

Abstract
14-3-3τ is frequently overexpressed in breast cancer; however, whether it contributes to breast cancer progression remains undetermined. Here, we identify a critical role for 14-3-3τ in promoting breast cancer metastasis, in part through binding to and inhibition of RhoGDIα, a negative regulator of Rho GTPases and a metastasis suppressor. 14-3-3τ binds Ser174-phosphorylated RhoGDIα and blocks its association with Rho GTPases, thereby promoting epidermal growth factor (EGF)-induced RhoA, Rac1, and Cdc42 activation. When 14-3-3τ is overexpressed in MCF7 breast cancer cells that express 14-3-3τ at low levels, it increases motility, reduces adhesion, and promotes metastasis in mammary fat pad xenografts. On the other hand, depletion of 14-3-3τ in MCF7 cells and in an invasive cell line, MDA-MB231, inhibits Rho GTPase activation and blocks breast cancer migration and invasion. Moreover, 14-3-3τ overexpression in human breast tumors is associated with the activation of ROCK (a Rho GTPase effector), high metastatic rate, and shorter survival, underscoring a clinically significant role for 14-3-3τ in breast cancer progression. Our work indicates that 14-3-3τ is a novel therapeutic target to prevent breast cancer metastasis.
INTRODUCTION
14-3-3 is a family of evolutionarily conserved proteins with the property of specific phosphoserine/threonine binding. Through their ability to bind phosphorylated protein ligands, they are involved in many different cellular processes, including mitogenesis, cell cycle control, DNA damage checkpoint, apoptosis, etc. (1). They are required for converting many phosphorylation events into subsequent biochemical or biological outcomes. In mammals, there are seven 14-3-3 isoforms (β, ε, γ, η, σ, τ, and ζ), each with distinct and sometimes overlapping functions (2). For example, 14-3-3τ binds to the cyclin-dependent kinase inhibitor p21Waf1/Cip1 and induces ubiquitin-independent proteasomal degradation of p21 to promote cell growth (3). Overexpression of 14-3-3τ is frequently seen in human breast cancer and is associated with shorter patient survival (3). 14-3-3τ is upregulated by tenascin-C (3, 4). Recent studies show that the expression of tenascin-C at the invasion border of early breast cancer significantly correlates with proliferative activity, higher risk of distant metastasis, and local recurrence (5). Thus, whether 14-3-3τ overexpression in breast cancer has any direct link to breast cancer metastasis deserves investigation.
Breast cancer is the most commonly diagnosed type of cancer in women. It represents 29% of all new cancer cases in 2013 among women in the United States (6) and is estimated to develop in one out of every eight women in the United States during their lifetimes (7). Although most breast cancers are diagnosed during relatively early stages, unfortunately nearly 30% of them will eventually develop metastasis in spite of treatment (8). Cancer metastasis comprises a series of sequential steps, including migration, invasion, dissemination through blood or lymphatic vessels, and repopulation (colonization) in distant sites (9). Thus, if there were a subset of master regulators that promote breast cancer metastasis, it would be the ones involved in many diverse signaling processes. There are a few master regulators that have been shown to promote breast cancer metastasis, including TWIST (10) and several proteins in the Rho GTPase pathways (11, 12).
Rho GTPases are small G proteins that transduce signals from cell surface receptors to a variety of intracellular responses. They control several essential cellular functions, including motility, adhesion, and proliferation, and are widely implicated in carcinogenesis and metastasis (13, 14). The Rho GTPase family is composed of five subfamilies, Rho, Rac, Cdc42, Rnd, and RhoBTB. Rho GTPases are controlled by three classes of regulators: GEFs (guanine nucleotide exchange factors), GAPs (GTPase activating proteins), and GDIs (GDP dissociation inhibitors). At steady state, most of the Rho GTPases are kept inactive in the cytosol by binding to GDIs, which inhibit the dissociation of GDP from Rho GTPase proteins and prevent GTPase activation by GEFs (15). While there are more than 20 Rho family members in humans, only three GDIs with proven biological functions have been found: RhoGDIα, RhoGDIβ, and RhoGDIγ (also known as GDI1, GDI2, and GDI3) (16). Among these three RhoGDIs, RhoGDIα is the most abundant one. Loss of RhoGDIα expression in breast cancer has been shown to enhance metastasis (17). RhoGDIβ (GDI2) also has been confirmed to be a metastasis suppressor gene in bladder cancer (18, 19). These data all support a metastasis suppressor role for RhoGDIs.
In this study, we investigated the role of 14-3-3τ in breast cancer metastasis. We show that 14-3-3τ binds to the phosphorylated RhoGDIα upon epidermal growth factor (EGF) stimulation, thereby releasing Rho GTPases and promoting EGF-induced RhoA, Rac1, and Cdc42 activation. This leads to increased breast cancer cell motility and invasion in cells and promotes metastasis in an in vivo breast cancer xenograft model. Additional analysis in human primary breast tumors shows that overexpression of 14-3-3τ is correlated with higher Rho GTPase activities and is associated with a higher incidence of breast cancer metastasis and shorter patient survival. Thus, these data suggest that 14-3-3τ is a novel regulator that promotes breast cancer metastasis.
MATERIALS AND METHODS
Cell culture and infection.
MCF7 cells, MDA-MB231 cells (both from ATCC), and HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (50 IU/ml), and streptomycin (50 μg/ml) in a humidified incubator with 5% CO2 at 37°C.
Retrovirus was produced in HEK293T cells. Twenty-four hours after transfection, the virus-containing medium was collected to infect MCF7 cells with the addition of 8 μg/ml Polybrene. Forty-eight hours after infection, the virus-containing medium was replaced by fresh culture medium. To establish stable cell lines, infected cells were selected with 1 μg/ml puromycin or 50 μg/ml hygromycin for 1 week.
Recombinant plasmids.
To construct pQCXIP-14-3-3τ-FLAG, human 14-3-3τ cDNA was amplified by PCR with the primers 5′-ACTTGGATCCACCATGGAGAAGACTGAGCTG-3′ and 5′-GCGCGAATTCTACTTATCGTCGTCATCCT-3′ to add FLAG tag at the C terminus of 14-3-3τ. The PCR product was digested with BamHI/EcoRI, cloned into pQCXIP, and verified by sequencing.
The pCMV6 constructs expressing His-tagged wild-type (WT) and S101A/S174A RhoGDIα were kindly provided by Celine DerMardirossian from The Scripps Research Institute. The S101A- and S174A-expressing vectors were constructed by replacing the N-terminal fragment (including the S101A site) or C-terminal fragment (including the S174A site) of S101A/S174A RhoGDIα cDNA with the corresponding fragment of wild-type RhoGDIα cDNA by HindIII/HpaI digestion. The murine stem cell virus luciferase PGK Hygro construct (deposited in Addgene by Scott Lowe) was purchased from Addgene. Two validated short hairpin RNAs (shRNAs) (1, TRCN0000078169; 2, TRCN0000078172) targeting human 14-3-3τ were purchased from Sigma.
GST pulldown assay, immunoprecipitation, and Western blotting.
Cells were harvested in NP-40 lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 10% glycerol, and 1% NP-40) with protease inhibitor cocktails and sonicated briefly. To detect in vitro binding, glutathione S-transferase (GST)–14-3-3τ protein was purified from Escherichia coli and coupled to glutathione-Sepharose beads (GE Healthcare) as described before (20). Cell lysates were incubated with GST or GST–14-3-3τ beads overnight at 4°C and then washed twice with cold phosphate-buffered saline (PBS). To detect cellular coimmunoprecipitation, cell lysates were incubated with control IgG or the indicated antibodies overnight and then incubated with protein A/G or protein G beads (Pierce) for 45 min at 4°C, followed by washing twice with NP-40 lysis buffer. In the in vitro reconstitution experiments, lysates prepared from 14-3-3τ-depleted cells were incubated with 200 ng of purified 14-3-3τ protein for 2 h at 4°C before addition of anti-RhoGDIα antibody for immunoprecipitation. Immunoprecipitates and input protein lysates were fractionated by SDS-PAGE and electrotransferred to an Immobilon-P membrane (Millipore).
The specific signals were detected with appropriate antibodies. The antibodies specific to RhoGDIα (A-20), RhoA (26C4), and GST (B-17) were purchased from Santa Cruz Biotechnology. The monoclonal antibody for 14-3-3τ (3B9), FLAG (F3135), and β-actin antibody (A2066) were purchased from Sigma. Anti-His antibody was purchased from Clontech Laboratories. Anti-pS174-RhoGDIα was purchased from Assay Biotechnology Company. Anti-ROCK2 antibody was purchased from Upstate. Anti-pS1366-ROCK2 antibody was purchased from GeneTex Inc. Anti-Rac1 and Cdc42 antibodies were purchased from BD Biosciences and Millipore, respectively.
Transwell cell migration and invasion assays.
14-3-3τ–FLAG-overexpressing or 14-3-3τ-depleted cells and their control cells were subjected to Transwell cell migration assays as described previously (21). A total of 25,000 cells were seeded in 0.1% bovine serum albumin (BSA)-containing medium in the top chamber of the Transwells. EGF (0.1 μg/ml for MCF7 cells and 0.01 μg/ml for MDA-MB231 cells) was added to the lower chamber, and cells were allowed to migrate through 8-μm-pore inserts for 6 h. After removal of the nonmigratory cells from the top chambers, cells that migrated to the fibronectin-coated bottom filter were fixed and stained with crystal violet. A total of 30 fields/sample were counted under the microscope using a 20× objective. The relative migratory rate was determined as the fold increase compared to that of unstimulated control cells.
In the invasion assays, 100,000 cells were seeded in serum-free medium in the top chamber of the Matrigel-coated inserts (BD Bioscience). Cells were allowed to migrate through 8-μm pores toward 5% serum-containing medium in the lower chamber for 24 h. Cells invading the bottom filter were stained and counted as described above. For ROCK kinase inhibition, MCF7 cells were preincubated with 10 μM ROCK inhibitor Y27632 (Sigma) for 2 h before trypsinization and then seeded in medium with 10 μM Y27632 in the Matrigel-coated inserts. For the RhoGDIα mutant, control and 14-3-3τ–FLAG-overexpressing stable MCF7 cells were transfected with either empty vector or His-RhoGDIα S174A mutant by Lipofectamine 2000. Twenty-four hours after transfection, cells were applied to Matrigel invasion assays as described above.
Cell adhesion and immunostaining of cytoskeleton.
MCF7 stable cells were treated with EGF for 20 to 60 min and then fixed. Polymerized actin stress fibers were stained with rhodamine-conjugated phalloidin, and focal adhesions were visualized using antivinculin antibody as described previously (21). For adhesion assays, cells were seeded in 2.5% serum-containing medium on the coverslips precoated with 20 μg/ml fibronectin (Sigma) and incubated at 37°C. Cells were fixed in 3.7% PBS-buffered paraformaldehyde at different time points and permeabilized with 0.1% Triton X-100. After blocking in 5% BSA-containing PBS, cells were incubated with antivinculin monoclonal antibody (Sigma) overnight at 4°C. Cells were washed and incubated with Alexa Fluor 488–rabbit anti-mouse IgG (Invitrogen) for 1 h at room temperature, followed by incubation with rhodamine-conjugated phalloidin (Cytoskeleton, Inc.) for 2 to 3 h at room temperature. Nuclei were stained with Hoechst 33258 (Sigma). After a final wash, the cells were mounted and images were captured using a Zeiss fluorescence microscope (Axio Observer inverted microscope).
Rho GTPase activity assays.
To examine EGF-induced Rho GTPase activation, GTP-bound RhoA or Rac1 was determined by specific binding to the Rho-binding domain of rhotekin (GST-RBD) or the p21-binding domain of PAK1 (GST-PBD), respectively. Cells were starved in 0.1% BSA-containing Dulbecco's modified Eagle medium (DMEM) overnight and then stimulated with 0.1 μg/ml EGF (for MCF7 cells) or 0.01 μg/ml EGF (for MDA-MB231 cells) for various times. For the PAK1 kinase inhibition assay, starved cells were preincubated with 10 μM IPA-3 (PAK inhibitor; Calbiochem) for 3 to 4 h before EGF treatment. Dimethyl sulfoxide (DMSO) was used as a vehicle control. For the RhoGDIα mutant, control and 14-3-3τ–FLAG-overexpressing stable MCF7 cells were transfected with either empty vector or the His-RhoGDIα S174A mutant by Lipofectamine 2000. Twenty-four hours after transfection, cells were starved before EGF treatment. Equal amounts of cell lysates were incubated with GST-RBD or GST-PBD-conjugated glutathione beads at 4°C for 2 to 3 h. The activated RhoA and Rac1 or Cdc42 was pulled down by GST-RBD and GST-PBD, respectively, and then resolved by SDS-PAGE. Immunoblotting was performed using an antibody specific to RhoA (Santa Cruz Biotechnology) or Rac1 (BD Biosciences).
Animal studies and imaging.
Five- to 6-week-old female athymic nude mice (Harlan Sprague-Dawley) were implanted with 17β-estradiol pellets (0.72 mg/90-day release; Innovative Research of America). MCF7 cells (5 × 106; vector control or 14-3-3τ–FLAG overexpressing) were injected into the mammary fat pad of mice. Tumors were measured with calipers, and multimodality imaging, including bioluminescence, was done every week as described previously (22). Briefly, tumors were visualized by tail vein injection of 3 mg VivoGlo luciferin (Promega) into the anesthetized mice. Mice were imaged immediately after injection. Optical and X-ray images were recorded by a Kodak in vivo multispectral system FX (Carestream Health Molecular Imaging). Computed tomography (CT) imaging was performed using a Siemens MicroCAT II SPECT/CT (Siemens Medical Solutions). Mice were killed when they were sick or tumor size reached 1.5 cm. Organ imaging was done immediately after mice were sacrificed. All experiments were performed under an Institutional Animal Care and Use Committee (IACUC)-approved protocol, and all experiments conformed to IACUC standards.
Frozen breast cancer tissue analysis.
The collection of frozen breast tumor tissues, under an institutional review board-approved protocol, had been described in prior publications (3, 23). The frozen tissues were minced and lysed in 1% SDS–60 mM Tris-HCl and boiled for 5 min. The lysates were briefly sonicated and clarified by centrifugation to remove insoluble tissues, followed by Western blotting. NIH ImageJ was used to quantify the intensity of the bands on Western blots.
Statistical analysis.
Kaplan-Meier analyses were performed using the R program. The Pearson correlation coefficient was calculated to evaluate correlations. The two-tailed probability (P values) for each Pearson correlation coefficient was also calculated. The t test (two tailed) was used to compare the differences between two experimental groups. P values of less than 0.05 were considered statistically significant.
RESULTS
14-3-3τ promotes cell invasiveness and reduces adhesion in MCF7 breast cancer cells.
Many primary human breast tumors overexpress 14-3-3τ at levels severalfold higher than that in MCF7 cells (Fig. 1A) (3). To investigate a role for 14-3-3τ in breast cancer cell motility and invasion, we established MCF7 cell lines stably expressing an empty control vector or 14-3-3τ–FLAG comparable to levels of endogenous 14-3-3τ seen in primary breast tumor cells (Fig. 1A). We then performed a Transwell migration assay in both cell lines in the absence or presence of epidermal growth factor (EGF). As shown in Fig. 1B, overexpression of 14-3-3τ–FLAG promoted MCF7 cell migration with or without EGF treatment. To investigate whether overexpression of 14-3-3τ–FLAG led to enhanced invasion activity, we next performed a Matrigel invasion assay. Indeed, 14-3-3τ–FLAG overexpression also enhanced MCF7 cell invasion (Fig. 1C). These results suggest that overexpression of 14-3-3τ promotes breast cancer cell motility and invasion. On the other hand, knockdown of 14-3-3τ by two different shRNAs attenuated MCF7 cell migration (Fig. 1D and E) and invasion (Fig. 1F). Since MCF7 is a poorly invasive cell line, we also tested the effect of 14-3-3τ depletion in another more motile and invasive breast cancer cell line, MDA-MB231. Indeed, the impact of 14-3-3τ depletion on cell migration and invasion was reproduced in MDA-MB231 cells with more pronounced effect (Fig. 1G to I). Taken together, these data support a role for 14-3-3τ in promoting breast cancer cell motility and invasiveness.
FIG 1.

14-3-3τ regulates breast cancer cell migration and invasion. (A) A stable MCF7 cell line expressing 14-3-3τ–FLAG or an empty vector (Vec) was established, and lysates were subjected to SDS-PAGE analysis side by side with four primary human breast tumor samples (5T, 37T, and 10T are from the 14-3-3τ overexpression group, whereas 9T is from the nonoverexpression group [3]). Total cell lysates were immunoblotted (IB) using anti-FLAG and anti-14-3-3τ antibodies, respectively, to verify the expression of 14-3-3τ–FLAG. The expression of β-actin serves as a loading control. (B) Overexpression of 14-3-3τ increased MCF7 cell migration. MCF7 stable cell lines were subjected to a Transwell cell migration assay. EGF was added to the lower chamber of Transwells, and cells were allowed to migrate for 6 h. The relative migration rate was defined as the fold increase of migrated cells compared to the level for unstimulated vector control cells. Data shown are the means ± standard errors of the means from four independent experiments. *, P < 0.05 (two-tailed t test). w/, with; w/o, without. (C) 14-3-3τ-overexpressing MCF7 cells had a higher invasion rate. Stable MCF7 cells were starved in serum-free DMEM for 24 h and then seeded on the upper chamber of a Matrigel-coated insert. Five percent serum-containing medium was added to the lower chamber, and cells were allowed to invade for 24 h. **, P < 0.001 (n = 3) (two-tailed t test). (D) MCF7 cells were infected with lentiviruses expressing either control scrambled shRNA (shScr) or one of the 14-3-3τ shRNAs (1 and 2) and selected with puromycin. Cell lysates were subjected to immunoblotting to confirm the knockdown of 14-3-3τ. (E) 14-3-3τ knockdown attenuated MCF7 cell migration rate. Stable MCF7 cell lines expressing an shScr or 14-3-3τ shRNAs were subjected to a Transwell cell migration assay in the absence or presence of EGF (0.1 μg/ml for 5 h) as described for panel B. *, P < 0.05 (n = 3) (two-tailed t test). (F) Cell invasion was reduced upon 14-3-3τ depletion. Stable MCF7 cell lines expressing an shScr or 14-3-3τ shRNAs were subjected to a Matrigel invasion assay as described for panel C. **, P < 0.001 (n = 4) (two-tailed t test). (G) MDA-MB231 cells were infected with lentiviruses expressing either control shScr or 14-3-3τ shRNA (1 and 2) and selected with puromycin. Cell lysates were subjected to immunoblotting to confirm the knockdown of 14-3-3τ. (H) MDA-MB231 cell lines expressing a scrambled shRNA or 14-3-3τ shRNAs were subjected to a Transwell cell migration assay in the absence or presence of EGF (0.01 μg/ml for 5 h). **, P < 0.001 (n = 3) (two-tailed t test). (I) 14-3-3τ-depleted MDA-MB231 cell lines were subjected to a Matrigel invasion assay as described for panel C. **, P < 0.001 (n = 3) (two-tailed t test).
Overexpression of 14-3-3τ also promoted EGF-induced actin cytoskeletal remodeling, with less stable focal adhesions and stress fiber formation but more actin rings at the leading edge, indicative of reduced adhesion and increased motility (Fig. 2A). Time-lapse imaging showed that 14-3-3τ-overexpressing MCF7 cells were more motile after EGF treatment (Fig. 2B; also see Videos S1 and S2 in the supplemental material). 14-3-3τ-overexpressing cells also had less adhesion ability in response to serum (Fig. 2C and D). We also seeded these cells on fibronectin-coated coverglasses in the presence of 2.5% fetal bovine serum and fixed the cells 2 and 4 h later, followed by the immunostaining of focal adhesions with an antivinculin antibody. As shown in Fig. 2E, while control MCF7 cells formed focal adhesions and spread out on the glass nicely, 14-3-3τ-overexpressing MCF7 cells remained rounded 4 h after seeding and had less stable focal adhesions.
FIG 2.

14-3-3τ overexpression promotes EGF-stimulated actin cytoskeletal reorganization and decreases cell adhesion ability. (A) 14-3-3τ overexpression promoted actin remodeling with less stable focal adhesions and increased membrane ruffling and protrusion upon EGF treatment. Stable MCF7 cells were starved in serum-free medium and then stimulated with EGF (0.1 μg/ml) for 20 to 60 min. Cells were then subjected to immunostaining using phalloidin and antivinculin antibody. A scale bar is shown in the bottom of each figure. (B) MCF7 cells stably expressing 14-3-3τ or an empty vector were transiently transfected with EGFP-TRIP6 (48), an indicator of focal adhesion complex, on glass-bottomed dishes. Time-lapse images of cell movement following EGF (0.1 μg/ml) stimulation were captured for 30 min and are shown in Videos S1 and S2 in the supplemental material. The images at 0, 10, 20, and 30 min are shown here. (C) 14-3-3τ overexpression decreased the cell adhesion rate in MCF7 cells. Equal numbers of control and 14-3-3τ-overexpressing MCF7 were seeded on tissue culture plates in the presence of 5% serum. After 2.5 h, nonadhered cells were washed away and the adhered cells were stained with crystal violet. Results shown are representative pictures. (D) To quantitatively measure the cell adhesion rate, equal numbers of control and 14-3-3τ-overexpressing MCF7 cells were seeded on 12-well culture plates in 10% serum-containing medium. Six hours later, nonadhered cells were washed away, and the adhered cells were stained with crystal violet and counted under a microscope. *, P < 0.05 (n = 4) (two-tailed t test) compared to the vector control. (E) MCF7 cells were seeded on fibronectin (FN)-coated slides and harvested at different time points for immunostaining with antivinculin antibody. The images were captured using a Zeiss fluorescence microscope under a 40× objective.
Overexpression of 14-3-3τ activates Rho GTPases.
Since Rho GTPase signaling plays a very important role in regulating cell motility and adhesion, we tested whether 14-3-3τ promoted cell migration and invasion through regulation of Rho GTPases. By pulling down active RhoA and Rac1 with GST-conjugated Rhotekin-RBD and PAK1-PBD beads, respectively, we found that 14-3-3τ overexpression increased RhoA and Rac1 activities before and after EGF treatment in MCF7 cells (Fig. 3A). On the contrary, depletion of 14-3-3τ inhibited these effects in MCF7 cells (Fig. 3B) and MDA-MB231 cells (Fig. 3C). 14-3-3τ knockdown also decreased Cdc42 activity during EGF treatment in MDA-MB231 cells (Fig. 3C). As a group of one of the main downstream effectors of Rho GTPase, Rho-associated kinases (also called ROCK), have been shown to play a crucial role in promoting cell migration by phosphorylating various substrates involved in actin assembly (24), we also examined ROCK activity in MCF7 cells by a phospho-S1366 ROCK2-specific antibody (pS1366), which reflects ROCK2 activity (25). Indeed, 14-3-3τ-overexpressing MCF7 cells had higher ROCK2 activity (Fig. 3A, lower), and 14-3-3τ-depleted MDA-MB231 cells had less ROCK2 activation during EGF treatment (Fig. 3C, lower). Consistent with this, a ROCK inhibitor, Y27632, inhibited cell invasion in 14-3-3τ-overexpressing MCF7 cells (Fig. 3D). These data demonstrate a role for 14-3-3τ in the regulation of Rho GTPase activity.
FIG 3.

14-3-3τ promotes EGF-induced Rho GTPase activation in breast cancer cells. (A) Overexpression of 14-3-3τ enhanced EGF-induced Rho GTPase activation in MCF7 cells. 14-3-3τ-overexpressing MCF7 and control cells were starved in 0.1% BSA-containing DMEM overnight and then stimulated with EGF (0.1 μg/ml) for the indicated duration. Equal amounts of cell lysates were incubated with GST-RBD- or GST-PBD-conjugated glutathione-Sepharose beads to pull down GTP-bound RhoA or Rac1, followed by SDS-PAGE and immunoblotting using anti-RhoA or anti-Rac1 antibody. Five percent input lysates were subjected to Western blot analysis. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) 14-3-3τ deficiency inhibited EGF-induced RhoA and Rac1 activation in MCF7 cells. 14-3-3τ was knocked down by two shRNA constructs in MCF7 cells. RhoA and Rac1 activity assays were performed as described for panel A. (C) 14-3-3τ was depleted by two shRNA constructs in MDA-MB231 cells. RhoA, Rac1, and Cdc42 activity assays were performed as described for panel A after EGF (0.01 μg/ml) treatment. short exp., a short exposure of the 14-3-3τ immunoblot. (D) ROCK inhibitor Y27632 blocked 14-3-3τ-enhanced cell invasion in MCF-7 cells. **, P < 0.001 (n = 3) (two-tailed t test).
14-3-3τ binds to S174-phosphorylated RhoGDIα to activate Rho GTPases.
We further investigated the mechanism by which 14-3-3τ regulates Rho GTPase signaling pathways. We found a conserved 14-3-3 consensus binding site around Ser174 of RhoGDIα (RGSYS174IKS) but not in RhoGDIβ or RhoGDIγ. RhoGDIα-Ser174 has been shown to be phosphorylated by PAK1 and protein kinase A (PKA) (26, 27), and this phosphorylation promotes its dissociation from either Rac1 or RhoA. Therefore, we speculated that 14-3-3τ could associate with RhoGDIα, and this binding led to the release of Rho GTPases from RhoGDIα, promoting the activation of Rho GTPases upon ligand stimulation. To investigate this possibility, we first tested whether 14-3-3τ could associate with RhoGDIα. Indeed, the GST pulldown assay showed that purified GST–14-3-3τ could pull down RhoGDIα derived from MCF7 cell lysates particularly after EGF treatment (Fig. 4A). Cellular coimmunoprecipitation showed that endogenous 14-3-3τ bound to RhoGDIα upon EGF treatment, and the 14-3-3τ-bound RhoGDIα could be recognized by a phospho-S174 RhoGDIα antibody (Fig. 4B). PAK1 phosphorylates RhoGDIα at both S101 and S174, leading to its dissociation of Rac GTPase (26). Therefore, we first examined the interaction of 14-3-3τ with S101A/S174A mutant RhoGDIα. While WT His-RhoGDIα interacted with 14-3-3τ and the interaction was enhanced by EGF treatment, His-S101A/S174A mutant RhoGDIα (labeled 2A in Fig. 4C) failed to interact with 14-3-3τ upon EGF stimulation. To further investigate the contribution of S101 and S174 individually to 14-3-3τ binding, we tested S101A and S174A mutant RhoGDIα, respectively. Indeed, S174A mutation alone was sufficient to completely block its interaction with 14-3-3τ (Fig. 4D), confirming a role for S174 phosphorylation in mediating 14-3-3τ binding. Interestingly, S101A mutation also attenuated its binding to 14-3-3τ despite having no effect on S174 phosphorylation. This observation is consistent with the observation that both S101 and S174 residues are important in the activation of Rho GTPases (26). It is possible that while phosphorylation of S101 does not directly involve the binding to 14-3-3τ, it facilitates 14-3-3τ binding through other unknown mechanisms, such as inducing structural changes.
FIG 4.

14-3-3τ binds RhoGDIα at phosphorylated S174 and activates Rho GTPases. (A) In vitro interaction between purified GST–14-3-3τ and RhoGDIα derived from EGF-treated cells. Starved MCF7 cells were stimulated with EGF for the indicated times. Cell lysates were then incubated with GST- or GST–14-3-3τ-conjugated glutathione-Sepharose beads. The amounts of cellular RhoDGIα pulled down by GST–14-3-3τ and total RhoGDIα present in MCF7 cell lysates were determined by Western blotting. (B) 14-3-3τ associated with S174-phosphorylated RhoGDIα upon EGF treatment in MCF7 cells. Endogenous 14-3-3τ was immunoprecipitated (IP) from the EGF-treated MCF7 cell lysates, followed by immunoblotting to detect the coimmunoprecipitated RhoGDIα and S174-phosphorylated RhoGDIα. Total cell lysates were subjected to immunoblotting to determine the amounts of input proteins (lower). (C) Double mutations of the Ser101/Ser174 residues abolished EGF-induced interaction between 14-3-3τ and RhoGDIα. MCF7 cells were transfected with His-tagged wild-type (WT) or S101A/S174A mutant (labeled as 2A) RhoGDIα. Cells were treated with EGF, and cell lysates were harvested for immunoprecipitation with anti-His antibody, followed by immunoblotting with the antibody specific to 14-3-3τ. Immunoblotting was also performed to determine the input levels of 14-3-3τ and His-tagged RhoDGIα in whole-cell lysates. (D) Mutation of Ser174 of RhoGDIα to Ala abolished its interaction with 14-3-3τ. MCF7 cells were transfected with either His-tagged WT or S101A or S174A mutant RhoGDIα. Cells were starved overnight and then treated with EGF. Cell lysates were subjected to coimmunoprecipitation to determine the association between His-RhoDIα and endogenous 14-3-3τ as described for panel C. Immunoblotting was also performed to determine the input levels of 14-3-3τ, endogenous and His-tagged RhoGDIα, pS174-RhoGDIα, and GAPDH in the whole-cell lysates.
Binding of 14-3-3τ to RhoGDIα releases and activates Rho GTPases.
We next investigated the role of 14-3-3τ in releasing Rho GTPases from RhoGDIα upon EGF treatment. We performed coimmunoprecipitation experiments between RhoGDIα and Rho GTPases in 14-3-3τ-overexpressing or 14-3-3τ-depleted MCF7 cells. Overexpression of 14-3-3τ inhibited RhoGDIα binding to RhoA and Rac1, particularly after EGF treatment (Fig. 5A). Conversely, RhoGDIα remained bound to RhoA and Rac1 even after EGF treatment in 14-3-3τ-depleted cells (Fig. 5B). This was not an indirect effect through changes of RhoGDIα S174 phosphorylation status, since EGF-induced S174 phosphorylation of RhoGDIα was not altered by 14-3-3τ overexpression or depletion (Fig. 5A and B, lower).
FIG 5.

Role for 14-3-3τ in promoting the dissociation of Rho and Rac1 from RhoGDIα. (A) Overexpression of 14-3-3τ enhanced the dissociation of both RhoA and Rac1 from RhoGDIα upon EGF treatment in MCF7 cells. The association between endogenous RhoGDIα and either RhoA or Rac1 was analyzed by coimmunoprecipitation experiments in 14-3-3τ-overexpressing or control MCF7 cells. (B) Knockdown of 14-3-3τ prevented the dissociation of both RhoA and Rac1 from RhoGDIα upon EGF treatment. The association of endogenous RhoGDIα with either RhoA or Rac1 was analyzed by coimmunoprecipitation experiments in 14-3-3τ-depleted MCF7 cells.
We also performed an in vitro rescue experiment to determine if 14-3-3τ physically interferes with the association between RhoGDIα and either RhoA or Rac1 directly. We first prepared the cell lysates from two 14-3-3τ-depleted cell lines and added purified recombinant 14-3-3τ protein. We then performed RhoGDIα immunoprecipitation. Indeed, the purified 14-3-3τ protein was able to bind RhoGDIα and release RhoA and Rac1 from RhoGDIα (Fig. 6A).
FIG 6.

Interaction between 14-3-3τ- and S174-phosphorylated RhoGDIα is responsible for dissociating Rho and Rac1 from RhoGDIα. (A) Addition of purified recombinant 14-3-3τ in 14-3-3τ-depleted MCF7 cell lysates outcompeted RhoA and Rac1 in RhoGDIα binding and facilitated their release from RhoGDIα upon EGF treatment. 14-3-3τ-depleted MCF7 cell lysates were incubated with purified recombinant 14-3-3τ protein for 2 h at 4°C before immunoprecipitation with anti-RhoGDIα antibody. The association between RhoGDIα and either 14-3-3τ, RhoA, or Rac1 in 14-3-3τ-depleted MCF7 or control cells was determined by coimmunoprecipitation as described in the legend to Fig. 5B. All input lysates were analyzed by Western blotting as indicated. The relative intensities of RhoA, Rac1, and 14-3-3τ were quantitated by ImageJ and are shown below each panel. (B) The effect of 14-3-3τ overexpression on dissociation of Rho GTPases from RhoGDIα was dependent on S174 phosphorylation. Control or 14-3-3τ–FLAG-overexpressing MCF7 cells were transfected with His-RhoGDIα (WT, S101A, S174A, or S101A/S174A) as indicated. After EGF treatment for 20 min, the association between His-RhoGDIα (WT, S101A, S174A, or S101A/S174A) and either RhoA, Rac1, or 14-3-3τ was determined by coimmunoprecipitation as described above. All immunoprecipitates and input lysates were analyzed by Western blotting as indicated. The relative intensities of RhoA, Rac1, and 14-3-3τ were quantitated by ImageJ and are shown below each panel.
To further investigate whether the activity of 14-3-3τ in blocking the interaction between RhoGDIα and either RhoA or Rac1 was dependent on the binding of 14-3-3τ to RhoGDIα, we also examined the effect of the S101A and S174A mutations on the release of RhoA and Rac1 from RhoGDIα upon EGF treatment. We transfected His-RhoGDIα (either WT, S101A, S101A/S174A, or S174A) into control MCF7 cells or 14-3-3τ–FLAG MCF7 cells, followed by EGF stimulation. As shown on the left in Fig. 6B, a coimmunoprecipitation experiment showed that the levels of WT or S101A RhoGDIα-bound RhoA and Rac1 were decreased upon EGF treatment in control MCF7 cells. In 14-3-3τ–FLAG MCF7 cells, RhoA and Rac1 were completely dissociated from WT or S101A His-RhoGDIα upon EGF treatment. This concurred with binding of endogenous 14-3-3τ or exogenously expressed 14-3-3τ–FLAG to His-RhoGDIα upon EGF treatment. In contrast, 14-3-3τ did not bind to S101A/S174A or S174A His-RhoGDIα (Fig. 6B, right). As a consequence, RhoA and Rac1 remained bound to S101A/S174A and S174A His-RhoGDIα in EGF-stimulated 14-3-3τ–FLAG MCF7 cells. These data provide compelling evidence that binding of 14-3-3τ to S174-phosphorylated RhoGDIα is responsible for the release and subsequent activation of RhoA and Rac1.
S174A RhoGDIα blocks 14-3-3τ activities in promoting cell invasion and activation of Rho GTPases.
To further demonstrate the effect of the interaction between 14-3-3τ and pS174-RhoGDIα on the function of 14-3-3τ in invasion, we tested whether overexpression of S174A RhoGDIα could act as a dominant-negative mutant to block 14-3-3τ activities. Indeed, overexpression of S174A RhoGDIα in MCF7 cells blocked the effect of 14-3-3τ in promoting cell invasion (Fig. 7A) as well as activation of RhoA, Rac1, and Cdc42 upon EGF treatment (Fig. 7B). Consistent with this, a PAK1 inhibitor could decrease S174 phosphorylation of RhoGDIα and its binding to 14-3-3τ (Fig. 7C); thus, it inhibited the release of RhoA, Rac1, and Cdc42 from RhoGDIα upon EGF treatment.
FIG 7.

RhoGDIα S174 phosphorylation is important for 14-3-3τ to affect the Rho GTPase signaling pathway. (A) RhoGDIα S174A mutant blocked 14-3-3τ-enhanced MCF7 cell invasion. Control and 14-3-3τ–FLAG MCF7 cells were transfected with either an empty vector or the His-RhoGDIα S174A mutant. Twenty-four hours after transfection, cells were harvested for Western blotting and Matrigel invasion assay as described for Fig. 1C. **, P < 0.001 (n = 3) (two-tailed t test). (B) RhoGDIα S174A mutant blocked 14-3-3τ-enhanced Rho GTPase activation in MCF7 cells. 14-3-3τ–FLAG MCF7 cells were transfected with either an empty vector or His-RhoGDIα S174A mutant. RhoA, Rac1, and Cdc42 activities were measured after EGF stimulation (0.1 μg/ml, 20 min) as described for Fig. 3A. long exp., long exposure. (C) A PAK inhibitor mitigated 14-3-3τ-promoted Rho GTPase dissociation from RhoGDIα protein in MCF-7 cells. Cells were prestarved in 0.1% BSA-containing DMEM overnight and preincubated with IPA-3 (PAK inhibitor) for 3 to 4 h before adding EGF. Cells were then stimulated with EGF (0.1 μg/ml) for 20 min and harvested for anti-RhoGDIα immunoprecipitation.
14-3-3τ promotes MCF7 xenograft metastasis and progression.
To investigate the in vivo activity of 14-3-3τ in breast cancer metastasis, we performed a live-animal longitudinal imaging study. We stably expressed the luciferase (Luc) gene in MCF7 cell lines expressing either 14-3-3τ–FLAG or an empty vector. We then injected the same amounts of cells to establish mammary fat pad xenografts in female nude mice, nine per group, and monitored the tumor growth and metastasis by bioluminescence–X-ray–CT imaging (Fig. 8). Our data show that overexpression of 14-3-3τ increased both tumor growth and metastasis. 14-3-3τ xenografts grew faster (Fig. 8A and B), invaded adjacent organs, such as the pelvic cavity (one mouse had radiographic features of bone invasion [Fig. 8A]), and metastasized to lymph nodes (Fig. 8C to F). Among the eight 14-3-3τ mice that were available for sensitive ex vivo imaging, two showed invasion and five were confirmed to have lymph node metastasis (by positive Luc imaging and hematoxylin and eosin [H&E] staining), whereas only one out of nine control mice developed metastasis. Overall, the mice with 14-3-3τ–FLAG xenografts had shorter overall survival (Fig. 8G) and higher rates of metastasis (Fig. 8H). As shown by H&E staining, some metastasized lymph nodes had been replaced by tumor cells to various extents (Fig. 8F). We harvested the primary and metastatic tumors and verified the expression of 14-3-3τ–FLAG (Fig. 8I and J). Consistent with this, 14-3-3τ–FLAG was present at various levels in these metastatic lymph nodes (Fig. 8I). For example, the lower level of 14-3-3τ–FLAG in metastatic lymph node number 247 is consistent with the patchy metastasis within the lymph node as shown by H&E staining (Fig. 8F, lymph node images, middle).
FIG 8.

14-3-3τ promotes MCF7 xenograft invasion and metastasis. (A) A representative image of a multimodality longitudinal imaging study of 14-3-3τ–FLAG or vector control MCF7 xenograft. X-ray and luciferase images are shown. Bar, 10.20 mm. (B) Tumor volumes of control and 14-3-3τ-overexpressing MCF7 xenografts (n = 9 for each group). (C) Bioluminescence–X-ray–CT imaging of MCF7-Luc mammary fat pad xenografts in nude mice. Bar, 7.25 mm. (D and E) Ex vivo imaging of primary and metastatic 14-3-3τ–FLAG MCF7 tumors. Bar (E), 9.61 mm. (F) H&E staining of metastatic lymph nodes from 14-3-3τ-overexpressing MCF7 xenograft mice and primary tumors from control and 14-3-3τ-overexpressing MCF7 xenografts (20× objective lens). L, lymphocytes; T, tumor cells. (G) Kaplan-Meier curves of survival of the tumor-bearing mice after injection of cells (n = 9 for both groups). The P value between both groups (log rank test) is indicated. (H) Kaplan-Meier curves of metastasis-free rate (n = 9 for control; n = 8 for 14-3-3τ–FLAG; one 14-3-3τ–FLAG xenograft-bearing mouse died over the weekend and was not available for ex vivo bioluminescence imaging). P value between both groups (log rank test) is indicated. (I) Immunoblots of primary (P) and metastatic (M) tumors. The number below each lane is the xenograft identification number in our experiment. LN, lymph nodes. (J) Immunoblots of primary tumors from both the control MCF7 group and the 14-3-3τ–FLAG-expressing MCF7 group. The number above each lane is the xenograft identification number in our experiment. (K) The pS1366-ROCK2 and ROCK2 signals from each xenograft were measured by densitometry using the NIH program ImageJ, and the pS1366-ROCK2 signal was normalized by its respective ROCK2 signal to reflect ROCK activity. *, P < 0.001 (two-tailed t test).
We next examined the ROCK activity in primary tumors by determining the expression of phospho-S1366 ROCK2. Indeed, 14-3-3τ-overexpressing tumors had higher ROCK activity (measured by p-ROCK2/ROCK2) than control tumors (Fig. 8J and K). Xenograft 236 (harvested at day 48 after mammary fat pad injection due to mouse sickness, but it exhibited no metastasis by ex vivo imaging) had a much lower level of expression of 14-3-3τ–FLAG and also had a relatively lower level of p-ROCK2. A repeated Western analysis of lysates from xenograft 236 confirmed the expression of 14-3-3τ–FLAG but found that it was at a lower level.
Taken together, these data demonstrate the in vivo activity of 14-3-3τ in promoting metastasis and progression of MCF7 xenografts.
High levels of 14-3-3τ are associated with ROCK activity in human breast carcinomas.
The Rho-associated protein kinases (ROCKs or Rho kinases) are the major downstream effectors of Rho GTPases, which regulate actin cytoskeletal rearrangement. Emerging evidence also supports ROCKs as important modulators in cancer invasion and metastasis. Somatic mutations in both ROCK genes (28, 29) and increased protein levels of ROCKs (30, 31), which result in elevated ROCK activity, have been described in several human cancers and are associated with cancer progression. Overexpression of 14-3-3τ has been reported in both lung cancer (32) and breast cancer tissues (3). Given the role of 14-3-3τ in the regulation of the Rho/ROCK pathway demonstrated in our studies, we next analyzed the correlation between the 14-3-3τ protein levels and ROCK activity in human breast carcinomas. We extracted proteins from frozen primary human breast tumor tissues (3) and examined the levels of 14-3-3τ protein, phosphorylated ROCK2 (pS1366), ROCK2, RhoGDIα, and β-actin (as a loading control) by Western blotting. In these samples, ROCK2 and RhoGDIα levels were relatively equal, but p-ROCK2 and 14-3-3τ levels varied among samples (Fig. 9; anonymous patient numbers are shown at the top of each sample). We then quantified the intensities of p-ROCK2, ROCK2, and 14-3-3τ from each immunoblot and analyzed the correlation between 14-3-3τ and ROCK2 activity (p-ROCK2/ROCK2 ratio). In both groups (n = 12 and 14, respectively), there is significant correlation between 14-3-3τ levels and ROCK2 activity (Pearson correlation coefficients [r] of 0.774 and 0.625) (Fig. 9, bottom graphs). Since ROCK2 levels are relatively comparable, the analysis between 14-3-3τ and p-ROCK2 levels also yielded similar correlations. We also analyzed both groups together after normalization of the levels between these two blots. We chose one sample from each blot (numbers 10 and 5) and performed Western blotting and densitometry analyses (Fig. 9C). 14-3-3τ and p-ROCK2 levels were then normalized accordingly. As shown in Fig. 9C, bottom graph, there is also a positive correlation between 14-3-3τ and p-ROCK2 levels (r = 0.717). These findings provide evidence supporting a role for 14-3-3τ in upregulating the Rho/ROCK pathway in human breast cancer.
FIG 9.

Higher levels of 14-3-3τ correlate with elevated ROCK activities in primary breast tumors. Proteins were extracted from 26 frozen primary human breast tumor tissues and analyzed by Western blotting (WB1 and WB2 in panels A and B, respectively). The intensities of p-ROCK2, ROCK2, and 14-3-3τ on each blot were quantified by densitometry. An analysis of the correlation between 14-3-3τ and ROCK2 activity (p-ROCK2 levels normalized by ROCK2) is shown in the bottom panels. (C) The relative protein levels in blot numbers 10 and 5 are compared side by side and serve as standards for protein normalization among all samples shown on the two different blots. 14-3-3τ and p-ROCK2 levels were then normalized accordingly and analyzed for Pearson correlation coefficient (r). The P values shown are two-tailed probability tests.
High levels of 14-3-3τ are associated with human breast cancer metastasis.
Our results presented so far have provided multiple lines of evidence for a prometastatic activity of 14-3-3τ overexpression in breast cancer. Lastly, we analyzed several published breast cancer data sets (33–38) to determine whether 14-3-3τ overexpression correlates with breast cancer metastasis. Using the Oncomine tool and NIH Gene Expression Omnibus (GEO) database, we stratified patients evenly according to their 14-3-3τ mRNA levels into low, intermediate 1 (int 1), intermediate 2 (int 2), and high groups and determined the metastasis rate of each group at 1, 3, and 5 years after diagnosis (Fig. 10A and Table 1). Apparently, high 14-3-3τ expression is associated with a high metastatic rate. We also chose van de Vijver et al. (38) and Desmedt et al. (33) data sets and performed Kaplan-Meier analysis of overall survival and metastasis-free survival (Fig. 10B). Consistent with our prior analysis on 14-3-3τ protein (3), high 14-3-3τ mRNA expression is associated with shorter overall survival. More importantly, it is also associated with early and more metastatic events in both data sets.
FIG 10.

Higher levels of 14-3-3τ are associated with a higher metastatic rate and shorter survival. (A) Several published breast cancer data sets (33–38) were analyzed to determine the correlation between 14-3-3τ expression and breast cancer metastasis. Patients were ranked according to 14-3-3τ expression levels in their breast tumors and were equally divided into four groups (14-3-3τ levels, low < int 1 < int 2 < high) or three groups (14-3-3τ levels, low < int < high). The metastasis rate of each group at 1 year, 3 years, and 5 years after diagnosis was correlated (raw values are presented in Table 1). (B) Kaplan-Meier analysis of breast cancer patients according to 14-3-3τ expression levels. The 14-3-3τ expression levels and clinical outcomes of van de Vijver et al. and Desmedt et al. data sets (33, 38) were analyzed. Patients were ranked according to 14-3-3τ expression levels in their breast tumors and then equally divided into four groups as described for panel A. The numbers (N) of patients in each group are indicated below. A Wilcoxon test was used to evaluate the significance. The P values for the differences between the high group and the other groups are indicated.
TABLE 1.
Metastasis events in each study according to 14-3-3τ expression levels
| Study (reference) and expression level (no. of patients) | No. of events by yr after diagnosis |
||
|---|---|---|---|
| 1 | 3 | 5 | |
| van de Vijver et al. (38) | |||
| Low (74) | 2 | 8 | 15 |
| Int 1 (74) | 2 | 11 | 17 |
| Int 2 (74) | 1 | 12 | 19 |
| High (73) | 4 | 21 | 27 |
| Minn et al. (35) | |||
| Low (23) | 0 | 2 | 4 |
| Int (23) | 1 | 4 | 8 |
| High (22) | 2 | 6 | 10 |
| Desmedt et al. (33) | |||
| Low (50) | 1 | 5 | 5 |
| Int 1 (50) | 1 | 2 | 6 |
| Int 2 (50) | 1 | 6 | 8 |
| High (48) | 2 | 10 | 16 |
| van't Veer et al. (37) | |||
| Low (33) | 0 | 8 | 12 |
| Int (32) | 1 | 8 | 16 |
| High (32) | 2 | 15 | 18 |
| Schmidt et al. (36) | |||
| Low (50) | 0 | 2 | 7 |
| Int 1 (50) | 1 | 4 | 4 |
| Int 2 (50) | 1 | 4 | 6 |
| High (48) | 4 | 9 | 11 |
| Loi et al. (34) | |||
| Low (28) | 0 | 1 | 1 |
| Int (27) | 0 | 3 | 5 |
| High (27) | 1 | 4 | 8 |
Taken together, our data support a model that 14-3-3τ interacts with phosphorylated RhoGDIα to block the inhibitory effect of RhoGDIα against Rho GTPases and leads to the activation of Rho GTPase signaling (Fig. 11). This mechanism contributes to the prometastatic activity of 14-3-3τ in breast cancer.
FIG 11.
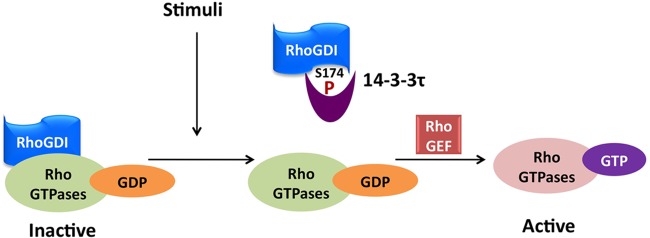
Model depicting how 14-3-3τ regulates Rho GTPases. In response to growth factor stimuli such as EGF, RhoGDIα is phosphorylated at Ser174 by kinases such as PAK1. Phosphorylation of Ser174 induces 14-3-3τ binding and leads to the release of RhoA, Rac1, and Cdc42. After dissociating from inhibitory RhoGDIα, RhoA, Rac1, and Cdc42 can then be activated by RhoGEF.
DISCUSSION
In this paper, we identify 14-3-3τ as one of the signaling molecules that drive breast cancer metastasis. Using a xenograft model, we show that overexpression of 14-3-3τ is able to greatly enhance breast cancer progression and metastasis. The mechanistic analysis shows that overexpression of 14-3-3τ increases cell mobility and invasiveness through activation of RhoA and Rac1 GTPases. We further demonstrate that 14-3-3τ binds to S174-phosphorylated RhoGDIα and blocks the inhibitory activity of RhoGDIα, leading to the release and activation of RhoA and Rac1 GTPases upon EGF stimulation. This regulation is clinically significant, since the levels of 14-3-3τ in human breast cancer tissues correlate with the activity of ROCK, one of the downstream effectors of Rho GTPases. Moreover, analyses of multiple breast cancer databases show that high levels of 14-3-3τ are strongly associated with breast cancer metastasis.
RhoGDI plays a key role in the control of Rho, Rac1, and Cdc42 GTPase activities in response to upstream signaling. RhoGDIα is phosphorylated by PAK1 at Ser101 and Ser174 and by PKA at Ser174 (26, 27), and the phosphorylation events lead to dissociation and subsequent activation of Rac1 and RhoA. However, it is unclear how phosphorylation of RhoGDIα leads to the release of these GTPases. In this study, we demonstrate that binding of 14-3-3τ to S174-phosphorylated RhoGDIα is responsible for the release and activation of Rac1, Cdc42, and RhoA upon EGF stimulation. The Ser174 residue is on the surface of the hydrophobic prenyl-binding pocket of RhoGDIα (39). Binding of 14-3-3τ to pSer174 might block the prenyl-binding pocket, releasing Rac1, Cdc42, and RhoA from RhoGDIα and leading to their activation and ultimately facilitating the dynamics of cell motility.
It is interesting that while pSer174 of RhoGDIα is the binding site for 14-3-3τ, phosphorylation of both Ser101 and Ser174 is required for releasing Rac1 (26). Consistent with a role for Ser101, we also found that S101A mutation attenuated RhoGDIα binding to endogenous 14-3-3τ (Fig. 4D). This is not because it affected phosphorylation of Ser174, since the EGF-induced pSer174 signals of the WT and S101A RhoGDIα were essentially the same (Fig. 4D). DerMardirossian and colleagues had noticed that Ser101 and Ser174 are very close to each other (only 9 Å apart) and proposed that repulsive force between negative charges of both residues causes a conformational change (26). We speculate that the repulsion generated by two closely opposed phosphorylation groups opens up space and facilitates 14-3-3τ binding to the pSer174 residue. It will be desirable to perform a detailed structural analysis of RhoGDIα-14-3-3τ complex in the future.
Our results show that the binding between 14-3-3τ and RhoGDIα leads to the release of RhoA, Rac1, and Cdc42. This is consistent with a general role for RhoGDIα in controlling RhoA, Rac1, and Cdc42. Expression of an S174A RhoGDIα mutant blocked activation of RhoA, Rac1, and Cdc42 upon EGF treatment (Fig. 7B). Consistent with this, treatment of PAK1 inhibitor in MCF7 blocked the release of three Rho GTPases, including RhoA, Rac1, and Cdc42, upon EGF treatment (Fig. 7C). DerMardirossian and colleagues reported that phosphorylation of RhoGDIα by expressing a catalytically active PAK1 C-terminal construct in 293T cells resulted in dissociation of Rac1 but not RhoA (26). Since the association between RhoGDIα and Rho GTPases is also regulated by posttranslational modifications on different Rho GTPases (40), which provide another layer of selective regulation, the discrepancy could be related to differences in experimental contexts and cell types (EGF-treated MCF7 and MDA-MB231 cells in this study versus active PAK1-transfected 293T cells in DerMardirossian et al.'s study). Thus, 14-3-3τ appears to be a general regulator for RhoA, Rac1, and Cdc42 via the action of GDIα.
Although the present study focused on breast cancer, 14-3-3τ is also upregulated in many other solid tumors according to TCGA and Oncomine databases. In fact, 14-3-3τ was identified as one of the genes upregulated in melanoma tumors that developed metastasis compared to its level in the nonmetastatic ones (41). Since the regulation of Rho GTPases by RhoGDIα is fundamental in all cells, the mechanistic link between 14-3-3τ and cancer cell motility and invasion via RhoGDIα may be applicable to many other types of cancer.
The association of 14-3-3 with breast cancer has also been noted in other isoforms. Overexpression of 14-3-3ζ is associated with a higher risk of breast cancer recurrence and metastasis (42, 43). 14-3-3ζ can cooperate with ErbB2 to induce epithelial-mesenchymal transition and promote breast cancer progression (44). High expression of 14-3-3γ is also found in many breast cancer patients and is associated with shorter overall survival (45). On the other hand, expression of 14-3-3σ is downregulated in breast cancer due to promoter hypermethylation (46, 47). Thus, different isoforms of 14-3-3 appear to play sometimes similar, but sometimes opposite, roles in cancer cells. Interestingly, 14-3-3τ overexpression alone was able to promote cell motility in MCF7 cells, as shown in our study, whereas overexpression of 14-3-3ζ had no effect on cell motility (44), suggesting different roles for these two 14-3-3 isoforms in breast cancer progression.
MCF7 cells are known to have low metastatic potential in xenograft models. The data that overexpression of 14-3-3τ alone greatly promotes its metastatic rate in xenografts underscores the important role for 14-3-3τ in metastasis. While we identify a mechanism for 14-3-3τ-mediated Rho GTPase activation via RhoGDIα binding, we cannot rule out the possibility of additional mechanisms for the metastasis-promoting activity of 14-3-3τ. However, given the established role for Rho GTPase signaling in cancer progression and metastasis (14), this mechanism is expected to contribute significantly to the metastasis promoted by 14-3-3τ. Moreover, the significant correlation between 14-3-3τ and ROCK activity in primary human breast samples strongly supports that 14-3-3τ is a key regulator of Rho activity in breast cancer. Analyses from multiple cohorts also show positive correlation between 14-3-3τ and breast cancer metastasis. Overexpression of 14-3-3τ also promoted the growth of MCF7 xenografts. With the pivotal role of 14-3-3 in many intracellular signaling pathways, the detailed mechanisms for the in vivo growth-promoting activity likely are multifactorial and deserve further investigation. Long-term activation of Rho GTPases, especially Rac, can promote breast cancer cell growth through the Rac/Pak/cell cycle regulator pathway (12). 14-3-3τ also regulates p21Waf1/Cip1 to promote cell growth (3). Overexpression of 14-3-3τ could regulate breast tumor growth via additional yet-to-be-defined mechanisms. In conclusion, our data suggest that 14-3-3τ serves as a therapeutic target to prevent breast cancer metastasis. Even before 14-3-3τ-specific inhibitors are developed, with several ROCK inhibitors currently available, it will be valuable to test these inhibitors in patients with 14-3-3τ-overexpressing breast cancers for prevention of future metastasis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Celine DerMardirossian for RhoGDIα constructs and Wen-Chen Xiong for the GST-PBD and GST-RBD expression vectors. This work was supported by the Department of Defense Breast Cancer Research Program (W81XWH-09-1-0338), the National Institutes of Health (RO1CA100857, RO1CA138641, and ARRA 3 P30CA125123-03S5), and the Pathology and Histology Core at the Baylor College of Medicine with funding from the NIH (NCI P30CA125123).
The manuscript was read and approved by all listed authors. We disclose that we have no financial interests that pose a conflict of interest regarding the article.
Footnotes
Published ahead of print 12 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00076-14.
REFERENCES
- 1.Gardino AK, Yaffe MB. 2011. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Biol. 22:688–695. 10.1016/j.semcdb.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu H, Subramanian RR, Masters SC. 2000. 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40:617–647. 10.1146/annurev.pharmtox.40.1.617 [DOI] [PubMed] [Google Scholar]
- 3.Wang B, Liu K, Lin HY, Bellam N, Ling S, Lin WC. 2010. 14-3-3τ regulates ubiquitin-independent proteasomal degradation of p21, a novel mechanism of p21 downregulation in breast cancer. Mol. Cell. Biol. 30:1508–1527. 10.1128/MCB.01335-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin D, Brown-Luedi M, Chiquet-Ehrismann R. 2003. Tenascin-C signaling through induction of 14-3-3 tau. J. Cell Biol. 160:171–175. 10.1083/jcb.200206109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jahkola T, Toivonen T, Virtanen I, von Smitten K, Nordling S, von Boguslawski K, Haglund C, Nevanlinna H, Blomqvist C. 1998. Tenascin-C expression in invasion border of early breast cancer: a predictor of local and distant recurrence. Br. J. Cancer 78:1507–1513. 10.1038/bjc.1998.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegel R, Naishadham D, Jemal A. 2013. Cancer statistics, 2013. CA Cancer J. Clin. 63:11–30. 10.3322/caac.21166 [DOI] [PubMed] [Google Scholar]
- 7.DeSantis C, Ma J, Bryan L, Jemal A. 2013. Breast cancer statistics, 2013. CA Cancer J. Clin. 64:52–62. 10.3322/caac.21203 [DOI] [PubMed] [Google Scholar]
- 8.O'Shaughnessy J. 2005. Extending survival with chemotherapy in metastatic breast cancer. Oncologist 10(Suppl 3):S20–S29. 10.1634/theoncologist.10-90003-20 [DOI] [PubMed] [Google Scholar]
- 9.Vanharanta S, Massague J. 2013. Origins of metastatic traits. Cancer Cell 24:410–421. 10.1016/j.ccr.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117:927–939. 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 11.Alan JK, Lundquist EA. 2013. Mutationally activated Rho GTPases in cancer. Small GTPases 4:159–163. 10.4161/sgtp.26530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, Kazanietz MG. 2012. Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell Signal. 24:353–362. 10.1016/j.cellsig.2011.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burridge K, Wennerberg K. 2004. Rho and Rac take center stage. Cell 116:167–179. 10.1016/S0092-8674(04)00003-0 [DOI] [PubMed] [Google Scholar]
- 14.Parri M, Chiarugi P. 2010. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 8:23. 10.1186/1478-811X-8-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olofsson B. 1999. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 11:545–554. 10.1016/S0898-6568(98)00063-1 [DOI] [PubMed] [Google Scholar]
- 16.DerMardirossian C, Bokoch GM. 2005. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 15:356–363. 10.1016/j.tcb.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 17.Barone I, Brusco L, Gu G, Selever J, Beyer A, Covington KR, Tsimelzon A, Wang T, Hilsenbeck SG, Chamness GC, Ando S, Fuqua SA. 2011. Loss of Rho GDIalpha and resistance to tamoxifen via effects on estrogen receptor alpha. J. Natl. Cancer Inst. 103:538–552. 10.1093/jnci/djr058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, Frierson HF, Conaway MR, Theodorescu D. 2002. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 62:6418–6423 http://cancerres.aacrjournals.org/content/62/22/6418 [PubMed] [Google Scholar]
- 19.Harding MA, Theodorescu D. 2007. RhoGDI2: a new metastasis suppressor gene: discovery and clinical translation. Urol. Oncol. 25:401–406. 10.1016/j.urolonc.2007.05.006 [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Liu K, Lin FT, Lin WC. 2004. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J. Biol. Chem. 279:54140–54152. 10.1074/jbc.M410493200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Lai YJ, Lin WC, Lin FT. 2004. TRIP6 enhances lysophosphatidic acid-induced cell migration by interacting with the lysophosphatidic acid 2 receptor. J. Biol. Chem. 279:10459–10468. 10.1074/jbc.M311891200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke S, Wang W, Qiu X, Zhang F, Yustein JT, Cameron AG, Zhang S, Yu D, Zou C, Gao X, Lin J, Yallampalli S, Li M. 2013. Multiple target-specific molecular agents for detection and image analysis of breast cancer characteristics in mice. Curr. Mol. Med. 13:446–458. 10.2174/1566524011313030014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu K, Bellam N, Lin HY, Wang B, Stockard CR, Grizzle WE, Lin WC. 2009. Regulation of p53 by TopBP1: a potential mechanism for p53 inactivation in cancer. Mol. Cell. Biol. 29:2673–2693. 10.1128/MCB.01140-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schofield AV, Bernard O. 2013. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit. Rev. Biochem. Mol. Biol. 48:301–316. 10.3109/10409238.2013.786671 [DOI] [PubMed] [Google Scholar]
- 25.Chuang HH, Yang CH, Tsay YG, Hsu CY, Tseng LM, Chang ZF, Lee HH. 2012. ROCKII Ser1366 phosphorylation reflects the activation status. Biochem. J. 443:145–151. 10.1042/BJ20111839 [DOI] [PubMed] [Google Scholar]
- 26.DerMardirossian C, Schnelzer A, Bokoch GM. 2004. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol. Cell 15:117–127. 10.1016/j.molcel.2004.05.019 [DOI] [PubMed] [Google Scholar]
- 27.Qiao J, Holian O, Lee BS, Huang F, Zhang J, Lum H. 2008. Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. Am. J. Physiol. Cell Physiol. 295:C1161–C1168. 10.1152/ajpcell.00139.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu P, Morrison C, Wang L, Xiong D, Vedell P, Cui P, Hua X, Ding F, Lu Y, James M, Ebben JD, Xu H, Adjei AA, Head K, Andrae JW, Tschannen MR, Jacob H, Pan J, Zhang Q, Van den Bergh F, Xiao H, Lo KC, Patel J, Richmond T, Watt MA, Albert T, Selzer R, Anderson M, Wang J, Wang Y, Starnes S, Yang P, You M. 2012. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 33:1270–1276. 10.1093/carcin/bgs148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lochhead PA, Wickman G, Mezna M, Olson MF. 2010. Activating ROCK1 somatic mutations in human cancer. Oncogene 29:2591–2598. 10.1038/onc.2010.3 [DOI] [PubMed] [Google Scholar]
- 30.Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, Oshima H. 2003. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin. Cancer Res. 9:2632–2641 http://clincancerres.aacrjournals.org/content/9/7/2632 [PubMed] [Google Scholar]
- 31.Kamai T, Yamanishi T, Shirataki H, Takagi K, Asami H, Ito Y, Yoshida K. 2004. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 10:4799–4805. 10.1158/1078-0432.CCR-0436-03 [DOI] [PubMed] [Google Scholar]
- 32.Qi W, Liu X, Qiao D, Martinez JD. 2005. Isoform-specific expression of 14-3-3 proteins in human lung cancer tissues. Int. J. Cancer 113:359–363. 10.1002/ijc.20492 [DOI] [PubMed] [Google Scholar]
- 33.Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y, d'Assignies MS, Bergh J, Lidereau R, Ellis P, Harris AL, Klijn JG, Foekens JA, Cardoso F, Piccart MJ, Buyse M, Sotiriou C. 2007. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin. Cancer Res. 13:3207–3214. 10.1158/1078-0432.CCR-06-2765 [DOI] [PubMed] [Google Scholar]
- 34.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, Klijn JG, Larsimont D, Buyse M, Bontempi G, Delorenzi M, Piccart MJ, Sotiriou C. 2007. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J. Clin. Oncol. 25:1239–1246. 10.1200/JCO.2006.07.1522 [DOI] [PubMed] [Google Scholar]
- 35.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. 2005. Genes that mediate breast cancer metastasis to lung. Nature 436:518–524. 10.1038/nature03799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kolbl H, Gehrmann M. 2008. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 68:5405–5413. 10.1158/0008-5472.CAN-07-5206 [DOI] [PubMed] [Google Scholar]
- 37.van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415:530–536. 10.1038/415530a [DOI] [PubMed] [Google Scholar]
- 38.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. 2002. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347:1999–2009. 10.1056/NEJMoa021967 [DOI] [PubMed] [Google Scholar]
- 39.Hoffman GR, Nassar N, Cerione RA. 2000. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell 100:345–356. 10.1016/S0092-8674(00)80670-4 [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Mata R, Boulter E, Burridge K. 2011. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 12:493–504. 10.1038/nrm3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alonso SR, Tracey L, Ortiz P, Perez-Gomez B, Palacios J, Pollan M, Linares J, Serrano S, Saez-Castillo AI, Sanchez L, Pajares R, Sanchez-Aguilera A, Artiga MJ, Piris MA, Rodriguez-Peralto JL. 2007. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 67:3450–3460. 10.1158/0008-5472.CAN-06-3481 [DOI] [PubMed] [Google Scholar]
- 42.Bergamaschi A, Frasor J, Borgen K, Stanculescu A, Johnson P, Rowland K, Wiley EL, Katzenellenbogen BS. 2013. 14-3-3ζ as a predictor of early time to recurrence and distant metastasis in hormone receptor-positive and -negative breast cancers. Breast Cancer Res. Treat. 137:689–696. 10.1007/s10549-012-2390-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, Danes CG, Guo H, Lan KH, Ensor J, Hittelman W, Hung MC, Yu D. 2009. 14-3-3ζ overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 69:3425–3432. 10.1158/0008-5472.CAN-08-2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, Ling C, Zhou X, Chen T, Chiao PJ, Feng X, Seewaldt VL, Muller WJ, Sahin A, Hung MC, Yu D. 2009. 14-3-3ζ cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell 16:195–207. 10.1016/j.ccr.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song Y, Yang Z, Ke Z, Yao Y, Hu X, Sun Y, Li H, Yin J, Zeng C. 2012. Expression of 14-3-3gamma in patients with breast cancer: correlation with clinicopathological features and prognosis. Cancer Epidemiol. 36:533–536. 10.1016/j.canep.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 46.Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal PA, Stampfer MR, Sukumar S. 2000. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 97:6049–6054. 10.1073/pnas.100566997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S. 2001. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene 20:3348–3353. 10.1038/sj.onc.1204438 [DOI] [PubMed] [Google Scholar]
- 48.Lai YJ, Chen CS, Lin WC, Lin FT. 2005. c-Src-mediated phosphorylation of TRIP6 regulates its function in lysophosphatidic acid-induced cell migration. Mol. Cell. Biol. 25:5859–5868. 10.1128/MCB.25.14.5859-5868.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.