

ABSTRACT
Successful adaptive immunity to virus infection often depends on the initial innate response. Previously, we demonstrated that Junín virus, the etiological agent responsible for Argentine hemorrhagic fever (AHF), activates an early innate immune response via an interaction between the viral glycoprotein and Toll-like receptor 2 (TLR2). Here we show that TLR2/6 but not TLR1/2 heterodimers sense Junín virus glycoprotein and induce a cytokine response, which in turn upregulates the expression of the RNA helicases RIG-I and MDA5. NF-κB and Erk1/2 were important in the cytokine response, since both proteins were phosphorylated as a result of the interaction of virus with TLR2, and treatment with an Erk1/2-specific inhibitor blocked cytokine production. We show that the Junín virus glycoprotein activates cytokine production in a human macrophage cell line as well. Moreover, we show that TLR2-mediated immune response plays a role in viral clearance because wild-type mice cleared Candid 1 (JUNV C1), the vaccine strain of Junín virus, more rapidly than did TLR2 knockout mice. This clearance correlated with the generation of Junín virus-specific CD8+ T cells. However, infected wild-type and TLR2 knockout mice developed TLR2-independent blocking antibody responses with similar kinetics. We also show that microglia and astrocytes but not neurons are susceptible to infection with JUNV C1. Although JUNV C1 infection of the brain also triggered a TLR2-dependent cytokine response, virus levels were equivalent in wild-type and TLR2 knockout mice.
IMPORTANCE Junín virus is transmitted by rodents native to Argentina and is associated with both systemic disease and, in some patients, neurological symptoms. Humans become infected when they inhale aerosolized Junín virus. AHF has a 15 to 30% mortality rate, and patients who clear the infection develop a strong antibody response to Junín virus. Here we investigated what factors determine the immune response to Junín virus. We show that a strong initial innate immune response to JUNV C1 determines how quickly mice can clear systemic infection and that this depended on the cellular immune response. In contrast, induction of an innate immune response in the brain had no effect on virus infection levels. These findings may explain how the initial immune response to Junín virus infection could determine different outcomes in humans.
INTRODUCTION
The Arenaviridae family constitutes a single genus that includes nearly 30 species, which, based on serologic, phylogenetic, and geographic differences, can be divided in Old World and New World arenaviruses (1). The New World arenaviruses can be further classified into clades A, B, A/B, and C (2). The Old World and clade B New World arenaviruses include important human pathogens. The Old World arenaviruses Lassa virus and Lujo virus together with the New World arenaviruses Junín virus, Machupo virus, Guanarito virus, and Sabia virus cause hemorrhagic fever in humans. Native rodents of the region where hemorrhagic fevers are endemic are the natural reservoirs of arenaviruses. Infection occurs when humans come into contact with contaminated urine, blood, or saliva from carrier rodents through skin abrasions or aerosol inhalation (3).
Infection with Junín virus, the etiological agent of Argentine hemorrhagic fever (AHF), has an incubation period ranging from 6 to 12 days (4). During the first week of infection, individuals develop high fever together with flu-like symptoms that include headache, myalgia, arthralgia, conjunctivitis, nausea, and diarrhea. Hemorrhagic manifestations such as gingival bleeding and petechia in the oral mucosa as well as in axillary regions may be present during the second week of infection. During this time, neurological manifestations such as mental confusion and a decreased reflex response can also become apparent (5). By the third week of infection, >80% of infected individuals generate a strong antibody response against Junín virus (6). The humoral response together with the derepression of cell-mediated immunity leads to virus clearance (7).
Although there is a prophylactic vaccine against Junín virus, the live attenuated strain JUNV C1 (8), current treatments for AHF are limited and consist of the early transfusion of neutralizing-antibody-containing plasma (6) and ribavirin (9). Ribavirin has shown mixed efficacy and significant side effects in some individuals. Moreover, although treatment with immune plasma reduces the overall mortality rate, approximately 10% of these treated patients develop late neurological syndrome (2). Thus, new treatments for Junín virus and other New World arenavirus infections are needed.
By understanding the interaction between Junín virus and the host immune response, it might be possible to design effective therapeutic agents. The initial targets of Junín virus infection are believed to be sentinel cells of the immune system, such as monocytes, macrophages, and dendritic cells (DCs) (10, 11). Typically, innate immune responses to virus infection by sentinel cells are characterized by the rapid induction of interferons (IFNs) and cytokines such as tumor necrosis factor alpha (TNF-α). Junín virus induces IFN-β expression in human macrophages, and acute infection with Junín virus is characterized by high serum levels of IFNs and TNF-α, suggesting that innate responses occur in humans (12–14). The role of these cytokines is to block viral replication in infected cells and cause neighboring uninfected cells to acquire an antiviral state, mediated by the expression of interferon-stimulated genes (ISGs). In addition, the expression of cytokines by sentinel cells such as macrophages and DCs plays an important role in the activation of B and T cells, thereby linking the innate and the adaptive immune responses needed to clear a viral infection (15). However, it has also been suggested that many of the Junín virus disease manifestations are caused by high levels of cytokines such as IFNs and TNF-α (3, 14, 16).
We previously showed that vaccine strain JUNV C1 as well as pseudoviruses bearing the glycoprotein complex (GPC) of the pathogenic Parodi strain induce the production of IFN-β and TNF-α in mouse macrophages through a Toll-like receptor 2 (TLR2)-dependent mechanism and that this induction occurs prior to virus replication (17). Here we further investigated the mechanism by which the GPCs of both JUNV C1 and the Parodi strain activate the innate immune response in macrophages and determined the signaling pathways used for this activation after interaction with TLR2. In addition, we assessed the role of the innate immune response in the subsequent activation of CD8+ T cell and antibody responses against JUNV C1 infection in mice. We show that JUNV C1 interacts with the TLR2/TLR6 complex and that this in turn triggers a series of signaling events leading to the activation of the transcription factors NF-κB and AP-1. We also demonstrate that prior activation of the TLR2-mediated innate immune response is necessary for mice to have an effective CD8+ T cell response against JUNV C1. However, the humoral response to JUNV C1 did not depend on TLR2, since wild-type mice as well as TLR2−/− mice generated blocking antibodies. Finally, we show that while TLR2-mediated activation is independent of the route of JUNV C1 inoculation, the consequences of this activation differ, namely, resulting in an antiviral cellular immune response only in the periphery.
MATERIALS AND METHODS
Ethics statement.
All mice were housed according to the policies of the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. The experiments performed with mice in this study were approved by this committee (IACUC protocol number 803700).
Cell lines and virus.
Vero cells; 293T cells; mouse macrophage cell lines NR-9456 (wild type [WT]), NR-9457 (TLR2−/−), Mφ TLR1−/−, Mφ TLR6−/−, and Mφ TLR7−/−; and mouse microglial cell lines NR-9460 (WT) and NR-9904 (Mal/MyD88−/−) were cultivated in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with glutamine, sodium pyruvate, 5% fetal bovine serum (FBS), and penicillin-streptomycin. The human monocyte cell line THP-1 was cultured in RPMI medium (Invitrogen) supplemented with glutamine, 5 μM β-mercaptoethanol, 10% fetal bovine serum, and penicillin-streptomycin. JUNV C1 was propagated in and titers were determined on Vero cells as described previously (17). The TLR1−/−, TLR6−/−, and TLR7−/− macrophage cell lines were kindly provided by Kate Fitzgerald, and JUNV C1 was kindly provided by Robert Tesh. Lymphocytic choriomeningitis virus (LCMV) (Armstrong) was provided by John Wherry. Vesicular stomatitis virus (VSV)-mCherry pseudotypes bearing the JUNV C1 and Parodi GPCs were generated by transfecting 293T cells with plasmids expressing the corresponding Junín virus GPCs, and at 24 h posttransfection, cells were infected with replication-competent VSV-mCherry lacking GPC (a kind gift from Paul Bates). At 16 h postinfection (hpi), supernatants were harvested, and pseudotype titers were determined on Vero cells.
Mice and generation of primary macrophage cultures.
C57BL/6 mice were obtained from the National Cancer Institute (Frederick, MD) and were bred at the University of Pennsylvania. MyD88−/−, TLR2−/−, and TLR4−/− (C57BL/6 background) mice were originally obtained from Shizuo Akira and Douglas Golenbock and have been bred at the University of Pennsylvania since 2000. Primary bone marrow-derived macrophages were isolated from the hind limbs of 8- to 10-week-old mice. Macrophages were seeded into 10-cm untreated dishes in DMEM supplemented with 10% FBS, 20% macrophage colony-stimulating factor (M-CSF) derived from L929 cells, sodium pyruvate, HEPES, and antibiotics. Cells were harvested at 7 days, and 1 × 106 cells/well were seeded into a 6-well plate for infection assays.
JUNV C1 infection of mouse and human macrophages and treatment with TLR agonists.
Mouse macrophages were infected with JUNV C1 at a multiplicity of infection (MOI) of 1, and after adsorption for 60 min at 37°C, unbound virus was washed off with phosphate-buffered saline (PBS), and cells were maintained in DMEM supplemented with 5% FBS. Cells were treated with the TLR agonists Pam3CSK4 (PAM) (TLR1/2; 100 ng/ml), FSL-1 (TLR2/6; 100 ng/ml), R848 (TLR7; 1 μg/ml), lipopolysaccharide (LPS) (TLR4; 100 ng/ml), and the LPS antagonist polymyxin B (50 μg/ml; Invivogen). THP-1 cells were differentiated into macrophages by treatment with 200 μM phorbol-12-myristate acetate (PMA; Sigma) for 24 h. Cells were washed with PBS, fresh RPMI medium was added, and cells were incubated at 37°C for 72 h. Cells were then infected with JUNV C1 as described above for mouse macrophages.
Quantification of virus isolated from organs.
Eight- to ten-week-old mice were infected by intraperitoneal (i.p.) (1 × 106 PFU) or intracranial (i.c.) (1 × 104 PFU) inoculation of JUNV C1; control mice received sham inoculations. At 1 week postinfection, spleens and brains from the i.p. and i.c. injected mice, respectively, were collected in PBS and homogenized by Dounce homogenization with the plunger of a 1-ml syringe. The homogenate was clarified by centrifugation at 4°C, and the supernatants were collected and stored at −70°C. Viral titers were quantified by plaque assays, as described previously (17). A portion of the homogenate was treated with RNAlater (Ambion), and RNA from spleens and brain were isolated by using an RNeasy kit (Qiagen) or TRIzol reagent (Ambion), respectively, according to the manufacturers' instructions.
RNA analysis.
Total RNA isolated from cell lines and mouse organs was reverse transcribed into cDNA by using the Quantitech reverse transcription kit (Qiagen) according to the manufacturer's specifications. JUNV C1, IFN-β, and TNF-α RNAs were quantified by real-time quantitative PCR (RT-qPCR) using a 7800HT sequence detector system (Applied Biosystems). Specific primer pairs to detect the S segment of JUNV C1 (5′-GGGGCAGTTCATTAGCTTCATGC-3′ and 5′-CAAAGGTAGGTCATGTGGATTGTTGG-3′), mouse IFN-β (5′-AAGAGTTACACTGCCTTTGCCACT-3′ and 5′-CACTGTCTGCTGGTGGAGTTCATC-3′), mouse TNF-α (5′-GCCACCACGCTCTTCTGTCT-3′ and 5′-GGTCTGGGCCATAGAACTGATG-3′), and human TNF-α (5′-CCCAGGCAGTCAGATCATCTTC-3′ and 5′-GCTGGTATCTCTCAGCTCCA-3′) were used. All RNA quantifications were normalized to values for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (5′-CCCCTTCATTGACCTCAACTACA-3′ and 5′-CGCTCCTGGAGGATGGTGAT-3′). For each primer pair, a no-template control was included, and each sample was run in triplicate. The Power Sybr green PCR kit (Applied Biosystems) was used to perform all RT-qPCR amplifications. The amplification conditions were 50°C for 2 min, followed by 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. The efficiency of amplification was determined for each primer pair by generating a standard curve with 10-fold serial dilutions of a known concentration of viral DNA. The slope values of the standard curves for the primer pair amplicons ranged from −3.5 to −3.2, indicating 90 to 100% efficiency. At the end of the RT-qPCR run, a dissociation curve was determined to ensure that each primer pair generated a single product of amplification.
Measurement of anti-Junín virus in vivo CD8+ T cell responses.
Eight- to ten-week-old mice were infected i.p. with 1 × 106 PFU of JUNV C1 or with 2 × 105 PFU of LCMV (Armstrong). One week after infection of mice, peripheral blood mononuclear cells (PBMCs) were obtained and costained with allophycocyanin-conjugated anti-CD8 antibody (BD Biosciences) and with NP205–212–phycoerythrin (PE)-labeled major histocompatibility complex (MHC) tetramers, kindly provided by Kate Mansfield, Douglas Dolfi, and John Wherry. Cells were analyzed by using a multicolor Calibur flow cytometer (BD Biosciences).
Antibody neutralization assay.
Mice infected with JUNV C1 or LCMV were bled at 2 weeks postinfection, and serum was stored at −70°C until use. 293T cells were seeded at 2.5 × 104 cells/well in a 96-well plate, and 2.5 × 103 PFU of JUNV C1 was added to serial dilutions of the serum (1 × 10−1 to 1 × 10−5). The serum-JUNV C1 mixture was added to cells, and 2 days after infection, the cells were fixed for 10 min with 2% paraformaldehyde, permeabilized in blocking buffer (PBS, 2% bovine serum albumin [BSA], 0.1% Triton X-100), and stained for virus with a monoclonal antibody specific for Junín virus nucleoprotein (NP) (SA02-BG12; BEI Resources), Alexa Fluor 488-coupled anti-mouse secondary antibody (Invitrogen), and 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). The plates were imaged with the 20× objective of an ImageXpress microscope (Molecular Devices). Nine random images were acquired per well in both the DAPI and 488-nm channels. The percentages of infection and DAPI-positive cells were calculated by using automated image analysis software (MetaXpress; Leica). In the absence of neutralizing serum, approximately 20% of the cells were infected; this proportion of infected cells was considered 100% infectivity. To determine the percent infection in the presence of neutralizing serum, the number of infected cells was divided by the average number of infected cells from nine random fields infected in the absence of neutralizing serum.
Western blot analysis.
Protein extracts were prepared from primary macrophages that had been previously treated with TLR agonists or infected with JUNV C1 in the presence or absence of the MEK1/2 inhibitor UO126 (10 μM 1 h prior to infection; Cell Signaling). RNA helicases were detected by using rabbit polyclonal antibodies specific for RIG-I (catalog number 4520; Cell Signaling) and MDA5 (catalog number ALX-210-352-R100; Enzo Life Sciences). Rabbit polyclonal antibodies were used to detect total (catalog number sc-372; Santa Cruz Biotechnology) and phosphorylated (catalog number 3039; Cell Signaling) forms of NF-κB. Different sets of antibodies were also used to detect total and phosphorylated forms of Erk1/2 (catalog numbers 9102 and 9106, respectively; Cell Signaling).
ELISAs.
Primary macrophages were treated with 10 μM the MEK1/2 inhibitor UO126 for 1 h. UO126-treated and untreated macrophages were incubated with 100 ng/ml LPS or infected with JUNV C1 at an MOI of 1. Cell supernatants were collected 6 h after infection with JUNV C1 or treatment with LPS. TNF-α was quantified from the medium by using the Quantikine enzyme-linked immunosorbent assay (ELISA) kit against mouse TNF-α (R&D Systems), according to the manufacturer's specifications.
Mouse brain sections and staining.
Four-week-old mice (6 WT and 4 TLR2−/− mice) were inoculated i.c. with JUNV C1 (1 × 104 PFU), and infection was allowed to progress for 1 week. Mice were sacrificed and perfused twice, once with ice-cold PBS and then with 4% paraformaldehyde. The brains were dissected and fixed in 4% paraformaldehyde for 24 h. Five-micrometer brain sections were treated with 9.4% citrate-based antigen unmasking solution (Vector Laboratories) and heated in a pressure cooker at 120°C for 10 min. Excess unmasking solution was removed by placing the samples under running water, followed by a 30-min incubation in PBS–0.5% Triton X-100 to permeabilize the tissue, and the slides were then incubated for 30 min in blocking solution (PBS, 0.5% Tween 20, and 10% normal goat serum). Slides were incubated overnight at 4°C in blocking solution containing monoclonal antibody NA05-AG12 (BEI Resources) to detect Junín virus NP together with an antibody specific for glial fibrillary protein (GFAP) (astroglia) (catalog number Z0334; Dako), ionized calcium binding adaptor molecule 1 (Iba-1) (microglia) (catalog number 019-19741; Wako), or neuronal nuclei (NeuN) (neurons) (catalog number ABN78; Millipore) to distinguish different brain cell types. After removal of the excess primary antibody with PBS–0.5% Tween 20, brain tissue was incubated with antibodies against mouse and rabbit antibodies labeled with the florescent markers Alexa 488 and Alexa 594, respectively. Coverslips were mounted onto the samples by using Vectashield (Vector Laboratories), and samples were observed under a fluorescence microscope (EFD-3; Nikon). For each brain, 10 sections at 15-μm intervals were stained with anti-Junín virus NP and either Iba-1, GFAP, or NeuN (approximately 3 sections for each marker).
Statistical analyses.
Statistical analyses were done by using Prism software. Methods used to calculate P values are described in the figure legends.
RESULTS
TLR2 recognition of Junín virus GPC leads to activation of the NF-κB and MAPK pathways.
Previously, we showed that TLR2 is important for the recognition of Junín virus by mouse macrophages and that these cells produced increased levels of TNF-α and IFN-β in response to the viral GPC (17). TLR2 signals through both the NF-κB and mitogen-activated protein kinase (MAPK) pathways, leading to the activation of the transcription factors NF-κB and AP-1, which are required for the expression of cytokines such as TNF-α (18). We first investigated whether JUNV C1 activated the NF-κB and MAPK pathways and if signaling occurred exclusively through TLR2. To test NF-κB activation, we incubated primary mouse macrophages with JUNV C1, and at 2 hpi, protein extracts were analyzed by Western blotting, using an antibody specific for the activated phosphorylated form of NF-κB (p65). In WT and TLR4−/− but not TLR2−/− macrophages, JUNV C1 caused NF-κB phosphorylation, the TLR2 and TLR4 ligands PAM and LPS induced phosphorylation only in cells bearing these receptors, and the LPS antagonist polymyxin B abrogated LPS- but not JUNV C1-mediated phosphorylation of NF-κB, demonstrating that the virus-mediated effect was not due to LPS contamination (Fig. 1A). Western blot analysis for phosphorylated Erk1/2 gave similar results: Erk1/2 became phosphorylated in WT and TLR4−/− but not in TLR2−/− macrophages (Fig. 1B). Erk1/2 phosphorylation was abrogated when cells were treated with the MAPK inhibitor UO126 1 h prior to infection with JUNV C1 (Fig. 1B).
FIG 1.

JUNV C1-mediated activation of the NF-κB and Erk1/2 pathways is dependent on TLR2 signaling. (A) Primary macrophages with deletions in TLR genes were infected with JUNV C1 or treated with LPS, LPS plus polymyxin B (Pmx), or PAM, and at 2 hpi, protein lysates were prepared and analyzed by Western blotting, using antibodies against total and phosphorylated (p-Ser486) NF-κB (p65). (B) Primary macrophages were treated with UO126 for 1 h, followed by infection with JUNV C1 or LPS. At 2 hpi, cell extracts were analyzed for phosphorylated Erk1/2 by Western blotting. (C and D) At 2 hpi, RNA was isolated from cells treated as indicated, and IFN-β (C) and TNF-α (D) RNA levels were quantified by RT-qPCR. (E) Supernatants from the same cells were used to quantify TNF-α levels by ELISA. The bar graphs represent the averages and standard deviations from three technical replicates. These experiments were performed twice, with similar results.
We also examined the effect of UO126 on the TLR2-dependent increase in IFN-β and TNF-α levels. UO126 completely blocked the induction of TNF-α and IFN-β RNA (Fig. 1C and D) and protein (Fig. 1E) production, most likely due to the lack of Erk1/2 phosphorylation and subsequent AP-1 activation (19). UO126 also blocked LPS-mediated Erk1/2 phosphorylation and TNF-α and IFN-β production by WT and TLR2−/− cells (Fig. 1B to E). Together, these data suggest that JUNV C1 activates both NF-κB and MAPK pathways via TLR2.
We showed previously that expression of the RNA helicases RIG-I and MDA5 was upregulated by JUNV C1 independent of virus replication (17). To determine if this upregulation was a secondary effect of TLR2-mediated cytokine production, we examined the levels of these RNA helicases in primary macrophages after infection with JUNV C1 for 2 and 24 h. At 2 hpi, low-level induction of RIG-I and MDA5 was detected (Fig. 2A), while at 24 hpi, there were increased expression levels of both RNA helicases in WT and TLR4−/− macrophages (Fig. 2B). However, RIG-I induction was not observed in TLR2−/− macrophages, and MDA5 showed only slight upregulation compared to WT or TLR4−/− macrophages (Fig. 2B). The TLR2 and TLR4 ligands PAM and LPS induced upregulation of RIG-I and MDA5 only in cells bearing these receptors. We also incubated macrophages with JUNV C1 and LPS in the presence of polymyxin B; only LPS- and not JUNV C1-mediated activation of RNA helicases was abrogated by polymyxin B. These data suggest that the increased cytokine levels produced as a result of TLR2 activation were required for initial induction of RIG-I and MDA5 expression.
FIG 2.
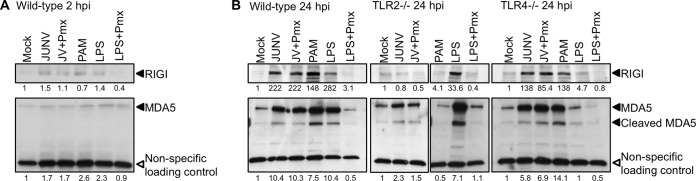
Upregulation of RIG-I and MDA5 expression by JUNV C1 is TLR2 dependent. RIG-I and MDA5 levels were analyzed by Western blotting of extracts from primary macrophages infected with JUNV C1 or treated with LPS, LPS plus polymyxin, or PAM. (A) Primary macrophages derived from wild-type mice were infected with JUNV C1, and at 2 hpi, protein lysates were analyzed by Western blotting, using antibodies against RIG-I and MDA5. (B) JUNV C1-infected macrophages derived from wild-type, TLR2−/−, and TLR4−/− mice were harvested at 24 hpi, and the proteins were analyzed by Western blotting with antibodies against RIG-I and MDA5. A cellular protein that is detected nonspecifically by the MDA5 antibody was used as a loading control. Shown below the lanes are the relative levels of protein in each sample relative to levels in mock-treated cells. Quantification of RIG-I and MDA5 levels was done by using ImageJ software. These experiments were done twice, with similar results.
Junín virus is recognized by TLR2/6 heterodimers.
TLR2 forms homo- and heterodimers with TLR1 and TLR6 and is able to discriminate between different pathogen-associated molecular patterns depending upon such associations. For instance, TLR1/2 heterodimers sense triacylated lipopeptides, while TLR2/6 complexes recognize diacylated lipopeptides (20, 21). To determine whether TLR2 heterodimerizes with TLR1 or TLR6 to sense Junín virus, we incubated WT or knockout mouse macrophage cell lines (TLR1, -2, -6, or -7) with JUNV C1 and quantified TNF-α and IFN-β RNA levels by RT-qPCR (Fig. 3A and B) and TNF-α protein levels by ELISA (Fig. 3C). JUNV C1 induced TNF-α expression/release in TLR1−/− but not TLR6−/− macrophage cells (Fig. 3A and C). IFN-β RNA expression was also induced by JUNV C1 in TLR1−/− but not TLR6−/− cells (Fig. 3B). Polymyxin B treatment abrogated the LPS response but did not affect JUNV C1-mediated cytokine induction. As controls, we treated macrophages with PAM and FSL-1, which are ligands for TLR1/2 and TLR2/6, respectively. As expected, TLR6−/− macrophages were activated only by PAM and not by FSL-1 (Fig. 3A and C), and IFN-β production was highly induced by FSL-1 in TLR1−/− macrophages (Fig. 3B). However, there was low-level induction of TNF-α in TLR1−/− cells treated with PAM, which is likely due to TLR2 homodimer formation (according to the manufacturer's specification sheet). TLR2−/− cells did not respond to JUNV C1 or to the ligands PAM and FSL-1 (Fig. 3A to C).
FIG 3.

Mouse macrophages sense Junín virus GPC through TLR2/6 heterodimers. (A to E) Mouse macrophage cell lines with deletions in different TLR genes were infected with JUNV C1 or treated with various TLR agonists (A to C) or infected with VSV pseudotypes bearing JUNV C1 GPC, Parodi GPC, or no GPC (Bald) (D and E). At 2 hpi, RNA was isolated, and IFN-β (A and D) and TNF-α (B and E) levels were quantified by RT-qPCR. At 6 hpi, medium from infected cells was collected and TNF-α levels were quantified by ELISA (C). The bar graphs represent the averages and standard deviations from three technical replicates. These experiments were performed three times independently, with similar results. (F) THP-1 human monocytes treated with PMA to induce macrophage differentiation were infected with JUNV C1. At 2 hpi, TNF-α RNA levels were quantified by RT-qPCR. Shown are data from duplicate experiments performed in triplicate under each condition. (G) PMA-differentiated macrophages were infected with VSV pseudotypes bearing JUNV C1 GPC, Parodi GPC, or no GPC, and TNF-α RNA levels were measured at 2 hpi. Each bar represents the average and standard deviation of data from an experiment performed in triplicate.
JUNV C1 has a single-stranded RNA genome and could potentially activate cytokine production via the single-strand RNA sensor TLR7. TNF-α and IFN-β RNA levels produced by TLR7−/− cells in response to JUNV C1 were similar to those produced by WT macrophages; these cells did not respond to the TLR7 ligand R848 (Fig. 3A and B). Although the amount of TNF-α protein in the medium was almost 10 times lower than that that in WT cells, this was also the case for TLR7−/− cells treated with PAM and FSL-1, indicating a general lower-level response to TLR2 ligands by this cell line (Fig. 3C). These results suggest that TLR2 and TLR6 form a complex that senses Junín virus in mouse macrophages and that JUNV C1 is not recognized by TLR7.
The JUNV C1 (attenuated) and Parodi (pathogenic) GPCs differ by 6 amino acids (22). We showed previously that the 2 GPCs had the same effect on TLR2-mediated induction of cytokines (17). To confirm that both GPCs were sensed by TLR2/TLR6 heterodimers, we also infected the macrophage lines with Junín virus GPC-VSV pseudotypes. Similar to replication-competent JUNV C1, the Parodi GPC pseudotypes showed no induction of IFN-β and TNF-α in TLR2−/− or TLR6−/− cells, and levels of cytokine production induced by JUNV C1 and Parodi pseudotypes were similar in wild-type and TLR1−/− macrophages (Fig. 3D and E). These results show that the JUNV C1 vaccine and Parodi pathogenic GPCs are sensed by TLR2/6 heterodimers and that both GPCs elicit the same cytokine response.
We also tested whether JUNV C1 or GPC pseudotypes would elicit a similar cytokine response in human macrophages. THP-1 cells were differentiated to macrophages with PMA, cultured for 72 h in medium without PMA, and then infected with JUNV C1 (MOI = 1). At 2 hpi, TNF-α RNA levels were quantified by RT-qPCR. Similar to mouse macrophages, JUNV C1 induced TNF-α RNA expression in human macrophages (Fig. 3F). We also infected human macrophages with VSV pseudotypes bearing the GPCs of the vaccine and pathogenic strains. Both GPCs induced TNF-α expression to the same extent, while bald virions had no effect (Fig. 3G). Thus, human and mouse macrophages respond similarly to the GPCs of both the pathogenic and vaccine strains of Junín virus.
TLR2−/− mice do not clear systemic JUNV C1 as rapidly as WT mice.
Previous studies have shown that IFN-α/β/γR−/− mice are more susceptible to Junín virus-associated pathogenesis than WT mice (23), although the mechanism behind this defect is not known. To determine whether the lack of JUNV C1/TLR2-mediated cytokine production in TLR2 knockout mice affects the ability of mice to clear virus, we injected mice i.p. with 1 × 106 PFU of JUNV C1 and analyzed viral RNA levels from spleens at 3, 7, 10, and 14 days postinfection (dpi). The TLR2−/− mice cleared infection more slowly than did the wild-type mice, and by 14 dpi, they still had significantly higher virus loads (Fig. 4A). We further examined the levels of JUNV C1 at the 7-dpi time point with additional mice; at this time point, viral RNA levels were 3 to 4 times higher in TLR2−/− mice than in WT mice (Fig. 4B), and the titers were 2 logs higher in TLR2−/− mice than in WT mice (Fig. 4C). This was not due to an inability of JUNV C1 to replicate in WT cells, since the kinetics of infection in WT and TLR2−/− macrophage cell lines were similar (Fig. 4D). These results showed that the initial TLR2-mediated innate immune response affected JUNV C1 replication in vivo but not in vitro.
FIG 4.
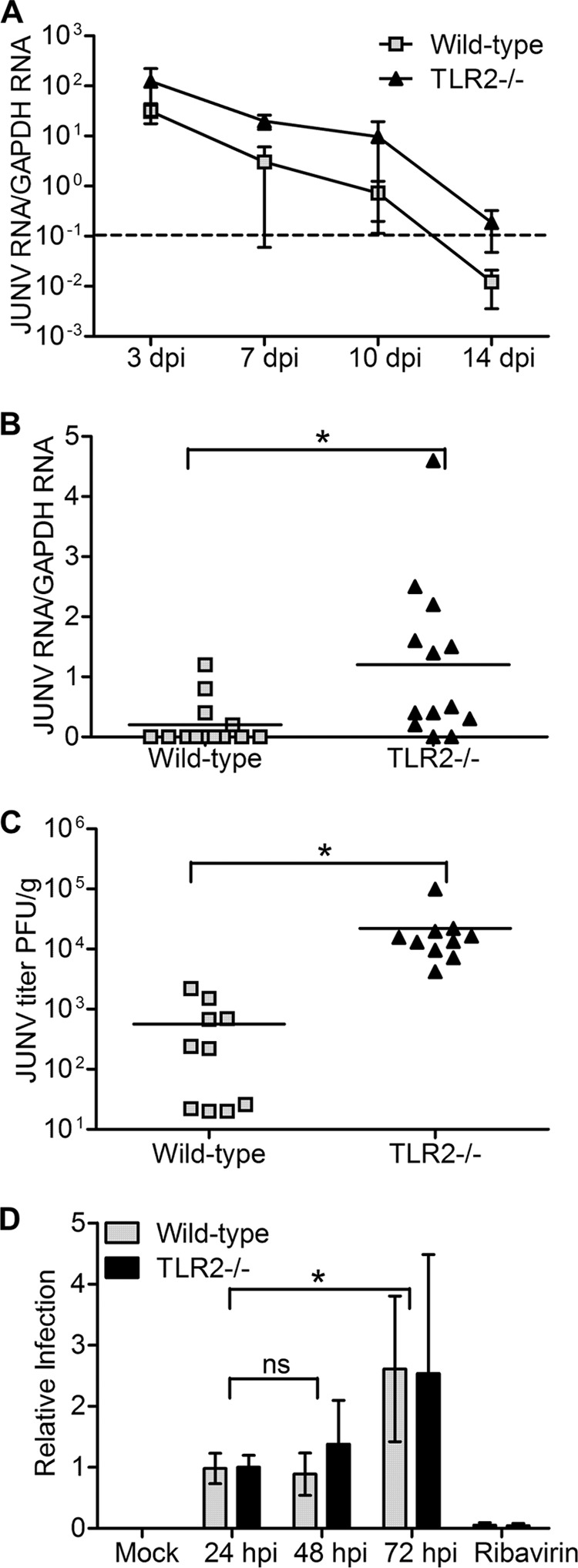
TLR2−/− mice are more highly infected with JUNV C1 than are wild-type mice. (A) Time course of infection. Spleens were harvested on the indicated days postinfection, and viral RNA was quantified by RT-qPCR. Five mice of the corresponding phenotypes were used for each time point. Kaplan-Meier analysis showed that there was statistical significance (P < 0.05) between WT and TLR2−/− mouse RNA level curves. The dashed line indicates the limit of detection for this RT-qPCR assay. (B and C) Additional mice were infected with JUNV C1, and at 7 days postinfection, viral RNA was quantified by RT-qPCR (B), and viral titers were determined (C). Each point represents an individual mouse. (D) WT (NR-9456) and TLR2−/− (NR-9457) mouse macrophage cells were infected with JUNV C1 at an MOI of 0.1 and harvested at 24, 48, and 72 hpi, and viral RNA was measured by RT-qPCR. As a control, infected cells were treated with ribavirin, and the viral RNA level was quantified at 48 hpi. Two-tailed Student's t test was used to determine significance. *, P < 0.05; ns, no statistical significance.
TLR2 signaling is important for CD8+ T cell but not humoral responses to JUNV C1.
It is well established that innate immune responses influence subsequent adaptive immune responses to viruses. Thus, the higher infection levels in the TLR2 knockout mice could be due to weaker T cell or humoral responses resulting from the lack of initial cytokine induction. We next tested whether mice infected with JUNV C1 established an antiviral CD8+ T cell response and if this response was diminished in TLR2−/− mice. Proliferation of CD8+ T cells specific to the peptide spanning residues 205 to 212 of LCMV NP (NP205–212) (YTVKYPNL) can be detected 1 week after infection of C57BL/6 mice (24). Comparison of the genome sequences of LCMV and Junín virus showed that the viruses share the same amino acid residues in their NPs. We infected mice i.p. with 1 × 106 or 2 × 105 PFU of JUNV C1 or LCMV, respectively, and at 7 dpi, CD8+ T lymphocytes isolated from peripheral blood were stained with NP205–212 tetramers. While infection of WT or TLR4−/− mice caused a 9-fold increase in the average percentage (Fig. 5A) or number (Fig. 5B) of CD8+ T cells/ml, in TLR2−/− mice, JUNV C1 induced a more modest 2.5-fold increase in the average percentage of NP205–212-specific cells.
FIG 5.

Increased levels of Junín virus-specific CD8+ T cells but not humoral responses require TLR2 signaling. Mice were infected with JUNV C1 or LCMV. At 1 week postinfection, mouse PBMCs were collected and stained for NP-specific CD8+ T cells. (A and B) Percentage (A) and total number (B) of Junín virus-specific CD8+ T cells (per ml of blood) from mice of different TLR backgrounds. Each point represents a single mouse. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, no statistically significant difference based on one-way analysis of variance. (C and D) Mice of the indicated backgrounds (WT [C] and TLR2−/− [D]) were infected with JUNV C1. At 2 weeks postinfection, serum was collected, and the indicated serial dilutions were incubated with JUNV C1 for 1 h prior to infection of Vero cells. Undiluted sera from mock- and LCMV-infected mice were used as negative controls. Each point represents an individual mouse.
We next determined if the humoral immune response was also dependent on TLR2 signaling. Sera were harvested from mice that had been infected with JUNV C1 for 2 weeks, and serial dilutions of the sera were tested for their ability to block JUNV C1 infection of Vero cells. There were no differences in the neutralizing antibody titers of wild-type (Fig. 5C), TLR2−/− (Fig. 5D), and TLR4−/− (data not shown) mice. These results suggest that although TLR2 activation is necessary for the activation of CD8+ T cells, the antibody response against JUNV C1 does not require this pathway.
Microglia and astrocytes are infected by JUNV C1 in vivo.
In addition to causing hemorrhagic fever, Junín virus can reach the central nervous system in animal models and may cause neurological manifestations in humans (25, 26). Moreover, JUNV C1 was generated by extensive passaging in newborn mouse brain (27). Previous studies have shown that after intracranial inoculation with the pathogenic Junín virus Gar strain in mice, neurons and astrocytes not only become infected but also shed viral particles (27, 28). In addition, other studies have demonstrated that Junín virus can activate the expression of inducible nitric oxide synthase (iNOS) in astrocytes (29). To determine which cell type was infected by JUNV C1 in vivo, we performed intracranial inoculations in mice. One week after infection, brain sections were costained with anti-NP, and anti-Iba-1, anti-GFAP, and anti-NeuN antibodies, to distinguish between microglia, astrocytes, and neurons, respectively. We found that both microglia (Fig. 6A) and astrocytes (Fig. 6C) were infected but that no NeuN-positive cells were stained with anti-Junín virus NP antibody (Fig. 6E). Similar results were observed for astrocytes and microglia cells in JUNV C1-infected TLR2−/− mice (data not shown).
FIG 6.

JUNV C1 targets microglia and astroglia cells in the brain. Mice that received intracranial inoculations of PBS or JUNV C1 were sacrificed at 7 dpi. Brain sections from JUNV C1-infected (A, C, and E) and mock-infected (B, D, and F) mice were stained with antibodies specific for Junín virus NP and the brain cell markers Iba-1 (microglia) (A and B), GFAP (astroglia) (C and D), and NeuN (neurons) (E and F). Brain cell makers were indirectly stained with a secondary antibody labeled with Alexa 564 (red), and viral NP was indirectly stained with an Alexa 488-labeled secondary antibody (green). Cell nuclei were stained with DAPI and are shown in the merged image. The selected region of the merged image is magnified 3× (bottom right quadrant of each panel).
TLR2 is required for cytokine induction in brain but does not affect JUNV C1 infection.
Junín virus infection in humans often results in neuropathology (26), and a recent study suggested that even JUNV C1 induces pathological changes when injected intracranially into newborn mice (30). First, we assessed whether microglia cells, the resident macrophages of the brain, had a similar cytokine induction response to that of JUNV C1. Since a TLR2−/− microglia cell line was unavailable, we tested JUNV C1 cytokine induction in cells lacking the downstream adapter molecules Mal and MyD88. We quantified IFN-β (Fig. 7A) and TNF-α (Fig. 7B) RNA levels in microglia cell lines incubated with JUNV C1. Similar to what was seen with macrophages, JUNV C1 incubation led to increased expression levels of both IFN-β and TNF-α in WT but not in Mal/MyD88−/− microglia cells. These data suggest that in the context of JUNV C1 infection, microglia behave similarly to macrophages and could be targets for Junín virus infection in the brain.
FIG 7.

JUNV C1 induces TLR2-dependent cytokine production in the brain which does not control infection. (A and B) WT and MyD88/Mal double-knockout microglia cell lines were infected with JUNV C1 or treated with various TLR agonists. Two hours after infection of cells, total RNA was isolated and used to quantify IFN-β (A) and TNF-α (B) levels by RT-qPCR. RNA values were normalized to that of GAPDH RNA. The bar graphs represent the averages and standard deviations from three technical replicates. These experiments were performed independently three times, with similar results. (C) Virus titers from the brains of wild-type mice that received intracranial inoculations of JUNV C1 and at 3, 5, 7, 10, and 14 dpi. (D and E) RNA isolated at 5 and 7 dpi from the brains of JUNV C1-infected WT and TLR2−/− mice was used to quantify IFN-β (D) and TNF-α (E) levels. (F) Virus titers from the brains of WT and TLR2−/− mice at 5 dpi. Each point represents an individual mouse. **, P < 0.01; ***, P < 0.001; ns, no statistically significant difference based on 2-tailed Student's t test.
We next monitored the kinetics of JUNV C1 infection in the brain. Mice received intracranial inoculations of JUNV C1, and at 3, 5, 7, 10, and 14 dpi, the brains were analyzed for viral RNA and titers. Viral titers at 7 dpi were about 1 order of magnitude higher than the titers at 3 dpi, suggesting active viral replication (Fig. 7C). By 10 dpi, the average titer was 2 orders of magnitude lower than that at 7 dpi, and by 14 dpi, no viral particles were detected (Fig. 7C).
We next examined the innate immune response in the brain in response to infection and whether, as in the periphery, TLR2−/− mice were defective in their ability to clear JUNV C1. WT and TLR2−/− adult mice were inoculated intracranially, and at 5 and 7 dpi, RNA isolated from their brains was analyzed for viral and cytokine RNAs by RT-qPCR. As we saw in the periphery, IFN-β (Fig. 7D) and TNF-α (Fig. 7E) RNAs were induced to higher levels in WT than in TLR2−/− mice, demonstrating that there is also an innate immune response to virus in the brain that depends on TLR2. JUNV C1 was also detected in the brains of both WT and TLR2−/− mice at 5 dpi (Fig. 7F), although in contrast to what was seen with peripheral infection, there was no significant difference in levels of infection between WT and TLR2−/− mice.
DISCUSSION
Here we report that the New World arenavirus Junín virus is sensed by the TLR2/TLR6 complex in macrophages, resulting in the early activation of innate immune responses prior to the onset of virus replication. While other viruses, such as respiratory syncytial virus (31) and hepatitis C virus (32), activate Toll-like receptor signaling via the TLR2/6 heterodimer complex, previous studies with the Old World arenavirus LCMV identified TLR2 but not the particular heterodimer required for activation (33). Importantly, we demonstrate that both attenuated JUNV C1 and pathogenic Parodi GPCs induce TLR2-mediated signaling, demonstrating that this response is not due to the amino acid changes in GPC that occurred during attenuation. Additionally, we show that JUNV C1 induced TNF-α expression in a human macrophage cell line, demonstrating that human and mouse cells respond similarly.
The transcription factors NF-κB and AP-1 both need to become activated for the transcription of cytokines such as TNF-α. Upon JUNV C1-mediated stimulation, TLR2/6 initiates a MyD88-dependent signaling cascade that leads to the activation of the TAB2-TAK1-IκB kinase (IKK) complex; this complex is responsible for IκB phosphorylation and targeting it for degradation, thereby allowing translocation of NF-κB into the nucleus. TAK1 is also involved in the phosphorylation of the MAPK pathway that leads to AP-1 activation and triggering of downstream gene transcription. Here we showed that JUNV C1 induced the phosphorylation of both NF-κB and the Erk1/2 kinase complex upstream of AP-1 shortly after infection and that this was abrogated in TLR2−/− macrophages. Moreover, when we treated cells with the selective MEK1/2 inhibitor UO126, it blocked JUNV C1-mediated phosphorylation of Erk1/2 and the production of TNF-α and IFN-β in WT macrophages (Fig. 1C to E).
We also showed that upregulation of the RIG-I and MDA5 RNA helicases depends on TLR2 signaling. Previously, we, as well as others, showed that levels of both RNA sensors were increased in macrophages infected with JUNV C1 (12, 17). However, whether this was the result of direct sensing of incoming viral RNA or the result of TLR2-mediated signaling was not known. It has been reported that there is a positive-feedback loop that activates both RIG-I and MDA5 transcription upon stimulation with IFN-α/β that is initially induced when the RNA helicases come into contact with viral RNA (34, 35). Indeed, a recent study by Huang et al. suggested that Junín virus-mediated activation of IFN-α expression and RIG-I RNA requires RIG-I signaling (12); it has also been suggested that the H5N1 strain of influenza virus (36) and the DV1 strain of dengue virus (37) upregulate RIG-I and MDA5 RNA levels in a RIG-I-dependent manner in human macrophages and hepatocytes, respectively. However, here we demonstrate that the increased expression levels of RIG-I and MDA5 result from the cytokine response triggered by Junín virus-mediated stimulation of TLR2 rather than by direct interaction with the RNA helicases, at least at the initial stages of infection. This suggests that when Junín virus is sensed by TLR2, the cytokines have an autocrine/paracrine effect on the infected cells, leading to the synthesis of RIG-I and MDA5. This in turn would activate the feedback loop in infected cells and explain why Junín virus encodes proteins that inactivate RIG-I in infected cells (38, 39).
Several studies have suggested that pathogenic Junín virus blocks innate immune responses (38–40). Moreover, Groseth and colleagues demonstrated previously that pathogenic Junín virus did not induce an innate immune response in human macrophages. However, their study examined cytokine induction at 4 days postinfection and likely depended on viral replication (41), unlike the responses detected here, which, as we previously showed, are initiated by initial virion contact with cells (17). We also showed previously that while JUNV C1 replicates in mouse macrophages, which is similar to what was seen by Groseth et al. for human macrophages, the cytokine response was not detected after 24 h, perhaps due to the ability of JUNV C1 to block innate immune responses. Indeed, both Z and NP of New World arenaviruses have been implicated in the blocking of innate immune responses (38, 40). Since the sequences of the NP and Z genes of JUNV C1 are identical to those of pathogenic Junín virus strains (8, 22, 42), the vaccine strain is likely to also block cytokine responses after the initiation of virus replication. The results presented here thus suggest that early IFN induction via TLR2 signaling by either pathogenic Junín virus or the vaccine strain, but not an ongoing innate immune response, is important for shaping the subsequent adaptive immune response. Interestingly, mice lacking type I and type II IFN receptors succumb to pathogenic Junín virus infection for unknown reasons, although mouse embryonic fibroblasts derived from these mice are no more infected than WT cells ex vivo (23). These results agree with studies showing that both JUNV C1 and pathogenic Junín virus are resistant to the antiviral effects of type I and type II IFNs in cultured cells (12). Since Junín virus suppresses postreplication but not initial TLR2-mediated innate responses, our data suggest that the pathogenesis and inability to control viral replication in IFN receptor (IFNR)-null mice may be the result of decreased adaptive immune responses rather than direct effects of the innate immune response on the virus.
The adaptive response to Junín virus has not been well characterized. However, the Old World arenavirus LCMV has been extensively used to delineate the immune response against viral infections in mice. Throughout the first week of infection, LCMV-specific CD8+ T cells proliferate vigorously; this response peaks at around 8 days postinfection (43, 44). This population of CD8+ T cells is made up of subsets of cells that recognize distinct peptide epitopes derived mainly from viral GPC and NP and that exhibit strong cytotoxic activity against infected cells (45, 46). We saw a similar induction of Junín virus-specific CD8+ T cells at 7 days after infection with JUNV C1, suggesting that there are kinetic similarities in the antiviral responses to both Old and New World arenaviruses in mice.
Our results suggest that a strong innate immune response is required for the induction of the adaptive immune response necessary to clear JUNV C1, since in the absence of TLR2, there was a decreased virus-specific CD8+ T cell responses and increased systemic infection (Fig. 5C). These findings are supported by several studies that have demonstrated the importance of type I IFNs in activating virus-specific CD8+ T cell responses and clearance of LCMV infection. In acute LCMV infections, the innate immune response is necessary for virus control. LCMV can induce sentinel cells to secrete IFN-α and IFN-β (47), which are associated with the upregulation of interferon-stimulated genes capable of constraining LCMV replication, such as ISG15 (48) and viperin (49). LCMV also activates sentinel cells, such as DCs, via IFN-α and IFN-β (50); these DCs then serve as LCMV antigen-presenting cells (APCs) that produce cytokines that activate LCMV-specific CD8+ T cells directly, without the participation of helper CD4+ T cells (51). Our results also suggest that JUNV C1 is able to stimulate sentinel cells, which in turn could serve as APCs to activate virus-specific CD8+ T cells. It has also been shown that LCMV induces the production of various chemokines in a TLR2-MyD88/Mal-dependent manner and that this activation is linked to an antiviral response (52). Our results also suggest that in the absence of TLR2, sentinel cells do not produce cytokines that activate the Junín virus-specific CD8+ T cells important for virus clearance.
In contrast, the antibody response to JUNV C1 was not dependent on TLR2 signaling, since sera from infected WT or TLR2−/− mice equivalently neutralized infection (Fig. 6). Although there have been numerous reports indicating that TLR signaling plays a role in antibody production (53–55), it is also known that in the presence of an adjuvant, B cells become active and secrete antibodies through a TLR-independent mechanism (56) and that LCMV-induced B cells have a robust antibody response through a MyD88-independent mechanism that is probably aided by helper CD4+ T cells (57). Indeed, although TLR2−/− mice had delayed virus clearance, they were able to control infection, most likely due to the humoral antibody response. These data may recapitulate what occurs in humans during Junín virus infection, where the humoral response is most important for virus clearance. Since the in vivo studies reported here were all done with attenuated JUNV C1, further experiments are required to determine the role of TLR2 in host innate and adaptive immune responses to pathogenic Junín virus in mice.
Because of the neuropathology associated with Junín virus infection, we also investigated whether TLR2-mediated cytokine signaling had an effect on the brain. We showed that JUNV C1 replicates in the brain and that it infects astroglia and microglia (Fig. 6A and C). Interestingly, although JUNV C1 induced higher cytokine levels in WT mice, unlike the periphery, infection in the brain was not reduced compared to that in TLR2−/− mice. The higher cytokine levels seen in WT mice could play a role in neuropathogenesis; studies to determine this are currently in progress in our laboratory. Although both pathogenic Parodi and JUNV C1 GPCs induced TLR2 responses in human and mouse macrophages similar to those induced by JUNV C1, we cannot rule out that JUNV C1, which was attenuated from a pathogenic strain of Junín virus by extensive passage in mouse brains (22), is an adapted virus that induces TLR2-mediated signaling in this tissue; studies to address this are ongoing.
In summary, we showed that macrophages sense JUNV C1 through TLR2/6 heterodimers and that this initiates signaling cascades leading to increased transcription levels of IFN-β and TNF-α. Secretion of these cytokines likely results in autocrine/paracrine responses, leading to the production of RNA helicases in infected as well as neighboring cells. In addition, IFN-β and TNF-α likely play a role in activating Junín virus-specific CD8+ T cells that have come into contact with viral antigens on APCs and help reduce virus loads. We are currently working on understanding the different pathways involved in JUNV C1 infection and what role the inflammatory response against the virus plays in central nervous system infection. Such studies should provide insights into the different pathologies caused by systemic versus neurologic Junín virus infection.
ACKNOWLEDGMENTS
We thank Kristin Blouch for help with the mouse studies and John Wherry for pointing out the sequence similarities in the NPs of LCMV and Junín virus. The following reagents were obtained through BEI Resources: NA05-AG12, SA02-BG12, NR-9456, NR-9457, NR-9460, and NR-9904.
This research was supported by MARCE grant U54 AI 057168. C.D.C. was supported by PHS grant T32-AI-055400.
Footnotes
Published ahead of print 23 April 2014
REFERENCES
- 1.Buchmeier MJ, de la Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, p 1791–1828 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Charrel RN, Coutard B, Baronti C, Canard B, Nougairede A, Frangeul A, Morin B, Jamal S, Schmidt CL, Hilgenfeld R, Klempa B, de Lamballerie X. 2011. Arenaviruses and hantaviruses: from epidemiology and genomics to antivirals. Antiviral Res. 90:102–114. 10.1016/j.antiviral.2011.02.009 [DOI] [PubMed] [Google Scholar]
- 3.Heller MV, Saavedra MC, Falcoff R, Maiztegui JI, Molinas FC. 1992. Increased tumor necrosis factor-alpha levels in Argentine hemorrhagic fever. J. Infect. Dis. 166:1203–1204. 10.1093/infdis/166.5.1203 [DOI] [PubMed] [Google Scholar]
- 4.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. 2012. Junin virus pathogenesis and virus replication. Viruses 4:2317–2339. 10.3390/v4102317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molinas FC, de Bracco MM, Maiztegui JI. 1981. Coagulation studies in Argentine hemorrhagic fever. J. Infect. Dis. 143:1–6. 10.1093/infdis/143.1.1 [DOI] [PubMed] [Google Scholar]
- 6.Enria DA, Briggiler AM, Fernandez NJ, Levis SC, Maiztegui JI. 1984. Importance of dose of neutralising antibodies in treatment of Argentine haemorrhagic fever with immune plasma. Lancet ii:255–256 [DOI] [PubMed] [Google Scholar]
- 7.Vallejos DA, Ambrosio AM, Feuillade MR, Maiztegui JI. 1989. Lymphocyte subsets alteration in patients with argentine hemorrhagic fever. J. Med. Virol. 27:160–163. 10.1002/jmv.1890270218 [DOI] [PubMed] [Google Scholar]
- 8.Goni SE, Iserte JA, Ambrosio AM, Romanowski V, Ghiringhelli PD, Lozano ME. 2006. Genomic features of attenuated Junin virus vaccine strain candidate. Virus Genes 32:37–41. 10.1007/s11262-005-5843-2 [DOI] [PubMed] [Google Scholar]
- 9.Enria DA, Briggiler AM, Levis S, Vallejos D, Maiztegui JI, Canonico PG. 1987. Tolerance and antiviral effect of ribavirin in patients with Argentine hemorrhagic fever. Antiviral Res. 7:353–359. 10.1016/0166-3542(87)90017-9 [DOI] [PubMed] [Google Scholar]
- 10.Ambrosio AM, Enria DA, Maiztegui JI. 1986. Junin virus isolation from lympho-mononuclear cells of patients with Argentine hemorrhagic fever. Intervirology 25:97–102. 10.1159/000149662 [DOI] [PubMed] [Google Scholar]
- 11.Ambrosio M, Vallejos A, Saavedra C, Maiztegui JI. 1990. Junin virus replication in peripheral blood mononuclear cells of patients with Argentine haemorrhagic fever. Acta Virol. 34:58–63 [PubMed] [Google Scholar]
- 12.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Poussard AL, Walker AG, Brasier AR, Zhao Y, Tian B, de la Torre JC, Paessler S. 2012. Junin virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl. Trop. Dis. 6:e1659. 10.1371/journal.pntd.0001659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levis SC, Saavedra MC, Ceccoli C, Falcoff E, Feuillade MR, Enria DA, Maiztegui JI, Falcoff R. 1984. Endogenous interferon in Argentine hemorrhagic fever. J. Infect. Dis. 149:428–433. 10.1093/infdis/149.3.428 [DOI] [PubMed] [Google Scholar]
- 14.Levis SC, Saavedra MC, Ceccoli C, Feuillade MR, Enria DA, Maiztegui JI, Falcoff R. 1985. Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J. Interferon Res. 5:383–389. 10.1089/jir.1985.5.383 [DOI] [PubMed] [Google Scholar]
- 15.Sen GC. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255–281. 10.1146/annurev.micro.55.1.255 [DOI] [PubMed] [Google Scholar]
- 16.Marta RF, Montero VS, Hack CE, Sturk A, Maiztegui JI, Molinas FC. 1999. Proinflammatory cytokines and elastase-alpha-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 60:85–89 [DOI] [PubMed] [Google Scholar]
- 17.Cuevas CD, Lavanya M, Wang E, Ross SR. 2011. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 85:11058–11068. 10.1128/JVI.05304-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda K, Akira S. 2004. TLR signaling pathways. Semin. Immunol. 16:3–9. 10.1016/j.smim.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 19.Ren MY, Wu XY. 2011. Toll-like receptor 4 signalling pathway activation in a rat model of Acanthamoeba keratitis. Parasite Immunol. 33:25–33. 10.1111/j.1365-3024.2010.01247.x [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. 2001. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13:933–940. 10.1093/intimm/13.7.933 [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169:10–14. 10.4049/jimmunol.169.1.10 [DOI] [PubMed] [Google Scholar]
- 22.Goni SE, Iserte JA, Stephan BI, Borio CS, Ghiringhelli PD, Lozano ME. 2010. Molecular analysis of the virulence attenuation process in Junin virus vaccine genealogy. Virus Genes 40:320–328. 10.1007/s11262-010-0450-2 [DOI] [PubMed] [Google Scholar]
- 23.Kolokoltsova OA, Yun NE, Poussard AL, Smith JK, Smith JN, Salazar M, Walker A, Tseng CT, Aronson JF, Paessler S. 2010. Mice lacking alpha/beta and gamma interferon receptors are susceptible to Junin virus infection. J. Virol. 84:13063–13067. 10.1128/JVI.01389-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Most RG, Murali-Krishna K, Whitton JL, Oseroff C, Alexander J, Southwood S, Sidney J, Chesnut RW, Sette A, Ahmed R. 1998. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology 240:158–167. 10.1006/viro.1997.8934 [DOI] [PubMed] [Google Scholar]
- 25.Lascano EF, Lerman GD, Blejer JL, Caccuri RL, Berria MI. 1992. Immunoperoxidase tracing of Junin virus neural route after footpad inoculation. Arch. Virol. 122:13–22. 10.1007/BF01321114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiztegui JI. 1975. Clinical and epidemiological patterns of Argentine haemorrhagic fever. Bull. World Health Organ. 52:567–575 [PMC free article] [PubMed] [Google Scholar]
- 27.Albarino CG, Ghiringhelli PD, Posik DM, Lozano ME, Ambrosio AM, Sanchez A, Romanowski V. 1997. Molecular characterization of attenuated Junin virus strains. J. Gen. Virol. 78(Part 7):1605–1610 [DOI] [PubMed] [Google Scholar]
- 28.Lascano EF, Berria MI. 1974. Ultrastructure of Junin virus in mouse whole brain and mouse brain tissue cultures. J. Virol. 14:965–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pozner RG, Collado S, Jaquenod de Giusti C, Ure AE, Biedma ME, Romanowski V, Schattner M, Gomez RM. 2008. Astrocyte response to Junin virus infection. Neurosci. Lett. 445:31–35. 10.1016/j.neulet.2008.08.059 [DOI] [PubMed] [Google Scholar]
- 30.Jaquenod De Giusti C, Alberdi L, Frik J, Ferrer MF, Scharrig E, Schattner M, Gomez RM. 2011. Galectin-3 is upregulated in activated glia during Junin virus-induced murine encephalitis. Neurosci. Lett. 501:163–166. 10.1016/j.neulet.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 31.Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, Kurt-Jones EA, Finberg RW. 2009. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 83:1492–1500. 10.1128/JVI.00671-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang S, Dolganiuc A, Szabo G. 2007. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 82:479–487. 10.1189/jlb.0207128 [DOI] [PubMed] [Google Scholar]
- 33.Zhou S, Cerny AM, Bowen G, Chan M, Knipe DM, Kurt-Jones EA, Finberg RW. 2010. Discovery of a novel TLR2 signaling inhibitor with anti-viral activity. Antiviral Res. 87:295–306. 10.1016/j.antiviral.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berghall H, Siren J, Sarkar D, Julkunen I, Fisher PB, Vainionpaa R, Matikainen S. 2006. The interferon-inducible RNA helicase, mda-5, is involved in measles virus-induced expression of antiviral cytokines. Microbes Infect. 8:2138–2144. 10.1016/j.micinf.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 35.Eisenacher K, Krug A. 2012. Regulation of RLR-mediated innate immune signaling—it is all about keeping the balance. Eur. J. Cell Biol. 91:36–47. 10.1016/j.ejcb.2011.01.011 [DOI] [PubMed] [Google Scholar]
- 36.Hui KP, Lee SM, Cheung CY, Mao H, Lai AK, Chan RW, Chan MC, Tu W, Guan Y, Lau YL, Peiris JS. 2011. H5N1 influenza virus-induced mediators upregulate RIG-I in uninfected cells by paracrine effects contributing to amplified cytokine cascades. J. Infect. Dis. 204:1866–1878. 10.1093/infdis/jir665 [DOI] [PubMed] [Google Scholar]
- 37.Nasirudeen AM, Wong HH, Thien P, Xu S, Lam KP, Liu DX. 2011. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 5:e926. 10.1371/journal.pntd.0000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan L, Briese T, Lipkin WI. 2010. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 84:1785–1791. 10.1128/JVI.01362-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martinez-Sobrido L, de la Torre JC, Kunz S. 2012. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKepsilon. J. Virol. 86:7728–7738. 10.1128/JVI.00187-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Sobrido L, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC. 2007. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 81:12696–12703. 10.1128/JVI.00882-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groseth A, Hoenen T, Weber M, Wolff S, Herwig A, Kaufmann A, Becker S. 2011. Tacaribe virus but not Junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl. Trop. Dis. 5:e1137. 10.1371/journal.pntd.0001137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephan BI, Lozano ME, Goni SE. 2013. Watching every step of the way: Junin virus attenuation markers in the vaccine lineage. Curr. Genomics 14:415–424. 10.2174/138920291407131220153526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchmeier MJ, Welsh RM, Dutko FJ, Oldstone MB. 1980. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv. Immunol. 30:275–331. 10.1016/S0065-2776(08)60197-2 [DOI] [PubMed] [Google Scholar]
- 44.Butz EA, Bevan MJ. 1998. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8:167–175. 10.1016/S1074-7613(00)80469-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. 1998. Counting antigen-specific CD8+ T cells: a reevaluation of bystander activation during viral infection. Immunity 8:177–187. 10.1016/S1074-7613(00)80470-7 [DOI] [PubMed] [Google Scholar]
- 46.Blattman JN, Sourdive DJ, Murali-Krishna K, Ahmed R, Altman JD. 2000. Evolution of the T cell repertoire during primary, memory, and recall responses to viral infection. J. Immunol. 165:6081–6090. 10.4049/jimmunol.165.11.6081 [DOI] [PubMed] [Google Scholar]
- 47.Sandberg K, Eloranta ML, Campbell IL. 1994. Expression of alpha/beta interferons (IFN-alpha/beta) and their relationship to IFN-alpha/beta-induced genes in lymphocytic choriomeningitis. J. Virol. 68:7358–7366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. 2004. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 10:1374–1378. 10.1038/nm1133 [DOI] [PubMed] [Google Scholar]
- 49.Hinson ER, Joshi NS, Chen JH, Rahner C, Jung YW, Wang X, Kaech SM, Cresswell P. 2010. Viperin is highly induced in neutrophils and macrophages during acute and chronic lymphocytic choriomeningitis virus infection. J. Immunol. 184:5723–5731. 10.4049/jimmunol.0903752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montoya M, Edwards MJ, Reid DM, Borrow P. 2005. Rapid activation of spleen dendritic cell subsets following lymphocytic choriomeningitis virus infection of mice: analysis of the involvement of type 1 IFN. J. Immunol. 174:1851–1861. 10.4049/jimmunol.174.4.1851 [DOI] [PubMed] [Google Scholar]
- 51.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4:1009–1015. 10.1038/ni978 [DOI] [PubMed] [Google Scholar]
- 52.Zhou S, Halle A, Kurt-Jones EA, Cerny AM, Porpiglia E, Rogers M, Golenbock DT, Finberg RW. 2008. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 194:70–82. 10.1016/j.jneuroim.2007.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banchereau J, Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392:245–252. 10.1038/32588 [DOI] [PubMed] [Google Scholar]
- 54.Pasare C, Medzhitov R. 2005. Control of B-cell responses by Toll-like receptors. Nature 438:364–368. 10.1038/nature04267 [DOI] [PubMed] [Google Scholar]
- 55.El Shikh ME, El Sayed RM, Szakal AK, Tew JG. 2009. T-independent antibody responses to T-dependent antigens: a novel follicular dendritic cell-dependent activity. J. Immunol. 182:3482–3491. 10.4049/jimmunol.0802317 [DOI] [PubMed] [Google Scholar]
- 56.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. 2006. Adjuvant-enhanced antibody responses in the absence of Toll-like receptor signaling. Science 314:1936–1938. 10.1126/science.1135299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jellison ER, Guay HM, Szomolanyi-Tsuda E, Welsh RM. 2007. Dynamics and magnitude of virus-induced polyclonal B cell activation mediated by BCR-independent presentation of viral antigen. Eur. J. Immunol. 37:119–128. 10.1002/eji.200636516 [DOI] [PubMed] [Google Scholar]