

ABSTRACT
Mountain lions (Puma concolor) throughout North and South America are infected with puma lentivirus clade B (PLVB). A second, highly divergent lentiviral clade, PLVA, infects mountain lions in southern California and Florida. Bobcats (Lynx rufus) in these two geographic regions are also infected with PLVA, and to date, this is the only strain of lentivirus identified in bobcats. We sequenced full-length PLV genomes in order to characterize the molecular evolution of PLV in bobcats and mountain lions. Low sequence homology (88% average pairwise identity) and frequent recombination (1 recombination breakpoint per 3 isolates analyzed) were observed in both clades. Viral proteins have markedly different patterns of evolution; sequence homology and negative selection were highest in Gag and Pol and lowest in Vif and Env. A total of 1.7% of sites across the PLV genome evolve under positive selection, indicating that host-imposed selection pressure is an important force shaping PLV evolution. PLVA strains are highly spatially structured, reflecting the population dynamics of their primary host, the bobcat. In contrast, the phylogeography of PLVB reflects the highly mobile mountain lion, with diverse PLVB isolates cocirculating in some areas and genetically related viruses being present in populations separated by thousands of kilometers. We conclude that PLVA and PLVB are two different viral species with distinct feline hosts and evolutionary histories.
IMPORTANCE An understanding of viral evolution in natural host populations is a fundamental goal of virology, molecular biology, and disease ecology. Here we provide a detailed analysis of puma lentivirus (PLV) evolution in two natural carnivore hosts, the bobcat and mountain lion. Our results illustrate that PLV evolution is a dynamic process that results from high rates of viral mutation/recombination and host-imposed selection pressure.
INTRODUCTION
Feline immunodeficiency viruses (FIVs) are complex retroviruses in the genus Lentivirus. Nine lineages of FIV infect members of the family Felidae in a species-specific pattern (1). The occurrence and monophyly of FIV across the Felidae resemble those of simian immunodeficiency virus (SIV) in African primates (2) and suggest that contemporary FIVs have all diverged from a common ancestor during feline speciation (1, 3).
It has been posited that FIV was introduced into New World cats with the arrival of migrating African cats during the Pleistocene era (3). The two FIV lineages infecting bobcats (Lynx rufus) and mountain lions (Puma concolor) are referred to as puma lentivirus (PLV) (or FIVPco), because both lineages (referred to here as PLV clade A [PLVA] and PLVB) were initially characterized in mountain lions. The two PLV clades are monophyletic with respect to the other feline lentiviruses, yet the genetic distance between PLVA and PLVB is equal to that separating viral lineages from different host species. PLVB isolates infect mountain lions throughout their entire geographic range, including most of North and South America, reflecting a long history of virus-host coevolution (3). In contrast, PLVA has been detected in only a small number of mountain lions in southern California and Florida. Franklin et al. hypothesized that the primary host of PLVA is the bobcat because it is the only FIV lineage detected in this species (8). Bobcat PLVA prevalence levels of 25 to 40% have been detected in Florida and southern California, but no PLVA (or other FIV) has been detected in bobcats outside these geographic areas (5).
Several previous reports have described the phylogenetic relationships among FIVs based upon nucleotide sequence alignments from short regions of the pol or env genes (4, 6–10). Additional studies comparing phylogenetic relationships among full-length sequences of lion lentivirus (LLV) isolates across Africa and PLVB isolates from central North America demonstrate that complex evolutionary histories cannot be fully resolved through single-gene analyses (11, 13, 14, 17). Since only one full-length PLVA sequence has been evaluated, no genomic evolutionary studies have been conducted on this clade (15).
We initiated a study to determine the genetic diversity and molecular evolution of PLVA and PLVB using new and existing full-length viral sequence data, including samples obtained from free-ranging North American bobcats and mountain lions. We describe here in detail (i) phylogenetic relationships among PLVs in bobcats and mountain lions, (ii) selective forces and constraints that shape PLV evolution, and (iii) patterns of recombination and their effect on genetic diversity. This work represents the most extensive analysis of FIV genome evolution to date and thus represents an important contribution toward furthering our understanding of virus-host dynamics, natural selection, and adaptation of retroviruses in natural-host populations.
MATERIALS AND METHODS
Sample collection and DNA extraction.
Mountain lion and bobcat samples were collected from five natural populations in three U.S. states (Fig. 1). Sampling locations included (i) two sites in coastal southern California (CA-south and CA-north), adjacent to one another but divided by the metropolitan area of Los Angeles; (ii) two sites in Colorado (CO-west and CO-east), separated by the Rocky Mountains; and (iii) one site in Florida (FL). Previously reported PLV sequences from mountain lions in Wyoming (WY), Montana (MT), British Columbia (BC), and FL were also included in our analyses (see Table SA1 in the supplemental material).
FIG 1.
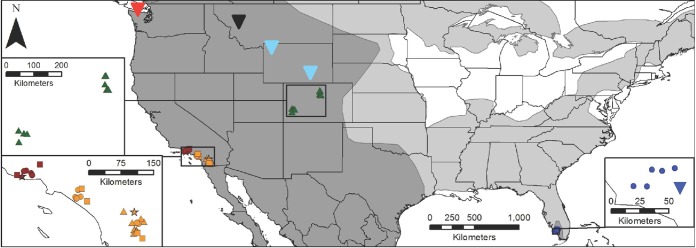
Host geographic ranges in North America and sample locations. Bobcat and mountain lion geographic ranges are depicted in light and dark gray shading, respectively (http://www.iucnredlist.org/). The dark shading also represents areas of sympatry because the geographic range of bobcats completely overlaps that of mountain lions within the map boundaries. Animal capture locations are indicated as follows: circles, bobcats (PLVA); squares, mountain lions (PLVA); small triangles, mountain lions (PLVB); stars, PLVA/PLVB-coinfected mountain lions. Large upside-down triangles represent approximate capture locations for previously reported PLV sequences included in this study (11, 15). The exact locations for Florida panther samples are not shown. Geographic regions are represented by the following symbol colors: orange, CA-south; dark red, CA-north; green, CO-west and CO-east; blue, FL; light blue, WY-west and WY-east; black, MT; bright red, BC.
All samples were collected from live, free-ranging animals captured by using baited cage traps or scent-trained tracking hounds, as previously described (16). All animal capture and handling protocols were done according to approved Animal Care and Use Committee guidelines and, where applicable, local government regulations. Blood samples were sent to Colorado State University for PLV detection by serology and sequence characterization, as described below.
DNA was extracted from whole blood or peripheral blood mononuclear cells (PBMCs) by using the standard DNeasy Blood and Tissue protocol (Qiagen Inc., Valencia, CA). Some fresh PBMC samples were cultured in vitro for up to 14 days to propagate the virus prior to DNA extraction, as previously described (17). Table SA1 in the supplemental material provides the sex, age, location, and collection date for the samples included in this study.
PCR amplification, sequencing, and assembly of viral genomes.
We designed a standardized set of PCR primers and protocols to amplify and sequence diverse viral isolates from each PLV clade. The majority of primers contained degenerate bases. The online program OligoCalc v. 3.26 was used to identify and avoid primer sequences likely to form problematic secondary structures (18). The genome coordinates and sequences for all PLVA and PLVB primers are listed in Table SA2 in the supplemental material, and a map of primer locations is depicted in Fig. SA1 in the supplemental material. PLVA and PLVB genomic coding regions (i.e., from the start of gag to the end of env) were amplified by using three subtype-specific overlapping nested PCRs (Table 1). The 5′ or 3′ portions of some PLVA isolates from mountain lions in California and Florida failed to be amplified by any primer pairs; thus, only partial genomes were sequenced for these individuals (see Table SA1 in the supplemental material).
TABLE 1.
PCR primer combinations used for amplification of the full protein-encoding region in PLVA and PLVB isolatesa
| PCR | Round | PLVA |
PLVB |
||||
|---|---|---|---|---|---|---|---|
| Forward primer | Reverse primer | Product length (kb) | Forward primer | Reverse primer | Product length | ||
| 1 | 1 | A3F | A16R | 4.2 | B3F | P2R | 3.3 kb |
| 2a | A4F | A12R | 2.2 | B4F | B9R | 500 bp | |
| 2b | A9F | A15R | 2.4 | B7F | P1R | 2.5 kb | |
| 2 | 1 | P1F | A26R | 5.7 | B14F | B39R | 6.2 kb |
| 2a | A14F | A21R | 3.7 | B15F | B25R | 2.6 kb | |
| 2b | A19F | A24R | 2.1 | B24F | B30R | 2.0 kb | |
| 2c | B29F | B38R | 2.0 kb | ||||
| 3 | 1 | A21F | A32R | 2.3 | B30F | B44R | 2.7 kb |
| 2 | A22F | A31R | 1.6 | B34F | B43R | 1.1 kb | |
Table SA2 in the supplemental material provides the sequence and genome location for each primer.
PCRs were conducted with Platinum Taq high-fidelity DNA polymerase (Invitrogen Inc., Carlsbad, CA) according to manufacturer-recommended protocols, modified to include twice the recommended units of enzyme. The amount of enzyme was increased to improve amplification of low-copy-number proviral DNA. Final reaction mixture volumes were 50 μl. Five microliters of DNA (100 ng to 250 ng) was used as the template for each first-round PCR. Second-round PCRs were performed by using 2 μl of first-round product as the template. All PCR protocols incorporated touchdown cycles in which the annealing temperature (Ta) was decreased from the optimal upper primer Ta to the optimal lower primer Ta in 0.5°C increments per cycle. These conditions were followed by additional cycles of amplification at the optimal lower Ta for a total of 40 cycles per reaction. Optimal primer Ta values were defined as 3°C to 5°C below the melting temperature (Tm) determined by the nearest-neighbor calculation in OligoCalc (18). The extension time for each PCR was adjusted for product length at a rate of 1 min/kb, as recommended by the manufacturer (Invitrogen Inc., Carlsbad, CA).
PCR products were separated by size via agarose gel electrophoresis and visualized with ethidium bromide. Specific PCR products were purified by using the QIAquick PCR purification kit (Qiagen Inc., Valencia, CA). If products contained multiple bands, the band at the correct fragment length was extracted and purified by using the QIAquick gel extraction kit (Qiagen Inc., Valencia, CA). PCR products were sequenced on an ABI 3130 XL genetic analyzer (Applied Biosystems Inc., Foster City, CA), using internal forward and reverse primers spanning approximately every 500 to 700 bp (see Table SA2 and Fig. SA1 in the supplemental material). To minimize sequencing errors, we sought to achieve at least 2× sequencing coverage at all positions in each viral genome. However, small regions in some viral genomes with high-quality chromatograms had single coverage.
All chromatogram files were manually screened to ensure that bases were scored correctly. Individual sequence files were trimmed and assembled into single consensus sequences by using default settings in Geneious Pro 5.6.4. Intrahost variation was observed in many individuals by the consistent presence of two or more chromatogram peaks at a single position across multiple sequencing reads. These positions were scored as a single base if the “highest-quality” option for handling ambiguous bases in Geneious resolved the ambiguity. Otherwise, the position was scored as ambiguous, using the appropriate degenerate character to indicate which bases cooccur at that site in a given sample. Thirty-eight ambiguous bases were present across 15 consensus genomes. No ambiguous bases were located within amino acids under positive selection or adjacent to recombination sites. Throughout the remaining text, proteins are referred to in plain text with the first letter capitalized (i.e., Gag), and open reading frames (ORFs) are written in italicized lowercase type (i.e., gag).
Sequence analysis.
The open reading frame predictor in Geneious and previously annotated sequences were used to identify the location of each open reading frame (15, 19). The gag, pol, env, and vif open reading frames could be identified with confidence in all PLVA and PLVB genomes (see Results, below), and therefore, the following analyses were performed on these four genetic regions. Putative cleavage sites within precursor proteins (Gag, Pol, and Env) were determined from conserved amino acid motifs identified in protein alignments with annotated FIV sequences (12, 20).
Each consensus sequence was trimmed into the four open reading frames gag, pol, vif, and env. For partially sequenced viral isolates, only complete open reading frames were analyzed (i.e., those sequenced from the start codon to the stop codon). Codon alignments and translated amino acid sequences were constructed for each open reading frame by using Muscle (21) with default parameters. All translated sequences contained intact open reading frames without premature stop codons. To characterize the levels of intra- and interclade genetic diversity at the protein level, pairwise identity and the proportion of invariant sites were estimated from amino acid alignments by using Geneious. The pairwise identity of each alignment reports the percentage of pairwise sites with identical amino acid residues. This value accounts for insertions and deletions (indels), as gap-versus-nongap comparisons are included.
Recombination breakpoints.
Codon alignments for each viral ORF were uploaded into Data Monkey, the online server of the HyPhy package (22). Recombination breakpoints within each ORF were identified by using the genetic algorithm for recombination detection (GARD), with a general discrete distribution of sites into two rate classes (23). Breakpoints identified by improved Akaike information criterion (AIC) values were considered significant only if subsequently supported by a Kishino-Hasegawa test result indicating significant topological incongruence between the trees constructed from the regions to the right and left of the breakpoint (alpha = 0.05). This is a conservative approach, since recombination does not always change phylogenetic relationships (24). An initial run of GARD with each full-length open reading frame was performed. Most analyses were stopped before the full search for all potential breakpoints was complete due to computational time constraints. Therefore, each alignment with at least one significant breakpoint was divided at a breakpoint and rerun in GARD as two shorter alignments. This process was repeated until all significant breakpoints were identified or GARD could run to completion without finding additional breakpoints.
Phylogenetic analyses.
A Bayesian phylogenetic tree of PLVA and PLVB sequences was constructed from a 1,439-bp alignment of a nonrecombinant region of pol by using Bayesian Evolutionary Analysis Sampling Trees v1.6 (BEAST) software (25). This region was chosen because it comprises the longest nonrecombinant region shared by all PLVA and PLVB isolates analyzed (see Results, below), and it overlaps the region of pol commonly used for FIV phylogenetic analyses of nondomestic cats (1, 7). The input file was compiled by using Bayesian Evolutionary Analysis Utility v1.6 (BEAUti). A normal distribution (mean = 1.7 million years ago [MYA]; standard deviation = 1) was used to calibrate the root height of the tree to correspond to the proposed timeline of FIV introduction into the Americas during the Pleistocene era (3). Model parameters included the SDR06 model of nucleotide substitution and a piecewise-linear coalescent Bayesian skyline tree model prior (26). An initial run of BEAST with 10 million Markov chain Monte Carlo (MCMC) iterations sampled every 1,000 iterations was performed by using default parameter settings in BEAUti. The first 10% of samples were discarded as burn-ins. Estimated model parameters were viewed in Tracer v1.5 (2007; A. Rambaut and A. Drummond [http://tree.bio.ed.ac.uk/software/tracer/]), and the results were used to modify the following model priors for the final analysis: relative rate parameters (0 to 10), relaxed-clock mean rate (0 to 10), skyline population size (0 to 10,000), gamma shape parameters (0 to 10), and transition-transversion initial value (codon positions 1 and 2 = 4; codon position 3 = 14). The final analysis ran for 20 million MCMC iterations, which provided sufficient sampling for model convergence (posterior parameter estimates supported by effective sample size values of >200). A maximum clade credibility (MCC) tree with median node heights was constructed from the sampled trees by using TreeAnnotator v1.6.
To investigate the effect of recombination on evolutionary histories and genetic diversity, neighbor-joining trees were constructed from all nonrecombinant genome regions by using default parameters in Geneious. Node support was estimated by using 1,000 bootstrap replicates. Isolates and lineages with incongruent phylogenetic relationships among trees (supported by bootstrap values of >70) were identified as putative recombinants. A comprehensive description of circulating recombinant forms was not the focus of this study and is currently in progress as a separate analysis.
Evolutionary hypothesis testing.
Specific hypotheses about the evolutionary history of PLV in bobcats and mountain lions were tested by using analyses available in Data Monkey (22). The mixed-effects model of evolution (MEME) was used to screen all open reading frames for amino acid residues under positive selection (nonsynonymous evolutionary changes [dN] > synonymous evolutionary changes [dS]; alpha = 0.05) (27). MEME has been shown to be more sensitive in detecting positive selection than fixed-effects methods yet has similar type I error rates (27). The fixed-effects likelihood (FEL) analysis was used to identify amino acid residues under negative selection (dN < dS; alpha = 0.05) (28). Neighbor-joining trees for all of the nonrecombinant genome fragments identified in GARD were used as the input in these analyses.
Nucleotide sequence accession numbers.
All viral sequences have been deposited in GenBank (29) (accession numbers KF906143 to KF906194).
RESULTS
Viral sequences.
Twenty-five full-length PLVA isolates (20 from bobcats and 5 from mountain lions) and 20 PLVB isolates (all from mountain lions) were sequenced (Fig. 1; see also Table SA1 in the supplemental material). All PLVA mountain lion isolates with full-length sequences were sampled in CA. Seven additional PLVA mountain lion isolates were partially sequenced (four from CA and three from FL). These 12 isolates represent 75% of the PLVA-positive (PLVA+) mountain lions detected to date (7, 8, 10). Thirteen bobcat isolates from CA and seven from FL were sequenced. PLVA was not amplified from 80 bobcats and 30 mountain lions from CO, which were screened for PLV by using primers that amplify both viral clades (5; our unpublished data). Of the PLVB isolates sequenced, 11 were from CA and 9 were from CO. Two viral sequences derived from cultures were compared to sequences derived from primary peripheral blood mononuclear cells (PBMCs) in order to identify genetic changes arising as an artifact ex vivo. No significant changes were observed in either case, indicating that short-term virus propagation in primary cell culture does not introduce bias into downstream genetic analyses.
We did not identify an open reading frame in the PLVA sequences that was consistent with the location and length of the orfA genes of other FIVs (13, 30). Several possibilities for the length, location, and organization of rev in PLV have been posited but not empirically proven (13). The potential variation in rev length due to multiple possible splice sites is large and would render analyses speculative. Thus, phylogenetic and evolutionary characteristics of these two gene regions were not included here.
Phylogenetic relationships.
Most of the genetic distance within the isolates evaluated in this study exists between PLV clades A and B, suggesting a long, separate evolutionary history, since these lineages diverged from a common ancestor (Fig. 2). Each clade has subsequently diversified into several distinct viral lineages that form well-supported phylogenetic clusters.
FIG 2.

Phylogenetic relationships among 26 PLVA and 32 PLVB isolates. (a) All isolates; (b) enlarged phylogram of PLVA; (c) enlarged phylogram of PLVB. This is a maximum-clade-credibility Bayesian phylogenetic tree of a 1,439-bp nonrecombinant region of pol. Branch lengths (genetic distances) represent the estimated history of coalescent events among isolates and between clades (see the scale in each panel). Isolates are color coded according to the geographic region from which they were sampled (Fig. 1). Tree clusters in panels b and c are labeled based on the geographic location from which the majority of isolates were sampled. Support values (posterior probability, >80) are also labeled on each node (**, >90; *, >80). Isolate names provide the following information: (i) PLV clade (clade A or B), (ii) host species (Lru, bobcat; Pco, puma), (iii) animal identification number (see Table SA1 in the supplemental material), (iv) sampling location, and (v) sample year (1987 to 2011).
Within PLVA, nearly all of the viruses sampled belong to one of three strongly supported clusters based on the geographic region from which they were collected (FL, CA-south, and CA-north) (Fig. 2b). One virus (Pco7), isolated from a mountain lion in CA-south, clusters generally with the other CA isolates but is paraphyletic to both the CA-south and CA-north groups. The PLVA CA-south cluster contains viral isolates from three mountain lions and eight bobcats. This cluster contains more genetic diversity than the other PLVA groups, which may reflect a sampling bias, as it comprises a larger sample size than the other clusters. However, the CA-south sampling area is also the largest from which PLVA isolates were collected, and thus, this level of diversity may represent the viral population structure within this larger geographic region.
Within CA-north, most of the genetic diversity is represented by a single divergent isolate (Lru2). The other five viruses, which include one mountain lion and four bobcat isolates, are highly similar. Mountain lion isolates cluster with bobcat isolates in both CA sites, recapitulating previously reported phylogenetic comparisons (8).
The FL cluster is highly divergent from the viruses sampled from animals in CA. Six of the seven FL isolates from bobcats exhibit very little divergence from one another, suggesting that a relatively homogeneous PLVA population circulates in this area (Fig. 1). However, these samples were collected from animals within a relatively close proximity to one another during a short time span, which may underrepresent the genetic diversity circulating in this region. Lru15 was sampled from a captive bobcat approximately 150 km away from the other FL isolates (Fig. 1). The genetic distance between this virus and the rest of the Florida cluster may therefore represent geographically distinct viral subpopulations that circulate in Florida. PLV14, an isolate from a relic Florida panther, was adapted to a domestic cat cell line in vitro prior to sequencing (15), and thus, the genetic distance between this isolate and the rest of the FL viruses may be artificially large. This isolate is included only as a reference for comparison with other previously reported PLV studies.
In contrast to the distribution of genetic diversity observed among PLVA isolates, the PLVB isolates exhibit less-strict geographic clustering (Fig. 2c). The Colorado cluster comprises seven isolates from CO (n = 3 for CO-west; n = 4 for CO-east) and two isolates from WY-west. Within this cluster, isolates sampled east and west of the Rocky Mountains are intermixed, suggesting that a relatively unstructured virus population circulates on both sides of the Continental Divide. The Wyoming cluster comprises seven isolates in three distinct subgroups sampled in four different areas: WY-west (n = 4), WY-east (n = 1), CO-west (n = 1), and CO-east (n = 1). The four viruses from MT are monophyletic in Fig. 2c, but closely related isolates from BC and WY have been sequenced (Fig. 3). The single PLVB isolate from CA-north coalesces with the reference sequence from BC (PLV1695), although there is a large genetic distance between the two viruses.
FIG 3.

PLVA and PLVB exhibit distinct evolutionary histories and different contemporary phylogeographic distributions. This is a maximum likelihood phylogenetic tree constructed from a 471-bp region of pol illustrating the relationship among the isolates analyzed in this study and previously reported PLV sequences (n = 80 isolates; 48 PLVB and 32 PLVA isolates). Bootstrap values (100 replicates) with >70% support are indicated on nodes. Branch lengths represent the numbers of substitutions per site (see the scale bar). Two domestic cat FIV sequences (chosen to provide continuity with data reported previously by Franklin et al. [8]) were included as an outgroup. Isolate names and colors are the same as those described in the legend of Fig. 2 (see also Fig. SA2 in the supplemental material). Sequences from geographic regions outside our study areas are depicted in purple and are labeled with the GenBank accession number and sample location (BC, British Columbia; AZ/NV, Arizona/Nevada; ID, Idaho; Mex, Mexico; Nic, Nicaragua; Bra, Brazil; TX, Texas).
Three divergent PLVB clusters (Colorado, Montana, and Wyoming) cocirculate in central U.S. mountain lion populations (Fig. 2c and 3). The Colorado and Montana clusters arose from the same lineage as viruses circulating in CA-south. The CA-south and Colorado clusters coalesce prior to the coalescence of the Montana cluster. However, this relationship changes when phylogenetic relationships are estimated for other genomic regions (see Fig. SA2 in the supplemental material). The Wyoming cluster does not coalesce with the other PLVB lineages until the basal node of the PLVB clade. The genetic diversity within the WY cluster is higher than that of any other PLVB cluster. The node that represents the divergence of the CA-north/BC cluster from the other PLVB isolates has low posterior support. Hence, the relationship of PLV1695 and Pco2 to the other PLVB isolates cannot be estimated with confidence for this region of the viral genome.
Protein diversity and natural selection. (i) Gag.
Among both PLVA and PLVB translation products, the Gag polyprotein has the highest pairwise identity and largest proportion of invariant sites (Table 2). Consistent with this, 53% of the amino acids in Gag are under purifying selection, while <1% are under positive selection. Among the products of Gag proteolytic cleavage, the capsid, matrix, and nucleocapsid proteins have increasing levels of genetic diversity within both PLV clades (Fig. 4; see also Table SA3 in the supplemental material). Capsid is the most conserved protein for both PLVA and PLVB, with the highest percentages of both invariant residues and sites under negative selection. No sites in capsid are under positive selection in either PLV clade; this is the only PLVA protein analyzed without at least one site under positive selection. The genetic diversity in matrix is intermediate compared to those in capsid and nucleocapsid, as are the proportions of sites under positive and negative selection. Nucleocapsid is the most variable protein in Gag, with approximately 30% more variant sites than capsid. Nucleocapsid is also the region of Gag with the highest percentage of sites under positive selection and the most length variation due to insertions and deletions (indels). These differences among the Gag proteins illustrate that amino acid homology and selection pressures can vary greatly among proteins translated from a single transcript.
TABLE 2.
Genetic diversity, natural selection, and recombination within each viral proteina
| Viral protein | Protein length (amino acids) |
% pairwise identity |
% invariant sites |
No. of recombination breakpoints |
No. of sites under positive selection |
No. of sites under negative selection |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLVA | PLVB | PLVA | PLVB | PLVA/PLVB | PLVA | PLVB | PLVA/PLVB | PLVA | PLVB | PLVA | PLVB | PLVA | PLVB | |
| Gag | 479–485 | 461–462 | 91.5 | 94.9 | 72.3 | 77.0 | 80.8 | 43.4 | 3 | 1 | 4 | 8 | 257 | 222 |
| Pol | 1,135–1,140b | 1,142–1,146 | 88.2 | 89.7 | 74.4 | 67.2 | 64.4 | 40.1 | 3 | 1 | 17 | 14 | 593 | 519 |
| Vif | 277 | 232–245 | 88.8 | 85.7 | 62.7 | 65.3 | 57.4 | 31.7 | 0 | 1 | 5 | 5 | 142 | 93 |
| Env | 834–841 | 809–813 | 83.2 | 87.4 | 63.2 | 55.2 | 57.8 | 25.2 | 4 | 6 | 16 | 23 | 400 | 325 |
Pairwise identity is the percentage of identical pairwise amino acids. Invariant sites are those columns in the alignment with a single amino acid shared across all sequences. Recombination breakpoints were identified by using GARD (23). Positively selected sites were identified by using MEME (27). Negatively selected sites were identified by using FEL (28). Table SA3 in the supplemental material provides additional details as well as results for all viral polyprotein cleavage products. The locations of recombination breakpoints and residues under selection are shown in Fig. 4.
The PLVA results shown exclude a unique 41-amino-acid insert present in the PLV14 pol sequence, which may have arisen during adaptation to cell culture.
FIG 4.

Recombination and positive selection contribute to the molecular evolution of PLVA and PLVB. The locations of recombination breakpoints and residues under selection are depicted for PLVA and PLVB genomes (see Table SA3 in the supplemental material). PLV14 and PLV1695 were used as the reference genomes for PLVA and PLVB, respectively (11, 15). Each genome is annotated with the four viral open reading frames studied (gag, pol, vif, and env) (top) as well as the protease cleavage products for the polyproteins (bottom). The scale below each genome represents base pairs starting at base 1 in the 5′ long terminal repeat (LTR).
(ii) Pol.
The translated Pol polyprotein is cleaved by the viral protease enzyme into five enzymes: protease (PR), reverse transcriptase (RT), RNase H, dUTPase (DU), and integrase (IN) (Fig. 4). Additionally, a small peptide (pre-PR) is produced from the 5′ end of the protein upon the N-terminal cleavage of PR. Pol exhibits intermediate levels of genetic diversity compared to the other PLV translation products (Table 2). Most of the Pol proteolytic cleavage products vary only slightly in pairwise identity and the percentage of invariant sites (see Table SA3 in the supplemental material). The exception to this is the pre-PR peptide, which has the lowest pairwise identity and percentage of invariant sites of any PLV gene region analyzed. Pol exhibits relatively little variation in length despite the fact that it is the longest open reading frame in the genome. Most of the observed length variation in both PLVA Pol and PLVB Pol results from truncated IN proteins due to premature stop codons in some isolates. More sites in Pol are under positive selection in PLVA than in PLVB. Of the six Pol protein domains, three in PLVA (pre-PR, PR, and RNase H) and two in PLVB (pre-PR and RT) have >1% of amino acids under positive selection.
(iii) Env.
The viral protease enzyme cleaves the Env polyprotein into leader (L), surface (SU), and transmembrane (TM) proteins (Fig. 4). Env exhibits high levels of genetic diversity, with <60% of residues being invariant within each PLV clade (Table 2). Similarly, a relatively large proportion of Env residues are under positive selection, and a small proportion of residues are under negative selection (see Table SA3 in the supplemental material). All Env protein domains contain indels, many of which are unique to individual viral isolates (alignments are available upon request). The L region is the least conserved of the Env cleavage products for both PLV clades, having only slightly more than 50% invariant residues. SU has a high proportion of sites under purifying selection and intermediate levels of positive selection compared to L or TM. Between viral clades, PLVA SU and TM have similar or slightly higher levels of genetic diversity than PLVB SU and TM. In light of this, it is interesting that SU and TM in PLVB have a higher proportion of residues under positive selection and a lower proportion of residues under negative selection than those in PLVA. This suggests that the marginally higher PLVA genetic diversity may be the result of neutrally evolving sites rather than higher positive selection in this clade.
(iv) Vif.
The diversity and selection characteristics of Vif vary between PLVA and PLVB more than any other protein (Table 2). Among PLVA isolates, Vif has no indels and is similar to Pol in both pairwise identity and the proportion of invariant sites. However, PLVB Vif has higher levels of genetic diversity (similar to that observed in PLVB Env) and varies in length more than any other protein analyzed. The length variations in PLVB Vif are due to additional residues at the 3′ end of the protein in four isolates previously described (13).
Recombination.
Recombination is surprisingly common within both PLVA and PLVB, although the distribution of recombination breakpoints differs greatly between the two clades. The PLVA isolates contain 10 breakpoints distributed relatively uniformly throughout the viral genome (Table 2 and Fig. 4). In contrast, the PLVB breakpoints occur almost exclusively in the 3′ half of the genome, with only one of nine breakpoints being located in the 5′ half. This finding is in agreement with a previous study of PLVB that analyzed the sequences from WY and MT (11).
DISCUSSION
PLVA and PLVB genomes exhibit high levels of genetic diversity resulting from the frequent mutation, recombination, and natural selection characteristics of lentiviruses. Both PLV lineages continue to adapt and evolve despite the putative long coexistence with their respective primary host species. These virus-host relationships likely reflect historic and contemporary host demographic patterns and mechanisms of retroviral evolution.
PLV geographic distribution and phylogenetic relationships.
The bicoastal pattern of the PLVA distribution reflects historical fluctuations in the geographical distribution of bobcats. Bobcats in the Midwestern United States (including CO) have a genetic signature reflecting secondary contact between distinct eastern and western bobcat lineages (31). Significant genetic structure further divides the two lineages into distinct populations. Together, these patterns are consistent with founder effects resulting from long-distance dispersers that recolonized the central and northern United States from a small number of Pleistocene refugia on each coast (31). Haplotype maps suggest that these refugia were most likely located in FL and the Pacific Northwest, as bobcats in these two locations share similar mitochondrial DNA (mtDNA) haplotypes, which were predicted to be ancestral to haplotypes from bobcats in the continental interior. The same founder effects may also explain the lack of PLVA in the Midwestern United States, if early colonizers of the region were PLV negative. This could have occurred due to stochastic effects, given that this region was likely recolonized by a small number of individuals and that gene flow among populations in this region has been relatively low since colonization (31). Additionally, even small fitness differences between infected and uninfected individuals could have increased the likelihood that long-distance dispersers were primarily PLV negative.
Similarly, the current distribution of PLVB lineages across North America appears to be in accordance with the estimated ancestry of mountain lions. Mountain lions span from Canada to southern Chile, but North American mountain lions are genetically distinct and relatively homogeneous compared to populations in Central and South America (32). Culver et al. (32) proposed previously that mountain lions were completely displaced from North America during the late Pleistocene era and that contemporary North American mountain lions have descended from a relatively small number of individuals that migrated north from Central America after the glaciers receded. However, one population of mountain lions in British Columbia carries a unique mtDNA haplotype most similar to the haplotype of mountain lions in Brazil, which was predicted by haplotype mapping to be the most ancestral population of the species (32). PLVB isolates from BC and Brazil cluster together in a lineage that forms the basal node of the PLVB clade in phylogenetic trees containing North, South, and Central American isolates (Fig. 3) (8). Therefore, ancestral genotypes among mountain lions, and among PLVB isolates, reside in British Columbia and Brazil. This observation is intriguing, as it suggests that, similarly to bobcats, a population of mountain lions infected with PLVB may have survived the most recent Ice Age in a Pacific Northwest refugium that has since recolonized parts of the West Coast of North America.
While past glaciation events may have similarly affected the distribution of the two North American felids and their lentiviruses, the contemporary geographic distributions and genetic diversities of PLVA and PLVB differ in several ways. The PLVA isolates sampled are highly spatially structured at both small and large geographic scales, reflecting the population dynamics of their primary host, the bobcat (31). Bobcats inhabit smaller home ranges, live at higher densities, and disperse shorter distances from their natal area than mountain lions (33, 34). Bobcat population structure has been detected at very fine scales where urban development decreases gene flow among nearby populations (4, 35). In contrast, while some PLVB isolates exhibit strict geographic structuring (i.e., CA-south), diverse PLVB isolates cocirculate in other areas, and genetically related viruses circulate in populations separated by thousands of kilometers (Fig. 2c and 3; see also Fig. SA2b in the supplemental material). These findings are consistent with the behavior and life history of mountain lions, which inhabit large home ranges and can disperse >1,000 km from their natal range (36).
The PLVB strains cocirculating in WY, CO, and MT arose from at least two genetically distinct lineages that diverged from one another early in the ancestry of this clade in North America (Fig. 2c; see also Fig. SA2b in the supplemental material). The Colorado and Montana clusters share ancestry with viruses circulating in CA-south. Viruses related to this lineage also circulate in BC and Mexico (Fig. 3). Therefore, viruses arising from this lineage circulate in mountain lion populations spanning all of western North America. Other viruses arising from the same lineage as the Wyoming cluster have been sampled in Idaho, Arizona, and Texas (Fig. 3). Given the distribution of this lineage across the current eastern limit of the mountain lion's geographic distribution in western North America, it is possible that this divergent lineage also circulated in mountain lion populations now absent from the eastern part of the continent.
Pco2 (CA-north) and PLV1695 (BC) are distinct from the viruses circulating in CA-south and the continental interior (Fig. 2c), and the evolutionary history shared by these two isolates varies across the genome (see Fig. SA2b in the supplemental material). Two other viruses from CA-north, which were partially sequenced (see Table SA1 in the supplemental material), also cluster with Pco2 and PLV1695 in a phylogenetic tree constructed from a nonrecombinant region of env (tree not shown). Therefore, PLVB strains circulating just north of Los Angeles share a more recent common ancestor with viruses circulating approximately 2,000 km north in British Columbia than with viruses circulating just south of Los Angeles, approximately 115 km away. This finding appears to be consistent with the genetic structure of mountain lion populations in southern California. Ernest et al. previously detected reduced gene flow between the lion populations separated by Los Angeles but observed that animals from north of Los Angeles to northern California form a continuous, albeit dispersed, population (37). Our results suggest that related viruses may also circulate in northern California, Oregon, and Washington. Anthropogenic influence via movement of animals or release of captive individuals into the wild is a possible confounding factor in the geographic distribution of viral phylogenies.
Genetic diversity, selection, and recombination.
Because the FIVs of nondomestic cats cause little known pathogenicity, it has been posited that coevolution has resulted in “stable” virus-host relationships (38). Supporting this idea, previously reported studies of PLVB concluded that pol, env, and vif evolve under purifying or neutral selection (6, 8). Only the nucleocapsid domain of gag has been found to be under positive selection (12). However, our results reveal that positive selection is an important force driving the evolution of PLV proteins. Some amino acid residues were found to be evolving under positive selection in all four genes analyzed for both clades. The discrepancy between our results and previously reported findings is likely due to differences in the samples analyzed and algorithms used for each analysis. Our analysis included genetically diverse isolates collected over a broad geographic range, enabling the detection of diversifying or directional evolution in multiple viral populations. We also used a recently developed algorithm (MEME), which has been demonstrated to have more power to detect residues under positive selection than previously employed methods (27). The level of positive selection that we detected in PLV is consistent with the evolutionary dynamics described previously for other lentiviruses (39, 40) and illustrates that despite apparently low pathogenicity, these viruses continue to evolve and adapt.
The PLV structural genes gag, pol, and env vary in levels of genetic diversity and selection, reflecting the different functions and host pressures on each protein. gag is the most conserved open reading frame, and the capsid protein is the most conserved region of the Gag polyprotein, with about 90% of capsid amino acid sites being invariant within each clade. Intrapopulation diversity in Gag is even lower (data not shown), corresponding to the pervasive negative selection detected in this gene. Together, these results suggest that nearly all nonsynonymous mutations in gag decrease viral fitness, and thus, circulating viruses are highly homogeneous at the protein level. This strong purifying selection likely maintains essential secondary and tertiary structural conformations vital for the role that Gag plays in the packaging, budding, and maturation of viral particles (41). However, some of the diversification in the PLVA and PLVB matrix and nucleocapsid protein domains resulted from positive selection. Therefore, the fitness landscape for Gag amino acid diversity appears to be characterized by steep peaks, with divergent virus populations evolutionarily constrained on different fitness peaks.
The pol gene, which encodes enzymes necessary for viral replication, exhibits levels of genetic diversity similar to those exhibited by gag. Changes in amino acid composition that alter the structure, charge, or hydrophobicity of protein domains could disrupt enzyme function, explaining the high levels of homology and negative selection in pol. However, the protein domain with the highest percentage of sites under positive selection in both clades is the short pre-PR peptide transcribed from within pol. Because the Pol polyprotein is translated via a ribosomal frameshift within the gag open reading frame, pre-PR entirely overlaps the 3′ end of gag. Therefore, mutations occurring in this region can affect both Gag and Pol polyproteins. Interestingly, while the PLVA and PLVB pre-PR products have approximately 10% of sites under positive selection, no Gag residues encoded from the same region are under positive selection in either PLV clade (Fig. 4). The level of negative selection among the overlapping Gag and pre-PR residues is the lowest of any region of the genome analyzed. This overlapping coding region is characterized by high levels of mutation, which evolve under neutral or purifying selection in the gag ORF but mostly neutral and positive selection in the pol ORF. The biological significance of this level of selection for pre-PR is unknown, as no function has been ascribed to this peptide among the lentiviruses. However, our results suggest that pre-PR may have important implications for viral fitness.
The Env polyprotein and its subunits were the least conserved proteins analyzed. The env open reading frame also has the highest numbers of indels and recombination breakpoints, illustrating that the genetic diversity observed for Env results from several different aspects of viral polymerase activity. Interestingly, the highest percentage of residues under positive selection among the Env protein domains of both clades is the L peptide. This domain has two putative functions, acting both as a signal to target the env transcript to the rough endoplasmic reticulum for translation and as the first exon of the accessory protein Rev (13, 42). Rev is encoded by all lentiviruses and is required for the export of unspliced and partially spliced mRNAs from the nucleus (43). This is accomplished by altering the host cell nuclear export mechanism, which, in the absence of Rev, allows the export of only fully spliced mRNAs from the nucleus (44). While both functions of the Env L domain rely on specific residue motifs, the diversity of Rev form and function may be evolutionarily advantageous. For example, highly efficient Rev proteins in equine infectious anemia virus (EIAV) rapidly produce virions during acute infection, but Rev proteins with low efficiency limit virus production and allow the virus to evade the host immune response during chronic infection (45). Unfortunately, no empirical studies on rev transcription, translation, or protein function have been performed with puma lentiviruses. Such work could elucidate the mechanisms underlying the selective forces acting on the highly variable L peptide domain of Env.
The SU and TM domains of Env were more conserved than those of the L peptide but exhibited higher levels of genetic diversity and positive selection than most other protein domains analyzed. This high level of genetic diversity, together with the low levels of negative selection, is consistent with the immune-driven antigenic variation observed for other lentiviruses (46–48). The SU and TM domains of lentiviral Env proteins are targets of the adaptive immune response, and this pressure often selects for viral diversity both within and between hosts. Our analysis suggests that PLV Env evolution may be similarly influenced by host immune pressures, resulting in the observed high levels of genetic diversity within and among viral populations.
The vif open reading frame encodes a lentiviral accessory protein that functions in primate and feline lentiviruses to overcome an antiviral intrinsic immune restriction factor known as APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like) (49, 50). In the absence of a functional Vif protein, APOBEC can create defective viruses through hypermutation during reverse transcription of the viral genome (51). The inability of host-adapted viral Vif proteins to counteract APOBEC activity in alternative hosts is thought to be an important factor limiting cross-species transmission events of lentiviruses (50, 52). It is therefore interesting that Vif diversity is higher among PLVB isolates than among PLVA isolates, given that the former infect one species and the latter infect two. The mountain lion and bobcat APOBEC proteins are highly homologous (50), and our findings suggest that PLVA Vif may successfully counteract both species' antiviral proteins. This is supported by the similar levels of positive selection detected in PLVA and PLVB Vif proteins, which are comparable to the level of positive selection observed for Env. Such a lack of host restriction may be one viral mechanism contributing to cross-species transmission of PLVA between bobcats and pumas. An understanding of differences in host-specific selection pressure, viral adaptation, and viral fitness in bobcats versus pumas will provide valuable insight into mechanisms of viral evolution during cross-species transmission.
Interestingly, PLVA appears to lack a single intact gene region encoding the OrfA protein. All lentiviruses encode a transactivator of proviral translation, which is considered essential for viral replication (43). OrfA, the transactivator protein of feline lentiviruses, is thought to be translated in PLVB from a single unspliced open reading frame overlapping the vif and env genes (Fig. 4) (13). It is possible that the orfA gene is present in PLVA but that it is translated from a spliced RNA or an internal ribosome entry site, as occurs in some other lentiviruses (i.e., HIV/EIAV) (43). In either case, this difference in genomic organization between PLVA and PLVB may have resulted from, or contributed to, the divergence of these clades. If orfA is indeed absent and is not necessary for productive PLVA infection, investigations of proviral transcription in the absence of this protein may reveal unique mechanisms of lentivirus replication.
Conclusion.
We have shown that high levels of genetic diversity, frequent recombination, and positive selection on viral proteins characterize PLV evolution in bobcats and mountain lions. These findings are in contrast to previous reports (6, 8, 12), which concluded that FIVs in nondomestic cats had reached a stable level of host-virus evolution (3, 38). These analyses were based primarily on studies of short gene fragments or isolates from single populations and/or tended to look at fewer isolates per species than this analysis. Our results illustrate that the long-term coevolution of lentiviruses and their hosts does not necessarily result in stable equilibrium. Analyzing intrahost diversity and quantifying viral loads among viral lineages could provide a better understanding of how the observed genetic diversity and selection ultimately relate to viral fitness.
PLVA and PLVB are highly divergent, and although historically classified as two clades of species-specific viruses infecting mountain lions, they are likely distinct viral species with different primary feline hosts. PLVA infects primarily bobcats, and the geographic distribution of regionally associated subtypes is consistent with the ecology and evolutionary history of this host species. Highly structured PLVA lineages circulate in distinct bobcat populations, and PLVA is likely absent from bobcats in central North America. PLVB is common in mountain lions throughout their geographic range, and in contrast to PLVA, distinct viral lineages cocirculate in some areas, corresponding to the ability of this virus to move long distances in its vagile host.
The prevailing hypothesis of FIV evolution assumes that species-specific viruses evolved from a common ancestor via host-virus cospeciation and historic cross-species transmission events. Our work also supports the possibility that viral genetic divergence may also occur contemporaneously via cross-species infections and subsequent adaptation to new host species. Factors that can alter population structure and contact rates, such as changing climactic conditions and habitat fragmentation, may contribute to increased numbers of interspecies transmission events. The remarkable capacity for generating genetic diversity documented here renders these viruses highly adaptable and would enable evolution to occur on a short time scale.
These findings represent the most robust characterization of feline lentiviral genetic evolution to date. Future studies investigating the genetic determinants of viral fitness will significantly enhance our understanding of PLV evolution and complement the existing body of literature describing the ecological and phylogenetic aspects of PLV.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an Ecology of Infectious Disease grant from the National Science Foundation and a Wildlife Fellowship grant from the Morris Animal Foundation.
Thanks to S. Carver and E. Schweickart for assistance with the production of Fig. 1 and 4, respectively.
Any use of trade, product, or firm names is for descriptive purposes only and does not imply an endorsement by the U.S. Government.
Footnotes
Published ahead of print 16 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00473-14.
REFERENCES
- 1.Troyer JL, Pecon-Slattery J, Roelke ME, Johnson W, VandeWoude S, Vazquez-Salat N, Brown M, Frank L, Woodroffe R, Winterbach C, Winterbach H, Hemson G, Bush M, Alexander KA, Revilla E, O'Brien SJ. 2005. Seroprevalence and genomic divergence of circulating strains of feline immunodeficiency virus among Felidae and Hyaenidae species. J. Virol. 79:8282–8294. 10.1128/JVI.79.13.8282-8294.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.VandeWoude S, Apetrei C. 2006. Going wild: lessons from naturally occurring T-lymphotropic lentiviruses. Clin. Microbiol. Rev. 19:728–762. 10.1128/CMR.00009-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pecon-Slattery J, Troyer JL, Johnson WE, O'Brien SJ. 2008. Evolution of feline immunodeficiency virus in Felidae: implications for human health and wildlife ecology. Vet. Immunol. Immunopathol. 123:32–44. 10.1016/j.vetimm.2008.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JS, Ruell EW, Boydston EE, Lyren LM, Alonso RS, Troyer JL, Crooks KR, VandeWoude S. 2012. Gene flow and pathogen transmission among bobcats (Lynx rufus) in a fragmented urban landscape. Mol. Ecol. 21:1617–1631. 10.1111/j.1365-294X.2012.05493.x [DOI] [PubMed] [Google Scholar]
- 5.Lagana DM, Lee JS, Lewis JS, Bevins SN, Carver S, Sweanor LL, Crooks KR, VandeWoude S. 2013. Characterization of regionally associated feline immunodeficiency virus (FIV) sequences in bobcats (Lynx rufus). J. Wildl. Dis. 49:718–722. 10.7589/2012-10-243 [DOI] [PubMed] [Google Scholar]
- 6.Biek R, Rodrigo AG, Holley D, Drummond A, Anderson CR, Jr, Ross HA, Poss M. 2003. Epidemiology, genetic diversity, and evolution of endemic feline immunodeficiency virus in a population of wild cougars. J. Virol. 77:9578–9589. 10.1128/JVI.77.17.9578-9589.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpenter MA, Brown EW, Culver M, Johnson WE, PeconSlattery J, Brousset D, Obrien SJ. 1996. Genetic and phylogenetic divergence of feline immunodeficiency virus in the puma (Puma concolor). J. Virol. 70:6682–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franklin SP, Troyer JL, Terwee JA, Lyren LM, Boyce WM, Riley SP, Roelke ME, Crooks KR, Vandewoude S. 2007. Frequent transmission of immunodeficiency viruses among bobcats and pumas. J. Virol. 81:10961–10969. 10.1128/JVI.00997-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimura Y, Goto Y, Yoneda K, Endo Y, Mizuno T, Hamachi M, Maruyama H, Kinoshita H, Koga S, Komori M, Fushuku S, Ushinohama K, Akuzawa M, Watari T, Hasegawa A, Tsujimoto H. 1999. Interspecies transmission of feline immunodeficiency virus from the domestic cat to the Tsushima cat (Felis bengalensis euptilura) in the wild. J. Virol. 73:7916–7921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olmsted RA, Langley R, Roelke ME, Goeken RM, Adgerjohnson D, Goff JP, Albert JP, Packer C, Laurenson MK, Caro TM, Scheepers L, Wildt DE, Bush M, Martenson JS, Obrien SJ. 1992. Worldwide prevalence of lentivirus infection in wild feline species: epidemiologic and phylogenetic aspects. J. Virol. 66:6008–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruen TC, Poss M. 2007. Recombination in feline immunodeficiency virus genomes from naturally infected cougars. Virology 364:362–370. 10.1016/j.virol.2007.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burkala E, Poss M. 2007. Evolution of feline immunodeficiency virus Gag proteins. Virus Genes 35:251–264. 10.1007/s11262-006-0058-8 [DOI] [PubMed] [Google Scholar]
- 13.Poss M, Ross H. 2008. Evolution of the long terminal repeat and accessory genes of feline immunodeficiency virus genomes from naturally infected cougars. Virology 370:55–62. 10.1016/j.virol.2007.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rigby MA, Holmes EC, Pistello M, Mackay A, Brown AJL, Neil JC. 1993. Evolution of structural proteins of feline immunodeficiency virus: molecular epidemiology and evidence of selection for change. J. Gen. Virol. 74:425–436. 10.1099/0022-1317-74-3-425 [DOI] [PubMed] [Google Scholar]
- 15.Langley RJ, Hirsch VM, O'Brien SJ, Adger-Johnson D, Goeken RM, Olmsted RA. 1994. Nucleotide sequence analysis of puma lentivirus (PLV-14): genomic organization and relationship to other lentiviruses. Virology 202:853–864. 10.1006/viro.1994.1407 [DOI] [PubMed] [Google Scholar]
- 16.Bevins SN, Carver S, Boydston EE, Lyren LM, Alldredge M, Logan KA, Riley SPD, Fisher RN, Vickers TW, Boyce W, Salman M, Lappin MR, Crooks KR, VandeWoude S. 2012. Three pathogens in sympatric populations of pumas, bobcats, and domestic cats: implications for infectious disease transmission. PLoS One 7:e31403. 10.1371/journal.pone.0031403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pecon-Slattery J, McCracken CL, Troyer JL, VandeWoude S, Roelke M, Sondgeroth K, Winterbach C, Winterbach H, O'Brien SJ. 2008. Genomic organization, sequence divergence, and recombination of feline immunodeficiency virus from lions in the wild. BMC Genomics 9:66. 10.1186/1471-2164-9-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kibbe WA. 2007. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 35:W43–W46. 10.1093/nar/gkm234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poss M, Ross HA, Painter SL, Holley DC, Terwee JA, VandeWoude S, Rodrig A. 2006. Feline lentivirus evolution in cross-species infection reveals extensive G-to-A mutation and selection on key residues in the viral polymerase. J. Virol. 80:2728–2737. 10.1128/JVI.80.6.2728-2737.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elder JH, Schnolzer M, Hasselkuslight CS, Henson M, Lerner DA, Phillips TR, Wagaman PC, Kent SBH. 1993. Identification of proteolytic processing sites within the Gag and Pol polyproteins of feline immunodeficiency virus. J. Virol. 67:1869–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. 10.1093/bioinformatics/btq429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23:1891–1901. 10.1093/molbev/msl051 [DOI] [PubMed] [Google Scholar]
- 24.Wiuf C, Christensen T, Hein J. 2001. A simulation study of the reliability of recombination detection methods. Mol. Biol. Evol. 18:1929–1939. 10.1093/oxfordjournals.molbev.a003733 [DOI] [PubMed] [Google Scholar]
- 25.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22:1185–1192. 10.1093/molbev/msi103 [DOI] [PubMed] [Google Scholar]
- 27.Kosakovsky Pond S, Murrell B, Fourment M, Frost SDW, Delport W, Scheffler K. 2011. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 28:3033–3043. 10.1093/molbev/msr125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kosakovsky Pond SL, Frost SDW. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22:1208–1222. 10.1093/molbev/msi105 [DOI] [PubMed] [Google Scholar]
- 29.Benson D, Karsch-Mizrachi I, Lipman D, Sayers E. 2011. GenBank. Nucleic Acids Res. 39:D32–D37. 10.1093/nar/gkq1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterji U, de Parseval A, Elder JH. 2002. Feline immunodeficiency virus OrfA is distinct from other lentivirus transactivators. J. Virol. 76:9624–9634. 10.1128/JVI.76.19.9624-9634.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reding DM, Bronikowski AM, Johnson WE, Clark WR. 2012. Pleistocene and ecological effects on continental-scale genetic differentiation in the bobcat (Lynx rufus). Mol. Ecol. 21:3078–3093. 10.1111/j.1365-294X.2012.05595.x [DOI] [PubMed] [Google Scholar]
- 32.Culver M, Johnson WE, Pecon-Slattery J, O'Brien SJ. 2000. Genomic ancestry of the American puma (Puma concolor). J. Hered. 91:186–197. 10.1093/jhered/91.3.186 [DOI] [PubMed] [Google Scholar]
- 33.Logan K, Sweanor L. 2001. Desert puma: evolutionary ecology and conservation of an enduring carnivore. Island Press, Washington, DC [Google Scholar]
- 34.Ruell EW, Riley SPD, Douglas MR, Pollinger JP, Crooks KR. 2009. Estimating bobcat population sizes and densities in a fragmented urban landscape using noninvasive capture-recapture sampling. J. Mammal. 90:129–135. 10.1644/07-MAMM-A-249.1 [DOI] [Google Scholar]
- 35.Riley SPD, Pollinger JP, Sauvajot RM, York EC, Bromley C, Fuller TK, Wayne RK. 2006. A southern California freeway is a physical and social barrier to gene flow in carnivores. Mol. Ecol. 15:1733–1741. 10.1111/j.1365-294X.2006.02907.x [DOI] [PubMed] [Google Scholar]
- 36.Thompson DJ, Jenks JA. 2005. Long-distance dispersal by a subadult male cougar from the Black Hills, South Dakota. J. Wildl. Manag. 69:818–820. 10.2193/0022-541X(2005)069[0818:LDBASM]2.0.CO;2 [DOI] [Google Scholar]
- 37.Ernest HB, Boyce WM, Bleich VC, May B, Stiver SJ, Torres SG. 2003. Genetic structure of mountain lion (Puma concolor) populations in California. Conserv. Genet. 4:353–366. 10.1023/A:1024069014911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Brien SJ, Troyer JL, Roelke M, Marker L, Pecon-Slattery J. 2006. Plagues and adaptation: lessons from the Felidae models for SARS and AIDS. Biol. Conserv. 131:255–267. 10.1016/j.biocon.2006.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross HA, Rodrigo AG. 2002. Immune-mediated positive selection drives human immunodeficiency virus type 1 molecular variation and predicts disease duration. J. Virol. 76:11715–11720. 10.1128/JVI.76.22.11715-11720.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans DT, O'Connor DH, Jing PC, Dzuris JL, Sidney J, da Silva J, Allen TM, Horton H, Venham JE, Rudersdorf RA, Vogel T, Pauza CD, Bontrop RE, DeMars R, Sette A, Hughes AL, Watkins DI. 1999. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nat. Med. 5:1270–1276. 10.1038/15224 [DOI] [PubMed] [Google Scholar]
- 41.Luttge BG, Freed EO. 2010. FIV Gag: virus assembly and host-cell interactions. Vet. Immunol. Immunopathol. 134:3–13. 10.1016/j.vetimm.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips TR, Lamont C, Konings DA, Shacklett BL, Hamson CA, Luciw PA, Elder JH. 1992. Identification of the Rev transactivation and Rev-responsive elements of feline immunodeficiency virus. J. Virol. 66:5464–5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed). 2007. Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 44.Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. 1989. The HIV-1 Rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 338:254–257 [DOI] [PubMed] [Google Scholar]
- 45.Belshan M, Baccam P, Oaks JL, Sponseller BA, Murphy SC, Cornette J, Carpenter S. 2001. Genetic and biological variation in equine infectious anemia virus rev correlates with variable stages of clinical disease in an experimentally infected pony. Virology 279:185–200. 10.1006/viro.2000.0696 [DOI] [PubMed] [Google Scholar]
- 46.Braun MJ, Clements JE, Gonda MA. 1987. The Visna virus genome: evidence for a hypervariable site in the env gene and sequence homology among lentivirus envelope proteins. J. Virol. 61:4046–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Payne SL, Fang FD, Liu CP, Dhruva BR, Rwambo P, Issel CJ, Montelaro RC. 1987. Antigenic variation and lentivirus persistence: variations in envelope gene sequences during EIAV infection resemble changes reported for sequential isolates of HIV. Virology 161:321–331. 10.1016/0042-6822(87)90124-3 [DOI] [PubMed] [Google Scholar]
- 48.Holmes EC, Zhang LQ, Simmonds P, Ludlam CA, Brown AJL. 1992. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc. Natl. Acad. Sci. U. S. A. 89:4835–4839. 10.1073/pnas.89.11.4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31. 10.1016/S0092-8674(03)00515-4 [DOI] [PubMed] [Google Scholar]
- 50.Zielonka J, Marino D, Hofmann H, Yuhki N, Lochelt M, Munk C. 2010. Vif of feline immunodeficiency virus from domestic cats protects against APOBEC3 restriction factors from many felids. J. Virol. 84:7312–7324. 10.1128/JVI.00209-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392–1396. 10.1016/j.cub.2004.06.057 [DOI] [PubMed] [Google Scholar]
- 52.VandeWoude S, Troyer J, Poss M. 2010. Restrictions to cross-species transmission of lentiviral infection gleaned from studies of FIV. Vet. Immunol. Immunopathol. 134:25–32. 10.1016/j.vetimm.2009.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.