectly, via complete linkage) Only rs6502998 is in linkage with rs1126667. rs6502997 is upstream of ALOX12.
The relationship of the SNPs to the NF-κB binding site to the promoter SNP might also provide an explanation for the effects observed. rs6502997 (C → A at bp −33472; miRN497-01) and close-by SNPs with high LD (rs311734, rs311735, rs311738, rs11740) do not appear to be near the NF-κB binding site (GGGACATCCC) in the promoter, which is at bp −539 to −535.
Increased levels of ALOX12 gene expression combined with increased levels of arachidonic acid in humans could signify a greater degree of protection against T. gondii infection. Arachidonic acid is a polyunsaturated fatty acid that is present in the phospholipid membranes of cells throughout the human body, including the brain, muscles, and liver (7). Arachidonic acid is cleaved from the cell membrane by phospholipase A2 (PLA2), where it can then be metabolized by cyclooxygenases, peroxidases, or lipoxygenases, including the 12-lipoxygenase encoded by ALOX12 (7). The eicosanoid product of arachidonic acid metabolism by 12-lipoxygenase, 12-hydroperoxyeicosatetraenoic acid (12-HPETE), which can be further converted to 12-HETE, has been shown previously to contribute to the production of reactive oxygen species (ROS) and the depletion of glutathione. This provides a mechanism whereby mononuclear cells can kill neighboring tumor, vascular, and endothelial cells (14). In developing oligodendrocytes (pre-OLs), the protein product of ALOX12, arachidonate 12-lipoxygenase (12-LOX), has been shown to play a major role in ROS generation and subsequent cell death (14, 15). Addition of arachidonic acid to pre-OLs induces oxidative damage and loss of cell viability, while inhibition of 12-lipoxygenase activity blocks arachidonic acid-induced cell death.
Further mechanisms of 12-HETE action include stimulating oxidative stress by increasing intramitochondrial ionized calcium concentrations in cardiac myocyte mitochondria and stimulating mitochondrial nitric oxide synthase activity, which goes on to induce cell apoptosis (16). A study of beta cells in the pancreases of diabetic patients showed that inflammatory cytokines can induce 12-lipoxygenase RNA and protein expression and that an increase in 12-HETE levels inhibits insulin secretion, reduces beta cell metabolic activity, and induces cell death in islet cells (17, 18). Furthermore, 12-HETE can greatly increase inflammatory cytokine production from macrophages (2, 19) and calcium release from neutrophils (20). It is possible that ALOX12 knockdown results in decreased local inflammation in macrophages and reduced oxidative stress, thus allowing T. gondii to invade and proliferate in host cells more aggressively. This is corroborated by our finding that ALOX12 knockdown led to increased expression of IL-6 (a cytokine that is anti-inflammatory in some circumstances and proinflammatory and protective in others), with concomitant inhibition of upregulation of TNF-α, a proinflammatory cytokine important in controlling intracellular T. gondii growth. Additionally, we observed inhibited upregulation of caspase-1 in T. gondii-infected MonoMac6 cells with ALOX12 knockdown. Caspase-1 plays a significant role in the activation of pyroptosis, a highly inflammatory cell death process often observed during infection with intracellular pathogens such as T. gondii (21). Thus, the upregulation of a sometimes anti-inflammatory cytokine, IL-6, and the inhibition of upregulation of caspase-1 and TNF-α in ALOX12 knockdown cells could contribute to the decreased rate of host cell death, because these effects could result in less-robust activation of the inflammatory immune responses that are important in inducing the death of infected cells. In wild-type monocytes, ALOX12 is able to convert arachidonic acid to its reactive oxygen products and to stimulate apoptosis as well as neighbor cell death, killing the invading T. gondii and preventing it from infecting nearby cells. However, because ALOX12 knockdown cells generate decreased levels of ROS and local inflammation, apoptosis and neighbor cell killing will decrease as well, resulting in increased parasite proliferation and spread.
While the mechanisms of T. gondii-induced arachidonic acid metabolism are still unclear, the arachidonic acid metabolite 12-HETE clearly plays an important role in both inflammation and cell death. Therefore, individuals who possess more active 12-lipoxygenase and more available arachidonic acid for conversion to 12-HETE could be better protected during T. gondii infection. Individuals with low levels of arachidonic acid may be more susceptible to T. gondii infection because of an inability to raise a strong 12-HETE-induced immune and cell death response. Further work will determine whether blocking arachidonic acid metabolism, in general, will produce the same effect as ALOX12 gene knockdown.
Previously, we investigated the role of another gene from the important TOXO1 gene region, NALP1, in T. gondii infection and found that it also plays a significant role in protection against T. gondii infection (10). The NALP1 gene encodes the NALP1 inflammasome, which recruits caspase-1 and caspase-5 to cleave and activate the cytokine IL-1β (10). In the present study, we found that ALOX12 knockdown inhibited IL-1β upregulation, with concomitant inhibition of caspase-1 upregulation, during infection with T. gondii. This suggests an interplay between the roles of ALOX12 and NALP1 in controlling T. gondii infection in human monocytic cells. Interestingly, IL-1β has also been shown to be an upregulator of ALOX12 expression in rat pancreatic beta cells (22). Earlier, Henderson and colleagues found that T. gondii altered eicosanoid release by human mononuclear phagocytes (23). Our work showed that these two genes from the TOXO1 region, NALP1 and ALOX12, may act in the same protection pathway in response to T. gondii infection. In fact, we will be studying additional genes from the TOXO1 susceptibility/resistance region that have similarly demonstrated associations with toxoplasmosis and could further contribute to protection against T. gondii infection. The success of T. gondii infection depends on the ability of the parasites to invade host cells and modulate many of the mechanisms that the host cell uses in trying to kill the parasite. Despite the fact that in vitro studies may not completely mimic the in vivo course of T. gondii pathogenesis, our study clearly provides evidence that the ALOX12 gene in humans activates some of the mechanisms that T. gondii attempts to overcome and demonstrates that ALOX12 plays a crucial role in controlling pathogenesis during T. gondii infection. Further, it is noteworthy that complete elucidation of the role of ALOX12 in human toxoplasmosis would require studies involving the full life cycle of T. gondii, including the encysted stage, which is not represented in the present study.
ACKNOWLEDGMENTS
We gratefully acknowledge the support of NIAID NIH RO1 AI27530 and AI071319-03, The Cornwell and Mann Family Foundation, the Engel, Taub, Musillami, Haider, Jensen, Morel, Samuel, and Rooney-Alden families, and S. Powers. Instrument acquisition at the Kansas Lipidomics Research Center was supported by the National Science Foundation (EPS 0236913, DBI 0521587, DBI 1228622), the Kansas Technology Enterprise Corporation, the Kansas IDeA Network of Biomedical Research Excellence (K-INBRE) of the National Institutes of Health (P20RR16475), and Kansas State University. S.P.M. is funded by an MRC career development fellowship grant (G1000567).
We thank Theodore Karrison and Kristen Wroblewski for assistance with biostatistics in the NCCCTS and for the statistical analysis of the lipidomics data in this report.
Footnotes
Published ahead of print 31 March 2014
REFERENCES
- 1.McLeod R, van Tubbergen C, Montoya JG, Petersen E. 2013. Human Toxoplasma infection and toxoplasmosis, p 100–147 In Weiss LM, Kim K. (ed), Toxoplasma gondii: the model apicomplexan: perspectives and methods, 2nd ed. Academic Press, Waltham, MA [Google Scholar]
- 2.Roizen N, Swisher C, Stein M, Hopkins J, Boyer K, Patel D, Stein M, Schey W, Meier P, Mets M, Beckman J, McLeod R. 1995. Neurologic and developmental outcome in treated congenital toxoplasmosis. Pediatrics 95:11–20 [PubMed] [Google Scholar]
- 3.Cavaillès P, Bisanz C, Papapietro O, Colacios C, Sergent V, Pipy B, Saoudi A, Cesbron-Delauw M-F, Fournié GJ. 2006. The rat Toxo1 locus controls the outcome of the toxoplasmic infection according to a Mendelian mode. Med. Sci. (Paris) 22:679–680 (In French.). 10.1051/medsci/20062289679 [DOI] [PubMed] [Google Scholar]
- 4.Cavaillès P, Sergent V, Bisanz C, Papapietro O, Colacios C, Mas M, Subra JF, Lagrange DD, Calise MM, Appolinaire SS, Faraut TT, Druet PP, Saoudi AA, Bessieres MH, Pipy B, Cesbron-Delauw MF, Fournié GJ. 2006. The rat Toxo1 locus directs toxoplasmosis outcome and controls parasite proliferation and spreading by macrophage-dependent mechanisms. Proc. Natl. Acad. Sci. U. S. A. 103:744–749. 10.1073/pnas.0506643103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson SE, Remington JS. 1974. Effect of normal and activated human macrophages on Toxoplasma gondii. J. Exp. Med. 139:1154–1174. 10.1084/jem.139.5.1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brash AR. 2001. Arachidonic acid as a bioactive molecule. J. Clin. Invest. 107:1339–1345. 10.1172/JCI13210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacape G, Daret D, Crockett R, Rigaud M, Larrue J. 1992. Dual metabolic pathways of 12-HETE in rat aortic smooth muscle cells. Prostaglandins 44:167–176 [DOI] [PubMed] [Google Scholar]
- 8.Phillis JW, Horrocks LA, Farooqui AA. 2006. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res. Rev. 52:201–243. 10.1016/j.brainresrev.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 9.Jamieson SE, de Roubaix LA, Cortina-Borja MM, Tan HK, Mui E, Cordell HJ, Kirisits MJ, Miller EN, Peacock CS, Hargrave AC, Coyne JJ, Boyer K, Bessieres MH, Buffolano W, Ferret N, Franck J, Kieffer F, Meier P, Nowakowska DE, Paul M, Peyron F, Stray-Pedersen B, Prusa AR, Thulliez P, Wallon M, Petersen E, McLeod R, Gilbert RE, Blackwell JM. 2008. Genetic and epigenetic factors at COL2A1 and ABCA4 influence clinical outcome in congenital toxoplasmosis. PLoS One 3:e2285. 10.1371/journal.pone.0002285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, Cavailles P, Bisanz C, Cesbron-Delauw M-F, Fournié GJ, McLeod R. 2011. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect. Immun. 79:756–766. 10.1128/IAI.00898-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Invitrogen. 29 June 2004. Gateway-adapted, lentiviral destination vector for high-level, regulated expression of shRNA in dividing and nondividing mammalian cells: user's manual. Invitrogen, Carlsbad, CA [Google Scholar]
- 12.Invitrogen. 21 June 2007. Gateway-adapted entry vector for regulated expression of shRNA in mammalian cells: user's manual. Invitrogen, Carlsbad, CA [Google Scholar]
- 13.Invitrogen. 29 September 2004. BLOCK-iT inducible H1 lentiviral RNAi system. Invitrogen, Carlsbad, CA [Google Scholar]
- 14.Maccarrone M, Melino G, Finazzi-Agro A. 2001. Lipoxygenases and their involvement in programmed cell death. Cell Death Differ. 8:776–784. 10.1038/sj.cdd.4400908 [DOI] [PubMed] [Google Scholar]
- 15.Li J, Wang H, Rosenberg PA. 2009. Vitamin K prevents oxidative cell death by inhibiting activation of 12-lipoxygenase in developing oligodendrocytes. J. Neurosci. Res. 87:1997–2005. 10.1002/jnr.22029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nazarewicz RR, Zenebe WJ, Parihar A, Parihar MS, Vaccaro M, Rink C, Sen CK, Ghafourifar P. 2007. 12(S)-Hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch. Biochem. Biophys. 468:114–120. 10.1016/j.abb.2007.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen M, Yang ZD, Smith KM, Carter JD, Nadler JL. 2005. Activation of 12-lipoxygenase in proinflammatory cytokine-mediated beta cell toxicity. Diabetologia 48:486–495. 10.1007/s00125-005-1673-y [DOI] [PubMed] [Google Scholar]
- 18.Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 2010. 12-Lipoxygenase products reduce insulin secretion and β-cell viability in human islets. J. Clin. Endocrinol. Metab. 95:887–893. 10.1210/jc.2009-1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen Y, Gu J, Vandenhoff GE, Liu X, Nadler JL. 2008. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. Am J. Physiol. Heart Circ. Physiol. 294:H1933–H1938. 10.1152/ajpheart.00260.2007 [DOI] [PubMed] [Google Scholar]
- 20.Reynaud D, Pace-Asciak CR. 1997. 12-HETE and 12-HPETE potently stimulate intracellular release of calcium in intact human neutrophils. Prostaglandins Leukot. Essent. Fatty Acids 56:9–12. 10.1016/S0952-3278(97)90518-4 [DOI] [PubMed] [Google Scholar]
- 21.Tschopp J, Martinon F, Burns K. 2003. NALPs: a novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 4:95–104. 10.1038/nrm1019 [DOI] [PubMed] [Google Scholar]
- 22.Bleich D, Chen S, Gu JL, Thomas L, Scott S, Gonzales N, Natarajan R, Nadler JL. 1995. Interleukin-1β regulates the expression of a leukocyte type of 12-lipoxygenase in rat islets and RIN m5F cells. Endocrinology 136:5736–5744. 10.1210/endo.136.12.7588331 [DOI] [PubMed] [Google Scholar]
- 23.Yong EC, Chi EY, Henderson WR., Jr 1994. Toxoplasma gondii alters eicosanoid release by human mononuclear phagocytes: role of leukotrienes in interferon gamma-induced antitoxoplasma activity. J. Exp. Med. 180:1637–1648. 10.1084/jem.180.5.1637 [DOI] [PMC free article] [PubMed] [Google Scholar]