

ABSTRACT
Prion diseases are characterized by a conformational change in the normal host protein PrPC. While the majority of mature PrPC is tethered to the plasma membrane by a glycosylphosphatidylinositol anchor, topological variants of this protein can arise during its biosynthesis. Here we have generated Drosophila transgenic for cytosolic ovine PrP in order to investigate its toxic potential in flies in the absence or presence of exogenous ovine prions. While cytosolic ovine PrP expressed in Drosophila was predominantly detergent insoluble and showed resistance to low concentrations of proteinase K, it was not overtly detrimental to the flies. However, Drosophila transgenic for cytosolic PrP expression exposed to classical or atypical scrapie prion inocula showed a faster decrease in locomotor activity than similar flies exposed to scrapie-free material. The susceptibility to classical scrapie inocula could be assessed in Drosophila transgenic for panneuronal expression of cytosolic PrP, whereas susceptibility to atypical scrapie required ubiquitous PrP expression. Significantly, the toxic phenotype induced by ovine scrapie in cytosolic PrP transgenic Drosophila was transmissible to recipient PrP transgenic flies. These data show that while cytosolic PrP expression does not adversely affect Drosophila, this topological PrP variant can participate in the generation of transmissible scrapie-induced toxicity. These observations also show that PrP transgenic Drosophila are susceptible to classical and atypical scrapie prion strains and highlight the utility of this invertebrate host as a model of mammalian prion disease.
IMPORTANCE During prion diseases, the host protein PrPC converts into an abnormal conformer, PrPSc, a process coupled to the generation of transmissible prions and neurotoxicity. While PrPC is principally a glycosylphosphatidylinositol-anchored membrane protein, the role of topological variants, such as cytosolic PrP, in prion-mediated toxicity and prion formation is undefined. Here we generated Drosophila transgenic for cytosolic PrP expression in order to investigate its toxic potential in the absence or presence of exogenous prions. Cytosolic ovine PrP expressed in Drosophila was not overtly detrimental to the flies. However, cytosolic PrP transgenic Drosophila exposed to ovine scrapie showed a toxic phenotype absent from similar flies exposed to scrapie-free material. Significantly, the scrapie-induced toxic phenotype in cytosolic transgenic Drosophila was transmissible to recipient PrP transgenic flies. These data show that cytosolic PrP can participate in the generation of transmissible prion-induced toxicity and highlight the utility of Drosophila as a model of mammalian prion disease.
INTRODUCTION
Prion diseases or transmissible spongiform encephalopathies are fatal neurodegenerative disorders of humans and various other mammalian species (1). These conditions include scrapie of sheep, bovine spongiform encephalopathy of cattle, Creutzfeldt-Jakob disease of humans, and chronic wasting disease of cervids. Susceptibility to prion disease requires expression of the host-encoded protein PrPC (2–5). Furthermore, prion diseases are associated with the conversion of PrPC, the normal form of the prion protein, into an abnormal conformer, PrPSc, in a template-directed manner (6, 7). Misfolding of PrP is associated with an increase in the β-sheet content of the protein, which accumulates principally in the central nervous system of an affected individual. There is now considerable evidence to suggest that the transmissible prion agent comprises PrPSc (8–14). However, despite intensive investigation, the molecular mechanisms of PrPC-to-PrPSc conversion and of prion-mediated neurodegeneration remain unknown.
Although PrPC is highly conserved among different mammalian species, its physiological functions remain elusive. PrPC is a glycosylphosphatidylinositol (GPI)-anchored sialoglycoprotein located principally in lipid rafts on the outer leaf of the cell membrane (15, 16). Nascent PrPC is synthesized as a preprotein approximately 250 amino acid residues in length. The N-terminal leader peptide is cleaved as PrP enters the endoplasmic reticulum (ER), and the C-terminal signal sequence is cleaved upon attachment of the GPI anchor that holds the protein to the membrane (15, 17, 18). Inside the ER lumen, PrPC undergoes additional posttranslational modification with the addition of carbohydrate structures at two asparagine residues (19). In addition, a disulfide bond forms within the C-terminal globular domain (20). During its biosynthesis, PrP may undergo aberrant translocation since leader peptide inefficiency prevents all of the nascent protein from entering the lumen of the ER. As a consequence, subpopulations of PrP are either retained fully in the cytosol (PrPcyt) or produced as a membrane-bound protein with either N- or C-terminal residues exposed to the cytosol (21–24). ER misfolded and aberrantly translocated proteins are targeted for degradation by the ubiquitin proteasome system, the major cellular proteolytic pathway, or via the autophagic/lysosomal system. While normal levels of cytosolic and aberrantly translocated PrP are usually metabolized by the cell, these forms of PrP have been reported to be neurotoxic when present in elevated amounts (25–28). Increased cellular levels of cytosolic PrP may arise as a consequence of PrPSc-mediated inhibition of the catalytic activity of the proteasome in cells (29–31). The role of cytosolic PrP in the generation of infectious prions has yet to be determined.
Ovine scrapie is an important model of prion disease, not only for the natural host but for mammalian species in general (32, 33). Polymorphisms within ovine PrPC correlate with susceptibility to different types of scrapie in sheep. Four major polymorphisms in the ovine prion protein, located at amino acid residues 136, 141, 154, and 171, are associated, in some cases relatively (34, 35), with susceptibility to two classifications of scrapie disease (36–38). Sheep that express A136L141R154Q171 (termed ARQ, where A, L, R, and Q stand for alanine, leucine, arginine, and glutamine, respectively) or V136L141R154Q171 (termed VRQ, where V stands for valine) ovine PrP are susceptible to classical scrapie, a transmissible prion disease within the natural host (39). In contrast, a different ovine prion disease, referred to as atypical or Nor98 scrapie, has been reported in classical scrapie-resistant PrP genotypes, including A136L141R154R171 (termed ARR), A136F141R154Q171 (termed AFRQ, where F stands for phenylalanine), and A136L141H154Q171 (termed AHQ, where H stands for histidine) (38). It is considered that atypical scrapie is a spontaneous disorder of PrP folding and/or metabolism (38, 40), although transmission by the oral route cannot be excluded (41–44). We have begun to model sheep scrapie in Drosophila in order to develop a more tractable model of mammalian prion disease. In doing so, we have previously generated Drosophila transgenic for polymorphic variants of ovine PrP expressed with a GPI anchor sequence [PrP(GPI)] (45). Furthermore, we have shown that Drosophila transgenic for AHQ(GPI) ovine PrP show a median survival time significantly shorter than that of flies transgenic for VRQ(GPI). It has yet to be established whether the toxic potential of AHQ prion protein is mediated by a cytosolic variant of this particular genotype of ovine PrP and whether cytosolic PrP per se can participate in prion-mediated toxicity.
Here we generated Drosophila transgenic for polymorphic variants of cytosolic ovine PrP in order to investigate for the first time its toxic potential in flies in the absence or presence of exogenous ovine prions. While cytosolic ovine PrP expressed panneuronally in Drosophila was predominantly detergent insoluble and showed resistance to low concentrations of proteinase K (PK), it was not overtly detrimental to the flies. In contrast, Drosophila transgenic for cytosolic PrP expression exposed to classical or atypical scrapie prion inocula showed a faster decrease in locomotor activity than similar flies exposed to scrapie-free material. The susceptibility to classical ovine scrapie was evident in Drosophila transgenic for panneuronal cytosolic PrP, whereas susceptibility to atypical ovine scrapie required ubiquitous expression. Significantly, the toxic phenotype induced by ovine scrapie in cytosolic transgenic Drosophila was transmissible to PrP transgenic recipient flies. These data show that while cytosolic ovine PrP is not inherently neurotoxic in Drosophila, this topological variant can participate in the generation of a transmissible toxicity induced by scrapie prion inocula. These novel observations highlight the utility of Drosophila as a model of mammalian prion disease.
MATERIALS AND METHODS
Fly stocks and generation of cytosolic ovine PrP transgenic Drosophila.
The UAS-PrP(GPI) fly lines w; M{AHQ-PrP(GPI), 3xP3-RFP.attP}ZH-51D and w; M{ARQ-PrP(GPI), 3xP3-RFP.attP}ZH-51D, which are transgenic for ovine A136H154Q171 and A136R154Q171 PrP, respectively, expressed with an N-terminal leader peptide and a C-terminal GPI signal sequence [AHQ(GPI) and ARQ(GPI), respectively] were generated by PhiC31 site-specific transformation as previously described (45). The UAS-PrP(cyt) fly lines generated here were w; M{AHQ-PrP, 3xP3-RFP.attP}ZH-51D, w; M{ARQ-PrP, 3xP3-RFP.attP}ZH-51D, and w; M{VRQ-PrP, 3xP3-RFP.attP}ZH-51D. The ovine PrP(cyt) transgenes for insertion into the Drosophila genome were prepared by a PCR that generated a DNA fragment encoding ovine PrP amino acid residues 25 to 232. PCR was carried out in the presence of Pfu DNA polymerase (Promega) by using substrate plasmid DNA that contained an insert encoding AHQ, ARQ, or VRQ ovine PrP amino acid residues 25 to 252 (45) and oligonucleotide primers P2 (forward; 5′ GATGA GAA TTC AAC ATG AAG AAG CGA CCA AAA CCT GGC 3′) and P4 (reverse; 5′ ACGATGAA CTC GAG CTA CCC CCT TTG GTA ATA AG 3′). PCR primers P2 and P4 contained EcoRI and XhoI restriction sites, respectively, that allowed directional cloning of the 658-bp PCR product into the Drosophila transgenesis vector pUASTattB. A Kozak translation site (46) was incorporated into the forward primer and a stop codon was incorporated into the reverse primer ahead of the XhoI restriction site. The PCR conditions comprised initial denaturation at 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 30 s, primer annealing at 55°C for 30 s, and primer extension at 75°C for 1 min and a final extension of the PCR product at 75°C for 10 min. PCR products that contained DNA encoding PrP(cyt) were subsequently ligated into pUASTattB and rescued by transformation in DH5α bacteria. Plasmid DNA was isolated from transformed bacteria by an alkaline lysis method with the Qiagen maxiprep kit and the PrP construct insert verified by DNA sequence analysis. Site-specific transformation of the pUASTattB-PrP constructs into the 51D fly line (y[1] M{vas-int.Dm}ZH-2A w[*]; M{3xP3-RFP.attP}ZH-51D) was performed by Bestgene Inc. F1 flies were balanced, and viable lines were maintained as balanced stocks by conventional fly crosses. DNA sequence analysis was performed on genomic DNA from each balanced fly line to confirm the presence of the correct PrP transgene at the 51D site. Cre-mediated removal of the red fluorescent protein (RFP) gene from the VRQ(cyt) PrP fly genome was performed by conventional fly crosses, and this fly line was used where stated in Fig. 5a and Fig. 7a. The Elav-GAL4 (P{w[+mW.hs]=GawB}elav[C155]), Actin-5C-GAL4 (y w; P{w[+mC]=Act5C-Gal4}25F01/CyO, y[+]), and GMR-GAL4 (w; wg[Sp-1]/CyO; GMR-GAL4, w+/TM6B) driver lines and the control 51D (w; M{3xP3-RFP.attP}ZH-51D) fly line were obtained from the Department of Genetics, University of Cambridge, Cambridge, United Kingdom. All of the fly lines were raised on standard cornmeal medium (47) at 25°C and maintained at low to medium density. Flies were used in the assays described below or harvested at various time points and then frozen at −80°C until required.
FIG 5.

Primary transmission of classical ovine scrapie in cytosolic VRQ PrP transgenic Drosophila. VRQ(cyt) PrP transgenic (squares) or 51D control flies (circles) crossed with either the Actin-GAL4 or Elav-GAL4 driver line were assessed for locomotor activity by a negative-geotaxis climbing assay following exposure at the larval stage to VRQ/VRQ scrapie-infected (filled symbols) or scrapie-free (open symbols and dashed lines) sheep brain homogenate. Actin-GAL4-VRQ(cyt) PrP flies did not contain the gene for RFP. The mean PI ± SD is shown for three groups of 15 flies of each genotype per time point (a total of 45 flies of each genotype). Statistical analysis of the linear regression plots was performed by one-way ANOVA and Tukey's honest significant difference test for post hoc analysis.
FIG 7.

Fly-to-fly transmission of the prion-induced toxic phenotype. VRQ(cyt) PrP (without the gene for RFP) transgenic flies crossed with the Actin-GAL4 driver line were assessed for locomotor activity by a negative-geotaxis climbing assay following exposure at the larval stage to a 10% (vol/vol) dilution of head homogenate derived from 30-day-old Drosophila exposed at the larval stage to either scrapie-infected (filled squares) or scrapie-free (open squares and dashed line) sheep brain homogenate. The mean PI ± SD is shown for three groups of 15 flies of each genotype per time point (a total of 45 flies of each genotype). Statistical analysis of the linear regression plots was performed by one-way ANOVA and Tukey's honest significant difference test for post hoc analysis.
Prion inoculation of Drosophila. (i) Primary passage of sheep scrapie (sheep to fly).
Drosophila larvae were exposed to brain homogenates from sheep confirmed to be scrapie positive or known to be scrapie negative. The classical scrapie-infected isolates were prepared from terminally scrapie-affected sheep identified by routine statutory surveillance (VRQ/VRQ isolate SE1848/0005; ARQ/ARQ isolate SE1848/0008) (48). The atypical scrapie prion-infected isolates (n = 2) were prepared from terminal AHQ/AHQ sheep challenged intracerebrally with atypical scrapie and that were confirmed to be positive for the disease (43). New Zealand-derived VRQ/VRQ (n = 1), ARQ/ARQ (n = 1), or AHQ/AHQ (n = 2) scrapie-free brain tissue was used as control material. Two hundred fifty microliters of a 1% brain homogenate prepared in phosphate-buffered saline (PBS; pH 7.4) was added to the top of the cornmeal that contained third-instar Drosophila larvae in 3-in. plastic vials. Flies were transferred to fresh, nontreated vials following eclosion.
(ii) Secondary passage of sheep scrapie (fly to fly).
Drosophila brain homogenates were prepared from 30-day-old flies that had been exposed at the larval stage to scrapie-positive or scrapie-negative sheep brain material. Two hundred fifty microliters of a 10% (vol/vol) dilution of the original fly brain homogenate was added to the top of the cornmeal that contained third-instar Drosophila larvae in 3-in. plastic vials. Flies were transferred to fresh, nontreated vials following eclosion.
Preparation of fly head homogenates.
Whole flies in an Eppendorf tube were frozen in liquid nitrogen for 10 min and then vortexed for 2 min. Individual fly heads were then isolated and placed in clean Eppendorf tubes with a paint brush. Homogenates were prepared by manual grinding of fly heads in Eppendorf tubes with sterilized plastic pestles. Homogenates for enzyme-linked immunosorbent assay (ELISA) or Western blotting without PK digestion of PrP were prepared by processing 20 fly heads in 20 μl of lysis buffer [50 mM Tris (pH 7.5), 100 mM NaCl, 0.5% (vol/vol) Nonidet P-40, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF)]. In the case of PK digestion of PrP, AEBSF was not added to the lysis buffer. Homogenates were prepared for conformation-dependent immunoassay (CDI) by processing 40 fly heads in 8 μl of 8 M guanidine HCl (GdnHCl), incubated at 18°C for 15 min, diluted 1:50 in assay buffer, and assessed as described previously (49). Homogenates for fly-to-fly transmission (secondary-passage samples) were prepared by processing 150 male and 150 female fly heads per group previously harvested at 30 days of age. Each group of 300 fly heads was added to 300 μl of PBS (pH 7.4) prior to homogenization.
Preparation of soluble and insoluble prion protein fractions.
PrP fractions were prepared from fly head homogenates by a method adapted from Fernandez-Funez et al. (50). A volume of fly head homogenate that was equivalent to 20 fly heads was mixed with 20 μl of 10% (wt/vol) Sarkosyl (pH 7.4). The sample was shaken at 225 rpm for 10 min at 37°C, 5 U of Benzonase was added, and the sample was shaken at 225 rpm for a further 10 min at 37°C. Sodium phosphotungstic acid (diluted in PBS [pH 7.4]) was added to the reaction mixture to give a 0.3% (wt/vol) final concentration, and the tubes were shaken at 225 rpm for 30 min at 37°C prior to centrifugation at 16,000 × g for 30 min at 4°C. To obtain the soluble and insoluble PrP fractions, the supernatant (soluble fraction, 40 μl) was transferred to a fresh tube and the pellet (insoluble fraction) was resuspended in 40 μl of 0.1% (wt/vol) Sarkosyl in PBS (pH 7.4).
PK digestion of fly head homogenate.
Fly head homogenates were prepared in 1.5-ml Eppendorf tubes by processing 10 fly heads in 9 μl of lysis buffer (50 mM Tris [pH 7.5], 100 mM NaCl, 0.5% Nonidet P-40) with plastic pestles. A 1-μl volume of PK at 10 times the required concentration was added to the homogenate, and the mixture incubated at 37°C for 15 min. Proteolysis was stopped by the addition of 1.1 μl of 10 mM AEBSF, and the samples were analyzed by SDS-PAGE and Western blot analysis to detect PrP.
SDS-PAGE and Western blot analysis.
Fly head homogenate was mixed with an equal volume of 2× Laemmli loading buffer, boiled for 10 min, cooled on ice, and then centrifuged at 13,000 × g for 5 min at 18°C to remove debris. Fly head homogenate was subjected to SDS-PAGE under reducing conditions and Western blot analysis as described in detail previously (48), except that the nitrocellulose membranes were probed with a 1:2,000 dilution of anti-PrP monoclonal antibody Sha31 (51).
Capture-detector ELISA.
Duplicate 40-μl aliquots of fly head homogenate were diluted to 100 μl with PBS (pH 7.4). PrP was quantified by a capture-detector ELISA carried out as described previously (52), except that the capture reagent was anti-PrP monoclonal antibody 245 (53) and the detector antibody was biotinylated anti-PrP monoclonal antibody SAF32 (51). The equivalent of 10 fly heads was assayed per well in duplicate.
CDI.
Head homogenate was prepared as described above, and PrP was quantified by CDI as described previously (49), except that the capture reagent was anti-PrP monoclonal antibody 245 (53) and the detector antibody was biotinylated anti-PrP monoclonal antibody SAF32 (51). The equivalent of 20 fly heads was assayed per well in duplicate.
Survival assay.
Newly eclosed flies were allowed to mature and mate for 24 h before the females were separated and collected for survival assays. One hundred flies of each genotype were housed in groups of 10, and the flies were flipped onto fresh food every 2 to 3 days. The number of dead flies was recorded three times a week (45). Survival curves were calculated by using Kaplan-Meier plots, and differences between them were analyzed by the log rank method with Prism (GraphPad Software Inc., San Diego, CA).
Locomotor assay.
The locomotor ability of flies was assessed in a negative-geotaxis climbing assay as described previously (54). Briefly, age-matched, premated female flies were placed in adapted plastic 25-ml pipettes that were used as vertical climbing columns. The flies were allowed to acclimatize for 30 min prior to the assessment of their locomotor ability. Flies were tapped to the bottom of the pipette (by using the same number and intensity of taps) and then allowed to climb for 45 s. At the end of the climbing period, the number of flies above the 25-ml mark, the number below the 2-ml mark, and the number between the 2- and 25-ml marks were recorded. This procedure was performed three times at each time point. The mean performance index (PI) of each group of flies ± the standard deviation (SD) was calculated as described previously (54).
Statistical analysis.
Statistical analysis of the data was performed by one-way analysis of variance (ANOVA), together with Tukey's honest significant difference test for post hoc analysis or the unpaired-sample t test with Prism (GraphPad Software Inc., San Diego, CA).
RESULTS
Cytosolic ovine PrP expression in Drosophila.
Here we generated Drosophila transgenic for polymorphic variants of cytosolic ovine PrP in order to investigate the toxic potential of intracellular PrP expression in the absence or presence of exogenous prions. The data in Fig. 1 show the detection of cytosolic PrP [PrP(cyt)] expression in Drosophila by Western blot analysis. The analysis in Fig. 1a shows that ARQ(cyt), AHQ(cyt), and VRQ(cyt) were all efficiently expressed at similar levels panneuronally in flies. The molecular mass of all three genotypes of PrP(cyt) was approximately 27 kDa, the same as that of nonglycosylated ovine recombinant PrP. The analysis in Fig. 1b shows that AHQ(cyt) and VRQ(cyt) transgenic Drosophila expressed significantly higher levels of PrP than flies that expressed AHQ or VRQ PrP with a GPI anchor sequence [PrP(GPI)]. The opposite trend was seen in Drosophila that expressed the ARQ PrP genotype.
FIG 1.
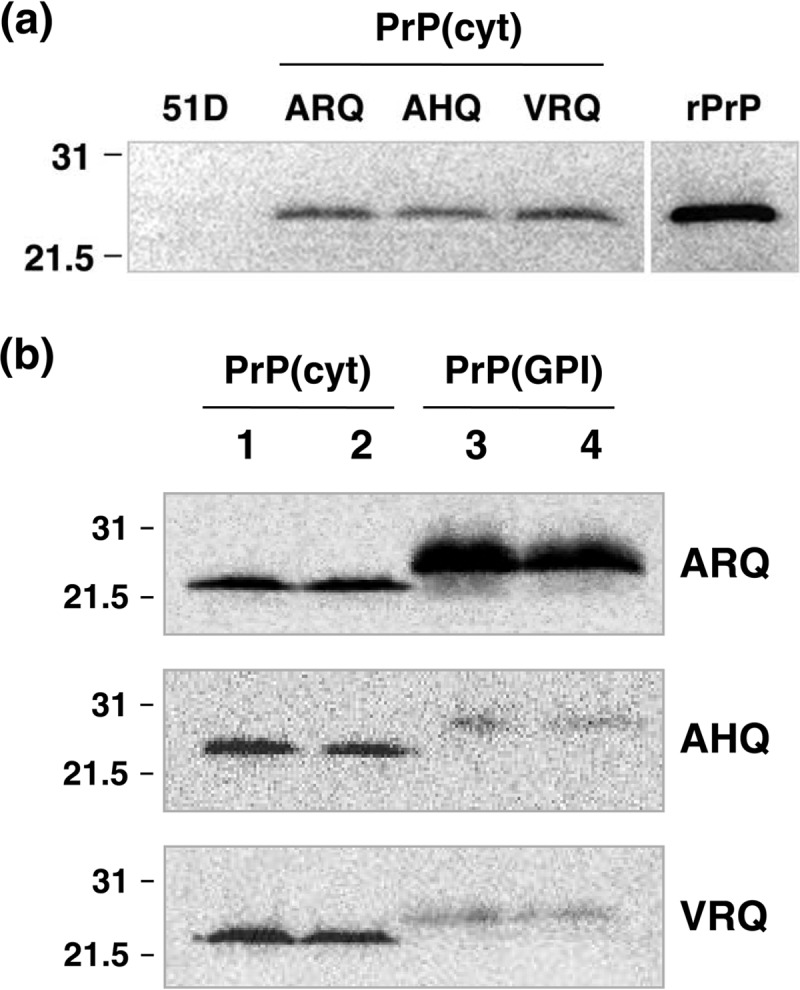
Western blot detection of cytosolic ovine PrP expression in Drosophila. Head homogenates were prepared from 5-day-old ovine PrP transgenic Drosophila or 51D control flies crossed with the Elav-GAL4 driver fly line. Samples were analyzed by SDS-PAGE and Western blot analysis with anti-PrP monoclonal antibody Sha31. (a) Molecular profile of ARQ(cyt), AHQ(cyt), and VRQ(cyt), all at the equivalent of 10 fly heads per track. Mature length ovine VRQ recombinant PrP (rPrP) was used at 10 ng. Molecular mass marker values (kDa) are shown on the left. (b) Comparison of ovine PrP(cyt) and PrP(GPI) expression in Drosophila. Tracks 1 and 2, PrP(cyt); tracks 3 and 4, PrP(GPI); tracks 1 and 3; male flies; tracks 2 and 4; female flies. The equivalent of five fly heads were run per track. Molecular mass marker values (kDa) are shown on the left. The ovine PrP genotype is indicated on the right.
We subsequently used a capture-detector ELISA with C-terminal specific anti-PrP monoclonal antibodies to quantify the level of each genotype of cytosolic ovine PrP expressed in Drosophila. The data in Fig. 2a show that significantly lower levels of panneuronally expressed ovine PrP(cyt) than ovine PrP(GPI) were recognized by the anti-PrP-specific ELISA. This observation suggested that cytosolic ovine PrP may adopt a conformation distinct from that of other forms of ovine PrP expressed in Drosophila that are recognized by this ELISA (45). In order to test this, we used a CDI whereby PrP(cyt) was denatured with guanidine prior to its recognition by capture-detector immunoassay (49). The data in Fig. 2b show that all of the genotypes of cytosolic ovine PrP expressed panneuronally in Drosophila were recognized by the denaturant-based CDI.
FIG 2.

Capture-detector immunoassay analysis of cytosolic ovine PrP expression in Drosophila. Head homogenates were prepared from 5-day-old ovine PrP transgenic Drosophila or 51D control flies crossed with the Elav-GAL4 driver fly line. Samples were analyzed by ELISA with anti-PrP monoclonal antibody 245 as the capture reagent and biotinylated anti-PrP monoclonal antibody SAF32 as the detector (a). The equivalent of 10 fly heads was measured per well, and the results are shown as the mean optical density (O.D.) at 415 nm ± SD of duplicate wells. (b) CDI. Fly head homogenates were treated with 8 M GdnHCl prior to immunoassay with anti-PrP monoclonal antibody 245 as the capture reagent and biotinylated anti-PrP monoclonal antibody SAF32 as the detector antibody. The equivalent of 20 fly heads was measured per well. Mature-length ovine ARQ recombinant PrP (rPrP) was used at 122 ng/well. The results are shown as average time-resolved fluorescence (TRF) counts per second (cps) ± SD for duplicate wells.
Cytosolic PrP is predominantly detergent insoluble and displays protease resistance.
We next investigated whether the immunobiochemical properties of PrP(cyt) expressed in Drosophila correlated with distinct conformers of the ovine prion protein. Figure 3 shows a comparison of ARQ(cyt) and ARQ(GPI) with respect to detergent solubility and relative resistance to proteolytic digestion. In order to determine the detergent solubility of cytosolic ovine PrP, we extracted fly head homogenates with Sarkosyl to prepare soluble and insoluble fractions for subsequent analysis by Western blot analysis with anti-PrP monoclonal antibody Sha31. The data in Fig. 3a show that while Elav-driven ARQ(cyt) and ARQ(GPI) flies both displayed a major band of approximately 27 kDa in detergent-soluble and insoluble head homogenate fractions, the proportion of PrP present in these fractions varied. The level of insoluble prion protein was greater than the level of soluble material in Elav-ARQ(cyt) flies than in Elav-ARQ(GPI) flies. The data in Fig. 3b show the Western blot analysis of PK-digested fly head homogenate from panneuronal ARQ(cyt) and ARQ(GPI) flies. Panneuronally expressed ARQ(GPI) PrP was readily cleaved by PK when treated with 3 to 9 μg/ml of proteolytic enzyme. In contrast, ARQ(cyt) was resistant to digestion with PK when treated with the proteolytic enzyme used in the same concentration range and was susceptible to complete digestion only when PK was used in excess of 27 μg/ml. All three polymorphic variants of cytosolic ovine PrP showed these trends (data not shown).
FIG 3.
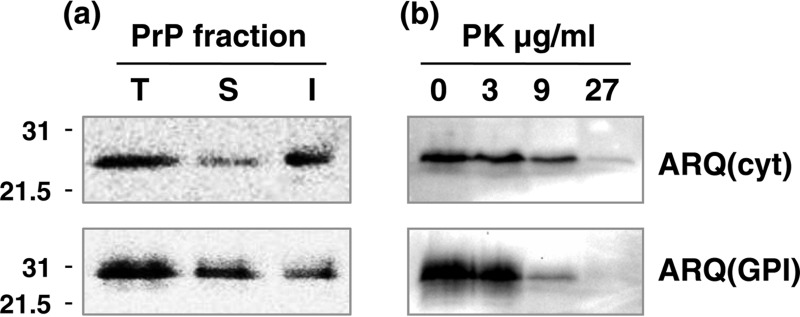
Cytosolic ovine PrP is characterized by reduced solubility and increased PK resistance. Head homogenates were prepared from 5-day-old ovine ARQ(cyt) or ARQ(GPI) PrP transgenic Drosophila crossed with the Elav-GAL4 driver fly line. After various treatments, fly head homogenate samples were analyzed by SDS-PAGE and Western blot analysis with anti-PrP monoclonal antibody Sha31. The equivalent of 10 fly heads was loaded per track. Molecular mass marker values (kDa) are shown on the left of each gel. (a) Total (T), soluble (S), and insoluble (I) fractions of PrP were prepared from fly heads as described in Materials and Methods. (b) Reaction products of fly head homogenates incubated with various concentrations of PK at 37°C for 30 min.
Survival of cytosolic ovine PrP transgenic Drosophila.
Cytosolic PrP accumulation is toxic to some neurons (26, 55) and may be part of the neurotoxic mechanism associated with prion diseases (25). It was important, therefore, to determine the effect of cytosolic PrP expression on the general well-being of Drosophila prior to prion infectivity studies of these fly lines.
The data in Fig. 4 show the survival curves for PrP transgenic fly lines that panneuronally expressed cytosolic ovine prion protein in comparison with the survival curve for the control Elav-51D fly line. Cytosolic PrP expression did not appear to be overtly detrimental to Drosophila since the survival curve of each of the ovine PrP transgenic fly lines showed a profile similar to that of the nontransgenic 51D control flies. However, log rank test analysis showed that the survival curves of all three genotypes of prion protein transgenic fly lines were significantly different from that of the 51D control flies (P ≤ 0.002), and this was reflected in differences in the median survival times, which were as follows: 51D, 86 days; AHQ(cyt), 81 days; ARQ(cyt), 79 days; VRQ(cyt), 76 days. The general lack of toxicity in Drosophila as a consequence of panneuronal cytosolic PrP expression was also evident when PrP(cyt) was expressed ubiquitously. For example, the survival percentages of Actin-driven and Elav-driven VRQ(cyt) PrP transgenic flies were similar at approximately 90 and 95%, respectively, when assessed at 50 days of age.
FIG 4.

Survival curves of cytosolic ovine PrP transgenic Drosophila. Groups of 100 age-matched Elav-PrP or control Elav-51D flies were selected for survival assays. The number of surviving flies was recorded three times a week as described in Materials and Methods. Survival curves were calculated by using Kaplan-Meier plots, and differences between them were analyzed by the log rank method with Prism (GraphPad Software Inc., San Diego, CA).
Cytosolic PrP transgenic Drosophila are susceptible to ovine prion inocula.
In order to assess whether cytosolic ovine PrP transgenic Drosophila were susceptible to the toxic effect of exogenous ovine prion inocula, flies at the larval stage of development were exposed to scrapie-infected sheep brain material and the locomotor activity of prion-exposed flies was assessed after eclosion (i.e., hatching). The prion inoculum used here was sheep brain homogenate derived from natural cases of VRQ/VRQ and ARQ/ARQ classical (48) or AHQ/AHQ experimental atypical (43) sheep scrapie. Genotype-matched, scrapie-free brain homogenates were used as control material, and 51D Drosophila were used as controls that were similarly exposed to scrapie-infected and scrapie-free sheep brain homogenates.
In order to assess the response to classical scrapie prion inocula, Actin- or Elav-driven VRQ(cyt) Drosophila were exposed at the larval stage to VRQ/VRQ scrapie-infected sheep brain homogenate. Figure 5 shows the climbing ability expressed as a PI of prion-exposed and control flies posteclosion. The data in Fig. 5a show that prion-exposed, Actin-driven VRQ(cyt) Drosophila displayed a significantly faster decline in locomotor activity than similar flies exposed to genotype-matched control brain homogenate (P < 0.001 over the whole assay). In contrast, Actin-driven 51D flies showed a similar decline in locomotor activity following exposure to scrapie-infected or genotype-matched control sheep brain homogenate. The data in Fig. 5b show that prion-exposed, Elav-driven VRQ(cyt) Drosophila also showed a significantly faster decline in locomotor activity than similar flies exposed to genotype-matched control brain homogenate (P < 0.05 between days 2 and 51 of the assay), which was somewhat slower than that seen in Actin-driven VRQ(cyt) Drosophila. In contrast, the PI of Elav-driven VRQ(cyt) PrP transgenic Drosophila exposed to ARQ/ARQ scrapie-infected sheep brain homogenate was not significantly different from that of similar flies exposed to scrapie-free ARQ/ARQ sheep brain homogenate (data not shown). Elav-driven 51D flies showed no difference in the decline of locomotor activity following exposure to scrapie-infected or genotype-matched control sheep brain homogenate.
We subjected head homogenates from prion-exposed flies to proteolytic digestion, followed by Western blot analysis with anti-PrP monoclonal antibody, to detect PK-resistant PrPSc. The data in Fig. 6 show that most of the panneuronally expressed VRQ(cyt) from prion-exposed and control treated flies was digested with PK at 10 to 30 μg/ml and with similar resultant molecular profiles. At 20 days of age, a greater fraction of the VRQ(cyt) was resistant to PK digestion at these concentrations of the proteolytic enzyme.
FIG 6.
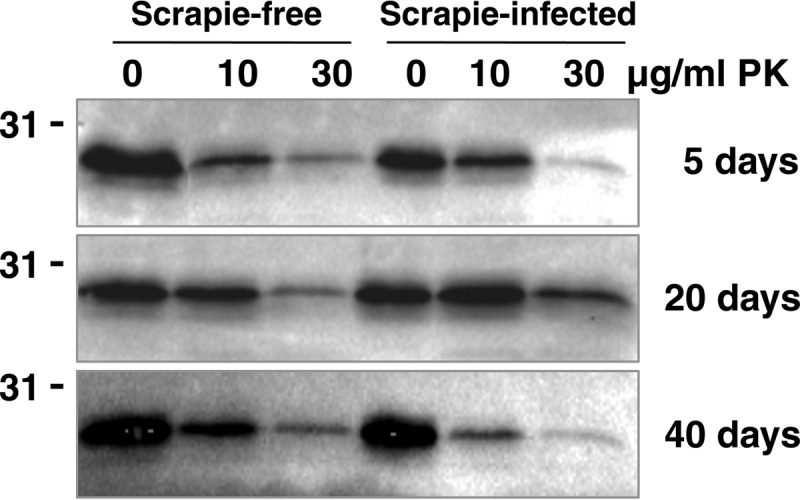
PK digestion of prion-exposed cytosolic VRQ fly head homogenate. Fly head homogenates were prepared from ovine VRQ(cyt) PrP transgenic Drosophila crossed with the Elav-GAL4 driver fly line following exposure at the larval stage to VRQ/VRQ scrapie-free (tracks 1 to 3) or scrapie-infected (tracks 4 to 6) sheep brain homogenate. Samples were incubated with 0, 10, or 30 μg/ml PK at 37°C for 15 min, and the reaction products were analyzed by SDS-PAGE and Western blot analysis with anti-PrP monoclonal antibody Sha31. The equivalent of 10 fly heads was loaded per track. Molecular mass marker values (kDa) are shown on the left of each gel. Ages of flies (in days) are shown on the right.
We subsequently investigated whether the toxic phenotype displayed by prion-exposed VRQ(cyt) Drosophila was transmissible. In order to do so, we prepared homogenates from the heads of 30-day-old Drosophila that had been exposed at the larval stage to either VRQ/VRQ prion-infected or genotype-matched scrapie-free sheep brain homogenate. Fly head homogenates were subsequently used to inoculate fresh batches of recipient VRQ(cyt) Drosophila larvae. After hatching, the locomotor activity of fly head homogenate-exposed Drosophila was assessed by a negative-geotaxis climbing assay. The data in Fig. 7 show that head homogenate from prion-exposed VRQ(cyt) Drosophila induced a significantly faster decline in locomotor activity than control fly head homogenates in VRQ(cyt) recipient flies (P < 0.01 over the whole assay) (Fig. 7a). In contrast, no significant differences were seen in the locomotor response of recipient VRQ(cyt) Drosophila to head homogenate prepared from nontransgenic 51D flies previously exposed to either scrapie-infected or scrapie-free sheep brain material (Fig. 7b).
In order to assess the response to atypical scrapie prion inocula, Actin- or Elav-driven cytosolic AHQ(cyt) ovine PrP transgenic Drosophila were exposed at the larval stage to AHQ/AHQ prion-infected or genotype-matched scrapie-free sheep brain homogenate. Actin- and Elav-driven AHQ(GPI) and Elav-driven ARQ(GPI) Drosophila, which both express PrP with a GPI anchor sequence, were included for comparison. The data in Fig. 8 show the climbing ability of Drosophila with ubiquitous AHQ expression after exposure to atypical scrapie-infected sheep brain homogenate. Actin-driven AHQ(cyt) flies showed a faster decline in locomotor activity following exposure to atypical scrapie-infected sheep brain homogenate than after exposure to control brain homogenate (P = 0.0226 between days 8 and 39) (Fig. 8a). Similarly, atypical-prion-exposed, Actin-driven AHQ(GPI) flies showed a significantly greater decline in locomotor activity than similar flies exposed to control inocula (P = 0.0278 over the whole assay) (Fig. 8b). Actin-driven ARQ(GPI) flies also showed a significantly greater decline in locomotor activity following exposure to AHQ/AHQ scrapie-infected brain homogenate than after exposure to control inocula (P = 0.0351 between days 8 and 39) (Fig. 8c). In contrast, Actin-driven 51D flies showed the same decline in locomotor activity following exposure to AHQ/AHQ scrapie-infected or genotype-matched scrapie-free sheep brain homogenate (Fig. 8d). Similar trends were seen with both atypical scrapie inocula (data not shown).
FIG 8.

Primary transmission of atypical scrapie in Actin-driven PrP transgenic Drosophila. PrP transgenic or 51D control flies crossed with the Actin-GAL4 driver line were assessed for locomotor activity by a negative-geotaxis climbing assay following exposure at the larval stage to AHQ/AHQ scrapie-infected (filled circles) or scrapie-free (open circles and dashed lines) sheep brain homogenate. The mean PI ± SD is shown for three groups of 15 flies of each genotype per time point (a total of 45 flies of each genotype). Statistical analysis of the scrapie-infected and scrapie-free linear regression plots for each fly line was compared by the unpaired-sample t test.
The data in Fig. 9 show the climbing ability of Elav-driven AHQ(cyt) and AHQ(GPI) Drosophila after exposure to atypical scrapie-infected and control sheep brain homogenates. Elav-driven AHQ(cyt) flies showed a similar decline in locomotor activity following exposure to AHQ/AHQ scrapie-infected or genotype-matched scrapie-free sheep brain homogenate or PBS (Fig. 9a). In a similar manner, Elav-driven AHQ(GPI) flies showed no difference in the decline of locomotor activity following exposure to AHQ/AHQ scrapie-infected brain homogenate or control inocula (Fig. 9b), although a response to one atypical scrapie inoculum was seen at day 33 (data not shown). In contrast to these data, Elav-driven ARQ(GPI) flies showed a significantly greater decline in locomotor activity following exposure to AHQ/AHQ scrapie-infected brain homogenate than after exposure to control inocula (P < 0.05 between days 7 and 40) (Fig. 9c). Elav-driven 51D flies showed the same decline in locomotor activity following exposure to AHQ/AHQ scrapie-infected sheep brain homogenate or control inocula (Fig. 9d). Similar trends were seen with both atypical scrapie inocula (data not shown).
FIG 9.

Lack of response by Elav-driven AHQ PrP transgenic Drosophila to atypical scrapie. PrP transgenic or 51D control flies crossed with the Elav-GAL4 driver line were assessed for locomotor activity by a negative-geotaxis climbing assay following exposure at the larval stage to AHQ/AHQ scrapie-infected (closed squares and continuous line) or scrapie-free (closed circles and dashed line) sheep brain homogenate or PBS (closed triangles and dotted line). The mean PI ± SD is shown for three groups of 15 flies of each genotype per time point (a total of 45 flies of each genotype). Statistical analysis of the linear regression plots was performed by one-way ANOVA and Tukey's honest significant difference test for post hoc analysis.
DISCUSSION
The pathogenesis that occurs during prion diseases is associated with the conformational change of PrPC into PrPSc and concomitant neurodegeneration (1). However, the mechanism of PrP conversion and its role in neurotoxicity are unknown. While PrPC is primarily attached by a GPI anchor to the external side of the cell membrane, topological variants of the protein can arise during its biogenesis and metabolism (25). Here we have shown that one such variant, namely, cytosolic PrP, can participate in the generation of a transmissible toxicity induced by ovine prion inocula.
To do so, we generated Drosophila that expressed cytosolic AHQ, ARQ, or VRQ ovine PrP. All three cytosolic PrP variants were expressed at similar levels in flies and comprised predominantly detergent-insoluble material that showed resistance to proteolytic digestion with relatively low concentrations of PK enzyme. In addition, epitopes normally exposed in ovine PrPC were either hidden or buried in PrP(cyt) since the latter required denaturation prior to its immunodetection by capture-detector immunoassay. The molecular profile and conformational properties of the PrP(cyt) variants expressed in Drosophila are distinct from those of the same polymorphic variants expressed with a GPI anchor in this host (45). This is likely to be due to the lack of posttranslational modifications experienced by PrP(cyt) as a consequence of its failure to enter the ER during biosynthesis. The modifications that PrPC normally experiences during its biogenesis include glycosylation and the introduction of a disulfide bond to the polypeptide chain, both of which influence protein folding and thermodynamic stability. Despite the acquisition of properties of misfolded prion protein, the panneuronal expression of PrP(cyt) in Drosophila was not overtly detrimental to the flies. We have previously shown that Drosophila transgenic for AHQ expressed with a GPI anchor sequence displayed a median life span that was significantly shorter than that of control 51D flies (45). Ovine AHQ PrP is associated with susceptibility to atypical scrapie in sheep, which is considered to be a spontaneous disorder of PrP folding and/or metabolism (38, 40) rather than an acquired condition (41–43). Our observation here that cytosolic AHQ does not induce a toxicity comparable to that of AHQ(GPI) suggests that the toxicity associated with AHQ targeted to the cell membrane is a consequence of this protein's trafficking through the secretory pathway of the cell. The expression of AHQ variants of ovine PrP in Drosophila provides a novel model system in which to investigate the potential spontaneous misfolding of this genotype of ovine prion protein.
We assessed the response of PrP(cyt) transgenic Drosophila to exogenous ovine prions in a negative-geotaxis climbing assay, a versatile and robust method used to assess locomotor defects in fly models of mammalian neurodegenerative conditions (54). Drosophila transgenic for VRQ(cyt) or AHQ(cyt) expression showed decreased climbing ability after exposure at the larval stage to classical or atypical scrapie-infected sheep brain homogenate, respectively. The toxic effect of classical and atypical scrapie in PrP(cyt) transgenic Drosophila is suggestive of a prion-mediated effect, as it was not induced by exposure to normal sheep brain homogenate and it was PrP dependent, since scrapie-infected sheep brain homogenate was not toxic to nontransgenic 51D flies. Importantly, we have shown that the toxic phenotype of prion-exposed PrP(cyt) flies was transmissible. Fly head homogenate from prion-exposed VRQ(cyt) PrP transgenic Drosophila efficiently induced a toxic phenotype in recipient flies of the same genotype. This was not due to carryover of scrapie-infected sheep brain inocula in the fly head homogenate, since no effect was induced in recipient PrP(cyt) flies by prion-exposed nontransgenic 51D fly head inocula. These observations are suggestive of the generation of an infectious moiety, analogous to prion replication, during the primary passage of scrapie in VRQ(cyt) flies (i.e., sheep-to-fly transmission) that was subsequently transmitted at the secondary passage (i.e., fly-to-fly transmission). However, we were unable to demonstrate an increase in PK-resistant VRQ(cyt) PrP in prion-exposed flies of this genotype. In other studies, we have shown that protein misfolding cyclic amplification can be used to detect PK-resistant PrPSc in prion-exposed VRQ(GPI) transgenic Drosophila but not in similarly treated VRQ(cyt) flies (A. M. Thackray et al., submitted for publication).
A feature of the response of AHQ(cyt) transgenic Drosophila to atypical scrapie toxicity was the requirement for ubiquitous PrP expression in flies. The lack of susceptibility of panneuronal AHQ(cyt) but not VRQ(cyt) transgenic Drosophila to scrapie prions does not appear to be due to the level of ovine PrP expressed in these flies since cytosolic PrP was expressed at a similar level in both Elav-driven fly lines. Furthermore, the resistance of Drosophila transgenic for panneuronal expression of AHQ(cyt) to atypical scrapie toxicity did not appear to be due to the topological expression of PrP in this fly line since Drosophila transgenic for panneuronal expression of AHQ(GPI) were also refractive to the same inocula. The need for ubiquitous PrP expression in AHQ PrP transgenic Drosophila for susceptibility to atypical scrapie toxicity may reflect a low infective titer in these particular prion-infected isolates compared to classical scrapie material. Alternatively, atypical scrapie infectivity may be more unstable than its classical scrapie counterpart. It is known that the PrPSc associated with atypical scrapie is less PK resistant than that associated with classical scrapie (56, 57). Whatever the case, ubiquitous expression of PrP in Drosophila may provide an environment for enhanced uptake and neuroinvasion of scrapie-infected material and generation of the toxic agent compared to panneuronal expression, which may be more important for the response to atypical scrapie prion inocula. In mammalian species, PrPC is ubiquitously expressed, a feature that plays an essential role in the transmission of prion infectivity in naturally acquired cases of prion disease (58), which may include atypical scrapie (41, 42). However, not all of the ovine PrP transgenic fly lines used here required ubiquitous expression of PrP in order to succumb to atypical scrapie prion inocula. Drosophila with panneuronal expression of ovine ARQ(GPI) showed susceptibility to AHQ/AHQ atypical scrapie-infected sheep brain homogenate, as they do to ARQ/ARQ and VRQ/VRQ classical scrapie prion inocula (59). The promiscuous susceptibility of ARQ(GPI) PrP flies to atypical and classical scrapie-induced toxicity correlates with the high level of ovine prion protein expressed by this fly line (45). It is known that the transmission barrier effect (1) can be circumvented by elevated levels of PrP expression. For example, tg338 mice that express high levels of ovine VRQ PrP are susceptible to atypical scrapie isolates, whereas VRQ/VRQ sheep are resistant (34, 42). Collectively, these observations suggest that Drosophila engineered for elevated levels of ubiquitous cell surface or cytosolic PrP expression will be susceptible to a greater diversity of scrapie prion isolates and potentially smaller quantities of associated toxicity. This suggests that PrP transgenic Drosophila could provide the basis of a new animal model to bioassay low levels of infectious toxicity in peripheral tissues and blood of prion-affected animals.
Our studies with cytosolic PrP transgenic Drosophila presented here begin to contribute to an understanding of the potential role of topological variants in prion-induced neurotoxicity. While the mechanism of prion toxicity remains to be defined, it is established that PrP expression is required for susceptibility to the neurotoxic agent. The essential requirement for PrP expression in prion-induced neurotoxicity may suggest an intermediate in the conversion of PrPC to PrPSc is the neurotoxic agent (60, 61). An alternative possibility is that neurotoxicity results from PrPSc interference with the normal biosynthesis and metabolism of PrPC (25). PrP can accumulate in the cytosol in a misfolded form when proteasomal activity is compromised (28, 31) and cytosolic PrP has been reported to be neurotoxic in some neurons (26, 55). However, the neurotoxicity of cytosolic PrP per se has been debated (62, 63). Our observations here have shown that while cytosolic PrP can adopt a conformation distinct from PrP targeted to the cell membrane, expression of PrP(cyt) in Drosophila does not result in the accumulation of a transmissible toxic moiety without prior exposure of these flies to exogenous prion inocula. Collectively, our data presented here suggest that cytosolic PrP is not overtly toxic to neurons per se but may participate in a toxicity mediated by scrapie prion inocula, possibly by acting as a substrate for the generation of PrP-dependent transmissible moiety that initiates or maintains repression of neuronal proteostasis (29, 30, 64–66). The tractable nature of Drosophila as a genetically and biochemically well-defined experimental model will allow us to test the validity of this hypothesis.
ACKNOWLEDGMENTS
We thank John Roote for useful discussion regarding fly genetics. We thank Marion Simmons and John Spiropoulos for the supply of sheep atypical scrapie samples.
This work was supported by funds from the China Scholarship Council and the Cambridge Overseas Trust, which provided Ph.D. studentship support for C.Z. We acknowledge funding support from the NC3Rs.
Footnotes
Published ahead of print 7 May 2014
REFERENCES
- 1.Prusiner SB. (ed). 2004. Prion biology and diseases, second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. 1996. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379:339–343. 10.1038/379339a0 [DOI] [PubMed] [Google Scholar]
- 3.Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. 10.1016/0092-8674(93)90360-3 [DOI] [PubMed] [Google Scholar]
- 4.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. 2003. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302:871–874. 10.1126/science.1090187 [DOI] [PubMed] [Google Scholar]
- 5.Ciccotosto GD, Cappai R, White AR. 2008. Neurotoxicity of prion peptides on cultured cerebellar neurons. Methods Mol. Biol. 459:83–96. 10.1007/978-1-59745-234-2_6 [DOI] [PubMed] [Google Scholar]
- 6.Collinge J. 2001. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24:519–550. 10.1146/annurev.neuro.24.1.519 [DOI] [PubMed] [Google Scholar]
- 7.Aguzzi A, Baumann F, Bremer J. 2008. The prion's elusive reason for being. Annu. Rev. Neurosci. 31:439–477. 10.1146/annurev.neuro.31.060407.125620 [DOI] [PubMed] [Google Scholar]
- 8.Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Jasanoff A, Lindquist S. 2009. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 63:438–450. 10.1016/j.neuron.2009.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sigurdson CJ, Nilsson KP, Hornemann S, Heikenwalder M, Manco G, Schwarz P, Ott D, Rulicke T, Liberski PP, Julius C, Falsig J, Stitz L, Wuthrich K, Aguzzi A. 2009. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc. Natl. Acad. Sci. U. S. A. 106:304–309. 10.1073/pnas.0810680105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watts JC, Giles K, Stohr J, Oehler A, Bhardwaj S, Grillo SK, Patel S, DeArmond SJ, Prusiner SB. 2012. Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc. Natl. Acad. Sci. U. S. A. 109:3498–3503. 10.1073/pnas.1121556109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saborio GP, Permanne B, Soto C. 2001. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813. 10.1038/35081095 [DOI] [PubMed] [Google Scholar]
- 12.Castilla J, Saa P, Hetz C, Soto C. 2005. In vitro generation of infectious scrapie prions. Cell 121:195–206. 10.1016/j.cell.2005.02.011 [DOI] [PubMed] [Google Scholar]
- 13.Deleault NR, Harris BT, Rees JR, Supattapone S. 2007. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U. S. A. 104:9741–9746. 10.1073/pnas.0702662104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, Wang X, Yuan CG, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135. 10.1126/science.1183748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. 1987. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240. 10.1016/0092-8674(87)90150-4 [DOI] [PubMed] [Google Scholar]
- 16.Taylor DR, Hooper NM. 2006. The prion protein and lipid rafts. Mol. Membr. Biol. 23:89–99. 10.1080/09687860500449994 [DOI] [PubMed] [Google Scholar]
- 17.Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. 1986. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46:417–428. 10.1016/0092-8674(86)90662-8 [DOI] [PubMed] [Google Scholar]
- 18.Turk E, Teplow DB, Hood LE, Prusiner SB. 1988. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176:21–30. 10.1111/j.1432-1033.1988.tb14246.x [DOI] [PubMed] [Google Scholar]
- 19.Haraguchi T, Fisher S, Olofsson S, Endo T, Groth D, Tarentino A, Borchelt DR, Teplow D, Hood L, Burlingame A, Lycke E, Kobata A, Prusiner SB. 1989. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 274:1–13. 10.1016/0003-9861(89)90409-8 [DOI] [PubMed] [Google Scholar]
- 20.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. 1997. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 413:282–288. 10.1016/S0014-5793(97)00920-4 [DOI] [PubMed] [Google Scholar]
- 21.Stewart RS, Harris DA. 2003. Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection. J. Biol. Chem. 278:45960–45968. 10.1074/jbc.M307833200 [DOI] [PubMed] [Google Scholar]
- 22.Kim SJ, Hegde RS. 2002. Cotranslational partitioning of nascent prion protein into multiple populations at the translocation channel. Mol. Biol. Cell 13:3775–3786. 10.1091/mbc.E02-05-0293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hay B, Barry RA, Lieberburg I, Prusiner SB, Lingappa VR. 1987. Biogenesis and transmembrane orientation of the cellular isoform of the scrapie prion protein. Mol. Cell. Biol. 7:914–920(Erratum, 7:2035.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. 1998. A transmembrane form of the prion protein in neurodegenerative disease. Science 279:827–834. 10.1126/science.279.5352.827 [DOI] [PubMed] [Google Scholar]
- 25.Chakrabarti O, Ashok A, Hegde RS. 2009. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 34:287–295. 10.1016/j.tibs.2009.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma J, Wollmann R, Lindquist S. 2002. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298:1781–1785. 10.1126/science.1073725 [DOI] [PubMed] [Google Scholar]
- 27.Grenier C, Bissonnette C, Volkov L, Roucou X. 2006. Molecular morphology and toxicity of cytoplasmic prion protein aggregates in neuronal and non-neuronal cells. J. Neurochem. 97:1456–1466. 10.1111/j.1471-4159.2006.03837.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J, Lindquist S. 2001. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc. Natl. Acad. Sci. U. S. A. 98:14955–14960. 10.1073/pnas.011578098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FW, Menendez-Benito V, Dantuma NP, Portis JL, Collinge J, Tabrizi SJ. 2007. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 26:175–188. 10.1016/j.molcel.2007.04.001 [DOI] [PubMed] [Google Scholar]
- 30.Deriziotis P, Andre R, Smith DM, Goold R, Kinghorn KJ, Kristiansen M, Nathan JA, Rosenzweig R, Krutauz D, Glickman MH, Collinge J, Goldberg AL, Tabrizi SJ. 2011. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J. 30:3065–3077. 10.1038/emboj.2011.224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Lindquist S. 2002. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 298:1785–1788. 10.1126/science.1073619 [DOI] [PubMed] [Google Scholar]
- 32.Bujdoso R, Thackray AM. 2013. The emergence of a Drosophila model to measure ovine prion infectivity. CAB Rev. 8:1–15. 10.1079/PAVSNNR20138058 [DOI] [Google Scholar]
- 33.Windl O, Dawson M. 2012. Animal prion diseases. Subcell. Biochem. 65:497–516. 10.1007/978-94-007-5416-4_18 [DOI] [PubMed] [Google Scholar]
- 34.Saunders GC, Cawthraw S, Mountjoy SJ, Hope J, Windl O. 2006. PrP genotypes of atypical scrapie cases in Great Britain. J. Gen. Virol. 87:3141–3149. 10.1099/vir.0.81779-0 [DOI] [PubMed] [Google Scholar]
- 35.Moum T, Olsaker I, Hopp P, Moldal T, Valheim M, Benestad SL. 2005. Polymorphisms at codons 141 and 154 in the ovine prion protein gene are associated with scrapie Nor98 cases. J. Gen. Virol. 86:231–235. 10.1099/vir.0.80437-0 [DOI] [PubMed] [Google Scholar]
- 36.Clouscard C, Beaudry P, Elsen JM, Milan D, Dussaucy M, Bounneau C, Schelcher F, Chatelain J, Launay JM, Laplanche JL. 1995. Different allelic effects of the codons 136 and 171 of the prion protein gene in sheep with natural scrapie. J. Gen. Virol. 76:2097–2101. 10.1099/0022-1317-76-8-2097 [DOI] [PubMed] [Google Scholar]
- 37.Goldmann W, Hunter N, Smith G, Foster J, Hope J. 1994. PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J. Gen. Virol. 75:989–995. 10.1099/0022-1317-75-5-989 [DOI] [PubMed] [Google Scholar]
- 38.Benestad SL, Arsac JN, Goldmann W, Noremark M. 2008. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet. Res. 39:19. 10.1051/vetres:2007056 [DOI] [PubMed] [Google Scholar]
- 39.Hunter N. 1997. PrP genetics in sheep and the applications for scrapie and BSE. Trends Microbiol. 5:331–334. 10.1016/S0966-842X(97)01081-0 [DOI] [PubMed] [Google Scholar]
- 40.Fediaevsky A, Maurella C, Noremark M, Ingravalle F, Thorgeirsdottir S, Orge L, Poizat R, Hautaniemi M, Liam B, Calavas D, Ru G, Hopp P. 2010. The prevalence of atypical scrapie in sheep from positive flocks is not higher than in the general sheep population in 11 European countries. BMC Vet. Res. 6:9. 10.1186/1746-6148-6-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons MM, Moore SJ, Konold T, Thurston L, Terry LA, Thorne L, Lockey R, Vickery C, Hawkins SA, Chaplin MJ, Spiropoulos J. 2011. Experimental oral transmission of atypical scrapie to sheep. Emerg. Infect. Dis. 17:848–854. 10.3201/eid1705.101654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andréoletti O, Orge L, Benestad SL, Beringue V, Litaise C, Simon S, Le Dur A, Laude H, Simmons H, Lugan S, Corbiere F, Costes P, Morel N, Schelcher F, Lacroux C. 2011. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 7:e1001285. 10.1371/journal.ppat.1001285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simmons MM, Konold T, Simmons HA, Spencer YI, Lockey R, Spiropoulos J, Everitt S, Clifford D. 2007. Experimental transmission of atypical scrapie to sheep. BMC Vet. Res. 3:20. 10.1186/1746-6148-3-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Dur A, Beringue V, Andréoletti O, Reine F, Lai TL, Baron T, Bratberg B, Vilotte JL, Sarradin P, Benestad SL, Laude H. 2005. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc. Natl. Acad. Sci. U. S. A. 102:16031–16036. 10.1073/pnas.0502296102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thackray AM, Muhammad F, Zhang C, Di Y, Jahn TR, Landgraf M, Crowther DC, Evers JF, Bujdoso R. 2012. Ovine PrP transgenic Drosophila show reduced locomotor activity and decreased survival. Biochem. J. 444:487–495. 10.1042/BJ20112141 [DOI] [PubMed] [Google Scholar]
- 46.Kozak M. 1986. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44:283–292. 10.1016/0092-8674(86)90762-2 [DOI] [PubMed] [Google Scholar]
- 47.Lewis EB. 1960. A new standard food medium. Drosophila Inf. Serv. 34:117–118 [Google Scholar]
- 48.Thackray AM, Hopkins L, Spiropoulos J, Bujdoso R. 2008. Molecular and transmission characteristics of primary-passaged ovine scrapie isolates in conventional and ovine PrP transgenic mice. J. Virol. 82:11197–11207. 10.1128/JVI.01454-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thackray AM, Hopkins L, Bujdoso R. 2007. Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem. J. 401:475–483. 10.1042/BJ20061264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez-Funez P, Zhang Y, Casas-Tinto S, Xiao X, Zou WQ, Rincon-Limas DE. 2010. Sequence-dependent prion protein misfolding and neurotoxicity. J. Biol. Chem. 285:36897–36908. 10.1074/jbc.M110.174391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Féraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Vilette D, Lehmann S, Grassi J. 2005. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J. Biol. Chem. 280:11247–11258. 10.1074/jbc.M407006200 [DOI] [PubMed] [Google Scholar]
- 52.Thackray AM, Fitzmaurice TJ, Hopkins L, Bujdoso R. 2006. Ovine plasma prion protein levels show genotypic variation detected by C-terminal epitopes not exposed in cell-surface PrPC. Biochem. J. 400:349–358. 10.1042/BJ20060746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thackray AM, Madec JY, Wong E, Morgan-Warren R, Brown DR, Baron T, Bujdoso R. 2003. Detection of bovine spongiform encephalopathy, ovine scrapie prion-related protein (PrPSc) and normal PrPc by monoclonal antibodies raised to copper-refolded prion protein. Biochem. J. 370:81–90. 10.1042/BJ20021280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White KE, Humphrey DM, Hirth F. 2010. The dopaminergic system in the aging brain of Drosophila. Front. Neurosci. 4:205. 10.3389/fnins.2010.00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Bowers SL, Wang F, Pu XA, Nelson RJ, Ma J. 2009. Cytoplasmic prion protein induces forebrain neurotoxicity. Biochim. Biophys. Acta 1792:555–563. 10.1016/j.bbadis.2009.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benestad SL, Sarradin P, Thu B, Schonheit J, Tranulis MA, Bratberg B. 2003. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet. Rec. 153:202–208. 10.1136/vr.153.7.202 [DOI] [PubMed] [Google Scholar]
- 57.Buschmann A, Biacabe AG, Ziegler U, Bencsik A, Madec JY, Erhardt G, Luhken G, Baron T, Groschup MH. 2004. Atypical scrapie cases in Germany and France are identified by discrepant reaction patterns in BSE rapid tests. J. Virol. Methods 117:27–36. 10.1016/j.jviromet.2003.11.017 [DOI] [PubMed] [Google Scholar]
- 58.Gough KC, Maddison BC. 2010. Prion transmission: prion excretion and occurrence in the environment. Prion 4:275–282. 10.4161/pri.4.4.13678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thackray AM, Muhammad F, Zhang C, Denyer M, Spiropoulos J, Crowther DC, Bujdoso R. 2012. Prion-induced toxicity in PrP transgenic Drosophila. Exp. Mol. Pathol. 92:194–201. 10.1016/j.yexmp.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 60.Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. 2011. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 470:540–542. 10.1038/nature09768 [DOI] [PubMed] [Google Scholar]
- 61.Zhou M, Ottenberg G, Sferrazza GF, Lasmezas CI. 2012. Highly neurotoxic monomeric alpha-helical prion protein. Proc. Natl. Acad. Sci. U. S. A. 109:3113–3118. 10.1073/pnas.1118090109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. 2005. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J. Biol. Chem. 280:11320–11328. 10.1074/jbc.M412441200 [DOI] [PubMed] [Google Scholar]
- 63.Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. 2003. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J. Biol. Chem. 278:40877–40881. 10.1074/jbc.M306177200 [DOI] [PubMed] [Google Scholar]
- 64.Morimoto RI. 2008. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22:1427–1438. 10.1101/gad.1657108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 66.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. 2012. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485:507–511. 10.1038/nature11058 [DOI] [PMC free article] [PubMed] [Google Scholar]