

Abstract
At the Tcrb locus, Vβ-to-Jβ rearrangement is permitted by the 12/23 rule but is not observed in vivo, a restriction termed the “beyond 12/23” rule (B12/23 rule). Previous work showed that Vβ recombination signal sequences (RSSs) do not recombine with Jβ RSSs because Jβ RSSs are crippled for either nicking or synapsis. This result raised the following question: how can crippled Jβ RSSs recombine with Dβ RSSs? We report here that the nicking of some Jβ RSSs can be substantially stimulated by synapsis with a 3′Dβ1 partner RSS. This result helps to reconcile disagreement in the field regarding the impact of synapsis on nicking. Furthermore, our data allow for the classification of Tcrb RSSs into two major categories: those that nick quickly and those that nick slowly in the absence of a partner. Slow-nicking RSSs can be stimulated to nick more efficiently upon synapsis with an appropriate B12/23 partner, and our data unexpectedly suggest that fast-nicking RSSs can be inhibited for nicking upon synapsis with an inappropriate partner. These observations indicate that the RAG proteins exert fine control over every step of V(D)J cleavage and support the hypothesis that initial RAG binding can occur on RSSs with either 12- or 23-bp spacers (12- or 23-RSSs, respectively).
INTRODUCTION
The first step in the assembly and diversification of antigen receptor genes is V(D)J recombination, a site-specific recombination reaction that joins variable (V), diversity (D), and joining (J) coding segments at immunoglobulin and T-cell receptor (TCR) loci. During B- and T-cell development, the recombination-activating gene 1 and 2 proteins (RAG1/2) initiate V(D)J recombination (1, 2) at sites specified by recombination signal sequences (RSSs), which flank each V, D, and J gene segment.
The biochemistry of V(D)J recombination can be divided into two phases: cleavage and joining (3, 4). The cleavage phase is catalyzed by a recombinase complex comprised of RAG1/2 (collectively, RAG) and the ubiquitous DNA binding/bending protein HMGB1 or the closely related HMGB2 protein (5). The first step in the cleavage phase is the binding of the recombinase complex to one RSS, forming a signal complex (SC). After SC formation, synapsis occurs, in which a partner RSS is captured to form the paired complex (PC) (6, 7). RAG catalyzes the nicking of DNA at the heptamer-RSS boundary, giving rise to a free hydroxyl group. Double-strand breaks are catalyzed in the PC via a transesterification reaction whereby the free hydroxyl created by nicking directly attacks the opposite strand (8). These four ends created by cleavage are resolved in the joining phase by the proteins of the nonhomologous end-joining (NHEJ) repair pathway to form a precise signal joint containing the RSSs and an imprecise coding joint containing the antigen receptor gene segments (reviewed in reference 9).
Each RSS is comprised of three sequence elements: a relatively well-conserved heptamer (consensus CACAGTG) and nonamer (consensus ACAAAAACC) and a less well-conserved spacer of either 12 or 23 bp (12-RSS and 23-RSS, respectively) (10). Recombination occurs only between gene segments where one segment is flanked by a 12-RSS and the other is flanked by a 23-RSS, a restriction known as the 12/23 rule. While the mechanisms underlying the 12/23 rule are poorly understood, the rule is enforced during the synapsis and hairpin formation steps (5, 6, 11, 12). Gel shift experiments performed by Jones and Gellert (6) showed that the preference for 12/23 synapsis was stronger if the RAG proteins were incubated first with the 12-RSS than if they first saw the 23-RSS. As a result, a capture model for RAG binding was proposed, stating that RAG initially binds to a 12-RSS and then captures a 23-RSS to form the PC. This model was supported by an in vivo analysis of patterns of RAG-mediated RSS nicking (13), but subsequent in vivo nicking and binding experiments argued that initial RAG binding can occur on either 12- or 23-RSSs (14, 15).
At the Tcrb locus (Fig. 1A), Vβ segments are flanked by 23-RSSs, and Jβ segments are flanked by 12-RSSs. The 12/23 rule would predict the occurrence of Vβ-Jβ recombination, but this is not seen in vivo (16, 17). This represents a restriction beyond the 12/23 rule (here, simplified to the “beyond 12/23” rule, or B12/23 rule) and suggests the existence of mechanisms that prohibit direct Vβ-to-Jβ rearrangement. Experiments using transgenic Tcrb miniloci or altered Tcrb knock-ins showed that this restriction is mediated by the Tcrb RSSs (18, 19). For example, mice homozygous for a knocked-in Tcrb allele where the 5′Dβ1 12-RSS was replaced by that of Jβ1.2 exhibited a drastic block in thymocyte development, consistent with an inability to assemble complete TCRβ polypeptides. T-cell hybridomas made from heterozygous littermates revealed that these alleles perform Dβ-to-Jβ recombination but failed to perform Vβ-to-DJβ rearrangement (18). Additional experiments showed that Vβ 23-RSSs can efficiently recombine only with 5′Dβ 12-RSSs and were almost entirely unable to recombine with Jβ 12-RSSs even when the Dβ RSSs were deleted from the locus (18, 20, 21).
FIG 1.

Tcrb locus. (A) The general structure of the Tcrb locus is depicted schematically and not to scale; rectangles represent gene segments, an oval represents the Eβ enhancer, shaded triangles represent 23-RSSs, and dotted triangles represent 12-RSSs. (B) Alignment of coding flank, heptamer, spacer, and nonamer sequences of the RSSs discussed in the text. For each RSS, any differences from consensus are underlined. Table 1 gives the full sequence of each oligonucleotide used. GenBank gene segment names (http://www.ncbi.nlm.nih.gov/gene/21577) are shown in parentheses for endogenous RSSs.
Extrachromosomal recombination substrates containing Tcrb RSSs exhibit B12/23-restricted rearrangement in nonlymphoid cell lines expressing the RAG proteins (21, 22), and the core RAG proteins and HMGB1 together recapitulate B12/23-regulated cleavage in vitro on short oligonucleotide substrates as well as on longer substrates containing RSSs in cis (21, 23, 24). Therefore, RAG1 and RAG2 are the only lymphoid-specific factors required for B12/23 regulation.
The mechanisms underlying the B12/23 rule are not yet clear. Preferential RAG binding to Dβ 12-RSSs over binding to Jβ 12-RSSs would explain why Vβ recombines only with DJβ, but no such preferential binding is observed by electrophoretic mobility shift assay (EMSA) (23, 24) or by chromatin immunoprecipitation (15). An alternative explanation is suggested by biochemical experiments from our group that showed that some Jβ RSSs nick poorly in the SC while others synapse inefficiently with the Vβ14 RSS (23). In contrast, the 5′Dβ1 12-RSS was found to support efficient nicking and synapsis with Vβ14. This study concluded that the Jβ 12-RSSs are functionally crippled for recombination compared to the 5′Dβ 12-RSSs, thereby explaining preferential recombination of Vβ RSSs to 5′Dβ RSSs. These results raise the following question: if Jβ RSSs are defective, how can Dβ-to-Jβ recombination occur efficiently?
One particular result from this previous study was suggestive: nicking of Jβ2.5 was almost undetectable in biochemical assays (23), and yet this J segment is one of the most frequently used in the mature TCRβ repertoire (25, 26). Because nicking is an absolute prerequisite for hairpinning and inclusion in a final variable region exon, this suggested that nicking can be stimulated by synapsis. Therefore, we hypothesized that, compared to the SC, RAG-mediated nicking can be stimulated in physiologically appropriate PCs (e.g., a Dβ-Jβ PC or a Vβ-Dβ PC) and not in inappropriate PCs (e.g., a Vβ-Jβ PC). Such stimulation would allow the otherwise crippled Jβ segments to recombine efficiently with Dβ segments. This hypothesis regarding Tcrb RSSs contrasts with a previous study (27) showing that nicking of consensus RSSs was not affected by synapsis.
We report here that the nicking of some RSSs can indeed be stimulated by synapsis with an appropriate partner RSS. Moreover, our data allow for the stratification of Tcrb RSSs into two categories: RSSs that nick slowly in the SC and RSSs that nick quickly in the SC. RSSs in the two categories display distinct nicking behaviors in the PC: slow-nicking RSSs are stimulated to nick more efficiently in appropriate PCs and inhibited or weakly stimulated in inappropriate PCs; fast-nicking RSSs appear to be strongly inhibited for nicking in inappropriate PCs, and their nicking can be stimulated, inhibited, or remain unaffected in appropriate PCs. Additionally, our data reveal an unexpected example of highly efficient nicking and synapsis followed by poor hairpinning, suggesting an additional level of regulation for RAG-mediated cleavage.
(This research was conducted by Joydeep K. Banerjee in partial fulfillment of the requirements for a Ph.D. from Yale University, New Haven, CT.)
MATERIALS AND METHODS
Expression and purification of coexpressed MBP-RAG1c and MBP-RAG2c.
Mammalian expression vectors cMR1 and cMR2 (kindly provided by Patrick Swanson) express amino acids (aa) 384 to 1040 of RAG1 and aa 1 to 387 of murine RAG2, respectively, each fused at its N terminus to maltose binding protein (MBP). Transfection and purification were performed similarly to Swanson's published protocol (28) but at larger scale and with a batch-binding/column-wash-and-elution format. For each batch of protein, 42 10-cm plates of 293T cells were each transfected with 25 μg of each plasmid by the calcium phosphate method. Cell pellets were resuspended in buffer A (10 mM NaPO4, pH 7.6, 500 mM NaCl, 1 mM dithiothreitol [DTT], 0.25% Tween 20) containing leupeptin, aprotinin, and pepstatin protease inhibitors (Sigma). Cells were lysed by 20 strokes in a Dounce homogenizer, and lysates were clarified by ultracentrifugation in an Sw55 Ti rotor (Beckman) at 30,000 rpm for 40 min. Clarified lysates were loaded onto a 1-ml bed volume of amylose resin (New England BioLabs) in a 50-ml conical tube and rocked for 1.5 h. The resin was then packed into a Polyprep chromatography column (Bio-Rad), and the lysate was allowed to flow through by gravity. The column was then washed with 5 ml of buffer A, followed by 5 ml of buffer A lacking Tween. Elution buffer (300 μl; buffer A lacking Tween and supplemented with 10 mM maltose) was then added to the column to push out some of the void volume. Protein was eluted with 1 ml of elution buffer and dialyzed against dialysis buffer (25 mM Tris, pH 8.0, 150 mM KCl, 2 mM DTT, 10% glycerol) for 3 h. Small aliquots were snap-frozen and stored at −80°C. Protein concentration was determined by SDS-polyacrylamide gel electrophoresis (PAGE) followed by a SYPRO Orange (Invitrogen) stain comparison to a bovine serum albumin (BSA) standard curve.
Purification of full-length HMGB1.
Full-length human HMGB1 (hHMGB1; aa 1 to 215) with an N-terminal 6×His tag was purified according to a previously published protocol (28) with some modifications. Four liters of HU− bacteria containing the pET11d-hHMGB1 plasmid was shaken at 37°C to an optical density at 600 nm (OD600) of 0.6 to 0.7 and induced to express protein by the addition of 245 mg of isopropyl-β-d-thiogalactopyranoside (IPTG). Bacteria were then shaken overnight at 25°C and harvested in the morning.
Bacterial pellets were resuspended in binding buffer (40 mM Tris-Cl, pH 8.0, 500 mM KCl, 20 mM imidazole) supplemented with DNase I (Sigma) and protease inhibitors (Sigma). Cells were lysed by sonication and clarified by centrifugation in a JA-17 (Beckman) rotor for 45 min at 17,000 rpm. Clarified lysates were loaded onto a 3-ml column of Ni-nitrilotriacetic acid (Ni-NTA) Superflow resin (New England BioLabs), washed with 12 ml of binding buffer and 9 ml of Ni-NTA wash buffer (40 mM Tris-Cl, pH 8.0, 65 mM KCl, 50 mM imidazole), and eluted with 24 ml of Ni-NTA elution buffer (Ni-NTA wash buffer containing 500 mM imidazole).
Ni-NTA eluate was then purified by ion exchange chromatography on an AKTA fast protein liquid chromatography (FPLC) system (GE Healthcare), first over a HiTrap SP HP 5-ml column. Fractions containing mostly full-length HMGB1 (as determined by SDS-PAGE and Coomassie staining) were purified over a HiTrap Q HP 1-ml column. Fractions containing only full-length HMGB1 were pooled and centrifugally concentrated in an Amicon Ultra concentrator (Millipore) and dialyzed overnight against dialysis buffer. Aliquots were snap-frozen the next morning and stored at −80°C. Protein was quantitated by SDS-PAGE and a Coomassie stain comparison to a BSA standard curve.
DNA substrate design.
DNA substrates were designed to match the strategy used by Drejer-Teel et al. (23). These substrates are shown in Fig. 1B and Table 1. We discovered that some of the sequences published in the work by Drejer-Teel et al. (23) differed slightly from those contained in GenBank. All experiments described here use the GenBank sequences. Oligonucleotides were designed with a coding flank of 16 nucleotides (nt); the 10 nt immediately adjacent to the heptamer were the same sequence as the endogenous gene segment, and the remaining 6 nt were the same in every RSS substrate. In addition, 15 nt of non-RSS sequence were appended to each substrate after the nonamer. Nonspecific sequences (ns3-12 and ns3-23) were taken from pJHSASNXB (21) by choosing regions of DNA that did not contain any sequence resembling an RSS heptamer or nonamer.
TABLE 1.
Complete sequences of oligonucleotides used in this study
| Oligonucleotide | Top-strand sequence (5′→3′) |
|---|---|
| 12RSS | GATCTGGCCTGGTCTGCACAGTGATACAGCCCTTAACAAAAACCTGCACTCGAGCGGAG |
| 5′Dβ1 | GATCTGCCCCTGTCCCCACAATGTTACAGCTTTATACAAAAAAGTGCACTCGAGCGGAG |
| Jβ2.5 | GATCTGTGTCTTGGTTCACAGCCCCAGAACCCAACACAAAAACTTGCACTCGAGCGGAG |
| Jβ2.7 | GATCTGTTCATAGGAGCACAGAGGCTCAACCCCACACACAAACCTGCACTCGAGCGGAG |
| Jβ2.7CF | GATCTGTTCATAGGAGCACAGCCCCAGGACCCAACACAAAAACTTGCACTCGAGCGGAG |
| Jβ1.1 | GATCTGTCTGTGTTTGCACAGTGCCATAGGATGAGGAGAAAAATTGCACTCGAGCGGAG |
| Jβ1.4 | GATCTGTCGTTGGAAACACAACATTAAAGCCTGGTGGTAAAACTTGCACTCGAGCGGAG |
| ns3-12 | TTGTCGCGCCAATCGAGCCATGTCGTCGTCGATCCTCTCATCGATGAGAGGATCGGCTC |
| 23RSS | GATCTGGCCTGTCTTACACAGTGATGGAAGCTCAATCTGAACTCTGACAAAAACCTGCACTCGAGCGGAG |
| 3′Dβ1 | GATCTGGACAGGGGGCCACGGTGATTCAATTCTATGGGAAGCCTTTACAAAAACCTGCACTCGAGCGGAG |
| Vβ14 | GATCTGCCTGGAGTCTCACACTGAGTAGGGTGGGGCAGACATCTGTGCAAAAACCTGCACTCGAGCGGAG |
| Vβ2 | GATCTGGCAGCCAAGACACAGCCCTGCAGAGCTCTTGCCTCCCTGTACACAAACCTGCACTCGAGCGGAG |
| ns3-23 | CCAAATTGATATAATTAAGCCCCAACCGCCTCTTCCCGCCTCATTTCAGCCTCACCACCATCATGGATAG |
Purification and labeling of oligonucleotide DNA substrates.
Oligonucleotides with the sequences shown in Table 1 were ordered PAGE purified from Integrated DNA Technologies. Reverse-complement oligonucleotides for each RSS were ordered with either 5′ biotin or 5′ C6-amino modifications and high-performance liquid chromatography (HPLC) purification or with no modification and PAGE purification. Lyophilized pellets were resuspended to 50 μM in TE buffer (10 mM Tris, pH 8, 1 mM EDTA). For substrates with no modification and for substrates with 5′ biotin modifications, top and bottom strands were mixed in equimolar concentrations, boiled for 5 min, and slowly cooled for 3 h. Bottom-strand stocks with 5′ C6-amino modifications were phosphorylated using T4 polynucleotide kinase (New England BioLabs) to ensure that any truncation impurities had a 5′ phosphoryl group, and excess ATP was removed by two passages through fresh G-50 gel filtration columns (GE Healthcare). Bottom strands thus treated were mixed with top strands in equimolar concentrations, boiled for 5 min, and slowly cooled for 3 h. Substrates with no modifications were not subjected to any further purification.
After slow annealing, all modified substrates were run on a 2.5-mm-thick 10% polyacrylamide gel (19:1) cast in Tris-borate-EDTA (TBE) using a Protean II xi electrophoresis system (Bio-Rad), and a guide lane was visualized by SYBR green I (Invitrogen). Excised bands were placed in 1.5-ml Eppendorf tubes and incubated in 550 μl of buffer EB (10 mM Tris, pH 8) at 37°C with shaking overnight. The next morning, the supernatant was removed, and 1.5 μl of 20 mg/ml glycogen, 150 μl of 3 M sodium acetate (NaOAc), pH 5, and 850 μl of 100% ethanol (EtOH) were added. The mixture was mixed well and placed at −80°C for several hours until frozen. The DNA was then pelleted, washed with 70% EtOH, and resuspended in 50 μl of TE buffer (10 mM Tris, pH 8, 1 mM EDTA). The concentration of this working stock was quantitated by NanoDrop (Thermo Scientific).
Substrates with bottom strands containing 5′ C6-amino modifications were radiolabeled by T4 polynucleotide kinase and [γ32P]ATP (Perkin-Elmer), heat inactivated at 65°C for 20 min, passed through a G-50 gel filtration column, boiled for 5 min, and slowly cooled for 3 h to ensure proper annealing. Radiolabeled substrates were quantitated by a SYBR green I stain comparison to a standard curve of a low-molecular-weight DNA ladder (New England BioLabs).
In-solution cleavage assays.
All RAG and HMGB1 preparations were first tested for maximal activity via diagnostic cleavage reactions according to a slight modification to the method of Bergeron et al. (28). Various concentrations of RAG and HMGB1 protein were incubated together on ice for 5 min before addition to 5 nM radiolabeled consensus 12-RSS in Mg2+ buffer (25 mM morpholinepropanesulfonic acid [MOPS], pH 7.0, 60 mM potassium glutamate, 100 μg/ml BSA, 1.5 mM MgCl2, 1 mM DTT) on ice. Reaction mixtures were then incubated on ice for 5 min, and a consensus 23-RSS was added to a final concentration of 25 nM. The final volume of these reaction mixtures was 20 μl. Reaction mixtures were incubated on ice for a further 5 min and placed at 37°C for 60 min. Reactions were stopped by the addition of 40 μl of loading buffer (95% formamide, 10 mM EDTA, 0.09% xylene cyanol, 0.09% bromophenol blue).
Reaction mixtures were boiled for 4 min and placed immediately on ice for a further 4 min before being loaded onto a 15% (19:1) acrylamide–7 M urea denaturing TBE gel in a Bio-Rad Protean II xi system. Before being loaded, this gel was prewarmed to 45°C by the use of a recirculating water bath and prerun at 12 W for 30 min. After the loading step, the gel was run at a constant 12 W for approximately 1.5 h. The gel apparatus was then disassembled, and the gel was dried and exposed to a PhosphorImager screen (Molecular Dynamics) overnight. The exposed screen was imaged by PhosphorImager and quantitated by ImageQuant, version 5.2, software.
Diagnostic reactions were used to determine the concentration of protein to use in subsequent reactions. Protein concentrations were chosen such that at least 50% of 12-RSS substrate was converted into hairpin product after 60 min. If no such concentration could be found, that batch of protein was discarded. The concentration of protein used in experimental reaction mixtures thus varied from batch to batch.
The experiments shown in Fig. 2A proceeded very similarly, except that a single predetermined concentration of protein was used, and the final reaction volume was 30 μl. This reaction mixture was placed at 37°C, and 5 μl was removed at the 0-, 5-, 10-, 15-, 30-, and 60-min time points and stopped by the addition of 10 μl of loading buffer.
FIG 2.

Development of a PC-only cleavage assay. (A) In-solution cleavage assays were performed with either 5′ top-strand-radiolabeled Jβ2.5 (Jβ2.5*) or radiolabeled consensus 12-RSS (12RSS*) in the presence or absence of the indicated partner oligonucleotide and for the indicated number of minutes. Reaction products were separated by denaturing PAGE. Uncleaved, hairpinned, and nicked products are indicated schematically and in respective order, top to bottom, by the symbols shown between lanes 18 and 19. (B) Quantitation of nicking from the experiment shown in panel A. (C) PC-only cleavage assay strategy. One partner oligonucleotide is biotinylated (Bio) on the 5′ end of the bottom strand, while the other is radiolabeled (32P) on the 5′ end of the top strand. (D) Representative experiment used to determine the 3% input threshold. Binding reactions were performed using 5 nM radiolabeled Jβ2.5 and the indicated concentration of either biotinylated 3′Dβ1 (3′Dβ1bio) or unbiotinylated 3′Dβ1 (3′Dβ1). Purification of PCs proceeded as described in Materials and Methods, and cleavage was allowed to occur for 60 min. Numbers above the bands indicate the percentage of the signal in the lane that is present in each band, and the bottom numbers indicate the percentage of the input DNA that was captured on the magnetic beads. (E) Quantitation of all PC-only assays on Jβ2.5*-3′Dβ1 PCs (logarithmic x axis). The 3% input threshold is indicated by a dotted line.
To ensure that maximum nicking would be observed at each RSS tested in the absence of a partner, a single RAG preparation was used in all RSS-alone time courses at a concentration previously determined to provide maximum nicking for all Jβ RSSs tested.
PC-only assays.
For each replicate time course experiment, 12 binding reactions were set up. All binding was done in Ca2+ buffer (25 mM MOPS, pH 7.0, 60 mM potassium acetate, 100 μg/ml BSA, 1.0 mM CaCl2, 1 mM DTT). Coexpressed MBP-tagged core RAGs were incubated with 50 ng/reaction HMGB1 for 5 min on ice before being added to tubes containing 5 nM radiolabeled RSS in Ca2+ buffer. Reaction mixtures were incubated for 5 min on ice before being placed at 37°C for 1 min. Reaction mixtures were removed from heat, and partner RSSs were added to a final concentration of 25 nM, bringing the reaction volume to 20 μl. Three binding reactions received nonbiotinylated partner, while the other nine received biotinylated partner. All 12 reaction mixtures were then incubated for 10 min at 37°C. Reaction mixtures were removed from the heat, and every three reaction volumes were pooled. Ten microliters of the nonbiotinylated reaction mixture was removed and mixed with 20 μl of loading buffer; this was later loaded on the denaturing gel for input. Ten microliters of Ca2+ buffer-equilibrated Dynabead streptavidin magnetic beads (Invitrogen) was then added to each of the four pooled reaction mixtures, and reaction mixtures were incubated at 37°C for another 15 min. Bead binding reaction were mixed by tapping when 11, 8, and 4 min remained in the incubation.
After 15 min, beads were separated on a magnetic stand, and supernatants were discarded. Beads were washed three times with 30 μl of cold Ca2+ buffer, placed on ice, and resuspended in 10 μl of cold Mg2+ buffer. Reaction mixtures were then placed at 30°C to allow cleavage to occur. Five microliters was removed from reaction mixtures at the desired time points, the reactions were stopped by the addition of 10 μl loading buffer, and reaction mixtures were run on denaturing PAGE gels as described above.
All bands were quantitated using ImageQuant, version 5.2, software, and any radioactivity observed in nonbiotinylated control lanes was subtracted as background from the bands representing the uncleaved DNA substrate in lanes containing biotinylated partners. The relative percentage of radioactivity in each band was calculated, as well as the percentage of input radioactivity found in each lane.
RESULTS
In-solution cleavage assays do not reliably report nicking in some PCs.
DNA substrates (Fig. 1B and Table 1) consisted of a 16-bp coding flank, the RSS (or nonspecific control) sequence, and a 15-bp nonamer flanking sequence (see Materials and Methods for details). Our initial experiments were in-solution cleavage assays performed similarly to our previous study (23), with one notable difference. The previous work used MBP-RAG1c (aa 384 to 1008) from bacteria, glutathione S-transferase (GST)–RAG2c (aa 1 to 383) from 293T cells, and a truncated form of HMGB2, whereas here we used a more active coexpressed MBP-RAG1 (aa 384 to 1040) and MBP-RAG2c (aa 1 to 387) preparation with full-length HMGB1. Some reaction mixtures included an unlabeled RSS substrate in addition to the radiolabeled RSS being interrogated; the unlabeled substrate was included at a 5-fold molar excess over the radiolabeled substrate to stimulate synapsis and coupled cleavage. These reaction mixtures were assembled as described in Materials and Methods and incubated at 37°C for a 60-min time course, and the products were separated by denaturing PAGE.
Nicking at the consensus 12-RSS was robust, and the addition of a partner consensus 23-RSS strongly stimulated hairpin formation (Fig. 2A, compare lanes 19 to 24 with lanes 25 to 30). Quantitation of the consensus 12-RSS time courses showed that the rate and magnitude of nicking were not changed by the addition of a partner consensus 23-RSS: in both scenarios, nearly all the radiolabeled 12-RSS was nicked after 15 min (Fig. 2B). This result agrees well with previously published data (27).
Experiments using Jβ2.5 as the radiolabeled substrate revealed a different pattern of nicking behavior (Fig. 2A, lanes 1 to 18). Nicking of Jβ2.5 was slow but was strongly stimulated, in both initial rate and overall magnitude, by the addition of 3′Dβ1. This stimulation was especially apparent at the 10-min time point (Fig. 2B). Because these reactions were performed in the presence of Mg2+, which supports hairpinning primarily in the PC, the appearance of hairpins in only the 12/23 context implies that a significant amount of synapsis occurred. In contrast, the addition of Vβ14 did not appreciably change the initial rate of Jβ2.5 nicking though it did somewhat increase the final magnitude of nicking at 60 min (Fig. 2A and B). Some hairpinning of Jβ2.5 was also observed in the presence of Vβ14 (Fig. 2A), suggesting that a low level of cleavage-competent PCs is formed with this pair.
Jβ2.7 exhibited a novel pattern of nicking in this assay. Jβ2.7 nicked very quickly in the SC context, with kinetics similar to that of the consensus 12-RSS (see below; also data not shown). When partner RSS was added (either 3′Dβ1 or Vβ14), the observed rate and magnitude of nicking dropped considerably (data not shown). Inhibition by 3′Dβ1 was surprising, given that this is the normal partner for Jβ cleavage. Interpretation of such an inhibition of nicking is not straightforward due to technical limitations associated with the in-solution cleavage assay. Because an excess of unlabeled partner is required to stimulate synapsis and coupled cleavage in this system, an observed decrease in activity at the radiolabeled RSS could be merely the result of competition for RAG binding. A similar phenomenon of an apparent decrease in activity upon addition of excess partner was also observed in our previous study (23) and by Zhang and Swanson (29). Furthermore, this system does not incisively interrogate the complex most relevant for this investigation (the PC). In particular, the in-solution reaction presumably contains three distinct species of protein-DNA complexes, a 12-SC, a 23-SC, and a 12/23-PC, and neither the relative amounts of these complexes nor the amount of nicking derived specifically from the PC can be directly determined. This suggested the need for a different assay that could directly interrogate PCs.
Development of a PC-only cleavage assay.
Based on previous studies (23, 30), we developed a biotin pulldown method that allows the purification of PCs containing any two RSSs and that can directly assay PC-only cleavage. This technique (diagrammed in Fig. 2C) involves radiolabeling one RSS and placing a biotin group on the other. Coexpressed MBP-RAGs and HMGB1 are incubated first with 5 nM radiolabeled RSS in a buffer containing CaCl2, allowing binding to occur without any cleavage. A 5-fold molar excess of biotinylated DNA is then added and allowed to bind. PCs are purified by pulldown with streptavidin magnetic beads and then incubated at 30°C in a buffer containing 1.5 mM MgCl2, allowing cleavage to occur. The reaction products are then separated by denaturing PAGE.
Because the radioactive label is on one RSS while the biotin label is on the other, any radioactive DNA that remains on the beads after washing should be part of a stable PC (after correcting for background, as described below). The magnitude of synapsis was measured in each experiment by running an input lane on the denaturing gel, representing the total radioactivity in the reaction mixture before the addition of streptavidin beads. The degree of nonspecific binding to the beads was also measured in each experiment by a control reaction in which the partner RSS was not biotin tagged; the radioactivity measured in this lane was considered the nonspecific background and was subtracted from the measurements derived from other lanes.
When applied to a Jβ2.5-3′Dβ1 PC, this method allowed for the highly specific purification of PCs. When nonbiotinylated 3′Dβ1 was used in the binding reaction, a very small amount of radioactive Jβ2.5 was pulled down, while biotinylated 3′Dβ1 was able to pull down at least 24% of input Jβ2.5 (Fig. 2D, lanes 2 and 3). After 60 min, Jβ2.5 SCs exhibited ∼10% nicking, while in a PC with 3′Dβ1, Jβ2.5 nicking increased to >60% at 60 min (Fig. 2D, lane 3) and had already exceeded 40% at 10 min (Fig. 3A).
FIG 3.

Jβ2.5 and 5′Dβ1 nicking is stimulated by partner substrates. (A) Quantitation of time course experiments on Jβ2.5. All reactions were repeated three times. The top graph shows percent hairpinning (HP) over time, while the bottom shows percent nicking over time. The table at the top of the panel provides the legend for the graphs and includes the mean and standard deviation (SD) of the percentage of input radioactivity recovered for the indicated RSS pair. In these and all similar plots, each data point represents the mean of three independent values and is plotted with an error bar that represents the standard error of the mean (SEM). In many cases, the error bar is so small that it is not easily visible. (B) Quantitation of time course experiments on 5′Dβ1.
Early optimization experiments revealed a modest correlation between decreases in the efficiency with which the PC was isolated and decreases in nicking activity across different RSS pairs (data not shown). We were concerned that this correlation reflected an artifact of the in vitro system in which inefficiently purified PCs exhibit poor nicking activity. To test this possibility, we purified Jβ2.5-3′Dβ1 PCs from binding reactions with various concentrations of 3′Dβ1 (Fig. 2D and E). If the biotin pulldown assay allowed for the unbiased assessment of cleavage potential of both efficiently and inefficiently synapsing RSS pairs, then the magnitude of nicking should remain constant as pulldown efficiency decreases. Instead, we observed an abrupt drop in the efficiency of nicking from ∼70% to ∼40% as the pulldown efficiency dropped below 3% (Fig. 2E). We were unable to find conditions that reliably improved the cleavage activity of PCs when they were purified with pulldown efficiencies below 3%. This is a caveat that must be considered in interpreting the behavior of RSS pairs that were recovered below this threshold (see Discussion).
Nicking is stimulated in some appropriate PCs.
With radiolabeled Jβ2.5 substrate, little nicking was detected after 60 min in the absence of partner RSS (Fig. 3A, lower graph, black line; an example of primary data is shown in Fig. 4). Note that reactions in the absence of partner were by necessity performed in solution, while other reactions reflect cleavage in purified PCs. Synapsis occurred efficiently with the appropriate partner RSS, 3′Dβ1, with more than 50% of total Jβ2.5 pulled down (Fig. 3A). Jβ2.5 was nicked much more quickly in the Jβ2.5-3′Dβ1 PC than in the Jβ2.5 SC, showing an ∼22-fold stimulation at the 10-min time point (Fig. 3A; data for all substrate pairs is summarized in Fig. 5), with nearly all nicked products converted to hairpins over the 60-min time course (Fig. 3A, compare lower and upper graphs). The inappropriate partner RSS Vβ14 also stimulated Jβ2.5 nicking, though to a smaller degree than 3′Dβ1, and Vβ2 did not affect nicking at Jβ2.5 at all (Fig. 3A). However, the extent of nicking in Jβ2.5-Vβ PCs must be interpreted as lower limits because both of these PCs were purified at efficiencies below the 3% threshold (Fig. 3A).
FIG 4.

Representative Jβ2.5 cleavage gels. Lanes 1 to 6 show a cleavage time course (in-solution reaction) of a radiolabeled (32P) Jβ2.5 RSS substrate with no partner RSS, while lanes 8 to 14 show cleavage in purified Jβ2.5-3′Dβ1 PCs. Lane 8 shows the nonbiotinylated partner control. Lane 7 shows an input lane, representing 33% of the total radiolabeled Jβ2.5 used in binding reactions. Percentages of input radioactivity pulled down (% input), hairpin product (% H), and nicked product (% N) are indicated.
FIG 5.

Summary of nicking alterations in purified PCs. The percentage of nicking at the 10-min time point for the indicated RSS pairs (n = 3) was divided by the average percentage of nicking at the 10-min time point for the indicated radiolabeled RSS in the absence of partner, and the quotient was plotted on the y axis (logarithmic). Points enclosed in a rectangle indicate an RSS pair that was, on average, purified above the 3% input threshold; one asterisk represents an RSS pair whose 10-min nicking ratio significantly differed from 1 (P < 0.05), and two asterisks represent a highly significant difference (P < 0.01).
We hypothesized that the notably inefficient nicking of Jβ2.5 in the absence of partner was due to its coding flank sequence 5′-TT-RSS-3′: previous studies using consensus RSSs had demonstrated that a run of Ts immediately 5′ of the heptamer inhibited V(D)J recombination in vivo (31), even a run as short as two (32), and that this effect operates specifically at the nicking step (33). We therefore assayed the nicking of a chimeric substrate, Jβ2.7CF, comprised of the Jβ2.7 coding flank (5′-AG-RSS-3′) and the Jβ2.5 heptamer, spacer, and nonamer (Fig. 6). In the absence of partner, Jβ2.7CF nicked much more efficiently than Jβ2.5, showing a 17.6-fold increase in nicking at the 10-min time point. This result confirms our previous findings in cleavage reaction mixtures containing hybrid Jβ2.5 substrates (23) and shows that Jβ2.5's coding flank is refractory to efficient nicking. Interestingly, replacing Jβ2.5's coding flank with that of Jβ2.7 increases nicking to nearly the same magnitude as that seen in a Jβ2.5-3′Dβ1 PC (Fig. 3A). We therefore conclude that the coding flank of Jβ2.5 restrains its ability to nick in the SC and that synapsis with 3′Dβ1 allows RAG to overcome this restraint.
FIG 6.
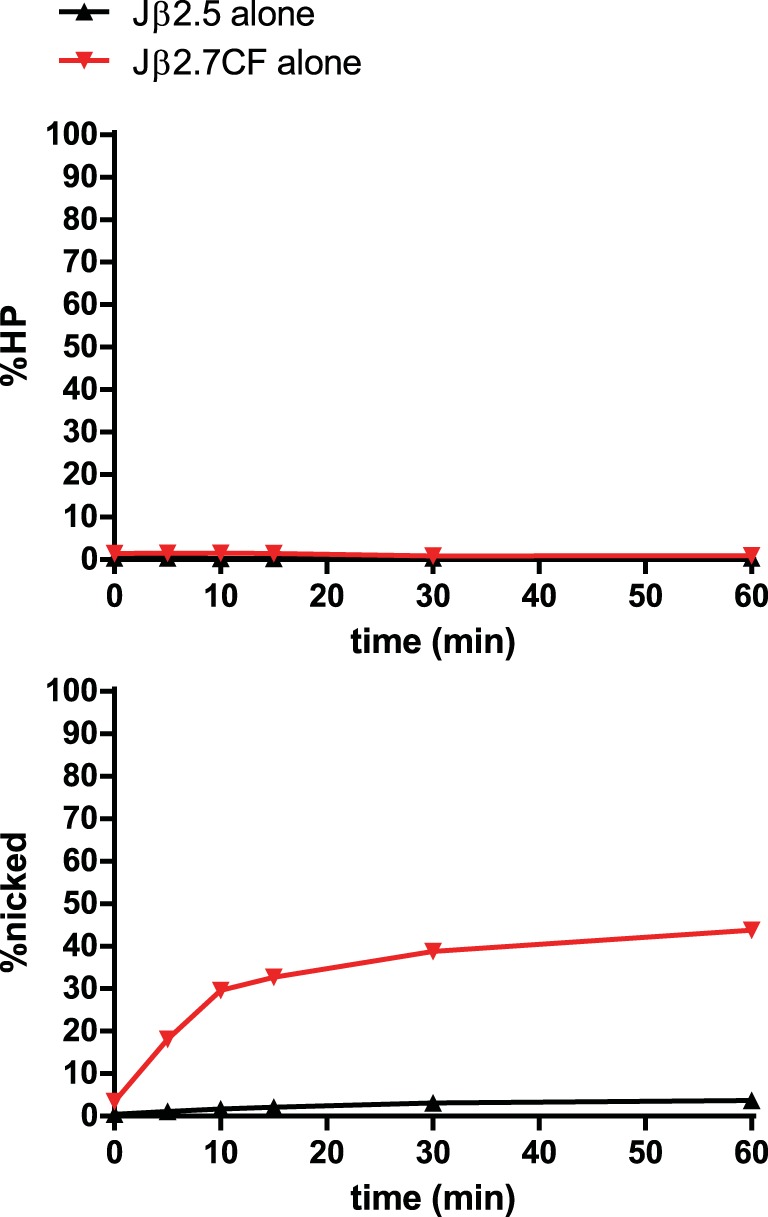
In-solution cleavage assay of Jβ2.5 and Jβ2.7CF. Jβ2.7CF is identical in sequence to Jβ2.5 except for its coding flank, which has been replaced by that of Jβ2.7.
When 5′Dβ1 12-RSS substrate was radiolabeled and used in these assays (Fig. 3B), nicking plateaued at 45% after 60 min. 5′Dβ1 PCs with a nonspecific partner control DNA were purified very inefficiently and exhibited the same nicking profiles as 5′Dβ1 SCs. In contrast, both Vβ14 and Vβ2 partner RSSs formed PCs with 5′Dβ1 that were purified above the 3% input threshold, with Vβ14 synapsing with 5′Dβ1 more efficiently than Vβ2 (Fig. 3B). 5′Dβ1 nicking and hairpinning were significantly stimulated by Vβ14 (Fig. 3B and Fig. 5), emphasizing the ability of synapsis with an appropriate partner to stimulate nicking. Some stimulation was also seen with the Vβ2 partner although this did not reach statistical significance.
Several interesting observations were made when labeled 5′Dβ1 was paired with biotinylated 3′Dβ1 (a pairing that could not occur in vivo due to the short distance between these RSSs). First, synapsis between this pair of Dβ RSSs was extremely efficient, as reflected by pulldown of more than 90% of the 5′Dβ1 substrate (Fig. 3B). Second, nicking of 5′Dβ1 in this 5′Dβ1-3′Dβ1 PC was much faster than in the SC, reaching a plateau of 90% after only 5 min. And third, despite this strong nicking and synapsis phenotype, strikingly little hairpin formation was detected in this PC (Fig. 3B). These results provide further evidence that nicking at 5′Dβ1 can be stimulated by synapsis and, together with data presented below, highlight an intriguing (and to our knowledge, unprecedented) example in which naturally occurring RSS substrates synapse and nick extremely efficiently but support double-strand break formation inefficiently.
The nicking behavior exhibited by Jβ2.5 and 5′Dβ1 confirmed our initial hypothesis that nicking at one RSS can be stimulated by synapsis with a partner RSS. Furthermore, with the exception of 5′Dβ1-3′Dβ1, this stimulation is most apparent with appropriate RSS pairs, such as Jβ2.5-3′Dβ1, and is much less pronounced with the inappropriate pair Jβ2.5-Vβ14.
Nicking can be inhibited in inappropriate PCs.
Radiolabeled Jβ2.7 (Fig. 7A and 8) nicked quickly and efficiently in the SC, in contrast to our previous study using less-active RAG proteins (23). Hairpin formation was significantly stimulated by the appropriate partner 3′Dβ1, and Jβ2.7-3′Dβ1 PCs were purified above the 3% threshold. Interestingly, Vβ14 and Vβ2 partner RSSs caused an inhibition of nicking of Jβ2.7 (Fig. 5 and 7A). However, these inappropriate Jβ2.7-Vβ PCs were purified at levels below threshold, making it unclear just how much of this inhibition is due to an intrinsic reduction of nicking in these PCs versus the low efficiency with which these PCs were recovered.
FIG 7.
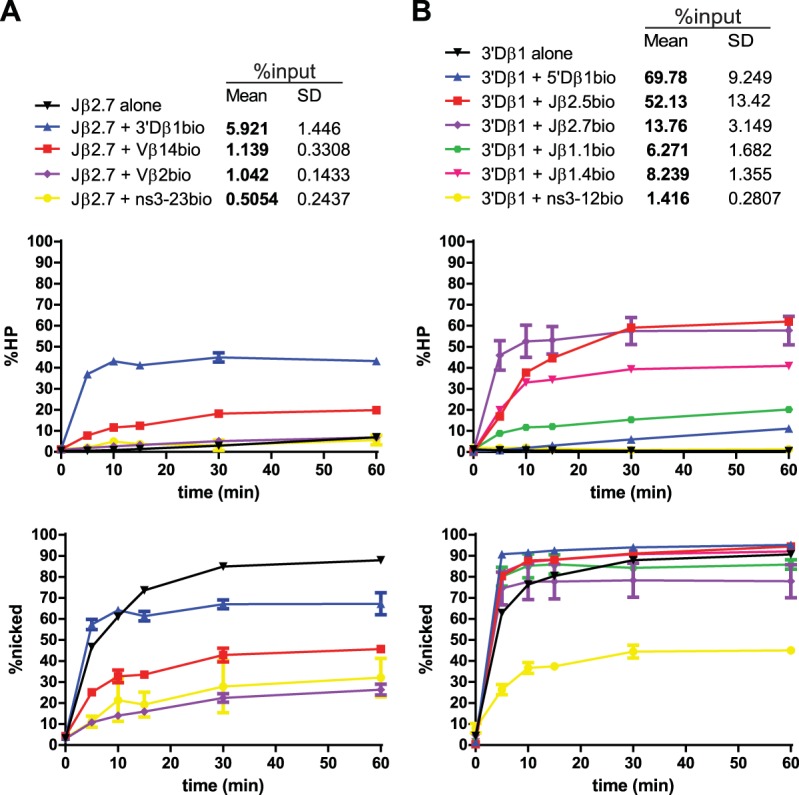
Jβ2.7 and 3′Dβ1 PC-only cleavage time courses. Data are presented as described in the legend to Fig. 3A. (A) Jβ2.7 nicking is inhibited by partner substrates. (B) 3′Dβ1 nicking is largely unaffected by partner substrates.
FIG 8.

Representative Jβ2.7 cleavage gels. Lanes 1 to 6 show a cleavage time course of a radiolabeled Jβ2.7 RSS substrate with no partner RSS, while lanes 8 to 14 show cleavage in purified Jβ2.7-Vβ14 PCs. Lane 8 shows the nonbiotinylated partner control. Lane 7 shows an input lane, representing 33% of the total radiolabeled Jβ2.7 used in binding reactions. Percentages of input radioactivity pulled down (% input), hairpin product (% H), and nicked product (% N) are indicated.
3′Dβ1 nicking is not affected by synapsis.
Radiolabeled 3′Dβ1 (Fig. 7B) nicked very efficiently in the SC context, at levels comparable to consensus RSSs (compare Fig. 7B and 2B) and was the most efficient of the naturally occurring RSS substrates tested. The initial rate of nicking of 3′Dβ1 was stimulated to a small but statistically significant extent by synapsis with RSS partners (Fig. 5), although this might reflect the delay in nicking that could be expected for an in-solution reaction compared to the biotin pulldown reaction in which all substrate is bound at the zero time point (t = 0). All 3′Dβ1 PCs were purified well above the 3% threshold, with the exception of the PC formed with the nonspecific partner ns3-12.
We also analyzed the 3′Dβ1-5′Dβ1 PC but now with 3′Dβ1 labeled. This PC was efficiently purified and displayed very fast nicking, but hairpinning was remarkably slow and inefficient (Fig. 7B, blue lines). These results closely match those obtained when 5′Dβ1 was labeled (Fig. 3B), and together they demonstrate that 3′Dβ1 and 5′Dβ1 form a tight synaptic complex within which nicking at both RSSs is very efficient but hairpin formation is largely blocked.
Jβ1.1 and Jβ1.4.
Radiolabeled Jβ1.1 (Fig. 9A) exhibited inefficient coupled cleavage, even with the appropriate partner 3′Dβ1. Jβ1.1-3′Dβ1 PCs were purified just above the 3% threshold, suggesting that these PCs are indeed poor substrates for coupled cleavage in vitro. These results are unexpected in light of the finding that Jβ1.1 is one of the most frequently used gene segments in the final TCRβ repertoire (26). Jβ1.1 nicking appeared to be inhibited by synapsis with any of the partner RSSs tested (Fig. 9A) though initial rates were unchanged by the appropriate partner 3′Dβ1, and inappropriate Jβ1.1-Vβ PCs were purified below threshold.
FIG 9.

Jβ1.1 and Jβ1.4 PC-only cleavage time courses. Data are presented as described in the legend to Fig. 3A. (A) Jβ1.1 nicking is inhibited by partner substrates. (B) Jβ1.4 nicking is moderately stimulated by synapsis with 3′Dβ1 and unaffected by synapsis with Vβ14 or Vβ2.
Radiolabeled Jβ1.4 (Fig. 9B) was purified above threshold when in a PC with 3′Dβ1, and these PCs supported hairpin formation, though to a lesser extent than most other appropriate pairs tested. Nicking of Jβ1.4 was somewhat stimulated by synapsis with 3′Dβ1; this stimulation was seen at early time points but did not reach statistical significance (Fig. 5). In contrast, synapsis with Vβ14 or Vβ2 did not appreciably change nicking at Jβ1.4 and did not stimulate hairpinning though, again, these inappropriate PCs were purified below threshold.
Vβ14 exhibits heterogeneous PC nicking behaviors.
Radiolabeled Vβ14 (Fig. 10A) synapsed well with the appropriate partner 5′Dβ1, and these PCs supported hairpin formation to levels of about 30% after 60 min. Vβ14 nicking was also significantly stimulated by synapsis with 5′Dβ1 (4.6-fold at 10 min) (Fig. 5 and 10A). Hairpinning was also apparently stimulated by Jβ2.7, but synapsis was very inefficient with this pair, with an average purification of only 0.6% of input. This extremely low efficiency of purification suggests, as we proposed previously (23), that recombination between these two gene segments is restricted largely through inefficient synapsis. Vβ14 synapsis with other Jβ RSSs either did not change or inhibited Vβ14 nicking. Two of these pairs were purified below threshold (Vβ14-Jβ1.1 and Vβ14-Jβ1.4) while one pair was purified well above the threshold (Vβ14-Jβ2.5) (Fig. 5 and Fig. 10A). Vβ14 thus provides an example of an RSS for which nicking is stimulated by its appropriate B12/23 partner and for which cleavage with inappropriate partners is limited by poor synapsis or nicking.
FIG 10.

Vβ14 and Vβ2 PC-only cleavage time courses. Data are presented as described in the legend to Fig. 3A. (A) Vβ14 nicking can be stimulated, unaffected, or inhibited by synapsis with various partner substrates. (B) Vβ2 nicking proceeds inefficiently and is unaffected by synapsis with any of the partner substrates tested.
Vβ2.
Lastly, radiolabeled Vβ2 (Fig. 10B) exhibited very little hairpinning or nicking activity, whether in an SC or PC. Vβ2 synapsed with 5′Dβ1, Jβ2.5, Jβ1.1, and Jβ1.4 with efficiencies exceeding the 3% threshold, but even in these cases, nicking and hairpin activity were very low. Hairpin formation activity measured in the Vβ2-5′Dβ1 PC when Vβ2 was labeled was only 5% at 60 min (Fig. 10B); this modest degree of hairpin formation is consistent with a recent quantitative determination of preselection Vβ gene segment usage that showed that Vβ2 exhibited a lower recombination frequency than most other Vβ segments (34).
Consistency of coupled cleavage.
Since hairpin formation is expected to occur in a coupled manner at the two RSSs in the PC (33, 35), we expected that the amount of hairpin formation would be approximately equal regardless of which RSS was labeled. For the most part, this was the case in PCs involving 3′Dβ1 and Jβ substrates (Fig. 11A). In 3′Dβ1-Jβ1.1 PCs, however, more hairpin formation was detected at 10 min when 3′Dβ1 was labeled (11.7%) than when Jβ1.1 was labeled (6.1%). This might reflect some uncoupling of hairpin formation at the two RSSs, or it might reflect the different efficiencies with which the PC was purified, depending on which RSS was labeled and incubated with RAG first. With labeled Jβ1.1, 3.7% was purified in a PC with 3′Dβ1, whereas 6.3% of labeled 3′Dβ1 was purified with Jβ1.1 (Fig. 11C). Because the former value is close to the 3% threshold and because the threshold was not determined precisely for every pair, it is possible that the activity of the PC containing labeled Jβ1.1 with 3′Dβ1 was somewhat inhibited. In 3′Dβ1-5′Dβ1 PCs, a statistically significant increase in hairpin formation was observed when 5′Dβ1 was labeled (Fig. 11A). However, because the absolute value of this difference is so small (0.74%), we do not believe that this represents a physiologically significant phenomenon.
FIG 11.

Comparison of hairpin formation and efficiencies of pulldown of PCs containing 3′Dβ1 or Vβ14. Radiolabeled RSSs (32P) were incubated with protein for 1 min before the addition of biotinylated (Bio) partner and subsequent incubation for 10 min, as described in Materials and Methods. P values were calculated by a two-tailed t test with Welch's correction. *, P < 0.05; **, P < 0.01; ***, P < 0.0001. (A) The magnitude of hairpin formation at 10 min is shown for the indicated substrate pairs containing 3′Dβ1. (B) The magnitude of hairpin formation at 10 min is shown for the indicated substrate pairs containing Vβ14. (C) The efficiency of radiolabeled RSS recovery (% input) is indicated for the indicated substrate pairs containing 3′Dβ1. (D) The efficiency of radiolabeled RSS recovery (% input) is indicated for the indicated substrate pairs containing Vβ14.
Similarly, in PCs involving Vβ14, hairpin formation at 10 min was approximately equal regardless of which RSS was labeled (Fig. 11B). The one exception was the Vβ14-Jβ2.7 PC, where more hairpin formation was detected when Vβ14 was labeled (25%) than when Jβ2.7 was labeled (12%). We note, however, that the pulldown efficiencies of these PCs were quite low (0.6% and 1.1%) (Fig. 11D), which suggests that the cleavage data for this pair might not be reliable.
RSS binding order can affect synapsis efficiency.
As described in Materials and Methods, binding reactions for the PC-only assay were performed by allowing RAG to bind the labeled RSS for 1 min prior to the addition of partner RSS. We observed that the binding order affected the efficiency of pulldown of several PCs. For example, when RAG was allowed to bind 3′Dβ1 before Jβ2.7, PCs were purified at an efficiency of 14%, but when RAG bound Jβ2.7 before 3′Dβ1, the efficiency of pulldown dropped to 6% (Fig. 11C). Similar asymmetries were observed with 5′Dβ1-3′Dβ1 PCs (Fig. 11C) and with Vβ14-Jβ PCs (Fig. 11D). In some cases, more efficient pulldown was seen when the 23-RSS was labeled, while in other cases, more efficient pulldown was seen when the 12-RSS was labeled. These data support the hypothesis that initial RAG binding in vivo can occur on either 12- or 23-RSSs (14, 36) and not only on 12-RSSs as previously suggested (6, 13).
DISCUSSION
Our previous study (23) showed that Vβ-5′Dβ recombination is preferred over Vβ-Jβ recombination in part because the Jβ RSSs are crippled for nicking and synapsis. Because Jβ2.5 is found in the TCRβ repertoire (26) yet nicks so inefficiently, it seemed plausible that nicking could be stimulated by synapsis with appropriate partner RSSs. We tested this hypothesis with a PC-only cleavage assay that allowed us to simultaneously measure the synapsis and nicking of substrates containing naturally occurring RSSs; to our knowledge, these experiments represent the first biochemical analysis of RAG activity on naturally occurring RSSs specifically within a PC. Our data support the hypothesis and also suggest that the nicking of some Tcrb RSSs can be inhibited by synapsis. These conclusions in turn suggest that the B12/23 rule is enforced in part by dynamic RAG-RSS interactions that promote Dβ-Jβ and Vβ-DJβ recombination through efficient nicking within appropriate PCs and inefficient nicking within inappropriate PCs.
Technical considerations.
The standard in-solution cleavage assay has technical limitations that make it difficult to interpret apparent inhibition of nicking by partner RSSs. To overcome this, we used a biotin pulldown method to purify and interrogate activity specifically within PCs. An alternative approach would have been the gel shift/in-gel cleavage method developed by Bergeron et al. (28), which has the advantage of ensuring that the protein-DNA complex(es) being interrogated is electrophoretically homogeneous. This approach, however, would require that the PC be sufficiently stable to be visualized by gel shift, and given the difficulty in detecting the SC with naturally occurring Tcrb RSSs (23, 24, 37), many of the PCs we investigated were unlikely to meet this criterion. A caveat associated with the biotin pulldown method is the possibility that the material interrogated contains, in addition to the PC, labeled RSS trapped in aberrant or nonfunctional complexes. Indeed, a low level of such nonfunctional complexes could help explain our observation that activity drops when complexes are purified with efficiencies below the 3% threshold.
In general, we found that the data gathered by our PC-only assay were internally consistent: hairpinning at one RSS in a purified PC followed similar kinetics as hairpinning at the other RSS (Fig. 11A and B). This consistency in coupled cleavage indicates that this system largely recapitulates proper regulation of RAG-mediated cleavage.
With the exception of the Jβ2.5 substrate, RSS nicking in the SC was higher, in some cases much higher, in the experiments reported here than in our previous study (23). This is likely explained by the different proteins used in our two experimental systems; it has long been known that the RAG proteins are more active when coexpressed and copurified (8), as was done here, and we (38) along with others (39) have shown that coexpressed MBP-RAGs bind to RSSs more efficiently than singly expressed RAGs.
Limitations associated with the 3% input threshold.
When a Jβ2.5-3′Dβ1 PC is purified inefficiently, Jβ2.5 nicking activity drops compared to when it is purified efficiently (Fig. 2E). This drop occurs at 3% of input. Our assay likely underestimates the cleavage activity of PCs purified with efficiencies below this threshold. This caveat does not affect our conclusion that nicking of some Tcrb RSSs can be stimulated upon synapsis with an appropriate partner because every such pair was purified well above the threshold (Fig. 5), and any tendency to underestimate cleavage in the PC would only lead us to underestimate the stimulation. We note, however, that technical differences between the in-solution SC assay and the purified PC assay might exaggerate the observed stimulation in nicking, particularly at early time points. In the in-solution SC assay, the recombinase begins binding to DNA at t = 0, but in purified PCs, the recombinase is already stably bound and can begin nicking at t = 0. We have attempted to minimize this problem by performing in-solution cleavage assays under optimal conditions (see Materials and Methods). In some cases, the stimulation of nicking is so large and persistent that this issue is unlikely to be significant (e.g., Jβ2.5).
The caveat associated with the 3% threshold, however, does limit the strength of conclusions that can be drawn regarding the observed inhibition of Tcrb RSS nicking in the PC. One of the most striking examples of nicking inhibition can be seen at Jβ2.7 (Fig. 5, 7A, and 8), where nicking is significantly decreased in PCs containing either Vβ14 or Vβ2. However, because both Jβ2.7-Vβ PCs were purified at only 1% of input, the observed efficiency of nicking in these PCs represents only a lower limit for their nicking activity.
Synapsis can stimulate nicking.
Several of the Tcrb RSSs we tested were stimulated for nicking upon synapsis with a partner RSS (Fig. 5). In particular, synapsis with 3′Dβ1 significantly stimulated the initial rate of nicking at Jβ2.5 (22-fold) and 5′Dβ1 (2-fold), synapsis with Vβ14 stimulated nicking at 5′Dβ1 (2-fold), and synapsis with 5′Dβ1 stimulated nicking at Vβ14 (4-fold). These data fit well with the findings of Franchini et al. (14), who incubated DNA substrates containing two Tcrb RSSs in cis with crude cell extracts containing RAG and observed that nicking occurred only in substrates containing a B12/23-appropriate pair of RSSs.
Two prior studies also examined the effect of synapsis on nicking. Yu and Lieber (27) performed cleavage experiments with bead-immobilized consensus RSS oligonucleotide substrates and found that that nicking was unaffected by synapsis, while Eastman and Schatz (40) used long DNA substrates containing consensus RSSs in cis and found that nicking could be stimulated approximately 2-fold by synapsis. The use of substrates with RSSs in cis in the latter study might have allowed faster synapsis and the detection of synapsis-stimulated nicking events at early time points. Together, these studies argue that nicking of consensus RSSs is at most modestly stimulated by synapsis, a conclusion supported by our finding, obtained with oligonucleotide substrates, that a consensus 23-RSS did not stimulate nicking at a consensus 12-RSS (Fig. 2B). In contrast, 5′Dβ1, Jβ2.5, and Vβ14 nicking were all stimulated by synapsis with appropriate partners (Fig. 5). Importantly, these PCs were all purified well above the 3% threshold. We conclude that RSS nicking can indeed be stimulated by synapsis, as previously proposed (40), and that the magnitude of this effect is highly dependent on the sequence of the RSS, its coding flank, and the partner RSS involved.
Potential mechanisms regulating nicking in the PC.
Endogenous RSSs (which typically differ from the consensus at multiple positions) and their coding flanks can present significant barriers to RAG-mediated binding and cleavage. In the Tcrb locus, these barriers are utilized to help enforce the B12/23 restriction, and our work demonstrates that nicking is one of the important steps at which this principle operates. We found that the inefficient nicking phenotype of Jβ2.5 is a function of its coding flank sequence (Fig. 6) and that this is overcome by synapsis with its appropriate partner 3′Dβ1 but much less so with inappropriate Vβ partners (Fig. 5). RAG binding to the RSS has been shown to perturb DNA structure near the site of cleavage (41), and data from RSS substrates containing mismatches in the heptamer supported the idea that such DNA distortion facilitates nicking (42), as has been shown for hairpin formation (43–47). Based on this, we propose that the Jβ2.5 coding flank inhibits a DNA distortion necessary for nicking at the coding flank-heptamer boundary and that this is overcome upon synapsis with 3′Dβ1. A similar mechanism might govern nicking stimulation in other PCs, such as that of 5′Dβ1 or Vβ14 in the 5′Dβ1-Vβ14 PC.
Our data suggesting that nicking can be inhibited by synapsis were unexpected. It will be interesting to determine whether reduced nicking correlates with structural changes in the PC indicative of less distortion near the site of cleavage.
Dissociation of hairpin formation from nicking and synapsis.
The 5′Dβ1-3′Dβ1 RSS pair supported the most efficient synapsis of all the naturally occurring RSS pairs tested, with recoveries of greater than 90% of radiolabeled 5′Dβ1 and ∼70% of radiolabeled 3′Dβ1 (Fig. 3B and 7B). Furthermore, nicking of both substrates in this PC was extremely rapid and efficient, reaching nearly maximal values of ∼90% at the earliest (5 min) time point measured (Fig. 3B and 7B, blue lines). Despite this, hairpinning was slow and inefficient, reaching levels of only 10% after 60 min, well below the levels observed in some other PCs containing these RSSs. It is clear from previous studies that, depending on the substrates involved, either nicking (33) or hairpin formation (48) can be the rate-limiting step in RAG-mediated cleavage. The 5′Dβ1-3′Dβ1 pair appears to represent an extreme example of the latter. Given the near identity between the 5′Dβ1 and 5′Dβ2 RSSs/coding flanks (Fig. 1B), it seems likely that 3′Dβ1 and 5′Dβ2 would similarly be very slow and inefficient in coupled cleavage. This would help suppress D-D fusion and unwanted deletion of the Jβ1-Cβ1 region due to recombination between these elements. In addition, our data with 5′Dβ1-3′Dβ1 are interesting in light of previous findings showing that hyperstable recombinase complexes (containing a very tight binding Saccharomyces cerevisiae HMG box protein in place of HMGB1) are defective for cleavage (49). The results of this study led to the hypothesis that cleavage requires a certain degree of flexibility within RAG-RSS complexes, and based on this, we speculate that restricted protein-DNA dynamics in the tightly synapsed 5′Dβ1-3′Dβ1 pair interferes with hairpin formation.
Regulated nicking and the B12/23 rule.
Nicking is an attractive step at which to regulate V(D)J recombination and enforce the B12/23 rule. Inefficient nicking in the SC appears advantageous given the potential of RAG-mediated nicks to trigger various forms of genome instability (50). Furthermore, because hairpin formation at one RSS requires nicking at its partner RSS (33), regulating nicking at one RSS provides control over catalytic events at both RSSs in the PC. Hence, when nicking at Jβ2.5 is activated by synapsis with 3′Dβ1, this enables hairpin formation at both RSSs in a B12/23-compliant context where the efficient DNA repair mechanisms associated with V(D)J recombination can operate.
Synapsis-dependent nicking inhibition, while yet to be definitely shown to occur, is also an appealing mechanism by which to reinforce the B12/23 rule, especially since evidence for it was observed only in B12/23-inappropriate PCs (e.g., Jβ2.7-Vβ14 and Jβ2.7-Vβ2) (Fig. 5). Such a mechanism would help address the unexpected behavior of Jβ2.7, whose rapid nicking in the SC is a challenge to genome stability and to the B12/23 rule. Suppression of Jβ2.7 nicking in PCs with Vβ RSSs would help address these challenges, and this could be facilitated further by mechanisms that suppress full engagement of the Jβ2.7 RSS prior to synapsis, such as nucleosome occupancy of some or all of the RSS (36).
Notably, the two RSSs within a given PC can display different nicking behavior patterns. For instance, Jβ2.5 nicking is strongly stimulated upon synapsis with 3′Dβ1, but 3′Dβ1 nicking is unaffected by synapsis with Jβ2.5 (Fig. 5). This suggests that the recombinase complex is capable of fine conformational changes that allow it to discriminate between different RSS pairs and tailor the catalysis at each RSS to the context of that specific PC. The heterogeneity of behaviors that we observe in Tcrb locus PCs suggests that the B12/23 rule emerges out of a combination of distinct mechanisms, with different Tcrb substrates tailored to take advantage of different mechanisms. Together with chromatin accessibility, such exacting discrimination is likely to be the foundation of the ordered assembly of the Tcrb variable region .
ACKNOWLEDGMENTS
We thank the members of the Schatz lab for their intellectual input.
This work was supported by National Institutes of Health grant AI32524 (D.G.S.). J.K.B. was supported in part by National Institutes of Health training grant T32 GM007233.
Footnotes
Published ahead of print 5 May 2014
REFERENCES
- 1.Schatz DG, Oettinger MA, Baltimore D. 1989. The V(D)J recombination activating gene, RAG-1. Cell 59:1035–1048. 10.1016/0092-8674(89)90760-5 [DOI] [PubMed] [Google Scholar]
- 2.Oettinger MA, Schatz DG, Gorka C, Baltimore D. 1990. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248:1517–1523. 10.1126/science.2360047 [DOI] [PubMed] [Google Scholar]
- 3.Gellert M. 2002. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71:101–132. 10.1146/annurev.biochem.71.090501.150203 [DOI] [PubMed] [Google Scholar]
- 4.Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. 2000. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol. 18:495–527. 10.1146/annurev.immunol.18.1.495 [DOI] [PubMed] [Google Scholar]
- 5.van Gent DC, Hiom K, Paull TT, Gellert M. 1997. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 16:2665–2670. 10.1093/emboj/16.10.2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones JM, Gellert M. 2002. Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J 21:4162–4171. 10.1093/emboj/cdf394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mundy CL, Patenge N, Matthews AGW, Oettinger MA. 2002. Assembly of the RAG1/RAG2 synaptic complex. Mol. Cell. Biol. 22:69–77. 10.1128/MCB.22.1.69-77.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. 1995. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 83:387–395. 10.1016/0092-8674(95)90116-7 [DOI] [PubMed] [Google Scholar]
- 9.Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79:181–211. 10.1146/annurev.biochem.052308.093131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsden DA, Baetz K, Wu GE. 1994. Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res. 22:1785–1796. 10.1093/nar/22.10.1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steen SB, Gomelsky L, Roth DB. 1996. The 12/23 rule is enforced at the cleavage step of V(D)J recombination in vivo. Genes Cells 1:543–553. 10.1046/j.1365-2443.1996.d01-259.x [DOI] [PubMed] [Google Scholar]
- 12.Hiom K, Gellert M. 1998. Assembly of a 12/23 paired signal complex: a critical control point in V(D)J recombination. Mol. Cell 1:1011–1019. 10.1016/S1097-2765(00)80101-X [DOI] [PubMed] [Google Scholar]
- 13.Curry JD, Geier JK, Schlissel MS. 2005. Single-strand recombination signal sequence nicks in vivo: evidence for a capture model of synapsis. Nat. Immunol. 6:1272–1279. 10.1038/ni1270 [DOI] [PubMed] [Google Scholar]
- 14.Franchini D-M, Benoukraf T, Jaeger S, Ferrier P, Payet-Bornet D. 2009. Initiation of V(D)J recombination by Dβ-associated recombination signal sequences: a critical control point in TCRβ gene assembly. PLoS One 4:e4575. 10.1371/journal.pone.0004575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. 2010. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell 141:419–431. 10.1016/j.cell.2010.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uematsu Y, Ryser S, Dembić Z, Borgulya P, Krimpenfort P, Berns A, von Boehmer H, Steinmetz M. 1988. In transgenic mice the introduced functional T cell receptor beta gene prevents expression of endogenous beta genes. Cell 52:831–841. 10.1016/0092-8674(88)90425-4 [DOI] [PubMed] [Google Scholar]
- 17.Ferrier P, Krippl B, Blackwell TK, Furley AJ, Suh H, Winoto A, Cook WD, Hood L, Costantini F, Alt FW. 1990. Separate elements control DJ and VDJ rearrangement in a transgenic recombination substrate. EMBO J. 9:117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bassing CH, Alt FW, Hughes MM, D'Auteuil M, Wehrly TD, Woodman BB, Gärtner F, White JM, Davidson L, Sleckman BP. 2000. Recombination signal sequences restrict chromosomal V(D)J recombination beyond the 12/23 rule. Nature 405:583–586. 10.1038/35014635 [DOI] [PubMed] [Google Scholar]
- 19.Sleckman BP, Bassing CH, Hughes MM, Okada A, D'Auteuil M, Wehrly TD, Woodman BB, Davidson L, Chen J, Alt FW. 2000. Mechanisms that direct ordered assembly of T cell receptor beta locus V, D, and J. gene segments. Proc. Natl. Acad. Sci. U. S. A. 97:7975–7980. 10.1073/pnas.130190597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hughes MM, Tillman RE, Wehrly TD, White JM, Sleckman BP. 2003. The B12/23 restriction is critically dependent on recombination signal nonamer and spacer sequences. J. Immunol. 171:6604–6610. 10.4049/jimmunol.171.12.6604 [DOI] [PubMed] [Google Scholar]
- 21.Jung D, Bassing CH, Fugmann SD, Cheng H-L, Schatz DG, Alt FW. 2003. Extrachromosomal recombination substrates recapitulate beyond 12/23 restricted VDJ recombination in nonlymphoid cells. Immunity 18:65–74. 10.1016/S1074-7613(02)00507-1 [DOI] [PubMed] [Google Scholar]
- 22.Tillman RE, Wooley AL, Khor B, Wehrly TD, Little CA, Sleckman BP. 2003. Cutting edge: targeting of Vβ to Dβ rearrangement by RSSs can be mediated by the V(D)J recombinase in the absence of additional lymphoid-specific factors. J. Immunol. 170:5–9. 10.4049/jimmunol.170.1.5 [DOI] [PubMed] [Google Scholar]
- 23.Drejer-Teel AH, Fugmann SD, Schatz DG. 2007. The beyond 12/23 restriction is imposed at the nicking and pairing steps of DNA cleavage during V(D)J recombination. Mol. Cell. Biol. 27:6288–6299. 10.1128/MCB.00835-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olaru A, Patterson DN, Villey I, Livák F. 2003. DNA-Rag protein interactions in the control of selective D gene utilization in the TCRβ locus. J. Immunol. 171:3605–3611. 10.4049/jimmunol.171.7.3605 [DOI] [PubMed] [Google Scholar]
- 25.Candéias S, Waltzinger C, Benoist C, Mathis D. 1991. The Vβ17+ T cell repertoire: skewed J beta usage after thymic selection; dissimilar CDR3s in CD4+ versus CD8+ cells. J. Exp. Med. 174:989–1000. 10.1084/jem.174.5.989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak F, Burtrum DB, Rowen L, Schatz DG, Petrie HT. 2000. Genetic modulation of T cell receptor gene segment usage during somatic recombination. J. Exp. Med. 192:1191–1196. 10.1084/jem.192.8.1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu K, Lieber MR. 2000. The nicking step in V(D)J recombination is independent of synapsis: implications for the immune repertoire. Mol. Cell. Biol. 20:7914–7921. 10.1128/MCB.20.21.7914-7921.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergeron S, Anderson DK, Swanson PC. 2006. RAG and HMGB1 proteins: purification and biochemical analysis of recombination signal complexes. Methods Enzymol. 408:511–528. 10.1016/S0076-6879(06)08032-3 [DOI] [PubMed] [Google Scholar]
- 29.Zhang M, Swanson PC. 2008. V(D)J recombinase binding and cleavage of cryptic recombination signal sequences identified from lymphoid malignancies. J. Biol. Chem. 283:6717–6727. 10.1074/jbc.M710301200 [DOI] [PubMed] [Google Scholar]
- 30.Nagawa F, Hirose S, Nishizumi H, Nishihara T, Sakano H. 2004. Joining mutants of RAG1 and RAG2 that demonstrate impaired interactions with the coding-end DNA. J. Biol. Chem. 279:38360–38368. 10.1074/jbc.M405485200 [DOI] [PubMed] [Google Scholar]
- 31.Gerstein RM, Lieber MR. 1993. Coding end sequence can markedly affect the initiation of V(D)J recombination. Genes Dev. 7:1459–1469. 10.1101/gad.7.7b.1459 [DOI] [PubMed] [Google Scholar]
- 32.Ezekiel UR, Engler P, Stern D, Storb U. 1995. Asymmetric processing of coding ends and the effect of coding end nucleotide composition on V(D)J recombination. Immunity 2:381–389. 10.1016/1074-7613(95)90146-9 [DOI] [PubMed] [Google Scholar]
- 33.Yu K, Lieber MR. 1999. Mechanistic basis for coding end sequence effects in the initiation of V(D)J recombination. Mol. Cell. Biol. 19:8094–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gopalakrishnan S, Majumder K, Predeus A, Huang Y, Koues OI, Verma-Gaur J, Loguercio S, Su AI, Feeney AJ, Artyomov MN, Oltz EM. 2013. Unifying model for molecular determinants of the preselection Vβ repertoire. Proc. Natl. Acad. Sci. U. S. A. 110:E3206–E3215. 10.1073/pnas.1304048110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eastman QM, Leu TM, Schatz DG. 1996. Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature 380:85–88. 10.1038/380085a0 [DOI] [PubMed] [Google Scholar]
- 36.Schatz DG, Ji Y. 2011. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 11:251–263. 10.1038/nri2941 [DOI] [PubMed] [Google Scholar]
- 37.Olaru A, Patterson DN, Cai H, Livák F. 2004. Recombination signal sequence variations and the mechanism of patterned T-cell receptor-beta locus rearrangement. Mol. Immunol. 40:1189–1201. 10.1016/j.molimm.2003.11.019 [DOI] [PubMed] [Google Scholar]
- 38.Ciubotaru M, Trexler AJ, Spiridon LN, Surleac MD, Rhoades E, Petrescu AJ, Schatz DG. 2013. RAG and HMGB1 create a large bend in the 23RSS in the V(D)J recombination synaptic complexes. Nucleic Acids Res. 41:2437–2454. 10.1093/nar/gks1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanson PC. 2002. A RAG-1/RAG-2 tetramer supports 12/23-regulated synapsis, cleavage, and transposition of V(D)J recombination signals. Mol. Cell. Biol. 22:7790–7801. 10.1128/MCB.22.22.7790-7801.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eastman QM, Schatz DG. 1997. Nicking is asynchronous and stimulated by synapsis in 12/23 rule-regulated V(D)J cleavage. Nucleic Acids Res. 25:4370–4378. 10.1093/nar/25.21.4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanson PC. 2004. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol. Rev. 200:90–114. 10.1111/j.0105-2896.2004.00159.x [DOI] [PubMed] [Google Scholar]
- 42.Santagata S, Besmer E, Villa A, Bozzi F, Allingham JS, Sobacchi C, Haniford DB, Vezzoni P, Nussenzweig MC, Pan ZQ, Cortes P. 1999. The RAG1/RAG2 complex constitutes a 3′ flap endonuclease: implications for junctional diversity in V(D)J and transpositional recombination. Mol. Cell 4:935–947. 10.1016/S1097-2765(00)80223-3 [DOI] [PubMed] [Google Scholar]
- 43.Cuomo CA, Mundy CL, Oettinger MA. 1996. DNA sequence and structure requirements for cleavage of V(D)J recombination signal sequences. Mol. Cell. Biol. 16:5683–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramsden DA, McBlane JF, van Gent DC, Gellert M. 1996. Distinct DNA sequence and structure requirements for the two steps of V(D)J recombination signal cleavage. EMBO J. 15:3197–3206 [PMC free article] [PubMed] [Google Scholar]
- 45.Grundy GJ, Hesse JE, Gellert M. 2007. Requirements for DNA hairpin formation by RAG1/2. Proc. Natl. Acad. Sci. U. S. A. 104:3078–3083. 10.1073/pnas.0611293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bischerour J, Lu C, Roth DB, Chalmers R. 2009. Base flipping in V(D)J recombination: insights into the mechanism of hairpin formation, the 12/23 rule, and the coordination of double-strand breaks. Mol. Cell. Biol. 29:5889–5899. 10.1128/MCB.00187-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishihara T, Nagawa F, Imai T, Sakano H. 2008. RAG-heptamer interaction in the synaptic complex is a crucial biochemical checkpoint for the 12/23 recombination rule. J. Biol. Chem. 283:4877–4885. 10.1074/jbc.M709890200 [DOI] [PubMed] [Google Scholar]
- 48.Yu K, Taghva A, Ma Y, Lieber MR. 2004. Kinetic analysis of the nicking and hairpin formation steps in V(D)J recombination. DNA Repair (Amst.) 3:67–75. 10.1016/j.dnarep.2003.09.006 [DOI] [PubMed] [Google Scholar]
- 49.Dai Y, Wong B, Yen Y, Oettinger MA, Kwon J, Johnson RC. 2005. Determinants of HMGB proteins required to promote RAG1/2-recombination signal sequence complex assembly and catalysis during V(D)J recombination. Mol. Cell. Biol. 25:4413–4425. 10.1128/MCB.25.11.4413-4425.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee GS, Neiditch MB, Salus SS, Roth DB. 2004. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell 117:171–184. 10.1016/S0092-8674(04)00301-0 [DOI] [PubMed] [Google Scholar]