

ABSTRACT
Despite its strong host tropism for erythroid progenitor cells, human parvovirus B19 (B19V) can also infect a variety of additional cell types. Acute and chronic inflammatory cardiomyopathies have been associated with a high prevalence of B19V DNA in endothelial cells of the myocardium. To elucidate the mechanisms of B19V uptake into endothelium, we first analyzed the surface expression of the well-characterized primary B19V receptor P antigen and the putative coreceptors α5β1 integrins and Ku80 antigen on primary and permanent endothelial cells. The receptor expression pattern and also the primary attachment levels were similar to those in the UT7/Epo-S1 cell line regarded as functional for B19V entry, but internalization of the virus was strongly reduced. As an alternative B19V uptake mechanism in endothelial cells, we demonstrated antibody-dependent enhancement (ADE), with up to a 4,000-fold increase in B19V uptake in the presence of B19V-specific human antibodies. ADE was mediated almost exclusively at the level of virus internalization, with efficient B19V translocation to the nucleus. In contrast to monocytes, where ADE of B19V has been described previously, enhancement does not rely on interaction of the virus-antibody complexes with Fc receptors (FcRs), but rather, involves an alternative mechanism mediated by the heat-sensitive complement factor C1q and its receptor, CD93. Our results suggest that ADE represents the predominant mechanism of endothelial B19V infection, and it is tempting to speculate that it may play a role in the pathogenicity of cardiac B19V infection.
IMPORTANCE Both efficient entry and productive infection of human parvovirus B19 (B19V) seem to be limited to erythroid progenitor cells. However, in vivo, the viral DNA can also be detected in additional cell types, such as endothelial cells of the myocardium, where its presence has been associated with acute and chronic inflammatory cardiomyopathies. In this study, we demonstrated that uptake of B19V into endothelial cells most probably does not rely on the classical receptor-mediated route via the primary B19V receptor P antigen and coreceptors, such as α5β1 integrins, but rather on antibody-dependent mechanisms. Since the strong antibody-dependent enhancement (ADE) of B19V entry requires the CD93 surface protein, it very likely involves bridging of the B19V-antibody complexes to this receptor by the complement factor C1q, leading to enhanced endocytosis of the virus.
INTRODUCTION
Human parvovirus B19 (B19V), discovered in 1975 (1), exhibits a marked tropism for erythroid progenitor cells (EPCs) in the bone marrow or the fetal liver (2, 3). It has therefore been assigned as a member of the genus Erythrovirus in the family Parvoviridae. B19V infections in childhood, primarily transmitted via aerosol droplets to the respiratory tract, are mainly associated with a mild disease termed erythema infectiosum (also known as fifth disease) (4). However, by causing a block in erythropoiesis, B19V infection can also lead to more severe clinical manifestations, like transient aplastic crisis, in patients with chronic hemolytic anemia or pure red-cell aplasia due to persistent infection in immunocompromised patients (5). After transplacental transmission, which occurs in about one-third of maternal infections (6), B19V may cause hydrops fetalis or fetal loss (7, 8). A number of reports during recent years have demonstrated associations between the presence of B19V and an increasing spectrum of additional clinical diseases, such as rheumatoid arthritis (9, 10), vasculitis (11, 12), meningoencephalitis (13), and hepatitis (14, 15). Furthermore, acute and chronic inflammatory cardiomyopathies have also been linked to B19V infection (16–18). In a screen for the presence of viral infections in endomyocardial biopsy specimens from patients with idiopathic dilated cardiomyopathy (DCM), B19V DNA could be detected in more than half of the cases (19). In contrast to enteroviruses, which are also capable of cardiac infections and are preferentially found in myocytes, B19V DNA has been detected in the endothelial cells (ECs) of small intramyocardial arterioles and postcapillary venules during acute infection and in ECs of the close-meshed capillary system of the myocardium in persistent infection (20, 21). ECs in placental villi have also been identified as potential B19V target cells after transplacental transmission (22).
The linear 5.6-kb single-stranded DNA genome of B19V contains two major open reading frames (ORF) for the nonstructural protein NS1 and the structural proteins, respectively. It is packaged into a nonenveloped, icosahedral capsid with a diameter of 22 to 25 nm, which is composed of 60 subunits. The structural proteins VP1 and VP2, encoded by the ORF in the 3′ part of the B19V genome, account for 5% and 95%, respectively, of these structural subunits. VP1 (83 kDa) and VP2 (58 kDa) are identical except for the so-called VP1 unique (VP1u) region of 227 amino acids at the VP1 amino-terminal end. VP1u harbors a conserved phospholipase A2 (PLA2)-like motif, which has been implicated in viral infectivity (23) and the inflammatory response to B19V in synoviocytes (24).
At least part of the tropism of B19V for EPCs seems to be determined by the tissue distribution of the primary B19V receptor, the glycosphingolipid globoside (globotetraosylceramide [Gb4Cer]), also known as P antigen (25). P antigen was detected not only on EPCs, but also on a variety of other cell types, including primary endothelial cells from umbilical cord blood (HUVEC) (26). In line with these data, experimental infection of HUVEC with detection of NS1 mRNA and small amounts of VP mRNAs has been reported (27). Whereas P antigen is required for primary B19V attachment to its target cells, it is not sufficient to trigger the internalization step (26). As a coreceptor involved in the entry step, the α5β1 integrins in their high-affinity conformation have been identified (28). Furthermore, Ku80 antigen expressed at the cell surface has been proposed to enhance B19V attachment in both P antigen-negative and -positive cells (29). The involvement of a complex binding/internalization process in B19V entry is further emphasized by the lack of B19V binding to membrane-associated P antigen in vitro (30).
In addition to B19V entry, a variety of intracellular processes, such as viral transcript maturation (31, 32), translation (33), genome replication (34, 35), and receptor signaling (36), also limit structural gene expression and virion formation to the erythroid lineage. A few specialized cell lines, such as the megakaryoblastoid cell lines MB-02 (37) and UT7/Epo (38), which support B19V entry and low-level genome replication in the presence of erythropoietin (Epo), have been established as in vitro model systems for B19V infection. However, a major amplification of input B19V genomes is observed only in EPCs, either after direct infection in vivo or after generation of a CD36+ EPC population from hematopoietic stem cells ex vivo (39).
As an alternative to interactions with specific receptor/coreceptor molecules on the target cells, some viruses have exploited antiviral antibodies for efficient entry. This phenomenon, termed antibody-dependent enhancement of viral infection (ADE), was first described for flaviviruses (40) and has been shown to be important for the pathogenesis of dengue disease (41). Meanwhile ADE has been identified in a variety of other virus-cell systems, such as HIV-1 (42), Ebola virus (43), and Aleutian mink disease parvovirus (44). Recently, ADE could also be demonstrated for B19V infection of the monocytic cell line U937, with both enhanced initial levels of B19V DNA and a time-dependent increase in copy numbers, indicative of DNA replication in the presence of anti-B19V antibodies (45). The most common mechanism for ADE, initially proposed by Halstead et al. (46) and first actually demonstrated for flaviviruses in macrophages (47), is the interaction of the virus-antibody complex through the Fc portion of the antibody with Fc receptors (FcRs) on the cell surface, leading to enhanced attachment of the virus. This mechanism of ADE was also described for uptake of B19V into monocytes (45). Whereas FcRs are expressed predominantly on immune cells, alternative ADE mechanisms involving complement receptors (CR) more widely distributed among different cell types have also been described (48, 49).
In view of the high prevalence of B19V DNA in endomyocardial biopsy specimens from patients with chronic cardiomyopathies, it was the aim of our study to assess possible B19V entry routes into endothelial cells. We could demonstrate that the classical pathways mediated by specific receptors and coreceptors obviously do not play a major role in B19V uptake into endothelial cells. More likely, an alternative pathway mediated by B19V-specific antibodies may be important for efficient internalization of the virus. This ADE pathway was mediated by the heat-sensitive complement factor C1q and its receptor, CD93.
MATERIALS AND METHODS
Cell lines and primary cells.
U937 is a human cell line derived from a diffuse histiocytic lymphoma and displays characteristics of monocytic cells. It was cultured in RPMI medium (Gibco BRL, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (FBS) (Gibco BRL, Karlsruhe, Germany) and 100 μg/ml each of penicillin and streptomycin (Sigma, Munich, Germany).
EA.hy926, a stable hybrid cell line derived from fusion of primary HUVEC with the human lung carcinoma cell line A549, was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 100 μg/ml of penicillin and streptomycin under selection with HAT (100 mM hypoxanthine, 0.4 mM aminopterin, 16 mM thymidine; Invitrogen, Karlsruhe, Germany).
Primary human coronary artery endothelial cells (HCAEC), human dermal microvascular endothelial cells (HDMEC), human pulmonary artery endothelial cells (HPAEC), human aortic endothelial cells (HAoEC), and HUVEC, obtained from PromoCell (Heidelberg, Germany), were cultured according to the manufacturer's recommendations in endothelial cell growth medium and endothelial cell growth medium MV2 (PromoCell).
UT7/Epo-S1 is an Epo-dependent cell line originating from UT7, a megakaryocytic leukemia cell line (38), and was kindly provided by Susan Wong (Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, USA). UT7/Epo-S1 cells were cultured in Iscove's modified Dulbecco's medium (Mediatech, Herndon, VA) supplemented with 10% FBS, 100 μg/ml each of penicillin and streptomycin, 2 mM l-glutamine (Invitrogen, Karlsruhe, Germany), and 2 units per ml of recombinant human Epo (PBL Biomedical Laboratories, USA).
All cell lines and primary cells were cultured at 37°C with 5% CO2.
Preparation of human IgG samples.
IgG fractions were purified from human sera of healthy volunteers by affinity chromatography with prepacked 1-ml protein G HiTrap columns on an Akta purifier system (GE Healthcare, Buckinghamshire, United Kingdom). Prior to loading on the column at a flow rate of 0.5 ml/min, the sera were filtered (0.45-μm pore size) and diluted 1:1 in binding buffer (0.02 M sodium phosphate buffer, pH 7.0). The columns were washed with a total volume of 20 ml of binding puffer at a flow rate of 1 ml/min, and IgG fractions were eluted with 0.1 M glycine buffer, pH 2.7, at the same flow rate. The eluted IgG fractions were immediately neutralized by the addition of 1 M Tris-HCl, pH 9.0. Anti-B19V antibody IgG titers in the purified IgG fractions were measured by using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Biotrin, Dietzenbach, Germany) and were depicted as index values according to the instructions provided. An index value below 0.9 was defined as negative, and an index value above 1.1 was defined as positive for the presence of anti-B19V antibodies. The anti-B19V IgG titer indices for the three negative probes used in this study were in the range of 0.2 to 0.6, while the three positive probes had values of 8.0 (serum 1), 6.0 (serum 2), and 5.2 (serum 3), respectively. The protein concentrations of the purified IgG samples were determined by using the Bio-Rad Protein Assay Kit (Bio-Rad, Munich, Germany) according to the manufacturer's instructions.
B19V infections.
B19V belonging to genotype 1 was purified from plasma samples from patients with acute B19V infection, kindly provided by Knut Gubbe (Institute of Transfusion Medicine and Immunohematology, German Red Cross, Plauen, Germany) or by Anna Maria Eis-Hübinger (Institute of Virology, University Clinic Bonn, Bonn, Germany). For purification, 2 ml of frozen phosphate-buffered saline (PBS) (20%) was overlaid with 10 ml of plasma in ultracentrifuge tubes. The tubes were filled with PBS, and samples were centrifuged in a Beckman SW-40 Ti rotor (Beckman Coulter Inc., Krefeld, Germany) at 100,000 × g for 2 h at 4°C. The virus pellet was resuspended in 100 μl PBS, and the amount of B19V DNA was quantified by real-time PCR.
Infections of the various primary endothelial cells or permanent cell lines with the purified B19V were performed in 24-well plates. Cells were incubated with B19V at a multiplicity of infection (MOI) of 1,000 or 10,000 genomic particles per cell in a final volume of 300 μl. For the assays in the presence of antibodies, B19V was preincubated with purified human IgG fractions at different concentrations for 1 h at 37°C and 5% CO2 prior to infection, also in a final volume of 300 μl. For the competition experiments, purified antibodies were added during the preincubation step as indicated in the respective figure legends. Unless otherwise indicated, infections were performed for 2 to 6 h, followed by processing of the cells for quantitative determination of B19V DNA by real-time PCR, either directly or after prior cell fractionation. Generally, for the determination of virus uptake, noninternalized virus was removed by trypsinization for 5 min at 37°C.
DNA isolation.
Genomic DNA was prepared from cells by using the Pure Link Genomic DNA minikit (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions.
Isolation of nuclear and cytoplasmic fractions.
Fractionation of mock- or B19V-infected cells seeded in 6-cm dishes (5 × 105 to 1 × 106 cells at the time of infection) was performed 48 h postinfection. The cells were washed with PBS and detached from the culture dishes by trypsinization. After centrifugation of the cells at 1,500 × g at 4°C for 5 min and repeated washing with PBS, the cell pellets were gently resuspended in 100 μl of hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT]) in the presence of protease inhibitors (0.1 M phenylmethylsulfonyl fluoride [PMSF], 1 mg/ml pepstatin, and 1.5 mg/ml aprotinin) and incubated on ice for 5 min. The samples were then mixed gently and centrifuged for 5 min at 15,000 rpm and 4°C. The first 50 μl of the supernatants was collected as cytoplasmic fractions in a separate tube, while the second 50 μl was discarded to avoid contamination of the cytoplasmic fraction with the pelleted nuclei. Pellets were extracted two more times with 100 μl of hypotonic buffer using the same procedure to obtain a total volume of 150 μl of cytoplasmic extract. Subsequently, the nuclear pellets were washed three times with hypotonic buffer and resuspended in 30 μl of high-salt buffer (20 mM HEPES, pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 25% glycerin, 0.1 M PMSF, 1 mg/ml pepstatin, 1.5 mg/ml aprotinin). The nuclear suspension was incubated on ice for 20 min and centrifuged at 15,000 rpm at 4°C. The supernatant (nuclear fraction) was collected, and both the cytoplasmic and nuclear fractions were stored at −20°C for further use.
Fab fragment preparation.
The generation and purification of antibody Fab fragments from whole IgG were performed with the Pierce Fab Preparation Kit (Thermo Scientific, Schwerte, Germany) following the manufacturer's instructions. Briefly, 0.25 to 4 mg of the purified IgG fractions was dialyzed with Thermo Scientific Zeba Desalting Spin Columns and digested with papain protease contained in an immobilized form on an agarose matrix, and subsequently, the Fc fragments and the undigested IgG were removed by their binding to an immobilized protein A matrix. The pure Fab fragments contained in the supernatants from the protein A column were then used at a concentration of 200 μg/ml in direct comparison to the intact IgGs in the B19V infection experiments.
Real-time PCR for B19V DNA.
The real-time PCRs were performed in a final volume of 25 μl containing 30 to 50 ng of genomic DNA, 12,5 μl TaqMan Gene Expression master mix (Applied Biosystems, Darmstadt, Germany), 5 nM minor-groove binder (MGB) probe (5′-ATGACCCAGAGCACC-3′; Applied Biosystems, Darmstadt, Germany), 8 μM forward primer (5′-CATTTTCYAGACAGTTTTTAATYCCA-3′; B19V nucleotides [nt] 3420 to 3445), and 8 μM reverse primer (5′-CTYGCTGCGGGAGAAAACAC-3′; B19V nt 3489 to 3470). All experiments were performed in duplicate on an ABI 7900 HT Fast real-time PCR system (Applied Biosystems, Darmstadt, Germany) under the following conditions: hot-start denaturation at 95°C for 10 min, followed by 38 cycles of a two-step program (denaturation at 95°C for 15 s and annealing/extension at 57°C for 1 min). B19V copy numbers were determined by means of a plasmid-based B19V standard curve and are depicted as copies per cell initially infected based on a value of 6 pg for the genomic-DNA content of a single human cell.
Flow cytometry.
For analysis of B19V (co)receptor expression on the cell surface, cells were harvested by scraping them into the medium, pelleted by centrifugation at 1,500 × g at 4°C for 5 min, and washed with 1× PBS. All subsequent washing steps were generally performed with 1× PBS, followed by centrifugation as described above. The cells were washed and blocked in PBS-2% endobolin for 10 min at room temperature. Sequential incubations with primary and secondary antibodies with an intermediate washing step were each performed for 1 h at 4°C. The primary antibodies were rabbit anti-globoside GL-4 (P antigen) (Matreya, Pleasant Gap, USA), mouse anti-human integrin β1 (P4C10; Millipore, Schwalbach, Germany), mouse anti-Ku80 N terminus Ab-7 (S10B1; Neomarkers, Fremont, CA, USA), and mouse anti-Ku80 C terminus Ab-2 (111; Neomarkers), all diluted 1:400 in PBS. The secondary antibodies were Alexa Fluor 588- or Alexa Fluor 594 (Invitrogen GmbH, Karlsruhe, Germany)-conjugated antibodies, also diluted 1:400. After washing of the cells and resuspension in fluorescence-activated cell sorter (FACS) buffer (PBS, 0.5% bovine serum albumin [BSA], 0.1% sodium azide), approximately 1 × 105 cells were analyzed by flow cytometry.
Analysis of Fc receptor expression was performed by a similar protocol. In this case however, the cells were detached using 0.25% trypsin-EDTA, and both the initial fixation step and the blocking step with PBS-2% endobolin were omitted. The cells were incubated at room temperature with fluorescein isothiocyanate (FITC)-labeled anti-CD64, anti-CD32, and anti-CD16 (BD Bioscience, Heidelberg, Germany/BioLegend, Fell, Germany) at a dilution of 1:5 for 30 min in the dark. In parallel, cells were incubated with a FITC-labeled isotype control (BD Bioscience, Heidelberg, Germany) under the same conditions. After washing of the cells with 1 ml PBS and resuspension in PBS-2% paraformaldehyde (PFA), 1 × 104 cells were analyzed by flow cytometry.
For flow cytometry a FACSCalibur device (Becton, Dickinson, Heidelberg, Germany) was used according to the manufacturer's protocol. A cutoff fluorescence level was set in such a way that less than 1.0% of the cells in the negative controls, corresponding either to incubation with fluorescence-labeled secondary antibodies only in the case of B19V (co)receptors or the FITC-labeled isotype control in the case of Fc receptors, scored positive. All cells above this fluorescence level were assigned as positive.
Western blot analysis.
For the assessment of the purity of nuclear and cytoplasmic fractions, 10-μl aliquots were lysed by the addition of 10 μl of protein sample buffer (1 mM EDTA, 50 mM Tris [pH 7.5], 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue), and incubated at 95°C for 5 min. Subsequently, 15 to 20 μl of the samples was separated on 10% polyacrylamide-SDS gels and transferred to nitrocellulose membranes by semidry blotting. After blocking with 6% milk-PBS the membranes were incubated with a mixture of anti-tubulin monoclonal mouse antibody (Calbiochem, Merck, Darmstadt, Germany) diluted 1:2,000, anti-mcm7 monoclonal mouse antibody (Santa Cruz) diluted 1:1,000, and horseradish peroxidase-conjugated secondary antibody, followed by enhanced chemiluminescence (ECL) detection.
RESULTS
B19V is poorly taken up into endothelial cells despite effective binding to the cell surface.
In view of the close association of cardiac diseases with the presence of B19V DNA in endothelial cells of small intramyocardial arterioles and postcapillary venules, the objective of our study was to more closely elucidate the mechanisms involved in B19V entry into these cells. For that purpose, we first analyzed the expression profiles of the well-characterized primary B19V receptor, blood group P antigen, and the reported coreceptors integrin α5β1 and Ku80 antigen on the surfaces of endothelial cells by FACS analysis. Several primary endothelial cells from different sources and the permanent endothelial hybrid cell line EA.hy926 were compared to the UT7/Epo-S1 megakaryocytic leukemia cell line, which is known to be semipermissive for B19V infection and to promote efficient B19V entry and gene expression. For P antigen, the highest surface levels were observed for the UT7/Epo-S1 line, with a 75% proportion of positive cells (Fig. 1A, first column from left). However, very similar expression levels were displayed by some of the primary endothelial cells, such as those from the pulmonary artery (HPAEC) or from the umbilical vein (HUVEC), with more than 50% positive cells. The permanent EA.hy926 cell line displayed about 50% positive cells. Of all the primary endothelial cells tested, only those from the aorta (HAoEC) scored less than 10% positive cells. The surface levels of α5β1 integrin, which has been described as an essential B19V coreceptor, were uniformly very high, with only minor variations (Fig. 1A, second column). Whereas Ku80 has been described as a second potential coreceptor for B19V, only very limited amounts of the antigen could be detected on the surfaces of the semipermissive UT7/Epo-S1 cell line and the endothelial cells, regardless of whether antibodies recognizing the Ku80 carboxy terminus (Fig. 1A, third column) or the amino terminus (Fig. 1A, fourth column) were used. In contrast, large amounts of intracellularly localized Ku80 could be detected with either antibody after permeabilization of the cells (data not shown). Thus, a role of Ku80 in the B19V entry process in the cells under investigation seemed rather unlikely. We then examined B19V binding and virus uptake after infection of the different cell types, as described in detail in the legends to Fig. 1B and C and Materials and Methods. We showed that during an initial incubation period at 4°C, all the primary endothelial cells bound amounts of the input B19V similar to those bound by the UT7/Epo-S1 control cells (Fig. 1B). Only the EA-hy926 cells showed a clear reduction of about 3-fold in B19V binding. In contrast, when we monitored B19V uptake, the amount of B19V DNA detected in the endothelial cells was almost negligible compared to that found in the UT7/Epo-S1 control cells (Fig. 1C). The finding that the total number of B19V particles entering the UT7/Epo-S1 cells exceeds those bound during the initial incubation period at 4°C can be explained by a continuous supply of virus from the medium during the subsequent incubation at 37°C. These experiments showed that, although no obvious major differences are present in the receptor and coreceptor patches and in the primary binding between UT7/Epo-S1 and endothelial cells, the latter are obviously deficient for B19V internalization.
FIG 1.

B19V uptake into endothelial cells is strongly limited by the internalization step. (A) Surface expression of primary B19V receptor P antigen and potential coreceptors β1-integrins and Ku80 assayed by FACS analysis. For the detection of Ku80, two different antibodies recognizing either the carboxy terminus (Ku80-C) or the amino terminus (Ku80-N) were used. The percentages of (co)receptor-positive cells, calculated as described in Materials and Methods, are indicated. (B and C) B19V binding and virus uptake after application of 1 × 104 genomic particles of B19V per cell. B19V binding was determined after incubation for 1 h on ice (B), whereas uptake of viral particles was scored after an additional 2 h of incubation at 37°C without prior washing of the cells (C). Trypsinization was used to remove noninternalized particles. B19V DNA is represented as copies per cell initially infected. The data are presented as means plus standard deviations.
B19V uptake into endothelial cells is strongly stimulated in the presence of anti-B19V antibodies.
Given the low entry rates of B19V into endothelial cells, as demonstrated above, it was hard to envisage how B19V could establish a persistent infection in these cells in vivo using the classical receptor-mediated route. As an alternative entry route, therefore, we considered ADE, which had recently been demonstrated for B19V in the U937 monocytic cell line (45). U937 cells were therefore used as a positive control and showed a several hundredfold increase in B19V uptake in the presence of an IgG fraction of a human serum positive for anti-B19V antibodies applied at a final protein concentration of 400 μg/ml (Fig. 2A; note the logarithmic scale in Fig. 2A to F). This strong enhancement resulted in an almost complete recovery of the input amount of 1,000 genomic particles (note that a 10-fold-reduced number of viral particles compared to the experiments shown in Fig. 1B and C was applied) of B19V per cell. In contrast, the addition of purified IgG from an antibody-negative donor at an identical concentration had only marginal effects on B19V uptake. Preincubation of B19V with the anti-B19V-positive IgG preparation also strongly augmented the amount of B19V DNA detected 6 h postinfection in the permanent EA.hy926 endothelial cell line (Fig. 2B) and in all primary endothelial cells investigated (Fig. 2C and D). Although the absolute levels of B19V uptake were about an order of magnitude lower than in U937 cells, the relative levels of ADE were quite comparable to those of U937 cells (677-fold), with values of 270-fold for HCAEC (Fig. 2C), 411-fold for HDMEC, 590-fold for HAoEC, and even almost a 4,000-fold increase in B19V uptake for HPAEC (Fig. 2D). ADE also contributed strongly to B19V entry in the semipermissive UT7/Epo-S1 cell line (Fig. 2D). B19V ADE in HCAEC (Fig. 2E) and UT7/Epo-S1 cells (Fig. 2F) was confirmed with purified IgG samples from additional donors, whereas various IgG samples from B19V antibody-negative donors were used in the subsequent experiments and generally had only negligible effects compared to the control in the absence of antibodies.
FIG 2.

Preincubation with anti-B19V antibodies strongly stimulates B19V uptake into the monocytic cell line U937 and endothelial cells. (A to F) Different cell lines and primary endothelial cells, as indicated, were either mock infected or infected with B19V at an MOI of 1,000 genomic particles per cell. After 6 h of incubation at 37°C, genomic-DNA preparations were assayed for the number of B19V genomes by quantitative PCR. B19V DNA is presented on a logarithmic scale as genomic copies per cell initially infected. For the bars marked “+ control-IgG” or “+ α-B19V-IgG,” the B19V particles were preincubated prior to infection with purified IgG preparations from selected human sera with anti-B19V titers (index numbers) of <0.9 (control) or 8.0 (α-B19V), respectively, for 1 h at 37°C. The final concentrations of immunoglobulins during infection were 400 μg/ml. (E and F) In addition to the anti-B19V antibody-positive IgG preparation from serum 1 used in panels A to D (index number, 8.0), two further preparations with index numbers of 6.0 (serum 2) and 5.2 (serum 3) were used. The data are presented as means plus standard deviations.
Antibody-mediated B19V uptake into endothelial cells requires only low IgG concentrations.
In the initial experiments to monitor antibody-enhanced uptake of B19V in endothelial cells (Fig. 2), an IgG concentration of 400 μg/ml during the preincubation step with B19V was used. Whereas this concentration is well below the total immunoglobulin concentration of about 10 mg/ml found in human sera, we further assessed the physiological relevance of the observed ADE by performing titration experiments with IgG concentrations ranging from 5 μg/ml to 800 μg/ml. As a representative panel for these and most of the subsequent experiments, the EA.hy926 permanent endothelial cell line, the primary HCAEC, and the U937 monocytic cells were chosen (Fig. 3). In U937 cells, ADE could already be detected at the lowest IgG concentration of 5 μg/ml and reached saturation levels at a concentration of 400 to 800 μg/ml (Fig. 3A). In EA.hy926 cells (Fig. 3B) and HCAEC (Fig. 3C), a clear enhancement of B19V uptake was first observed at an IgG concentration of 10 μg/ml. Whereas ADE in HCAEC showed a continuous increase up to the maximum IgG concentration of 800 μg/ml, uptake into EA.hy926 cells was largely saturated at concentrations of 50 to 100 μg/ml. One possible explanation for this observation may be a limiting expression of the cellular receptor(s) recognizing the virus-antibody complexes in EA.hy926 cells.
FIG 3.

Concentration dependency of antibody-mediated B19V uptake into endothelial cells. (A to C) The indicated cell lines and primary endothelial cells were either mock infected (lanes marked control) or infected with B19V at an MOI of 1,000 genomic particles per cell. Where indicated, B19V was preincubated with increasing amounts (5- to 800-μg/ml final concentrations) of human serum-derived IgG preparations either negative (index number, <0.9) or positive (index number, 8.0) for anti-B19V antibodies. Genomic DNA extracted 6 h postinfection was assayed by quantitative PCR for the number of B19V genomes, which are presented on a logarithmic scale as genomic copies per cell initially infected. The data are presented as means plus standard deviations (**, P < 0.01; ***, P < 0.001).
The presence of antibodies enhances B19V uptake at the level of virus internalization.
To elucidate the stage of the B19V uptake process that is stimulated by the presence of antibodies, we analyzed the binding of B19V to the cell surface and the subsequent internalization step separately (Fig. 4). B19V virions at a final MOI of 1,000 genomic particles per cell were first incubated with the cells for 1 h at 4°C after prior preincubation of selected samples with IgG fractions from either B19V antibody-negative or -positive sera at a final concentration of 400 μg/ml. B19V binding was scored directly after the 4°C incubation step, which should have largely excluded possible internalization of the virus. In assays performed in parallel, the fraction of internalized virus was determined by extensive washing at 4°C to eliminate unbound virus, followed by further incubation for 2 h at 37°C for the internalization step. Finally, the noninternalized virus was released by trypsinization, and cells were processed for quantitative B19V DNA analysis. In the control U937 cells, the presence of anti-B19V antibodies increased the number of B19V virions bound to the cell surface by 204-fold (Fig. 4A), while the amount of internalized viral DNA was increased by a factor of 1,217 (Fig. 4B). In contrast to the strong stimulation of B19V binding by anti-B19V antibodies found in U937 cells, the binding of B19V to the endothelial EA.hy926 cells and HCAEC was only marginally affected, with increments of 2.8- and 2.0-fold, respectively (Fig. 4C and E). However, the presence of anti-B19V antibodies strongly increased the fraction of the bound virus actually internalized, leading to increases in B19V uptake of 117- and 46-fold, respectively (Fig. 4D and F). The lower absolute numbers of B19V particles entering the cells compared to the experiments shown in Fig. 2 were most probably due to the shorter time available for virus internalization (2 h versus 6 h) and the fact that in the experiments shown in Fig. 2 there was a continuous supply of virus from the medium during the 6 h of incubation. Thus, in endothelial cells, the enhancing effects of anti-B19V antibodies on B19V uptake for the most part are mediated at the level of virus internalization, whereas in the monocytic U937 cells, the antibodies predominantly, but not exclusively, stimulate virus binding.
FIG 4.

Enhancement of B19V uptake by antibodies is mediated at the level of virus internalization. The monocytic cell line U937 (A and B), the permanent endothelial cell line EA.hy926 (C and D), and primary HCAEC (E and F) were either mock infected or infected with B19V at an MOI of 1,000 genomic particles per cell in the presence or absence of anti-B19V antibodies, as described in the legend to Fig. 2. (A, C, and E) For determination of B19V binding to the cell surface, cells were directly processed after an initial incubation step at 4°C for 1 h. (B, D, and F) For the internalization assay, cells were incubated after extensive washing for a further 2 h at 37°C, followed by trypsinization to remove the noninternalized virus. Cell-associated B19V genomes were determined by PCR-based quantitative DNA analysis and are presented on a logarithmic scale as genomic copies per cell initially infected. The data are presented as means plus standard deviations.
Whereas antibody-mediated uptake of B19V into endothelial cells requires the Fc portion of the antibodies, it is not mediated by Fc receptors on the cell surface.
To further elucidate the determinants for enhanced cellular entry of the B19V-antibody complexes compared to noncomplexed virus, we first prepared the corresponding Fab fragments by papain protease digestion from the B19V antibody-positive IgG fractions. After removal of undigested IgG and Fc fragments on immobilized protein A, the effects of the purified Fabs in the B19V uptake assay were compared to those of intact IgG antibodies at identical protein concentrations of 200 μg/ml. ADE was completely lost for the Fab fragments in all three cell types tested, the monocytic control cell line U937 (Fig. 5A), the permanent endothelial cell line EA.hy926 (Fig. 5B), and the primary HCAEC (Fig. 5C). Binding of the virus-antibody complex to cellular FcRs through the Fc portion of the antibody is the most common ADE mechanism, identified for a variety of viruses. FcRs are predominantly expressed on cells of the immune system, but in some instances have also been described on endothelial cells (50). When we monitored the expression levels of the Fcγ type I, II, and III receptors (Fcγ-RI, -RII, and -RIII), we found high levels of Fcγ-RII, with 99.5% positive cells, and intermediate levels of the Fcγ-RI receptor, with 36.7% positive cells, in the U937 cell line (Fig. 5D). In endothelial cells, the surface levels of either receptor were much lower, with strongly reduced Fcγ-RII and no detectable Fcγ-RI expression in EA.hy926 cells and HCAEC. Of note, the endothelial cells from the pulmonary artery (HPAEC), which had shown the highest levels of ADE (compare Fig. 2D), had the lowest surface levels of Fcγ-RII and were completely negative for Fcγ-RI and Fcγ-RIII. Considering the low levels of Fcγ-R expression on the surfaces of endothelial cells, it seemed rather unlikely that the ADE of B19V entry in these cells would be mediated by the receptors. In line with this argument, the enhancing effect of the α-B19V antibodies in EA.hy926 or HPAEC could not be blocked by the addition of a mixture of antibodies against all three Fcγ receptors (Fig. 5E and F). For the monocytic cell line U937, a minor inhibition of antibody-mediated B19V uptake of about 2-fold in the presence of anti-Fcγ receptor antibodies was observed (Fig. 5G), which is in line with the findings of Munakata et al. (45). In contrast to the experiments of Munakata et al., however, we also observed inhibition with the isotype control antibodies, so no unambiguous conclusions for the monocytic cells can be drawn.
FIG 5.

The Fc portion of the anti-B19V antibodies is required for ADE in endothelial cells despite low surface levels of Fcγ receptors. (A to C) The monocytic cell line U937 (A), the permanent endothelial cell line EA.hy926 (B), and primary HCAEC (C) were either mock infected or infected with B19V at an MOI of 1,000 genomic particles in the absence or presence of either whole IgG preparations from a B19V antibody-positive serum or purified Fab fragments derived therefrom. (D) Surface levels of Fcγ receptors Fcγ-RI (CD64), Fcγ-RII (CD32), and Fcγ-RIII (CD16) on monocytic U937 cells, endothelial EA.hy926 cells, and primary HCAEC and HPAEC, as indicated, assayed by FACS analysis with specific antibodies. Nontreated cells and an isotype control are included. The percentages of cells that scored above the highest value of the isotype controls for the specific antibodies are indicated. The permanent endothelial cell line EA.hy926 (E), primary HPAEC (F), and the monocytic cell line U937 (G) were either mock infected or infected with B19V at an MOI of 1,000 genomic particles in the presence of IgG preparations from either B19V antibody-negative or -positive sera. Where indicated, a mixture of antibodies against all three classes of Fcγ-R (α-FcR) or isotype control antibodies was added during the infection. The data are presented as means plus standard deviations.
Blockage of the CD93 receptor for the complement factor C1q reduces B19V ADE in endothelial cells.
To identify possible alternative surface structures involved in antibody-mediated B19V uptake in endothelial cells, we first examined whether these structures could be blocked by excess nonspecific immunoglobulins. If B19V ADE were mediated by interaction of the virus-antibody complexes with cellular immunoglobulin binding structures other than Fcγ-R, a decrease in ADE would be expected. B19V was preincubated with increasing concentrations of the IgG fraction from a B19V antibody-negative serum in the presence of a constant amount of B19V antibody-positive IgG fraction (100-μg/ml final concentration). However, no competition of ADE was observed after the addition of nonspecific antibodies up to a final concentration of 1,600 μg/ml (Fig. 6A to C). In fact, whereas no major changes were observed in the U937 monocytic cells, increasing concentrations of nonspecific immunoglobulin led to a clear enhancement of ADE in the endothelial cell line EA.hy926 (Fig. 6B) and the primary HCAEC (Fig. 6C), with 4.3- and 47.7-fold increases in B19V uptake, respectively, at the highest concentration of 1,600 μg/ml. As we never observed any major effects of the B19V antibody-negative IgG preparations in the absence of α-B19V antibodies (compare Fig. 3), we hypothesized that one or more soluble factors may have been copurified in protein G affinity chromatography, due to their close association with antibodies, and that these factors may have been responsible for this further enhancement in the presence of α-B19V antibodies.
FIG 6.

Excess control IgG does not compete for B19V ADE in endothelial cells. (A to C) The indicated cell lines and primary endothelial cells were either mock infected or infected with B19V at an MOI of 1,000 genomic particles per cell. For some samples, B19V was preincubated either solely with IgG preparations from B19V antibody-negative or -positive sera or with a mixture of IgG from B19V antibody-positive serum (100 μg/ml) and increasing amounts of IgG from B19V antibody-negative serum, as indicated. Genomic DNA extracted 6 h postinfection was assayed by quantitative PCR for the number of B19V genomes, which are presented on a logarithmic scale as genomic copies per cell initially infected. The data are presented as means plus standard deviations (*, P < 0.05).
In addition to Fc receptors, complement receptors have also been implicated in ADE for some viruses (51). Therefore, components of the complement system were predicted as candidates for such soluble factors. The complement system has been shown to be inactivated by heat under conditions where the activity of antibodies is preserved. Heat inactivation of the α-B19V IgG preparation for 30 min at 56°C markedly reduced the antibody-mediated uptake in EA.hy926 cells (Fig. 7A, left). Furthermore, the additional effect of the nonspecific IgG preparation in the presence of the untreated α-B19V IgG was also abolished by heat inactivation of the former (Fig. 7A, right). Heat inactivation of the α-B19V IgG preparation could be compensated for by addition of an excess amount of nonspecific IgG, whereas heat inactivation of both α-B19V IgG and nonspecific IgG led to a strong reduction in ADE (Fig. 7A, right). Results very similar to those in the permanent EA.hy926 cell line were obtained in the primary HCAEC (Fig. 7B). Taken together, these results pointed to the possible involvement of a complement factor(s) in B19V ADE. A heat-labile component of the complement system shown to interact with the Fc portion of antibodies is the C1 complex, consisting of C1q and the two serine protease proenzymes C1r and C1s. Ca2+-dependent association of C1q with C1r and C1s is the initial step in the activation of the classical complement activation pathway and can be inhibited by addition of EGTA. However, separation of C1r and C1s can lead to increased binding of C1q to its own cell surface receptors, which include the CD93 molecule strongly expressed on endothelial cells (52). When we monitored the influence of EGTA as an inhibitor of the classical complement pathway on B19V ADE in HCAEC, we observed no significant effect (Fig. 7C, last column). In contrast, the addition of antibodies against the C1q receptor (CD93) strongly reduced B19V uptake (Fig. 7C). These results indicate that C1q is at least one of the heat-labile factors that mediate B19V ADE in endothelial cells, together with virus-specific antibodies. Hence, the mechanism seems to be very similar to the one determined for Ebola Zaire virus infection (43), where C1q bridges the immune complex of the virus and antibody, with the C1q receptor on the cell surface.
FIG 7.

Heat inactivation of IgG preparations or blockage of the CD93 receptor for the heat-labile complement factor C1q reduces B19V ADE in endothelial cells. (A and B) The permanent endothelial cell line EA.hy926 (A) and primary HCAEC (B) were either mock infected or infected with B19V at an MOI of 1,000 genomic particles after preincubation with the indicated IgG preparations, either in native form or after heat inactivation (h.i.) for 30 min at 56°C. (C) Primary HCAEC were either mock infected or infected with B19V at an MOI of 1,000 genomic particles after preincubation with IgG preparations (final concentration, 400 μg/ml) either negative (control-IgG) or positive for α-B19V antibodies. Selected samples positive for α-B19V antibodies were additionally preincubated either with a monoclonal antibody against the C1q CD93 receptor or with 50 mM EGTA as a general inhibitor of complement activation. (A to C) Genomic DNA extracted 6 h postinfection was assayed by quantitative PCR for the number of B19V genomes, which are presented as genomic copies per cell initially infected. The data are presented as means plus standard deviations (*, P < 0.05; ***, P < 0.001).
Despite efficient translocation of the viral DNA to the nucleus, B19V infection in the presence of antibodies is abortive.
We demonstrated that α-B19V antibodies dramatically increased B19V uptake into endothelial cells in the presence of heat-labile serum factors, such as C1q. An important issue that remained to be clarified, however, was whether the virus internalized by this route was efficiently translocated to the nucleus and could subsequently initiate a productive infection. To monitor nuclear translocation, cytoplasmic and nuclear fractions from U937 and EA.hy926 cells and HCAEC infected with B19V were assayed for the number of B19V genomes. Infections were performed with or without prior incubation with IgG fractions from anti-B19V-negative or -positive sera, as described previously. For cell fractionation, special emphasis was put on avoiding possible contamination of the nuclei with residual cytoplasmic fraction by repeated washing of the nuclei with hypotonic buffer. In line with these considerations, no tubulin, which was used as a cytoplasmic marker protein, was detectable in the nuclear fractions by Western blot analysis (Fig. 8A to C, bottom right). Whereas contamination of the nuclear fraction by cytoplasmic proteins could largely be ruled out in the Western analysis, the MCM7 replication protein used as a nuclear marker protein was also found in the cytoplasmic fraction, especially in U937 cells (Fig. 8A to C, bottom left). However, despite the resulting overestimation of the amount of B19V DNA present in the cytoplasm, more than 99% of the B19V DNA internalized in the presence of anti-B19V antibodies was actually found in the nuclear fraction in the monocytic U937 cell line, and also in the EA.hy926 and HCAEC endothelial cells (Fig. 8A to C, top, compare the cytoplasm with the nucleus). Thus, the B19V virions internalized by the antibody-mediated route, or at least the associated genomes, seem to be very efficiently transported to the nucleus.
FIG 8.
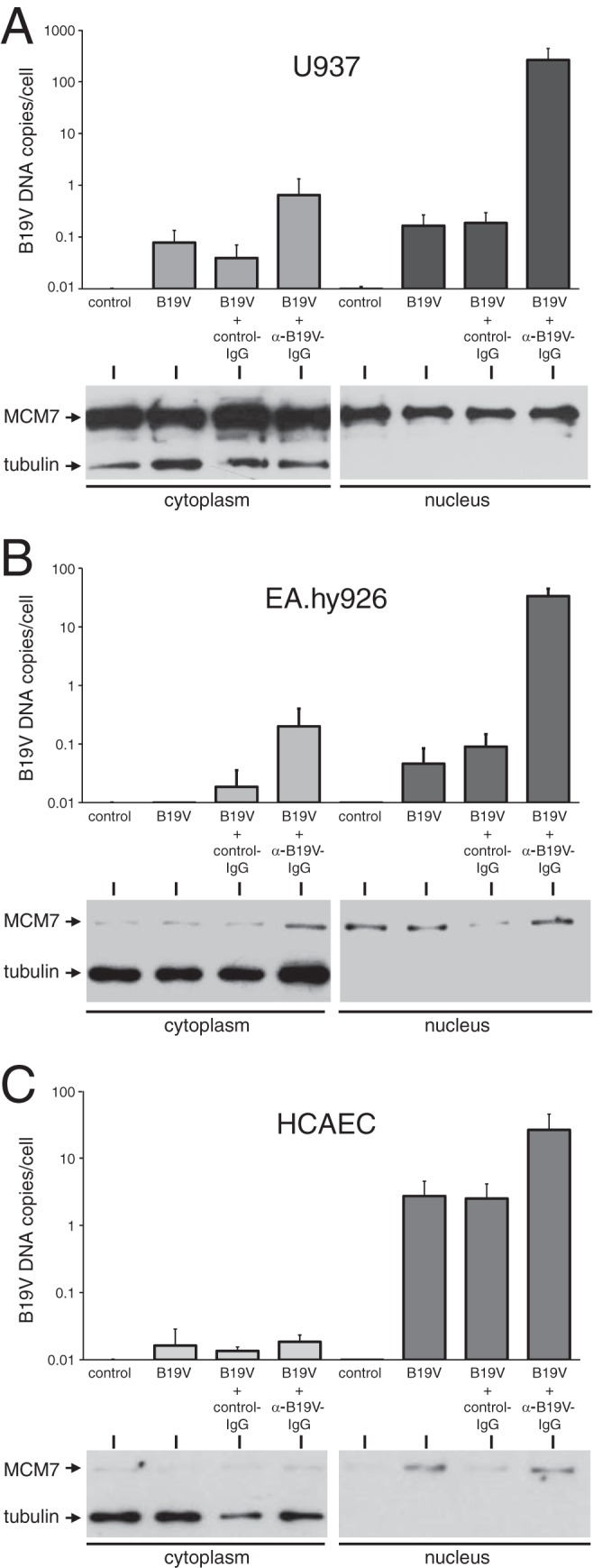
B19V internalized in the presence of antibodies is mainly found in the nuclear fraction. The monocytic cell line U937 (A), the permanent endothelial cell line EA.hy926 (B), and primary HCAEC (C) were either mock infected or infected with B19V at an MOI of 1,000 genomic particles per cell in the presence or absence of anti-B19V antibodies, as described in the legend to Fig. 2. After 6 h of incubation at 37°C, the cells were fractionated into a cytoplasmic and a nuclear fraction by hypotonic extraction, followed by high-salt extraction of the pelleted nuclei. At the bottom of each panel is shown Western blot analysis of equal proportions of the cytoplasmic and nuclear extracts for the presence of tubulin as a cytoplasmic marker protein and the replication factor MCM7 as a nuclear marker. Genomic DNA isolated from the remaining portions of the cytoplasmic and nuclear extracts was assayed by quantitative PCR for the number of B19V genomes, which are presented above the blots on a logarithmic scale as genomic copies per cell. The data are presented as means plus standard deviations.
To analyze whether the large amounts of B19V DNA, which accumulated in the nuclei of endothelial cells in the presence of anti-B19V antibodies, could initiate a productive B19V replication cycle, we first analyzed transcripts for the B19V nonstructural protein NS1, as well as for the capsid protein VP2, in HCAEC 72 h post-B19V infection by quantitative reverse transcriptase PCR (qRT-PCR). The low levels of NS1 transcripts found after B19V infection of endothelial cells were not raised by the presence of either nonspecific antibody or α-B19V antibodies (data not shown). Furthermore, no VP2 transcripts could be detected after infection with the B19V stock used for the ADE experiments at an MOI of 104 either in the absence or in the presence of α-B19V antibodies. Similar results were obtained with the EA.hy926 cell line (data not shown). Experiments with a second virus preparation that was also used in the initial uptake experiments in the absence of immunoglobulin (Fig. 1) led to barely detectable VP2 mRNA levels at least 104-fold lower than those found in the semipermissive UT7/Epo-S1 cells (data not shown). In line with the low to undetectable structural gene expression, no evidence of B19V replication was found after antibody-enhanced uptake of B19V in EA.hy926 cells and HCAEC, either by Southern blotting for replicative intermediates or by monitoring the total amount of B19V DNA over a 3-week period (data not shown). Thus, although antibodies can lead to strongly elevated levels of B19V DNA in the nuclei of endothelial cells, a productive infection cycle is not initiated.
DISCUSSION
Associations between B19V infection and acute and chronic inflammatory cardiomyopathies have been described, with the identification of myocardial endothelial cells as B19V target cells (20, 21). Whereas endothelial cells internalize B19V very poorly in vitro, the results of our present study suggest that in vivo B19V infection of endothelial cells may be subject to ADE. For a variety of endothelial cells from different tissues, the addition of immunoglobulin preparations positive for anti-B19V antibodies strongly increased B19V uptake at the level of virus internalization. B19V ADE led to accumulation of B19V DNA in the nuclei of the infected cells. The enhanced B19V uptake in the presence of anti-B19V antibodies, however, had no major impact on viral-gene expression and genome replication.
ADE of B19V in endothelial cells differs in several important respects from that recently described for the monocytic cell line U937 (45). For U937 cells, addition of B19V antibodies strongly stimulated the binding of B19V to the cell surface, whereas the effects of B19V antibodies on this initial step of the B19V entry process were only marginal in endothelial cells, and ADE was mediated almost exclusively at the level of virus internalization. One of the most common mechanisms for ADE of viral infection, which has been demonstrated not only for several members of the flavivirus family (47, 53–55), but also for a number of other viruses, including foot-and-mouth disease virus (56), coxsackievirus B4 (57), and human immunodeficiency virus (HIV) (58), is the binding of virus-antibody complexes to FcRs. For HIV, it has been demonstrated that ADE might be due to increased virus binding to the cell surface through the interaction between the Fc portion of the antibodies and the FcRs (59). The results of Munakata et al. (45), who were able to compete the B19V ADE in U937 cells with anti-FcR antibodies, suggested that such a mechanism may also apply to the antibody-mediated uptake of B19V into monocytic cells. Though we were not able to fully corroborate these finding in our experimental setting, the strong stimulation of B19V binding to the cell surface for U937 cells strongly argues for at least partial involvement of such a mechanism. As confirmed by our results, substantial levels of FcR are limited to cells of the immune system, such as B cells, monocytes/macrophages, neutrophils, and granulocytes (60). In line with the low levels of Fc receptors on endothelial cells, we demonstrated that antibody-mediated B19V uptake into endothelial cells is mediated by an alternative mechanism, the direct interaction of antibody-bound complement factor C1q with its receptor, CD93, on the cell surface. Such a direct mode of action of C1q in ADE without the requirement for further complement factors was first demonstrated for Ebola virus infection (43), and a detailed model has been proposed. It suggests that two or more molecules of the monomeric antibodies bind to closely adjacent viral epitopes, allowing C1q to bind to the Fc portion of the antibodies. Based on our results, the interaction of C1q with the CD93 receptor on the endothelial cell surface then strongly promotes the endocytosis of the virus-antibody complexes. We demonstrated that B19V DNA accumulates in the nucleus in the presence of antibodies and that both endosomal escape and nuclear translocation of the virus may be mediated by the VP1u-associated phospholipase A2 activity, which has been implicated in these processes in parvoviral infection of other cell systems (61, 62). The CD93 receptor is predominantly expressed in endothelial cells (63), but it has also been found in cells of the myeloid lineage and platelets (52). The initial binding of B19V to endothelial cells seems to be only marginally affected by the presence of antibodies, and we therefore propose that it may be mediated by interaction with the primary B19V receptor, P antigen. Substantial amounts of P antigen are expressed on the surfaces of endothelial cells, and we observed no major differences in initial B19V binding between the endothelial cells and the UT7/Epo-S1 cells regarded as permissive for B19V uptake.
Although we have not formally shown that our purified immunoglobulin preparations contain the C1q complement component, it seems very likely in view of the known properties of the factor. C1q is present in serum and plasma at relatively high concentrations of 70 to 80 μg/ml. Immobilized IgG present on the protein G affinity column during the purification step has been shown to form tightly associated complexes with C1q, which are stable in the presence of several reagents known to dissociate other protein-protein interactions (64). Whereas C1q would probably dissociate from the complex during elution of the IgG at acidic pH, the presence of soluble C1q in the purified fraction may also explain the stimulatory effect of an excess amount of anti-B19V-negative immunoglobulins on B19V uptake in the presence of anti-B19V antibodies. Interestingly, C1q has been shown to bind with higher affinity to polymeric IgM than to IgG (65). Thus, it is tempting to speculate that during the acute phase of the B19V infection cycle in the erythroid progenitor cells of the bone marrow, anti-B19V IgM antibodies may contribute to the spread of the virus to other tissues, such as synovial joints, the skin, or the endothelial cells of the myocardium.
Despite enhanced uptake of B19V in the presence of antibodies, the B19V infection of endothelial cells remained abortive, with only low levels of the NS1 mRNA and no detectable VP2 transcripts or DNA replication. It has been shown previously that endothelial cells are susceptible to B19V infection in vitro, with detection of low levels of NS1 and VP1 transcripts, but no evidence for a productive infection, such as an increase in B19V DNA amounts or detectable levels of VP1 or VP2 capsid proteins, was found (27). In vivo, B19V infection of endothelial cells has been demonstrated in myocardium (20) and skin (66), but again without any hints of a productive infection. However, both B19V nucleic acids and viral proteins have been detected in the fetal capillary endothelium in placental villi from a case of intrauterine fetal death caused by B19V (22). Interestingly, no P antigen expression could be found in the placental capillary endothelium (6), suggestive of alternative entry mechanisms. It is possible that under specific conditions in vivo, such as hypoxia or stimulation with growth factors or cytokines, endothelial cells may support the synthesis of viral proteins and viral-genome replication. Super- or coinfection with other viruses could also stimulate B19V replication. We recently demonstrated that infection with adenovirus or expression of early adenoviral functions activates B19V capsid gene expression and leads to the formation of putative replicative intermediates in certain endothelial cells (67). Human herpesvirus 6 (HHV-6) is another possible candidate to promote B19V replication in otherwise nonpermissive endothelial cells, since coinfections with B19V and HHV-6 have been found in over 10% of endomyocardial biopsy specimens from patients with dilated cardiomyopathy (19), and, similar to adenovirus, HHV-6 can act as a helper for the helper-dependent parvoviruses, the adeno-associated viruses (AAV) (68). Two possible explanations can be envisaged for our finding that no increase in B19V gene expression or genome replication could be observed despite highly elevated amounts of B19V DNA in the nuclei of infected cells in the presence of antibodies. On one hand, the lack of intracellular factors involved in transcriptional and/or posttranscriptional regulation of B19V gene expression in fully permissive cells (32, 34) may strongly limit these processes in endothelial cells despite large amounts of B19V DNA present. This view is supported by our recent findings that by providing large numbers of B19V genomes in transcription-competent form through transfection of an infectious B19V plasmid clone (pB19-M20) we could not enhance capsid protein synthesis in endothelial cells compared to B19V infection (67). As shown here, this leads to the uptake of only a small number of B19V genomes. On the other hand, we have not assessed whether the B19V particles taken up in the presence of antibodies are efficiently unpackaged, so this may represent a limiting step requiring a time frame beyond the one employed in our experiments.
In summary, we demonstrated for the first time that anti-B19V antibodies present in human sera can strongly enhance B19V uptake into endothelial cells by a mechanism dependent upon the CD93 surface protein, which acts as a cellular receptor for the complement factor C1q. Thus, in addition to the Fc receptor-mediated mechanism already described for the B19V ADE in monocytes, an additional mechanism of ADE exists for nonimmune cells. These findings offer a good explanation for the high prevalence of B19V in endothelial cells from a variety of tissues and may also be related to the spread of B19V to other cell types. Since C1q contributes to a number of inflammatory biological functions, its association with B19V-bound antibodies may also be involved in the inflammatory processes associated with some of the pathogenic effects of B19V, such as rheumatoid arthritis or chronic inflammatory cardiomyopathies.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Sonderforschungsbereich (SFB)/Transregio 19.
We thank Knut Gubbe (German Red Cross, Dresden, Germany) and Anna Maria Eis-Hübinger (Institute of Virology, University Clinic Bonn, Bonn, Germany) for providing human plasma containing parvovirus B19 (B19V). Furthermore, we thank Eva Hammer for technical assistance.
Footnotes
Published ahead of print 7 May 2014
REFERENCES
- 1.Cossart YE, Field AM, Cant B, Widdows D. 1975. Parvovirus-like particles in human sera. Lancet i:72–73 [DOI] [PubMed] [Google Scholar]
- 2.Ozawa K, Kurtzman G, Young N. 1986. Replication of the B19 parvovirus in human bone marrow cell cultures. Science 233:883–886. 10.1126/science.3738514 [DOI] [PubMed] [Google Scholar]
- 3.Srivastava A, Lu L. 1988. Replication of B19 parvovirus in highly enriched hematopoietic progenitor cells from normal human bone marrow. J. Virol. 62:3059–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young NS, Brown KE. 2004. Parvovirus B19. N. Engl. J. Med. 350:586–597. 10.1056/NEJMra030840 [DOI] [PubMed] [Google Scholar]
- 5.Anderson MJ, Higgins PG, Davis LR, Willman JS, Jones SE, Kidd IM, Pattison JR, Tyrrell DA. 1985. Experimental parvoviral infection in humans. J. Infect. Dis. 152:257–265. 10.1093/infdis/152.2.257 [DOI] [PubMed] [Google Scholar]
- 6.Broliden K, Tolfvenstam T, Norbeck O. 2006. Clinical aspects of parvovirus B19 infection. J. Intern. Med. 260:285–304. 10.1111/j.1365-2796.2006.01697.x [DOI] [PubMed] [Google Scholar]
- 7.Brown T, Anand A, Ritchie LD, Clewley JP, Reid TM. 1984. Intrauterine parvovirus infection associated with hydrops fetalis. Lancet ii:1033–1034 [DOI] [PubMed] [Google Scholar]
- 8.Knott PD, Welply GA, Anderson MJ. 1984. Serologically proved intrauterine infection with parvovirus. Br. Med. J. 289:1660. 10.1136/bmj.289.6459.1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerr JR. 2000. Pathogenesis of human parvovirus B19 in rheumatic disease. Ann. Rheum. Dis. 59:672–683. 10.1136/ard.59.9.672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore TL. 2000. Parvovirus-associated arthritis. Curr. Opin. Rheumatol. 12:289–294. 10.1097/00002281-200007000-00010 [DOI] [PubMed] [Google Scholar]
- 11.Dingli D, Pfizenmaier DH, Arromdee E, Wennberg P, Spittell PC, Chang-Miller A, Clarke BL. 2000. Severe digital arterial occlusive disease and acute parvovirus B19 infection. Lancet 356:312–314. 10.1016/S0140-6736(00)02512-5 [DOI] [PubMed] [Google Scholar]
- 12.Finkel TH, Leung DYM, Harbeck RJ, Gelfand EW, Török TJ, Zaki SR, Anderson LJ, Ferguson PJ, Saulsbury FT, Durigon EL, Hollister JR. 1994. Chronic parvovirus B19 infection and systemic necrotising vasculitis: opportunistic infection or aetiological agent? Lancet 343:1255–1258. 10.1016/S0140-6736(94)92152-0 [DOI] [PubMed] [Google Scholar]
- 13.Barah F, Vallely PJ, Chiswick ML, Cleator GM, Kerr JR. 2001. Association of human parvovirus B19 infection with acute meningoencephalitis. Lancet 358:729–730. 10.1016/S0140-6736(01)05905-0 [DOI] [PubMed] [Google Scholar]
- 14.Drago F, Semino M, Rampini P, Rebora A. 1999. Parvovirus B19 infection associated with acute hepatitis and a purpuric exanthem. Br. J. Dermatol. 141:160–161. 10.1046/j.1365-2133.1999.02943.x [DOI] [PubMed] [Google Scholar]
- 15.Yoto Y, Kudoh T, Haseyama K, Suzuki N, Chiba S. 1996. Human parvovirus B19 infection associated with acute hepatitis. Lancet 347:868–869. 10.1016/S0140-6736(96)91348-3 [DOI] [PubMed] [Google Scholar]
- 16.Bultmann BD, Klingel K, Sotlar K, Bock CT, Kandolf R. 2003. Parvovirus B19: a pathogen responsible for more than hematologic disorders. Virchows Arch. 442:8–17. 10.1007/s00428-002-0732-8 [DOI] [PubMed] [Google Scholar]
- 17.Nigro G, Bastianon V, Colloridi V, Ventriglia F, Gallo P, D'Amati G, Koch WC, Adler SP. 2000. Human parvovirus B19 infection in infancy associated with acute and chronic lymphocytic myocarditis and high cytokine levels: report of 3 cases and review. Clin. Infect. Dis. 31:65–69. 10.1086/313929 [DOI] [PubMed] [Google Scholar]
- 18.Schowengerdt KO, Ni J, Denfield SW, Gajarski RJ, Bowles NE, Rosenthal G, Kearney DL, Price JK, Rogers BB, Schauer GM, Chinnock RE, Towbin JA. 1997. Association of parvovirus B19 genome in children with myocarditis and cardiac allograft rejection: diagnosis using the polymerase chain reaction. Circulation 96:3549–3554. 10.1161/01.CIR.96.10.3549 [DOI] [PubMed] [Google Scholar]
- 19.Kuhl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller W, Kandolf R, Schultheiss HP. 2005. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 111:887–893. 10.1161/01.CIR.0000155616.07901.35 [DOI] [PubMed] [Google Scholar]
- 20.Bultmann BD, Klingel K, Sotlar K, Bock CT, Baba HA, Sauter M, Kandolf R. 2003. Fatal parvovirus B19-associated myocarditis clinically mimicking ischemic heart disease: an endothelial cell-mediated disease. Hum. Pathol. 34:92–95. 10.1053/hupa.2003.48 [DOI] [PubMed] [Google Scholar]
- 21.Klingel K, Sauter M, Bock CT, Szalay G, Schnorr JJ, Kandolf R. 2004. Molecular pathology of inflammatory cardiomyopathy. Med. Microbiol. Immunol. 193:101–107. 10.1007/s00430-003-0190-1 [DOI] [PubMed] [Google Scholar]
- 22.Pasquinelli G, Bonvicini F, Foroni L, Salfi N, Gallinella G. 2009. Placental endothelial cells can be productively infected by Parvovirus B19. J. Clin. Virol. 44:33–38. 10.1016/j.jcv.2008.10.008 [DOI] [PubMed] [Google Scholar]
- 23.Filippone C, Zhi N, Wong S, Lu J, Kajigaya S, Gallinella G, Kakkola L, Soderlund-Venermo M, Young NS, Brown KE. 2008. VP1u phospholipase activity is critical for infectivity of full-length parvovirus B19 genomic clones. Virology 374:444–452. 10.1016/j.virol.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu J, Zhi N, Wong S, Brown KE. 2006. Activation of synoviocytes by the secreted phospholipase A2 motif in the VP1-unique region of parvovirus B19 minor capsid protein. J. Infect. Dis. 193:582–590. 10.1086/499599 [DOI] [PubMed] [Google Scholar]
- 25.Brown KE, Anderson SM, Young NS. 1993. Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science 262:114–117. 10.1126/science.8211117 [DOI] [PubMed] [Google Scholar]
- 26.Weigel-Kelley KA, Yoder MC, Srivastava A. 2001. Recombinant human parvovirus B19 vectors: erythrocyte P antigen is necessary but not sufficient for successful transduction of human hematopoietic cells. J. Virol. 75:4110–4116. 10.1128/JVI.75.9.4110-4116.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zakrzewska K, Cortivo R, Tonello C, Panfilo S, Abatangelo G, Giuggioli D, Ferri C, Corcioli F, Azzi A. 2005. Human parvovirus B19 experimental infection in human fibroblasts and endothelial cells cultures. Virus Res. 114:1–5. 10.1016/j.virusres.2005.05.003 [DOI] [PubMed] [Google Scholar]
- 28.Weigel-Kelley KA, Yoder MC, Srivastava A. 2003. Alpha5beta1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of beta1 integrin for viral entry. Blood 102:3927–3933. 10.1182/blood-2003-05-1522 [DOI] [PubMed] [Google Scholar]
- 29.Munakata Y, Saito-Ito T, Kumura-Ishii K, Huang J, Kodera T, Ishii T, Hirabayashi Y, Koyanagi Y, Sasaki T. 2005. Ku80 autoantigen as a cellular coreceptor for human parvovirus B19 infection. Blood 106:3449–3456. 10.1182/blood-2005-02-0536 [DOI] [PubMed] [Google Scholar]
- 30.Kaufmann B, Baxa U, Chipman PR, Rossmann MG, Modrow S, Seckler R. 2005. Parvovirus B19 does not bind to membrane-associated globoside in vitro. Virology 332:189–198. 10.1016/j.virol.2004.11.037 [DOI] [PubMed] [Google Scholar]
- 31.Brunstein J, Soderlund-Venermo M, Hedman K. 2000. Identification of a novel RNA splicing pattern as a basis of restricted cell tropism of erythrovirus B19. Virology 274:284–291. 10.1006/viro.2000.0460 [DOI] [PubMed] [Google Scholar]
- 32.Liu JM, Green SW, Shimada T, Young NS. 1992. A block in full-length transcript maturation in cells nonpermissive for B19 parvovirus. J. Virol. 66:4686–4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pallier C, Greco A, Le Junter J, Saib A, Vassias I, Morinet F. 1997. The 3′ untranslated region of the B19 parvovirus capsid protein mRNAs inhibits its own mRNA translation in nonpermissive cells. J. Virol. 71:9482–9489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallinella G, Manaresi E, Zuffi E, Venturoli S, Bonsi L, Bagnara GP, Musiani M, Zerbini M. 2000. Different patterns of restriction to B19 parvovirus replication in human blast cell lines. Virology 278:361–367. 10.1006/viro.2000.0673 [DOI] [PubMed] [Google Scholar]
- 35.Guan W, Cheng F, Yoto Y, Kleiboeker S, Wong S, Zhi N, Pintel DJ, Qiu J. 2008. Block to the production of full-length B19 virus transcripts by internal polyadenylation is overcome by replication of the viral genome. J. Virol. 82:9951–9963. 10.1128/JVI.01162-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen AY, Guan W, Lou S, Liu Z, Kleiboeker S, Qiu J. 2010. Role of erythropoietin receptor signaling in parvovirus B19 replication in human erythroid progenitor cells. J. Virol. 84:12385–12396. 10.1128/JVI.01229-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munshi NC, Zhou S, Woody MJ, Morgan DA, Srivastava A. 1993. Successful replication of parvovirus B19 in the human megakaryocytic leukemia cell line MB-02. J. Virol. 67:562–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimomura S, Komatsu N, Frickhofen N, Anderson S, Kajigaya S, Young NS. 1992. First continuous propagation of B19 parvovirus in a cell line. Blood 79:18–24 [PubMed] [Google Scholar]
- 39.Wong S, Zhi N, Filippone C, Keyvanfar K, Kajigaya S, Brown KE, Young NS. 2008. Ex vivo-generated CD36+ erythroid progenitors are highly permissive to human parvovirus B19 replication. J. Virol. 82:2470–2476. 10.1128/JVI.02247-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hawkes RA. 1964. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls. Aust. J. Exp. Biol. Med. Sci. 42:465–482. 10.1038/icb.1964.44 [DOI] [PubMed] [Google Scholar]
- 41.Halstead SB, O'Rourke EJ. 1977. Antibody-enhanced dengue virus infection in primate leukocytes. Nature 265:739–741. 10.1038/265739a0 [DOI] [PubMed] [Google Scholar]
- 42.Robinson WE, Jr, Montefiori DC, Mitchell WM. 1988. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet i:790–794 [DOI] [PubMed] [Google Scholar]
- 43.Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. 2003. Antibody-dependent enhancement of Ebola virus infection. J. Virol. 77:7539–7544. 10.1128/JVI.77.13.7539-7544.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanno H, Wolfinbarger JB, Bloom ME. 1993. Aleutian mink disease parvovirus infection of mink macrophages and human macrophage cell line U937: demonstration of antibody-dependent enhancement of infection. J. Virol. 67:7017–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munakata Y, Kato I, Saito T, Kodera T, Ishii KK, Sasaki T. 2006. Human parvovirus B19 infection of monocytic cell line U937 and antibody-dependent enhancement. Virology 345:251–257. 10.1016/j.virol.2005.09.040 [DOI] [PubMed] [Google Scholar]
- 46.Halstead SB, O'Rourke EJ, Allison AC. 1977. Dengue viruses and mononuclear phagocytes. II. Identity of blood and tissue leukocytes supporting in vitro infection. J. Exp. Med. 146:218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peiris JS, Gordon S, Unkeless JC, Porterfield JS. 1981. Monoclonal anti-Fc receptor IgG blocks antibody enhancement of viral replication in macrophages. Nature 289:189–191. 10.1038/289189a0 [DOI] [PubMed] [Google Scholar]
- 48.Cardosa MJ, Porterfield JS, Gordon S. 1983. Complement receptor mediates enhanced flavivirus replication in macrophages. J. Exp. Med. 158:258–263. 10.1084/jem.158.1.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.June RA, Schade SZ, Bankowski MJ, Kuhns M, McNamara A, Lint TF, Landay AL, Spear GT. 1991. Complement and antibody mediate enhancement of HIV infection by increasing virus binding and provirus formation. AIDS 5:269–274 [DOI] [PubMed] [Google Scholar]
- 50.Nimmerjahn F, Ravetch JV. 2008. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8:34–47. 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- 51.Takada A, Kawaoka Y. 2003. Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev. Med. Virol. 13:387–398. 10.1002/rmv.405 [DOI] [PubMed] [Google Scholar]
- 52.Nepomuceno RR, Tenner AJ. 1998. C1qRP, the C1q receptor that enhances phagocytosis, is detected specifically in human cells of myeloid lineage, endothelial cells, and platelets. J. Immunol. 160:1929–1935 [PubMed] [Google Scholar]
- 53.Peiris JS, Porterfield JS. 1979. Antibody-mediated enhancement of Flavivirus replication in macrophage-like cell lines. Nature 282:509–511. 10.1038/282509a0 [DOI] [PubMed] [Google Scholar]
- 54.Daughaday CC, Brandt WE, McCown JM, Russell PK. 1981. Evidence for two mechanisms of dengue virus infection of adherent human monocytes: trypsin-sensitive virus receptors and trypsin-resistant immune complex receptors. Infect. Immun. 32:469–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlesinger JJ, Brandriss MW. 1983. 17D yellow fever virus infection of P388D1 cells mediated by monoclonal antibodies: properties of the macrophage Fc receptor. J. Gen. Virol. 64:1255–1262. 10.1099/0022-1317-64-6-1255 [DOI] [PubMed] [Google Scholar]
- 56.Mason PW, Baxt B, Brown F, Harber J, Murdin A, Wimmer E. 1993. Antibody-complexed foot-and-mouth disease virus, but not poliovirus, can infect normally insusceptible cells via the Fc receptor. Virology 192:568–577. 10.1006/viro.1993.1073 [DOI] [PubMed] [Google Scholar]
- 57.Hober D, Chehadeh W, Bouzidi A, Wattre P. 2001. Antibody-dependent enhancement of coxsackievirus B4 infectivity of human peripheral blood mononuclear cells results in increased interferon-alpha synthesis. J. Infect. Dis. 184:1098–1108. 10.1086/323801 [DOI] [PubMed] [Google Scholar]
- 58.Takeda A, Tuazon CU, Ennis FA. 1988. Antibody-enhanced infection by HIV-1 via Fc receptor-mediated entry. Science 242:580–583. 10.1126/science.2972065 [DOI] [PubMed] [Google Scholar]
- 59.Lund O, Hansen J, Soorensen AM, Mosekilde E, Nielsen JO, Hansen JE. 1995. Increased adhesion as a mechanism of antibody-dependent and antibody-independent complement-mediated enhancement of human immunodeficiency virus infection. J. Virol. 69:2393–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fanger NA, Fanger MW, Guyre PM. 1998. Fc receptors, p 886–892 In Roitti I, Delves PJ. (ed), Encyclopedia of immunology, 2nd ed. Academic Press, San Diego, CA [Google Scholar]
- 61.Farr GA, Zhang LG, Tattersall P. 2005. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. U. S. A. 102:17148–17153. 10.1073/pnas.0508477102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zadori Z, Szelei J, Lacoste MC, Li Y, Gariepy S, Raymond P, Allaire M, Nabi IR, Tijssen P. 2001. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 1:291–302. 10.1016/S1534-5807(01)00031-4 [DOI] [PubMed] [Google Scholar]
- 63.Fonseca MI, Carpenter PM, Park M, Palmarini G, Nelson EL, Tenner AJ. 2001. C1qR(P), a myeloid cell receptor in blood, is predominantly expressed on endothelial cells in human tissue. J. Leukoc. Biol. 70:793–800 [PubMed] [Google Scholar]
- 64.Kaul M, Loos M. 1997. Dissection of C1q capability of interacting with IgG. Time-dependent formation of a tight and only partly reversible association. J. Biol. Chem. 272:33234–33244 [DOI] [PubMed] [Google Scholar]
- 65.Meri S, von Bonsdorff C-H. 1998. Compement fixation test, p 617–619 In Roitt I, Delves PJ. (ed), Encyclopedia of immunology, 2nd ed. Academic Press, San Diego, CA [Google Scholar]
- 66.Magro CM, Nuovo G, Ferri C, Crowson AN, Giuggioli D, Sebastiani M. 2004. Parvoviral infection of endothelial cells and stromal fibroblasts: a possible pathogenetic role in scleroderma. J. Cutan. Pathol. 31:43–50. 10.1046/j.0303-6987.2003.0143.x [DOI] [PubMed] [Google Scholar]
- 67.Pozzuto T, von Kietzell K, Bock T, Schmidt-Lucke C, Poller W, Zobel T, Lassner D, Zeichhardt H, Weger S, Fechner H. 2011. Transactivation of human parvovirus B19 gene expression in endothelial cells by adenoviral helper functions. Virology 411:50–64. 10.1016/j.virol.2010.12.019 [DOI] [PubMed] [Google Scholar]
- 68.Thomson BJ, Weindler FW, Gray D, Schwaab V, Heilbronn R. 1994. Human herpesvirus 6 (HHV-6) is a helper virus for adeno-associated virus type 2 (AAV-2) and the AAV-2 rep gene homologue in HHV-6 can mediate AAV-2 DNA replication and regulate gene expression. Virology 204:304–311. 10.1006/viro.1994.1535 [DOI] [PubMed] [Google Scholar]