

ABSTRACT
We recently reported that herpes simplex virus 1 (HSV-1) protein kinase Us3 phosphorylated viral dUTPase (vdUTPase) at serine 187 (Ser-187) to upregulate its enzymatic activity, which promoted HSV-1 replication in human neuroblastoma SK-N-SH cells but not in human carcinoma HEp-2 cells. In the present study, we showed that endogenous cellular dUTPase activity in SK-N-SH cells was significantly lower than that in HEp-2 cells and that overexpression of cellular dUTPase in SK-N-SH cells increased the replication of an HSV-1 mutant with an alanine substitution for Ser-187 (S187A) in vdUTPase to the wild-type level. In addition, we showed that knockdown of cellular dUTPase in HEp-2 cells significantly reduced replication of the mutant vdUTPase (S187A) virus but not that of wild-type HSV-1. Furthermore, the replacement of Ser-187 in vdUTPase with aspartic acid, which mimics constitutive phosphorylation, and overexpression of cellular dUTPase restored viral replication to the wild-type level in cellular dUTPase knockdown HEp-2 cells. These results indicated that sufficient dUTPase activity was required for efficient HSV-1 replication and supported the hypothesis that Us3 phosphorylation of vdUTPase Ser-187 upregulated vdUTPase activity in host cells with low cellular dUTPase activity to produce efficient viral replication.virus.
IMPORTANCE It has long been assumed that dUTPase activity is important for replication of viruses encoding a dUTPase and that the viral dUTPase (vdUTPase) activity was needed if host cell dUTPase activity was not sufficient for efficient viral replication. In the present study, we showed that the S187A mutation in HSV-1 vdUTPase, which impaired its enzymatic activity, reduced viral replication in SK-N-SH cells, which have low endogenous cellular dUTPase activity, and that overexpression of cellular dUTPase restored viral replication to the wild-type level. We also showed that knockdown of cellular dUTPase in HEp-2 cells, which have higher dUTPase activity than do SK-N-SH cells, reduced replication of HSV-1 with the vdUTPase mutation but had no effect on wild-type virus replication. This is the first report, to our knowledge, directly showing that dUTPase activity is critical for efficient viral replication and that vdUTPase compensates for low host cell dUTPase activity to produce efficient viral replication.
INTRODUCTION
DNA viruses and a subset of retroviruses are known to encode homologs of host cell enzymes involved in nucleotide metabolism (e.g., thymidine kinase [TK], ribonucleotide reductase, uracil-DNA glycosylase, and/or dUTPase), which are mostly not essential for viral replication in cell cultures (1–7). Of these, viral dUTPase (vdUTPase) is of special interest because it is the homolog most widely encoded by viruses (3, 7–9). dUTPases catalyze the hydrolysis of dUTP to dUMP and pyrophosphate (10, 11). Since DNA polymerases are known to readily misincorporate dUTP into replicating DNA, which causes point mutations and strand breakage, dUTP hydrolysis by dUTPases is necessary for accurate DNA replication (11–14). dUTPase also plays a role in providing a substrate for thymidylate synthase, which converts dUMP to TMP, a major biosynthetic pathway for TTP (10, 11). dUTPases are present in a wide variety of eukaryotic and prokaryotic organisms, including mammals, plants, Drosophila melanogaster, and Escherichia coli. This ubiquity suggests the importance of dUTPase for DNA replication.
vdUTPases are encoded by a number of viruses, including herpesviruses, poxviruses, adenoviruses, D-type retroviruses, and African swine fever virus (ASFV) (8, 9). It has long been assumed that dUTPase activity is critical for the replication of viruses encoding a dUTPase and that vdUTPase activity compensates for it if there is not sufficient host cell dUTPase activity for efficient viral replication, e.g., in resting and differentiated cells, such as neurons and macrophages, where cellular dUTPase activity has been suggested to be low (4, 15). In support of this hypothesis, it has been reported that a herpes simplex virus 1 (HSV-1) mutant with a null mutation in its vdUTPase was less virulent than wild-type virus and replicated less well in the central nervous system (CNS) in a mouse model of HSV-1 infection (16). Furthermore, replication of recombinant ASFV and D-type retroviruses with mutations in each of their vdUTPases was significantly reduced in nondividing cells in vitro, whereas replication in actively dividing cells was only minimally decreased (4, 17–20). However, experimental evidence to directly prove this widely accepted hypothesis still has not been reported, e.g., data that knockdown of cellular dUTPase reduces the replication of viruses with mutations in vdUTPase and that overexpression of cellular dUTPase compensates for the reduction in replication of viruses carrying mutations in vdUTPase. Thus, definitive roles for dUTPase activity in viral replication and for viral dUTPases in compensating for low host cell dUTPase activity remain to be determined.
We recently reported that the HSV-1 protein kinase Us3 phosphorylated vdUTPase at serine 187 (Ser-187) and that this phosphorylation upregulated the enzymatic activity of vdUTPase in infected cells (21). We also showed that Us3 phosphorylation of vdUTPase at Ser-187 promoted viral replication in human neuroblastoma SK-N-SH cells but not in human carcinoma HEp-2 cells (21) and promoted viral pathogenicity in the CNS of mice but not at peripheral sites, including the eyes and vagina (22). These observations, together with the hypothetical role of dUTPases encoded by the various viruses described above, led us to hypothesize that (i) dUTPase activity was critical for efficient HSV-1 replication; (ii) Us3 phosphorylation of vdUTPase at Ser-187 upregulated its enzymatic activity to compensate for insufficient host cell dUTPase activity for efficient viral replication, such as in SK-N-SH cells; and (iii) this phosphorylation played no role in viral replication in cells with sufficient cellular dUTPase activity, such as HEp-2 cells. In agreement with this hypothesis, in the present study, we have shown that the enzymatic activity of cellular dUTPase in SK-N-SH cells was significantly lower than that in HEp-2 cells and that overexpression of cellular dUTPase in SK-N-SH cells restored viral replication of a mutant HSV-1 virus with an alanine substitution for Ser-187 (S187A) in vdUTPase to the wild-type level. We have also presented data further supporting the hypothesis that knockdown of endogenous cellular dUTPase in infected HEp-2 cells significantly reduced replication of the mutant virus with vdUTPase (S187A) but had no effect on replication of wild-type virus. In addition, a phosphomimetic mutation in vdUTPase Ser-187 and overexpression of cellular dUTPase restored mutant virus replication to the wild-type level in cellular dUTPase knockdown HEp-2 cells. This is the first report, to our knowledge, directly showing that dUTPase activity is critical for efficient viral replication and that virally encoded dUTPase compensates for low cellular dUTPase activity if it is not sufficient for efficient viral replication.
MATERIALS AND METHODS
Cells and viruses.
Simian kidney epithelial Vero, human carcinoma HEp-2, and human neuroblastoma SK-N-SH cells were described previously (23, 24), as was HSV-1 wild-type strain HSV-1(F) (25). Recombinant virus strain YK750, with a vdUTPase null mutation (ΔvdUTPase); recombinant virus strain YK751, with a vdUTPase S187A mutation (vdUTPaseS187A); recombinant virus strain YK752, in which the vdUTPase S187A mutation in YK751 was repaired (vdUTPaseΔ/SA-repair); recombinant virus strain YK753, with a vdUTPase S187D mutation (vdUTPaseS187D); and recombinant virus strain YK754, in which the vdUTPase S187D mutation in YK753 was repaired (vdUTPaseS187D-repair), were described previously (21) (Fig. 1). YK752 (vdUTPaseΔ/SA-repair) was the repaired virus for both YK750 (ΔvdUTPase) and YK751 (vdUTPaseS187A), based on a sequential construction strategy for recombinant viruses described previously (21).
FIG 1.
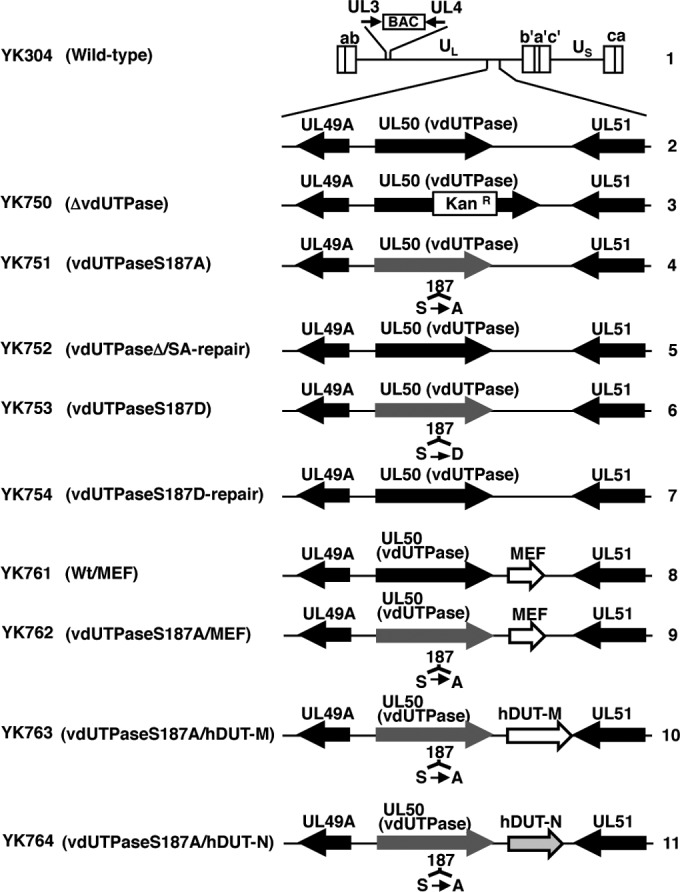
Schematic diagrams of the genome structure of the wild-type and recombinant HSV-1 strains used in this study. Line 1, wild-type HSV-1(YK304) genome carrying a bacmid (BAC) in the intergenic region between UL3 and UL4; line 2, domain carrying the UL49A, UL50 (vdUTPase), and UL51 open reading frames; lines 3 to 7, recombinant viruses with a mutation in the UL50 (vdUTPase) gene; lines 8 to 11, recombinant viruses in which a foreign gene expression cassette was inserted into the intergenic region between UL50 (vdUTPase) and UL51. MEF represents an MEF tag with myc and Flag epitopes and a TEV protease cleavage site. hDUT-M and hDUT-N represent the mitochondrial and nuclear isoforms of human dUTPase, respectively.
Plasmids.
To generate a stable cell line expressing short hairpin RNA (shRNA) against the 3′ untranslated region (UTR) of cellular dUTPase mRNA, pSSCH-hdUTPase was constructed, as follows. Oligonucleotides shown in Table 1 were annealed and cloned into the BbsI and SalI sites of pmU6 (26). The BamHI-SalI fragment of the resultant plasmid, containing the U6 promoter and the sequence encoding shRNA against the 3′ UTR of cellular dUTPase mRNA, was cloned into the BamHI and SalI sites of pSSCH (27), which is a derivative of retrovirus vector pMX containing a hygromycin B resistance gene, to produce pSSCH-hdUTPase. Plasmid pSSCH-Luc, encoding shRNA against firefly luciferase (Luc) mRNA, was constructed by using the same procedure as that used for the construction of pSSCH-hdUTPase, except using oligonucleotides shown in Table 1. To generate a retrovirus vector expressing human dUTPase isoform 2 (hDUT-N), pMX-hDUT-N was constructed by amplifying the hDUT-N open reading frame (ORF) by PCR from cDNA synthesized from total RNA of HEp-2 cells and cloning it into pMxs-puro (28). Total RNA was isolated from HEp-2 cells with a High Pure RNA isolation kit (Roche), and cDNA was synthesized from the isolated RNA with a Transcriptor First Strand cDNA synthesis kit (Roche), according to the manufacturer's instructions.
TABLE 1.
Oligonucleotide sequences for construction of plasmids and recombinant viruses
| Plasmid or recombinant virus | Oligonucleotide sequence (5′–3′) |
|---|---|
| Plasmids | |
| pSSCH-hdUTPase | TTTGTTTTTGCTTCAAGTGTTTTGGCTTCCTGTCACCAAAACACTTGAAGCAAAAACTTTTTTG |
| AATTCAAAAAAGTTTTTGCTTCAAGTGTTTTGGTGACAGGAAGCCAAAACACTTGAAGCAAAAA | |
| pSSCH-Luc | TTTGTCAAATGGCGATTACCGTTGGCTTCCTGTCACCAACGGTAATCGCCATTTGACTTTTTTG |
| AATTCAAAAAAGTCAAATGGCGATTACCGTTGGTGACAGGAAGCCAACGGTAATCGCCATTTGA | |
| pBS-EGRp-MEF-polyA-Kan | GCACTAGTATGGAGCAAAAGCTCATTTC |
| GCACTAGTTTAATCTTTGTCATCGTCGTCCT | |
| pBS-hDUT-M-Kan | CGGCATGCAGCTCCGCTTTGCCCGGAGGATGACGACGATAAGTAGGG |
| GCGCATGCCGCCCACCTCCGCAGGCCAACCAATTAACCAATTCTGATTAG | |
| Viruses | |
| YK761 (Wt/MEF) | TATCTCATCTTTCCTGTGTGTAGTTGTTTCTGTTGGAGGCCTGTGGGTTATGCGCCGACCCGGAAACGCC |
| TTCATCCAACCCGTGTGTTCTGTGTTTGTGGGATGGAGGGGCGGGTTAATGGACAAGTGTCCCGTTTTTT | |
| YK762 (vdUTPaseS187A/MEF) | AAGCGTGACTCCGGCCCTACCGGCGCGACGCCGAGGGCGGGCCCTCGTCTATGCCGGCGAAGGATGACGACGATAAGTAGGG |
| GTTCCGTCTGAACCGGCGTCAGCTCGCCGGCATAGACGAGGGCCCGCCCTCGGCGTCGCGCAACCAATTAACCAATTCTGATTAG | |
| YK763 (vdUTPaseS187A/hDUT-M) | GCCAGCTTCCGGTCGAGGTACCTAGGCTAGAACTAGTACCATGACTCCCCTCTGCCCTCGCCCCG |
| TGATATCGAATTCCTGCAGCCCGGGGGATCCACTAGTTTAATTCTTTCCAGTGGAACCAAAACC | |
| YK764 (vdUTPaseS187A/hDUT-N) | GCCAGCTTCCGGTCGAGGTACCTAGGCTAGAACTAGTATGCCCTGCTCTGAAGAGACACCCGCCAAGGATGACGACGATAAGTAGGG |
| GCAGGCCGGGCCCGCTTACTGGGTGAAATGGCGGGTGTCTCTTCAGAGCAGGGCATACTAGTTCTAGCCAACCAATTAACCAATTCTGATTAG |
pBS-EGRp-MEF-polyA-Kan and pBS-hDUT-M-Kan, used to generate recombinant viruses in the two-step Red-mediated mutagenesis procedures described below, were constructed as follows. To construct pBS-EGRp-MEF-polyA-Kan, the pGEM-MEF domain (28), encoding the I-SceI site, an MEF (Myc epitope–tobacco etch virus [TEV] protease cleavage site–Flag epitope) tag (29) and carrying the kanamycin resistance gene, was amplified by PCR from pGEM-MEF by using primers shown in Table 1 and cloned into the SpeI site of pRB5160, which contained the Egr-1 promoter region, a multicloning site, and bidirectional polyadenylation signals of the HSV-1 UL21 and UL22 genes (30). pBS-hDUT-M was constructed by amplifying the entire coding sequence of human cellular dUTPase isoform 1 (hDUT-M) by PCR from cDNA synthesized from total RNA of HEp-2 cells, as described above, and cloning it into pBluescript II KS(+) (Stratagene). pBS-hDUT-M-Kan was generated by amplifying the domain of pEPkan-S (31) carrying the I-SceI site and the kanamycin resistance gene by PCR from pEPkan-S using primers shown in Table 1 and cloning it into the SphI site of pBS-hDUT-M.
Mutagenesis of viral genomes and generation of recombinant HSV-1.
All recombinant HSV-1 strains used in this study were constructed by the two-step Red-mediated mutagenesis procedure in E. coli harboring wild-type or mutant HSV-1 genomes, as described previously (31, 32). To generate YK761 carrying an expression cassette consisting of the Egr-1 promoter, an MEF tag, and bidirectional polyadenylation signals of the HSV-1 UL21 and UL22 genes (EGRp-MEF-polyA) in the intergenic region between the UL50 and UL51 genes (Wt/MEF) (Fig. 1), a two-step Red-mediated mutagenesis procedure was carried out by using the primers listed in Table 1, pBS-EGRp-MEF-polyA-Kan, and E. coli GS1783 harboring pYEbac102, as described previously (31, 32). pYEbac102 contained a complete HSV-1(F) sequence with the bacterial artificial chromosome (BAC) sequence inserted into the HSV intergenic region between UL3 and UL4 (23). As a result of the two-step Red-mediated mutagenesis procedure, an E. coli clone (YEbac761) harboring the mutant HSV-1 BAC (pYEbac761), in which the EGRp-MEF-polyA expression cassette was inserted into the intergenic region between the UL50 and UL51 genes, was obtained. pYEbac761 was isolated from YEbac761, and YK761 (Wt/MEF) was generated by transfection of pYEbac761 into rabbit skin cells, as described previously (31, 32). Recombinant virus strain YK762 with an S187A mutation in vdUTPase and the EGRp-MEF-polyA expression cassette (vdUTPaseS187A/MEF) (Fig. 1) was generated by using the same procedure as that used to generate YK761 (Wt/MEF), except using primers listed in Table 1 and an E. coli clone (YEbac761) harboring the mutant HSV-1 BAC (pYEbac761). Recombinant virus strain YK763 with an S187A mutation in vdUTPase and an expression cassette consisting of the Egr-1 promoter, the human hDUT-M ORF, and bidirectional polyadenylation signals of the HSV-1 UL21 and UL22 genes (vdUTPaseS187A/hDUT-M) (Fig. 1) was generated by using the same procedure as that used to generate YK761 (Wt/MEF), except using the primers listed in Table 1, pBS-hDUT-M-Kan, and an E. coli clone (YEbac762) harboring the mutant HSV-1 BAC (pYEbac762). Recombinant virus strain YK764 with an S187A mutation in vdUTPase and an expression cassette consisting of the Egr-1 promoter, the human hDUT-N ORF, and bidirectional polyadenylation signals of the HSV-1 UL21 and UL22 genes (vdUTPaseS187A/hDUT-N) (Fig. 1) was generated by using the same procedure as that used to generate YK761 (Wt/MEF), except using the primers listed in Table 1 and an E. coli clone (YEbac763) harboring the mutant HSV-1 BAC (pYEbac763). The genotype of each recombinant virus was confirmed by sequencing (data not shown).
Antibodies.
Rabbit polyclonal antibodies to UL50 and UL51 were described previously (21, 33). Mouse monoclonal antibodies to human dUTPase (F6) and β-actin (AC15) were purchased from Santa Cruz Biotechnology and Sigma, respectively.
Generation of recombinant retroviruses.
Recombinant retroviruses were generated as described previously (34). Briefly, Plat-GP cells, a 293T-derived murine leukemia virus-based packaging cell line, were cotransfected with pMDG, encoding the vesicular stomatitis virus envelope G protein (28), and pSSCH-hdUTPase, pSSCH-Luc, or pMX-hDUT-N. Retrovirus-containing supernatants of Plat-GP cells were harvested at 2 days posttransfection.
Establishment of cell lines stably expressing shRNA against cellular dUTPase and firefly luciferase.
HEp-2 cells were infected with retrovirus-containing supernatants of Plat-GP cells that had been cotransfected with pMDG (28) and pSSCH-hdUTPase or pSSCH-Luc and selected with 50 μg hygromycin B/ml. This led to the isolation of sh-hDUT-HEp-2 and sh-Luc-HEp-2 cells.
Establishment of sh-hDUT-HEp-2 cells expressing hDUT-N exogenously.
sh-hDUT-HEp-2 cells were infected with retrovirus-containing supernatants of Plat-GP cells that had been cotransfected with pMD-G (28) and pMX-hDUT-N and selected with 1 μg puromycin/ml and 50 μg hygromycin B/ml. Single colonies of sh-hDUT-HEp-2 cells transduced with pMX-hDUT-N were isolated and screened by immunoblotting with an anti-human dUTPase antibody, which led to the isolation of sh-hDUT-HEp-2/hDUT-N(+) cells.
Assay for cell viability.
The viability of sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT-HEp-2/hDUT-N(+) cells was determined by using Cell Counting kit 8 (Dojindo) according to the manufacturer's instructions.
Immunoblotting.
Immunoblotting was performed as described previously (30). The amount of protein in immunoblot bands was quantitated by using the Dolphin Doc image capture system with Dolphin-1D software (Wealtec).
dUTPase enzyme assay.
dUTPase activity was assayed as described previously (16, 21, 35), with minor modifications. Briefly, SK-N-SH, HEp-2, sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT-HEp-2/hDUT-N(+) cells in 6-well plates were harvested and lysed in 200 μl NP-40 buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1.0% Nonidet P-40 [NP-40]). After a brief centrifugation, 0.2 μl of each supernatant was mixed with 200 μl of reaction buffer (50 mM Tris-HCl [pH 8.0], 2 mM β-mercaptoethanol, 1 mM MgCl2, 0.1% bovine serum albumin, 2 mM p-nitrophenylphosphate, 0.24 nM [3H]dUTP [28.8 Ci/mmol; PerkinElmer]). The reaction was allowed to proceed for 30 min at 37°C and then terminated by spotting the reaction mixture onto DE81 circle discs (Whatman). The discs were washed three times for 5 min each with washing solution (1 mM ammonium formate, 4 M formic acid), followed by one wash with 95% ethanol for 3 min. The discs were air dried and assayed for radioactivity by using an LS3801 scintillation counter (Beckman).
Statistical analysis.
Differences in the relative amount of hDUT-N, relative dUTPase activity, and relative cell viability were statistically analyzed by using the two-tailed Student t test. Differences in virus titers were statistically analyzed by using Holm's sequentially rejective Bonferroni multiple-comparison test.
RESULTS
Endogenous cellular dUTPase activity in SK-N-SH and HEp-2 cells.
To test the hypothesis described above, we first compared the endogenous cellular dUTPase activity in SK-N-SH cells with that in HEp-2 cells. The human dUTPase gene encodes both a nuclear isoform (hDUT-N) and a mitochondrial isoform (hDUT-M) of dUTPase with isoform-specific transcripts expressed by using alternative 5′ exons (36, 37). hDUT-N is the dominant isoform and is localized predominately in the nucleus, while hDUT-M is associated mainly with mitochondria (36, 37). In a denaturing gel, hDUT-M is detected as a band migrating more slowly than hDUT-N (36, 37). In agreement with a previous report (36, 37), cellular dUTPase from HEp-2 cells was detected as two bands in a denaturing gel, with the faster-migrating band (i.e., hDUT-N) being the dominant band (Fig. 2A). Interestingly, the amount of protein from SK-N-SH cells in the faster-migrating band was significantly smaller than that in the band from HEp-2 cells (Fig. 2A and B). In contrast, the amount of protein from HEp-2 cells in the slower-migrating band (i.e., hDUT-M) was similar to that from SK-N-SH cells. Consistent with this result, endogenous dUTPase activity in SK-N-SH cells was significantly lower than that in HEp-2 cells (Fig. 2C).
FIG 2.
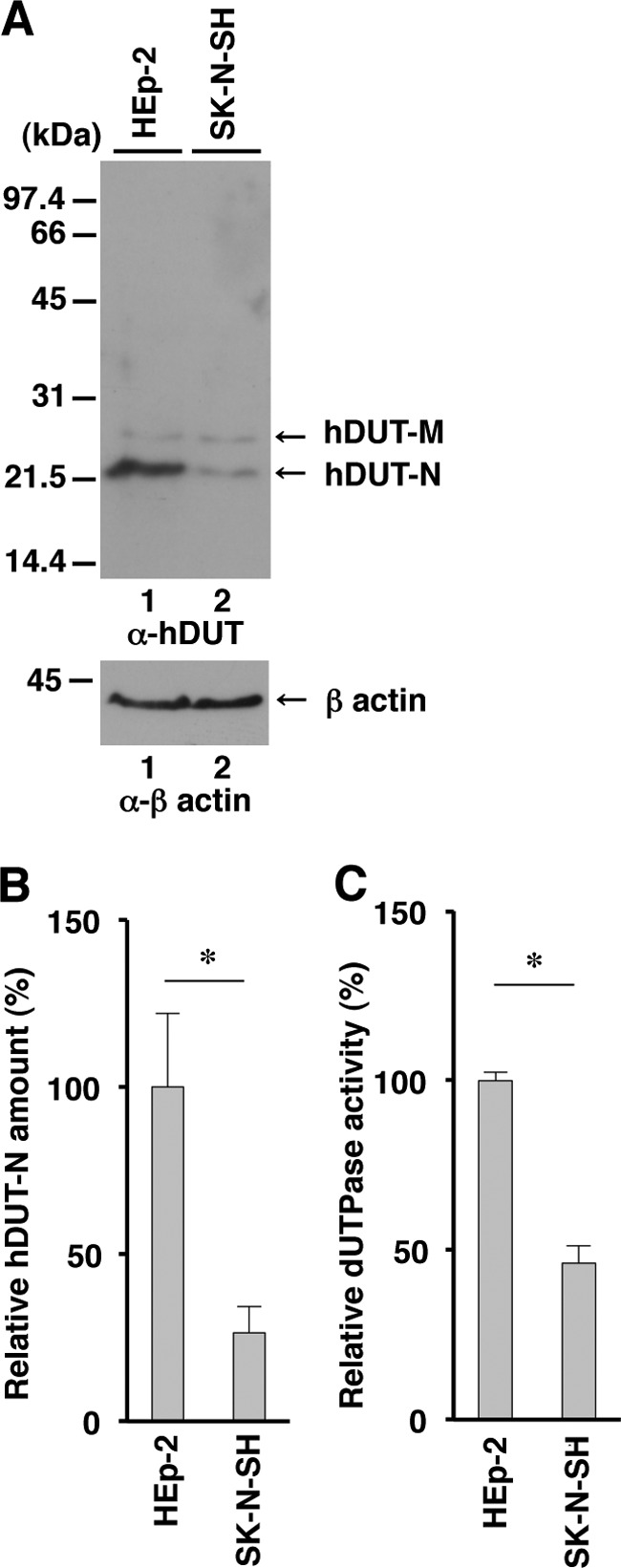
Expression and enzymatic activity of endogenous cellular dUTPase in SK-N-SH cells. (A) Expression of cellular dUTPase protein in HEp-2 (lane 1) and SK-N-SH (lane 2) cells analyzed by immunoblotting with anti-cellular dUTPase mouse monoclonal antibody (top) and anti-β-actin mouse monoclonal antibody (bottom). Molecular mass markers (in kilodaltons) are shown on the left. (B) Amount of hDUT-N in the SK-N-SH and HEp-2 cells shown in panel A (top) relative to that of β-actin shown in panel A (bottom). Each value is the mean ± standard error from triplicate experiments and is expressed relative to the mean value for HEp-2 cells, which was normalized to 100%. (C) Cellular dUTPase activity in SK-N-SH and HEp-2 cells. Each value is the mean ± standard error from triplicate experiments and is expressed relative to the mean value for HEp-2 cells, which was normalized to 100%. Asterisks indicate significant differences (*, P < 0.05) by the two-tailed Student t test. Data are representative of three independent experiments.
Effect of overexpression of cellular dUTPase on replication of HSV-1 with the vdUTPase S187A mutation.
To investigate the effect of overexpression of cellular dUTPase on the replication of HSV-1 with the S187A mutation in vdUTPase, which was reported previously to impair its dUTPase activity (21), we constructed three recombinant viruses: YK761, carrying the expression cassette for an MEF tag consisting of myc and Flag epitopes and a TEV protease cleavage site (29) in the intergenic region between UL50 and UL51 (Wt/MEF); YK762, with an S187A mutation in vdUTPase and carrying the MEF tag expression cassette in the intergenic region between UL50 and UL51 (vdUTPaseS187A/MEF); and YK764, with an S187A mutation in vdUTPase and carrying a cellular hDUT-N expression cassette (vdUTPaseS187A/hDUT-N) (Fig. 1). We used the MEF tag gene for a control foreign gene, which was unrelated to the hDUT-N gene. We previously reported that insertion of foreign genes into the intergenic region between UL50 and UL51 had no effect on viral replication in cell cultures (38). In agreement with that report, the growth curves of these three recombinant viruses were almost identical to that of wild-type HSV-1(F) in Vero cells infected at multiplicities of infection (MOIs) of 5 and 0.01 (Fig. 3A and B). Furthermore, insertion of these foreign genes into the intergenic region between UL50 and UL51 in the three recombinant viruses had no effect on the expression of UL50 and UL51, based on the observation that wild-type HSV-1(F), YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), and YK764 (vdUTPaseS187A/hDUT-N) produced similar levels of UL50 and UL51 in infected Vero cells (Fig. 3C). Overexpression of hDUT-N in Vero cells infected with YK764 (vdUTPaseS187A/hDUT-N) was confirmed by the observation that the anti-human dUTPase antibody used in this study, which could not react with simian endogenous dUTPase, did react with hDUT-N in the lysate of Vero cells infected with YK764 (vdUTPaseS187A/hDUT-N) (Fig. 3D).
FIG 3.
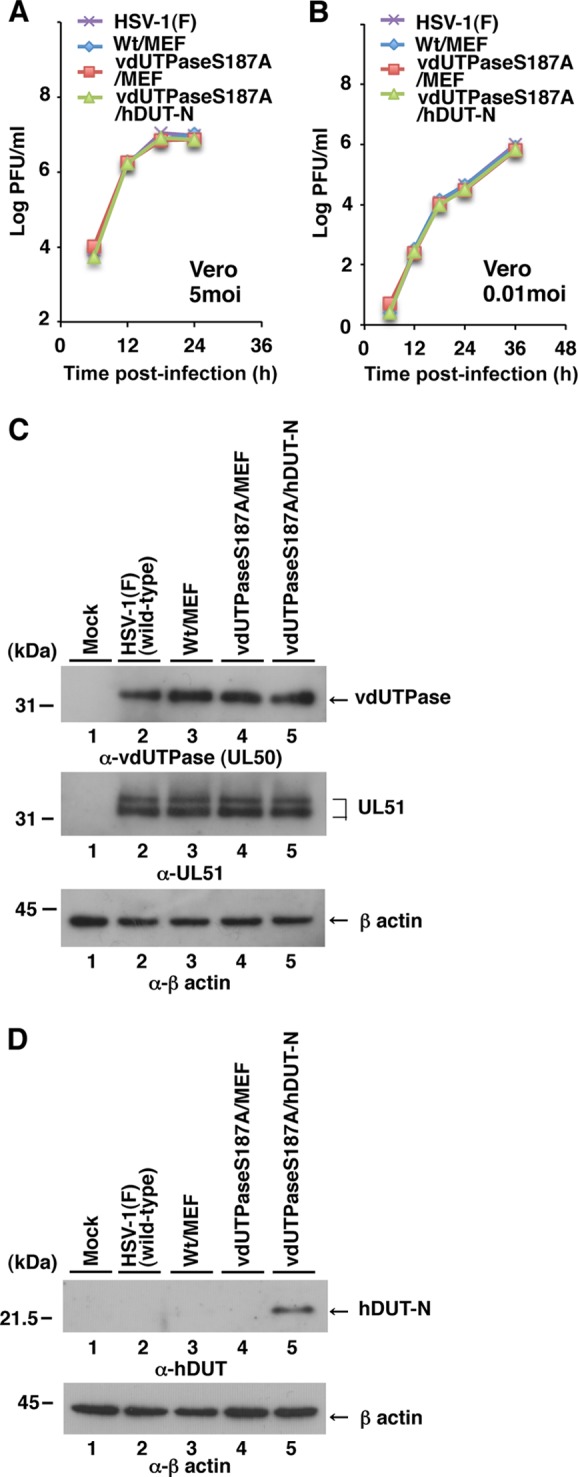
Characterization of the recombinant HSV-1 strains used in this study. (A) Expression of cellular dUTPase in Vero cells analyzed by immunoblotting with anti-cellular dUTPase mouse monoclonal antibody (top) and anti-β-actin mouse monoclonal antibody (bottom). (B) Expression of vdUTPase and UL51 in Vero cells analyzed by immunoblotting with anti-vdUTPase rabbit polyclonal antibody (top), anti-UL51 rabbit polyclonal antibody (middle), and anti-β-actin mouse monoclonal antibody (bottom). Molecular mass markers (in kilodaltons) are shown on the left. (C and D) Vero cells were infected at an MOI of 5 (C) or 0.01 (D) with wild-type strain HSV-1(F), YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N). Total virus from cell culture supernatants and infected cells was harvested at the indicated times and was assayed on Vero cells. Data are representative of three independent experiments.
SK-N-SH and HEp-2 cells were then infected with YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N) at an MOI of 5, and viral titers were assayed at 36 h postinfection. In agreement with our previous report that an HSV-1 strain with the vdUTPase S187A mutation replicated less efficiently than wild-type HSV-1 in SK-N-SH cells (21), the progeny virus titer (1.6 × 106 PFU/ml) in SK-N-SH cells infected with YK762 (vdUTPaseS187A/MEF) was significantly lower than that (4.2 × 106 PFU/ml) in these cells infected with YK761 (Wt/MEF) (Fig. 4B). In contrast, the progeny virus titer in SK-N-SH cells infected with YK764 (vdUTPaseS187A/hDUT-N) was similar to that in these cells infected with YK761 (Wt/MEF) (Fig. 4B). However, the progeny virus titers were similar in HEp-2 cells infected with YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N) (Fig. 4A). These results indicated that overexpression of cellular hDUT-N in SK-N-SH cells increased viral replication from the lower level caused by the vdUTPase S187A mutation to the wild-type level.
FIG 4.
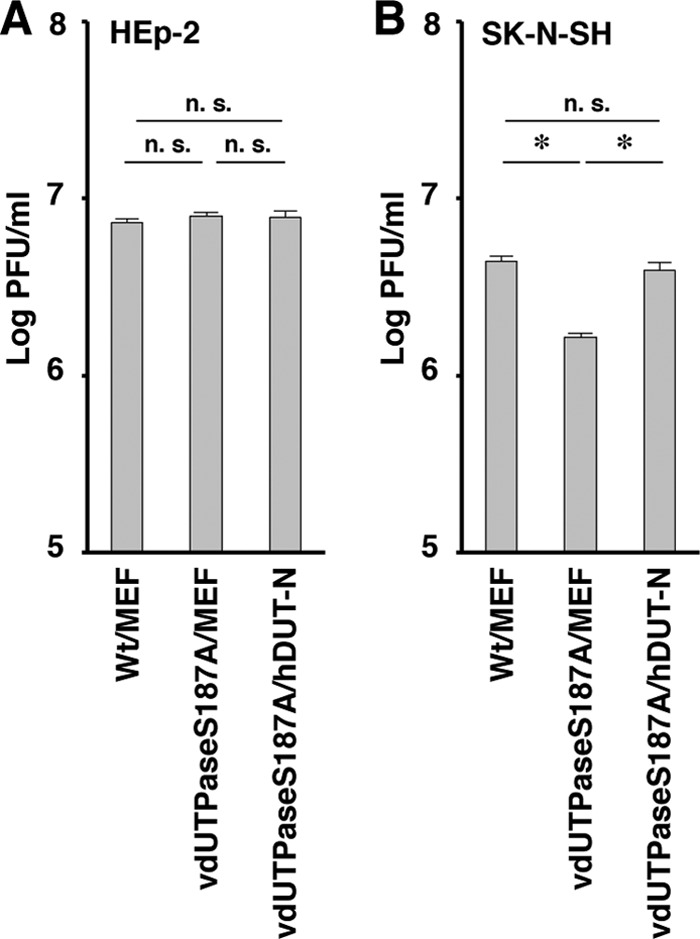
Effect of cellular dUTPase overexpression on HSV-1 replication in SK-N-SH cells. HEp-2 (A) and SK-N-SH (B) cells were infected with YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N) at an MOI of 5. Total virus from cell culture supernatants and infected cells was harvested at 36 h postinfection, and virus titers were assayed on Vero cells. Each value is the mean ± standard error from six duplicate experiments. Asterisks indicate significant differences (*, P < 0.0167) by Holm's sequentially rejective Bonferroni multiple-comparison test. n.s., not significant. Data are representative of three independent experiments.
Effect of endogenous cellular dUTPase knockdown on replication of HSV-1 with the vdUTPase S187A mutation.
To investigate the effect of cellular dUTPase knockdown in HEp-2 cells on replication of YK751 (vdUTPaseS187A), we generated a HEp-2 cell line stably expressing short hairpin RNA (shRNA) against the 3′-UTR regions of hDUT-N and hDUT-M mRNAs (sh-hDUT-HEp-2 cells) and a control cell line expressing shRNA against the ORF in firefly luciferase mRNA (sh-Luc-HEp-2 cells). In agreement with the results shown in Fig. 2A, hDUT-N and hDUT-M proteins in sh-Luc-HEp-2 cells were detected as two bands in a denaturing gel, with the faster-migrating band (i.e., hDUT-N) being the dominant band (Fig. 5A). In contrast, both hDUT-N and hDUT-M proteins were barely detectable in sh-hDUT-HEp-2 cells (Fig. 5A). Consistent with this result, endogenous dUTPase activity in sh-hDUT-HEp-2 cells was significantly lower than that in sh-Luc-HEp-2 cells (Fig. 5B). However, the viability of sh-hDUT-HEp-2 cells was similar to that of sh-Luc-HEp-2 cells (Fig. 5C).
FIG 5.
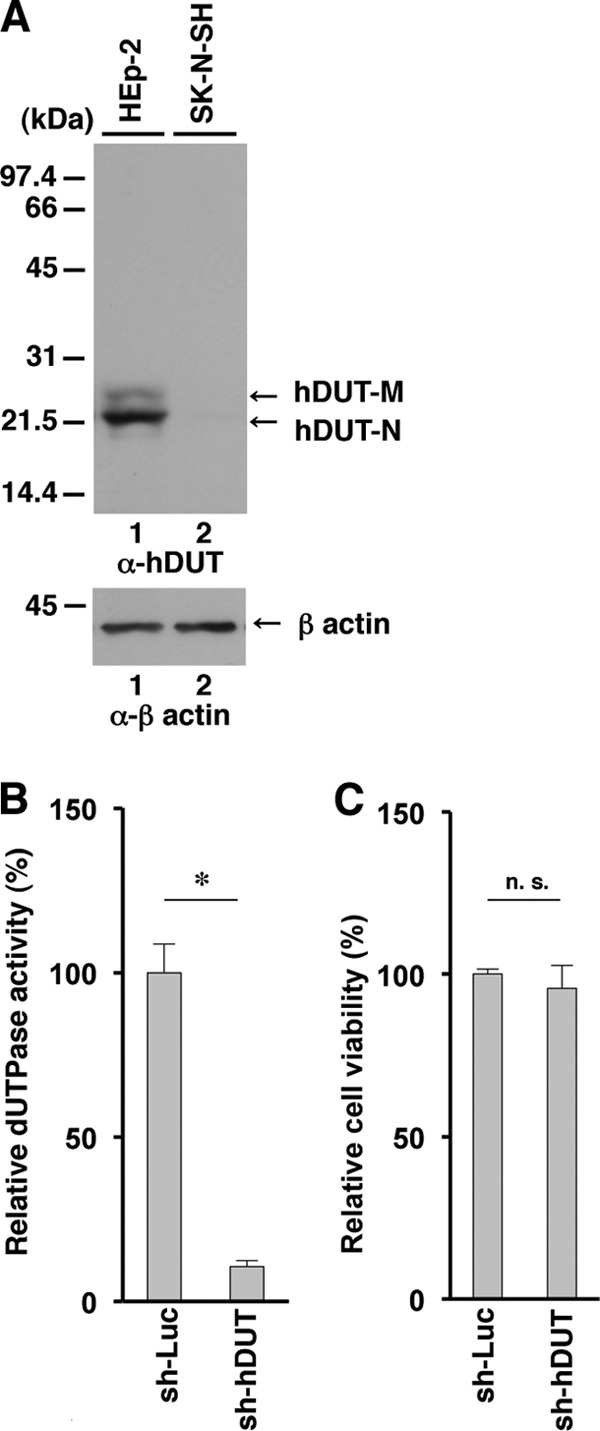
Effect of knockdown of endogenous cellular dUTPase in HEp-2 cells. (A) Expression of cellular dUTPase in sh-Luc-HEp-2 and sh-hDUT-HEp-2 cells analyzed by immunoblotting with anti-cellular dUTPase mouse monoclonal antibody (top) and anti-β-actin mouse monoclonal antibody (bottom). Molecular mass markers (in kilodaltons) are shown on the left. (B and C) Relative dUTPase activity (B) and cell viability (C) of sh-Luc-HEp-2 and sh-hDUT-HEp-2 cells. Each value is the mean ± standard error of data from triplicate experiments and is expressed relative to the mean for sh-Luc-HEp-2 cells, which was normalized to 100%. Asterisks indicate significant differences (*, P < 0.05) by the two-tailed Student t test. n.s., not significant. Data are representative of three independent experiments.
To further investigate the effect of dUTPase knockdown on viral replication, sh-hDUT-HEp-2 and sh-Luc-HEp-2 cells were infected with wild-type HSV-1(F); YK750 (ΔvdUTPase), a vdUTPase-null mutant virus; YK751 (vdUTPaseS187A); YK752 (vdUTPaseΔ/SA-repair), the repaired YK751 virus; YK753 (vdUTPaseS187D), a mutant virus carrying a phosphomimetic mutation in Ser-187; or YK754 (vdUTPaseS187D-repair), the repaired YK753 virus, at an MOI of 5 or 0.01, and viral titers were assayed at 36 and 60 h postinfection. As shown in Fig. 6A and C, the progeny virus titers of all these mutant viruses were similar to that of wild-type HSV-1(F) in sh-Luc-HEp-2 cells, as reported previously (21). In contrast, the titers of the YK750 (ΔvdUTPase) and YK751 (vdUTPaseS187A) viruses in sh-hDUT-HEp-2 cells were significantly lower than those of repaired virus strain YK752 (vdUTPaseΔ/SA-repair) and wild-type strain HSV-1(F) (Fig. 6B and D). Furthermore, the wild-type virus titer was restored in sh-hDUT-HEp-2 cells infected with YK753 (vdUTPaseS187D) carrying a phosphomimetic mutation in vdUTPase Ser-187, which has been reported to mimic constitutive phosphorylation (34, 39) and increase vdUTPase activity to the wild-type level (21) (Fig. 6B and D).
FIG 6.

Effect of endogenous cellular dUTPase knockdown in HEp-2 cells on HSV-1 replication. sh-Luc-HEp-2 (A and C) and sh-hDUT-HEp-2 (B and D) cells were infected with wild-type HSV-1(F), YK750 (ΔvdUTPase), YK751 (vdUTPaseS187A), YK752 (vdUTPaseΔ/SA-repair), YK753 (vdUTPaseS187D), or YK754 (vdUTPaseS187D-repair) at an MOI of 5 (A and B) or 0.01 (C and D). Total virus from the cell culture supernatants and infected cells was harvested at 36 h (A and B) and 60 h (C and D) postinfection (pi), and virus titers were assayed on Vero cells. Each value is the mean ± standard error from quadruplicate experiments. Asterisks indicate a significant difference in the mean (*, P < 0.0083; **, P < 0.0167) by Holm's sequentially rejective Bonferroni multiple-comparison test. n.s., not significant. Data are representative of three independent experiments.
To examine whether the phenotype(s) observed in sh-hDUT-HEp-2 cells was due to an off-target effect(s) of the anti-cellular dUTPase shRNA, two sets of experiments were performed. First, we generated sh-hDUT-HEp-2/hDUT-N(+) cells, in which cellular hDUT-N was expressed exogenously, by transduction of sh-hDUT-HEp-2 cells with a retrovirus vector expressing hDUT-N. The levels of hDUT-N expression and cellular dUTPase activity in sh-hDUT-HEp-2/hDUT-N(+) cells were comparable to those in sh-Luc-HEp-2 cells but considerably higher than those in sh-hDUT-HEp-2 cells (Fig. 7A and B). In contrast, the viability of sh-hDUT-HEp-2/hDUT-N(+) cells was similar to those of sh-Luc-HEp-2 and sh-hDUT-HEp-2 cells (Fig. 7C). sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT-HEp-2/hDUT-N(+) cells were then infected with wild-type strain HSV-1(F), YK750 (ΔvdUTPase), or YK751 (vdUTPaseS187A) at an MOI of 5 or 0.01, and viral titers were assayed at 36 and 60 h postinfection. In agreement with the results shown in Fig. 6B and D, the progeny virus titers of YK750 (ΔvdUTPase) and YK751 (vdUTPaseS187A) in sh-hDUT-HEp-2 cells were significantly lower than those in sh-Luc-HEp-2 cells (Fig. 8B, C, E, and F). However, the progeny virus yields of YK750 (ΔvdUTPase) and YK751 (vdUTPaseS187A) in sh-hDUT-HEp-2/hDUT-N(+) cells increased significantly compared to those in sh-hDUT-HEp-2 cells and were comparable to those in sh-Luc-HEp-2 cells (Fig. 8B, C, E, and F). The virus titers of wild-type strain HSV-1(F) in sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT-HEp-2/hDUT-N(+) cells were similar (Fig. 8A and D). Second, sh-Luc-HEp-2 cells and sh-hDUT-HEp-2 cells were infected with wild-type strain HSV-1(F), YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N) at an MOI of 5, and viral titers were assayed at 36 h postinfection. Consistent with the results for wild-type strain HSV-1(F), YK751 (vdUTPaseS187A), and YK752 (vdUTPaseΔ/S187A-repair) (Fig. 6B and 8C), the progeny viral titer in sh-hDUT-HEp-2 cells infected with YK762 (vdUTPaseS187A/MEF) was significantly lower than the titers in these cells infected with wild-type strain HSV-1(F) or YK761 (Wt/MEF) (Fig. 9B). In contrast, the progeny viral titer in sh-hDUT-HEp-2 cells infected with YK764 (vdUTPaseS187A/hDUT-N) was similar to those in these cells infected with wild-type HSV-1(F) or YK761 (Wt/MEF) (Fig. 9B). The titers of these viruses in sh-Luc-HEp-2 cells were similar (Fig. 9A). These results eliminated the possibility that the reduced replication of the vdUTPase mutant virus in sh-hDUT-HEp-2 cells was due to an off-target effect(s) of the shRNA against cellular dUTPase.
FIG 7.
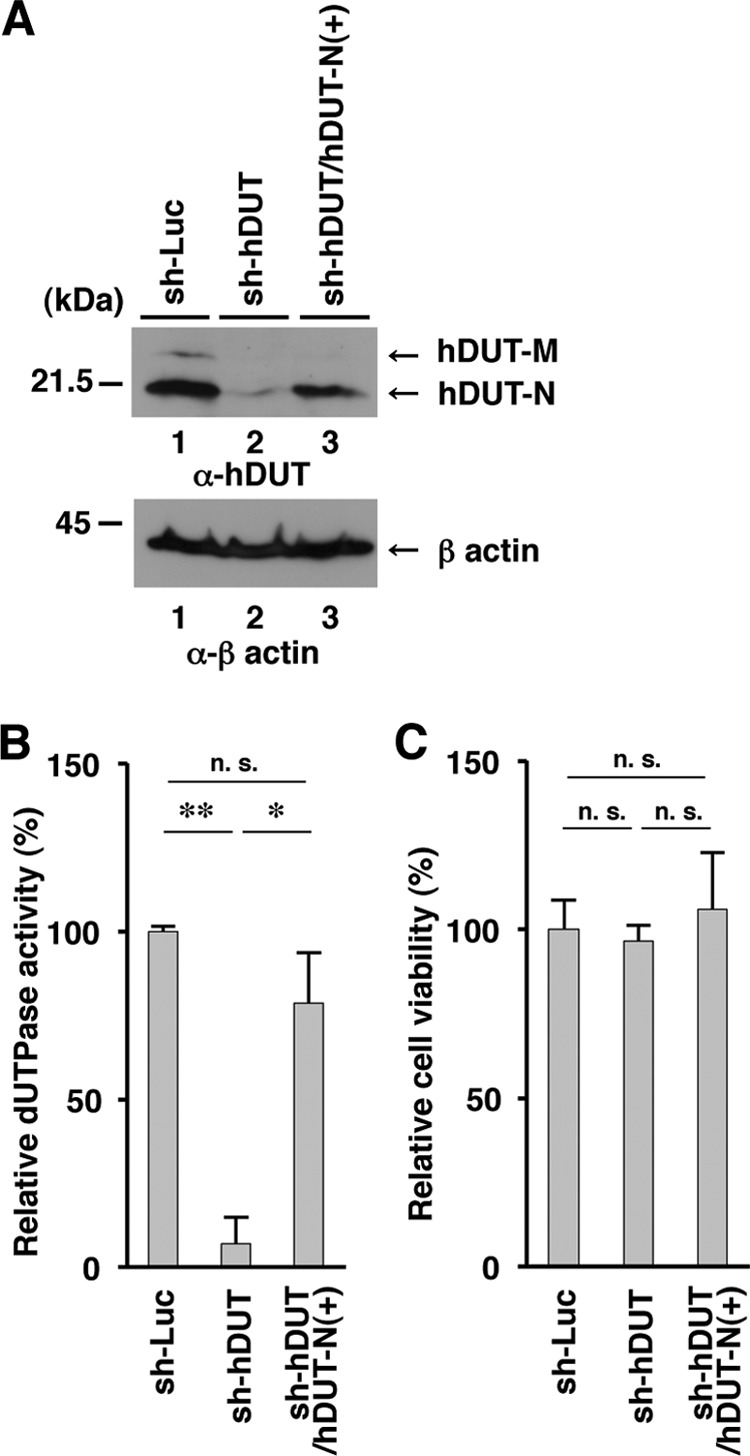
Expression of exogenous hDUT-N in sh-hDUT-HEp-2 cells. (A) sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT/hDUT-N(+)-HEp-2 cells were analyzed by immunoblotting with anti-cellular dUTPase mouse monoclonal antibody (top) and anti-β-actin mouse monoclonal antibody (bottom). Molecular mass markers (in kilodaltons) are shown on the left. (B and C) Relative dUTPase activity (B) and cell viability (C) of sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT/hDUT-N(+)-HEp-2 cells. Each value is the mean ± standard error from triplicate experiments and is expressed relative to the mean for sh-Luc-HEp-2 cells, which was normalized to 100%. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by the two-tailed Student t test. n.s., not significant. Data are representative of three independent experiments.
FIG 8.
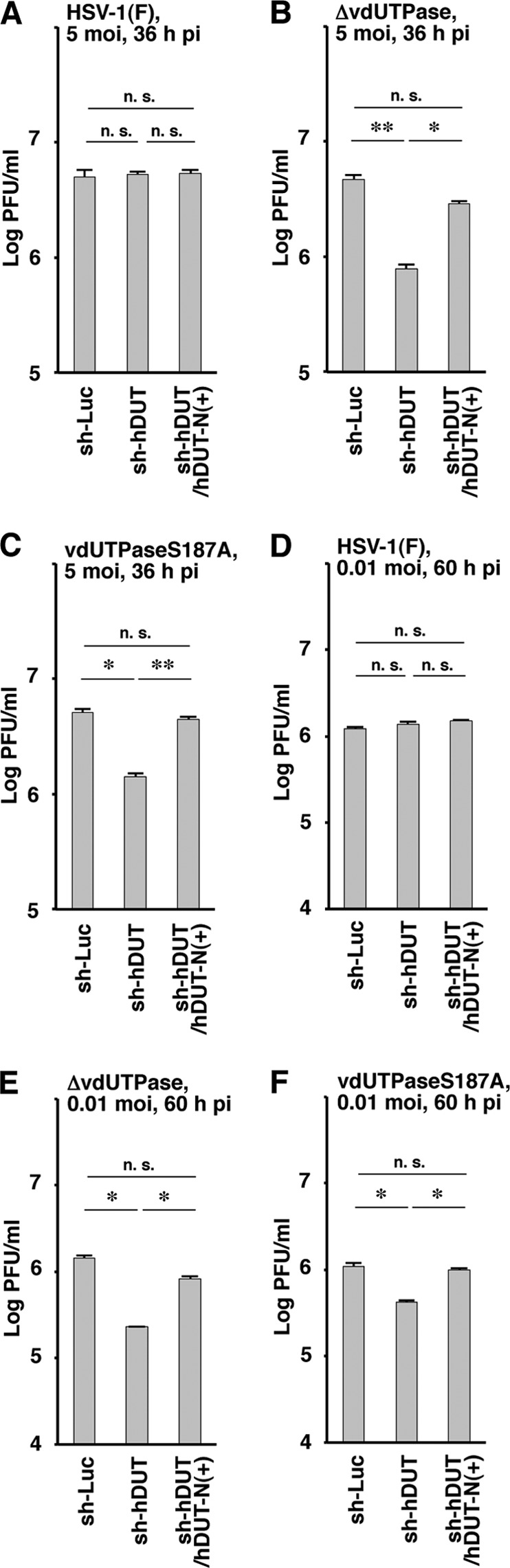
Effect of exogenous hDUT-N expression in sh-hDUT-HEp-2 cells on HSV-1 replication. sh-Luc-HEp-2, sh-hDUT-HEp-2, and sh-hDUT/hDUT-N(+)-HEp-2 cells were infected with wild-type HSV-1(F) (A and D), YK750 (ΔvdUTPase) (B and E), or YK751 (vdUTPaseS187A) (C and F) at an MOI of 5 (A to C) or 0.01 (D to F). Total virus from the cell culture supernatants and infected cells was harvested at 36 h (A to C) and 60 h (D to F) postinfection, and virus titers were assayed on Vero cells. Each value is the mean ± standard error from triplicate experiments. Asterisks indicate a significant difference in the mean (*, P < 0.0167; **, P < 0.025) by Holm's sequentially rejective Bonferroni multiple-comparison test. n.s., not significant. Data are representative of three independent experiments.
FIG 9.
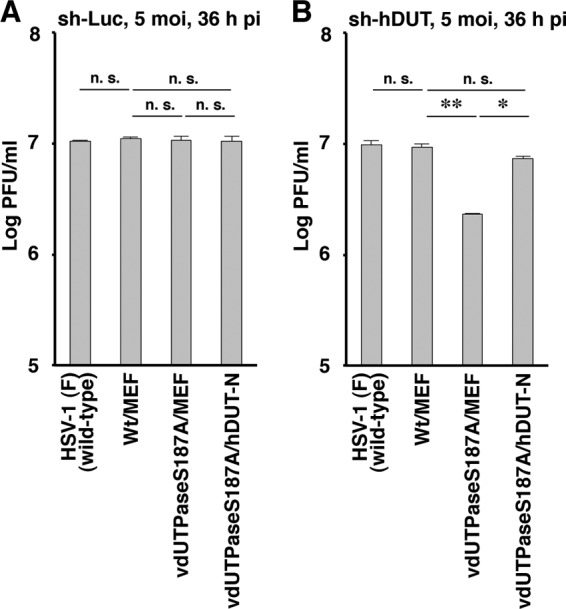
Effect of cellular dUTPase overexpression on HSV-1 replication in sh-hDUT-HEp-2 cells. sh-Luc-HEp-2 (A) and sh-hDUT-HEp-2 (B) cells were infected with wild-type strain HSV-1(F), YK761 (Wt/MEF), YK762 (vdUTPaseS187A/MEF), or YK764 (vdUTPaseS187A/hDUT-N) at an MOI of 5. The cells were grown and viruses were assayed as described in the legend of Fig. 4. Each value is the mean ± standard error from triplicate experiments. Asterisks indicate a significant difference in the mean (*, P < 0.0083; **, P < 0.0167) by Holm's sequentially rejective Bonferroni multiple-comparison test. n.s., not significant. Data are representative of three independent experiments.
Taken together, these results indicated that the level of endogenous cellular dUTPase activity in HEp-2 cells was sufficient for efficient replication of HSV-1 mutant viruses carrying the null or S187A mutation in vdUTPase and that phosphorylation of vdUTPase Ser-187 was required for efficient viral replication in cells in which endogenous dUTPase activity was low.
DISCUSSION
In the present study, we tested the hypothesis that dUTPase activity was important for replication of viruses that encode a vdUTPase and that the vdUTPase was able to compensate if the endogenous host cell dUTPase activity was not sufficient for efficient viral replication. This hypothesis had been based on observations that low endogenous cellular dUTPase activity was linked to low viral replication and to a reduced virulence of viruses with a mutation in vdUTPase (4, 17–20). In this study, we obtained the following direct experimental data supporting this hypothesis. (i) Although the endogenous cellular dUTPase activity in SK-N-SH cells was significantly lower than that in HEp-2 cells (Fig. 2B), overexpression of cellular dUTPase in SK-N-SH cells restored the replication of a virus with the vdUTPase S187A mutation, which was reported previously to prevent Us3 phosphorylation of vdUTPase Ser-187 and to impair its enzymatic activity (21) to the wild-type virus level (Fig. 4B). (ii) Knockdown of cellular dUTPase in HEp-2 cells, which also caused the downregulation of its enzymatic activity, significantly reduced the replication of the vdUTPase S187A mutant virus (Fig. 6B and D and 9B). (iii) A phosphomimetic substitution at vdUTPase Ser-187, which was reported previously to restore vdUTPase activity to the wild-type level (21), had the wild-type level of viral replication in cellular dUTPase knockdown HEp-2 cells (Fig. 6B and D). Collectively, these results suggested that a particular level of dUTPase activity is required for efficient HSV-1 replication and that Us3 phosphorylation of vdUTPase at Ser-187, which was reported previously to upregulate its enzymatic activity (21), is able to compensate for cellular endogenous dUTPase activity if it is too low for efficient viral replication. This is the first report, to our knowledge, directly showing that dUTPase activity is critical for efficient viral replication and that virally encoded dUTPase compensates for low cellular dUTPase activity if it is not sufficient for efficient viral replication.
We recently reported that Us3 phosphorylation of vdUTPase Ser-187 was required for efficient viral replication and virulence in the CNS of mice after intracranial inoculation, whereas it played no role in viral replication and pathogenicity in the eyes and vaginas of mice after ocular and vaginal inoculation, respectively (22). Therefore, these results in mice, like those in the present study with cultured cells, suggested that Us3 phosphorylation of vdUTPase Ser-187 is required to compensate for low cellular dUTPase activity to provide sufficient dUTPase for efficient viral replication in the CNS of mice, since epithelial cells in the eyes and vagina are actively dividing but most cells in the CNS are not (40, 41). There has been an analogous suggestion for HSV-1 thymidine kinase (vTK), another viral homolog of a host cell enzyme involved in nucleotide metabolism, which has been reported to compensate for endogenous cellular TK in ganglia in vivo, based on observations that (i) vTK was required for viral replication in ganglia of mice following ocular inoculation and for reactivation from latency following ganglionic explant and (ii) replacement of vTK with human thymidine kinase (hTK) was able to fulfill the vTK function in the ganglia of mice (42). A similar strategy could be used to investigate the role of Us3 phosphorylation of vdUTPase at Ser-187 in compensating for a cellular dUTPase activity that was not sufficient for efficient viral replication and virulence in the CNS of mice. We are now currently investigating the effect of the overexpression of cellular vdUTPase on viral replication in the CNS and on the virulence of recombinant viruses carrying the S187A mutation in vdUTPase in mice following intracranial inoculation.
An important question that remains to be answered is how dUTPase activity contributes to efficient HSV-1 replication. As described above, dUTPase is known to prevent misincorporation of dUTP into replicating DNA, which is necessary for accurate DNA replication. Therefore, dUTPase activity may function as an antimutator and may play a role in HSV-1 replication by increasing the fidelity of viral DNA replication. In agreement with this hypothesis, it has been reported that the mutation frequency of a feline immunodeficiency virus (FIV) mutant lacking vdUTPase that was integrated into the DNA of T lymphocytes was significantly lower than the mutation frequency of the FIV mutant integrated into primary macrophages, where endogenous cellular dUTPase activity was lower, and the FIV mutant replicated less efficiently in primary macrophages than in T lymphocytes (18, 43). In addition, loss of HSV-1 vdUTPase has been reported to significantly increase the mutation frequency of the viral genome in NIH 3T3 cells (44). However, we note that the loss of HSV-1 vdUTPase had no effect on viral replication in NIH 3T3 cells (44), and therefore, it has not been determined whether the high mutation frequency in the viral genome in cells infected with an HSV-1 mutant lacking vdUTPase is related to the low level of replication of the FIV mutant. Further studies are needed to answer this question, and we are currently investigating the effect of knockdown and overexpression of cellular dUTPase on the mutation frequency in recombinant viruses with the S187A mutation in vdUTPase in cell cultures and/or in the CNS of mice.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and grants for scientific research from the Japan Society for the Promotion of Science (JSPS); a contract research fund for the program of the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID); a grant for scientific research on innovative areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan; and a grant from the Takeda Science Foundation.
Footnotes
Published ahead of print 23 April 2014
REFERENCES
- 1.Boehmer PE, Lehman IR. 1997. Herpes simplex virus DNA replication. Annu. Rev. Biochem. 66:347–384. 10.1146/annurev.biochem.66.1.347 [DOI] [PubMed] [Google Scholar]
- 2.Beaud G. 1995. Vaccinia virus DNA replication: a short review. Biochimie 77:774–779. 10.1016/0300-9084(96)88195-8 [DOI] [PubMed] [Google Scholar]
- 3.Elder JH, Lerner DL, Hasselkus-Light CS, Fontenot DJ, Hunter E, Luciw PA, Montelaro RC, Phillips TR. 1992. Distinct subsets of retroviruses encode dUTPase. J. Virol. 66:1791–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Payne SL, Elder JH. 2001. The role of retroviral dUTPases in replication and virulence. Curr. Protein Pept. Sci. 2:381–388. 10.2174/1389203013381008 [DOI] [PubMed] [Google Scholar]
- 5.Chen R, Wang H, Mansky LM. 2002. Roles of uracil-DNA glycosylase and dUTPase in virus replication. J. Gen. Virol. 83:2339–2345 http://vir.sgmjournals.org/content/83/10/2339.long [DOI] [PubMed] [Google Scholar]
- 6.Lembo D, Brune W. 2009. Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 34:25–32. 10.1016/j.tibs.2008.09.008 [DOI] [PubMed] [Google Scholar]
- 7.Basta HA, Cleveland SB, Clinton RA, Dimitrov AG, McClure MA. 2009. Evolution of teleost fish retroviruses: characterization of new retroviruses with cellular genes. J. Virol. 83:10152–10162. 10.1128/JVI.02546-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldo AM, McClure MA. 1999. Evolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts. J. Virol. 73:7710–7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McClure MA. 2001. Evolution of the DUT gene: horizontal transfer between host and pathogen in all three domains of life. Curr. Protein Pept. Sci. 2:313–324. 10.2174/1389203013381062 [DOI] [PubMed] [Google Scholar]
- 10.Shlomai J, Kornberg A. 1978. Deoxyuridine triphosphatase of Escherichia coli. Purification, properties, and use as a reagent to reduce uracil incorporation into DNA. J. Biol. Chem. 253:3305–3312 [PubMed] [Google Scholar]
- 11.Vertessy BG, Toth J. 2009. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 42:97–106. 10.1021/ar800114w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bessman MJ, Lehman IR, Adler J, Zimmerman SB, Simms ES, Kornberg A. 1958. Enzymatic synthesis of deoxyribonucleic acids. III. The incorporation of pyrimidine and purine analogues into deoxyribonucleic acids. Proc. Natl. Acad. Sci. U. S. A. 44:633–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sedwick WD, Brown OE, Glickman BW. 1986. Deoxyuridine misincorporation causes site-specific mutational lesions in the lacI gene of Escherichia coli. Mutat. Res. 162:7–20. 10.1016/0027-5107(86)90066-7 [DOI] [PubMed] [Google Scholar]
- 14.Kunz BA, Kohalmi SE. 1991. Modulation of mutagenesis by deoxyribonucleotide levels. Annu. Rev. Genet. 25:339–359. 10.1146/annurev.ge.25.120191.002011 [DOI] [PubMed] [Google Scholar]
- 15.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott-Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 16.Pyles RB, Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J. Virol. 66:6706–6713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Threadgill DS, Steagall WK, Flaherty MT, Fuller FJ, Perry ST, Rushlow KE, Le Grice SF, Payne SL. 1993. Characterization of equine infectious anemia virus dUTPase: growth properties of a dUTPase-deficient mutant. J. Virol. 67:2592–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerner DL, Wagaman PC, Phillips TR, Prospero-Garcia O, Henriksen SJ, Fox HS, Bloom FE, Elder JH. 1995. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. U. S. A. 92:7480–7484. 10.1073/pnas.92.16.7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turelli P, Petursson G, Guiguen F, Mornex JF, Vigne R, Querat G. 1996. Replication properties of dUTPase-deficient mutants of caprine and ovine lentiviruses. J. Virol. 70:1213–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliveros M, Garcia-Escudero R, Alejo A, Vinuela E, Salas ML, Salas J. 1999. African swine fever virus dUTPase is a highly specific enzyme required for efficient replication in swine macrophages. J. Virol. 73:8934–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J. Virol. 88:655–666. 10.1128/JVI.02710-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 2014. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J. Virol. 88:2775–2785. 10.1128/JVI.03300-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211. 10.1128/JVI.02681-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364. 10.1099/0022-1317-2-3-357 [DOI] [PubMed] [Google Scholar]
- 26.Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, Kimura S, Ito T, Yahagi N, Ichinose M, Omata M, Iba H. 2005. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene 24:5471–5481. 10.1038/sj.onc.1208716 [DOI] [PubMed] [Google Scholar]
- 27.Haraguchi T, Ozaki Y, Iba H. 2009. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 37:e43. 10.1093/nar/gkp040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. 10.1038/nature09420 [DOI] [PubMed] [Google Scholar]
- 29.Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B, Jazag A, Ohta M, Imamura J, Ikenoue T, Ijichi H, Kawabe T, Isobe T, Omata M. 2006. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 25:633–642 [DOI] [PubMed] [Google Scholar]
- 30.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 33.Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, Nishiyama Y. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 79:6947–6956. 10.1128/JVI.79.11.6947-6956.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250–261. 10.1128/JVI.01451-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams MV, Cheng Y. 1979. Human deoxyuridine triphosphate nucleotidohydrolase. Purification and characterization of the deoxyuridine triphosphate nucleotidohydrolase from acute lymphocytic leukemia. J. Biol. Chem. 254:2897–2901 [PubMed] [Google Scholar]
- 36.Ladner RD, McNulty DE, Carr SA, Roberts GD, Caradonna SJ. 1996. Characterization of distinct nuclear and mitochondrial forms of human deoxyuridine triphosphate nucleotidohydrolase. J. Biol. Chem. 271:7745–7751. 10.1074/jbc.271.13.7745 [DOI] [PubMed] [Google Scholar]
- 37.Ladner RD, Caradonna SJ. 1997. The human dUTPase gene encodes both nuclear and mitochondrial isoforms. Differential expression of the isoforms and characterization of a cDNA encoding the mitochondrial species. J. Biol. Chem. 272:19072–19080 [DOI] [PubMed] [Google Scholar]
- 38.Morimoto T, Arii J, Akashi H, Kawaguchi Y. 2009. Identification of multiple sites suitable for insertion of foreign genes in herpes simplex virus genomes. Microbiol. Immunol. 53:155–161. 10.1111/j.1348-0421.2008.00104.x [DOI] [PubMed] [Google Scholar]
- 39.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 85:9599–9613. 10.1128/JVI.00845-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin-Belmonte F, Perez-Moreno M. 2012. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 12:23–38. 10.1038/nrc3169 [DOI] [PubMed] [Google Scholar]
- 41.Jakobsson J, Lundberg C. 2006. Lentiviral vectors for use in the central nervous system. Mol. Ther. 13:484–493. 10.1016/j.ymthe.2005.11.012 [DOI] [PubMed] [Google Scholar]
- 42.Patel R, Harper DR. 1998. Subclinical herpes virus reactivation and latency. Curr. Opin. Infect. Dis. 11:31–35. 10.1097/00001432-199802000-00008 [DOI] [PubMed] [Google Scholar]
- 43.Wagaman PC, Hasselkus-Light CS, Henson M, Lerner DL, Phillips TR, Elder JH. 1993. Molecular cloning and characterization of deoxyuridine triphosphatase from feline immunodeficiency virus (FIV). Virology 196:451–457. 10.1006/viro.1993.1501 [DOI] [PubMed] [Google Scholar]
- 44.Pyles RB, Thompson RL. 1994. Mutations in accessory DNA replicating functions alter the relative mutation frequency of herpes simplex virus type 1 strains in cultured murine cells. J. Virol. 68:4514–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]