

ABSTRACT
Following mucosal human immunodeficiency virus type 1 transmission, systemic infection is established by one or only a few viral variants. Modeling single-variant, mucosal transmission in nonhuman primates using limiting-dose inoculations with a diverse simian immunodeficiency virus isolate stock may increase variability between animals since individual variants within the stock may have substantial functional differences. To decrease variability between animals while retaining the ability to enumerate transmitted/founder variants by sequence analysis, we modified the SIVmac239 clone to generate 10 unique clones that differ by two or three synonymous mutations (molecular tags). Transfection- and infection-derived virus stocks containing all 10 variants showed limited phenotypic differences in 9 of the 10 clones. Twenty-nine rhesus macaques were challenged intrarectally or intravenously with either a single dose or repeated, limiting doses of either stock. The proportion of each variant within each inoculum and in plasma from infected animals was determined by using a novel real-time single-genome amplification assay. Each animal was infected with one to five variants, the number correlating with the dose. Longitudinal sequence analysis revealed that the molecular tags are highly stable with no reversion to the parental sequence detected in >2 years of follow-up. Overall, the viral stocks are functional and mucosally transmissible and the number of variants is conveniently discernible by sequence analysis of a small amplicon. This approach should be useful for tracking individual infection events in preclinical vaccine evaluations, long-term viral reservoir establishment/clearance research, and transmission/early-event studies.
IMPORTANCE Human immunodeficiency virus type 1 transmission is established by one or only a few viral variants. Modeling of limited variant transmission in nonhuman primates with a diverse simian immunodeficiency virus isolate stock may increase the variability between animals because of functional differences in the individual variants within the stock. To decrease such variability while retaining the ability to distinguish and enumerate transmitted/founder variants by sequence analysis, we generated a viral stock with 10 sequence-identifiable but otherwise genetically identical variants. This virus was characterized in vitro and in vivo and shown to allow discrimination of distinct transmission events. This approach provides a novel nonhuman primate challenge system for the study of viral transmission, evaluation of vaccines and other prevention approaches, and characterization of viral reservoirs and strategies to target them.
INTRODUCTION
Historically, nonhuman primate (NHP) models of human immunodeficiency virus (HIV) transmission have used large-dose intravenous (i.v.) and large-dose mucosal (intrarectal [i.r.] or intravaginal) simian immunodeficiency virus (SIV) challenges that lead to nearly 100% infection rates with rapid and relatively consistent viral load profiles (reviewed in references 1 and 2). However, in recent years, we and others have used single-genome amplification (SGA) technology to provide molecular evidence that in the vast majority of HIV-infected patients, systemic infection stems from a single transmitted viral variant (3–9). In light of this finding and the fact that human-to-human HIV transmission most often occurs via unprotected sexual exposure, investigators have sought to more authentically model mucosal HIV-1 transmission through the use of limited-dose SIV infections with virus stock titrations and by using SGA methods to precisely determine the number of variants initiating systemic infection within each challenged animal to achieve conditions that consistently yield transmission of only one or only a few variants (10–13). Importantly, challenge stocks of SIV isolates generally contain sufficient maximum Env diversity (ranging from 1 to 3%) to distinguish even closely related variants by SGA sequence analyses, thereby allowing the enumeration of T/F variants (10, 14). The evidence that infection of rhesus macaques can be initiated with a single SIV variant was established initially with an i.r. challenge model where infection rates were limited to ∼10% per exposure (10). Since that initial study, researchers have performed mucosal infections via the i.r., intravaginal, and penile routes of exposure with both repeated, specific-titer challenges and single, limited-dose challenges, providing models that more closely recapitulate HIV infection in terms of numbers of T/F variants (10–13). The ability to initiate SIV infection in rhesus macaques with one or only a few variants and to confirm this by SGA provides an invaluable tool for preclinical vaccine trials and basic research studies, recapitulating a key feature of clinically relevant HIV transmission.
While SGA is a powerful tool for precisely determining the nucleotide sequences contained within a viral population and definitively enumerating the number of variants establishing systemic infection, it is time-consuming and costly. Currently, the use of SGA to enumerate T/F variants requires PCR amplification of large portions of the viral genome (e.g., Pol or Env) to have sufficient genetic resolution to distinguish one variant from another. These large amplicons, typically, 2.5 to 3.5 kb in size, require approximately 10 Sanger sequencing reactions for complete coverage of each amplicon. Power calculations using Env sequences reveal that more than 30 individual sequences are required for 95% confidence that a virus representing at least 10% of the total population would be detectable (6). For both HIV and SIV, the number of sequences one can examine by SGA is limited by the time and resources required for such an in-depth sequence analysis. Previously, we have performed next-generation sequencing to identify transmitted but minor variants, but this approach requires ample viral template amounts and substantial computational analyses (15).
Although procedural refinements of experimental design and the use of new technologies have allowed investigators to more closely model the inefficient nature of mucosal HIV transmission, the viral stocks and stock production methods used for these experiments have changed little over the years since SIVmac was first identified as a cross-species transmission from a naturally infected African sooty mangabey to the Asian rhesus macaque (16). The major viral lineages currently used in pathogenic SIV infection models include the molecular clone SIVmac239 and the closely related uncloned isolate, SIVmac251, as well as a divergent viral lineage represented by the SIVsmE543-3 molecular clone and the SIVsmE660 isolate. These and a few other viruses constitute the current challenge stocks used to model the global diversity of HIV-1 for both homologous and heterologous NHP vaccine study challenges, representing both clonal inocula and diverse isolates. Although virus stock diversity, like that present in an uncloned viral isolate after multiple rounds of viral replication, is critical for distinguishing and enumerating T/F variants, this type of stock diversity can also diminish the degree of experimental control and reproducibility for in vivo infection studies. In addition to generating genetically distinct viral variants, in vitro expansion of viral isolates can also generate a virus population with remarkable phenotypic diversity. Recently, the phenotypes of various Env clones within the SIVsmE660 isolate were examined in detail and profound differences between variants within the same population were identified in both infectivity and neutralization sensitivity (17). Furthermore, expansion of the same nominal isolate in different laboratories under various conditions results in virus stocks with the same name but substantially different viral genome constituents (14). While most of these differences are likely functionally irrelevant in a large-dose challenge model, where the most fit variants of any lineage would have the greatest opportunity for success, with limiting-dose inocula, these differences within and between different isolates may be magnified as each animal becomes infected with a genetically and potentially phenotypically different virus. These disparities may require excessively large group sizes to allow statistically meaningful differences to be detected, increasing the resource intensiveness for preclinical vaccine trials, and may complicate comparisons between studies where different specific virus stock expansions were used. Moreover, unlike HIV transmission that originates from a population of virus under constant immune and replicative selection pressure in vivo, SIV stocks expanded in vitro are influenced by other selective pressures of uncertain clinical relevance.
To improve experimental reproducibility and control while also limiting the costs and time associated with the enumeration of viral variants and tracking of individual viral lineages, we have developed a synthetic swarm of molecularly tagged viruses within an otherwise isogenic background, providing the benefits of both a clonal population and sequence-discriminable variants to allow the enumeration of distinct transmission events. By modifying two or three bases in the conserved integrase gene of SIVmac239 without altering the amino acid sequence, we have generated a population of viruses easily distinguishable by the sequencing of a short genome fragment that demonstrates minimal functional heterogeneity. Here, we describe the development of this synthetic swarm, its in vitro characteristics, the generation of infectious stocks by both transfection and infection, and their in vivo infectivity in rhesus macaques. Sequence-based discrimination of independent infection events with biologically identical viruses revealed disparate proportions of each variant at or near peak viremia, suggesting some number of nonviral determinants either augment or suppress successful replication. This approach to the generation of a synthetic swarm is generalizable to many different viruses and models and should be especially useful for (i) preclinical vaccine development, where consistency between groups is essential for successful evaluation of vaccine modalities; (ii) the establishment of long-term viral reservoirs, where tracking of unique infection events is essential; and (iii) studies of viral transmission and systemic dissemination spread by expanding the range of questions that can be effectively addressed in NHP models.
MATERIALS AND METHODS
Molecular tag cloning.
Two or three synonymous base changes were introduced into the integrase gene of the Nef-open, infectious molecular clone SIVmac239 (p239SpXFL) (18–20) with the QuikChange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Changes were introduced at positions with known polymorphisms in published SIV sequences to avoid introducing changes that might subtly alter viral fitness. Nine unique clones, designated SIVmac239A through SIVmac239I, were generated and sequenced throughout the viral genome to confirm the changes introduced and that the sequences were identical to that of wild-type (WT) SIVmac239 at all other positions throughout the genome. The specific mutations in the clones are shown in Fig. 1.
FIG 1.

Comparisons of the molecular tags (A), infectivity (B), and replication kinetics (C) of each molecularly tagged SIVmac239 variant with those of WT SIVmac239. (A) Alignment of the nucleotide sequences of SIVmac239 (WT) and nine molecularly tagged SIVmac239 clones (designated variants A through I) for a portion of the integrase gene indicated by the genome schematic at the bottom. Each new variant is identical to the parental virus over the entire genome, except for the two or three synonymous mutations indicated here. Dashes represent identical nucleotides, and substitutions are indicated. All clones encode the identical amino acid sequence over the entire genome. LTR, long terminal repeat. (B) Transfection-derived virus from each variant was tested for infectivity titer in a single-cycle TZM-bl reporter cell assay expressed as dilution-normalized IU per milliliter. (C) Titers represent mean values for all dilution-normalized titers that fell within the linear range of the dilution series. Viral replication (one of three representative experiments shown) was determined in primary rhesus PBMCs with virus from each variant normalized for infectivity. Viral production was determined by p27 enzyme-linked immunosorbent assay on days 3, 6, 10, and 14 postinfection.
Virus stocks.
Individual infectious stocks of SIVmac239A through SIVmac239I and WT SIVmac239 were generated by transfecting 293T cells with the corresponding full-length viral molecular clone plasmid for 24 h with the TransIT HEK293 transfection reagent (Mirus Bio) according to the manufacturer's instructions. Culture medium was changed at 24 h posttransfection and again at 48 h posttransfection. At 72 h posttransfection, virus-containing supernatant was clarified by centrifugation, sterile filtered through a 0.45-μm filter, aliquoted, and stored at −80°C. To generate a pooled stock, termed SIVmac239X, containing all nine molecularly tagged variants (SIVmac239A through SIVmac239F) plus WT SIVmac239, 293T cells were transfected as described above with a pool containing each of the molecularly tagged full-length viral clone plasmids. A series of small-scale cotransfections and subsequent sequencing analyses to determine the relative proportion of each tagged variant in the virus pool were used to identify the relative input proportion of each plasmid in the DNA cotransfection pool that would yield roughly equal proportions of tagged viruses in the SIVmac239X stock. To generate an infection-derived SIVmac239X stock, peripheral blood mononuclear cells (PBMCs) from 11 specific-pathogen-free, Indian-origin rhesus macaques were pooled and infected with transfection-derived SIVmac239X 2 days after stimulation with 5 μg/ml phytohemagglutinin (PHA). The infected culture was maintained in 100 U/ml interleukin-2 (IL-2) with a complete medium change 24 h prior to harvest. On day 8 postinfection, virus-containing supernatants were clarified by centrifugation, sterile filtered through a 0.45-μm filter, aliquoted, and stored in liquid nitrogen.
In vitro infectious titer.
Virus titers were determined with TZM-bl reporter cells (reference no. 8129; NIH AIDS Research and Reference Reagent Program), which contain a Tat-inducible luciferase and a β-galactosidase gene expression cassette. Infectious titers were measured by counting the individual β-galactosidase-expressing cells per well in cultures infected with serial 3-fold dilutions of virus as previously described (21). Wells containing dilution-corrected blue cell counts within the linear range of the virus dilution series were averaged to generate an infectious titer in infectious units (IU) per milliliter.
In vitro virus replication.
To evaluate the replication of each of the molecularly tagged SIVmac239 variants and WT SIVmac239, pooled, CD8-depleted PBMCs from three rhesus macaque (Macaca mulatta) donors cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (RPMI-Complete) were stimulated for 3 days with 5.0 μg/ml PHA and IL-2 (100 U/ml) and then infected with each virus in independent cultures at a nominal multiplicity of infection of 0.01. Following overnight incubation at 37°C, cells were washed twice with phosphate-buffered saline and once with RPMI-Complete to remove excess virus. Viral replication was monitored by measuring the viral p27 protein content in culture supernatants with a commercial SIV p27 antigen capture assay according to the manufacturer's recommendations (ABL).
Animals and in vivo virus challenges.
Twenty-nine purpose-bred Indian-origin rhesus macaques (Macaca mulatta) were housed and cared for in accordance with American Association for Accreditation of Laboratory Animal Care (AAALAC) guidelines in an AAALAC-accredited facility, and all animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of the National Cancer Institute or by the Oregon National Primate Research Center Institutional Animal Care and Use Committee under the standards of the NIH Guide for the Care and Use of Laboratory Animals. All animals were free of cercopithecine herpesvirus 1, D-type simian retrovirus, simian T-lymphotrophic virus type 1, and SIV at study initiation. Animals were genotyped for common major histocompatibility complex (MHC) class 1 alleles such as Mamu-A*01, -A*02, -B*08, and -B*17 by sequence-specific priming PCR performed as previously described (22). For all mucosal infections, animals were placed at an ∼20° down angle in an inverted Trendelenburg position (e.g., the animal's pelvis was elevated above its head with its sternum against the table) and the challenge was performed with a 1-ml slip tip syringe (BD Biosciences) with a small amount of nonbacteriostatic, single-use, sterile lubricant. Four macaques were challenged i.r. with 3 × 104 IU of transfection-derived SIVmac239X, and six macaques, two of which had been previously challenged but not infected with 3 × 104 IU, were challenged i.r. with 3 × 105 IU of virus. With infection-derived SIVmac239X, animals were challenged either i.r. or i.v. For i.r. challenges, 12 animals were each serially challenged up to 12 times at biweekly intervals with 100 IU of virus until they became detectably viremic; animals that remained uninfected after 12 challenges were then serially challenged up to three times at biweekly intervals with 300 IU of virus. For i.v. challenges with infection-derived SIVmac239X, three animals were challenged once with 5 IU, three animals were challenged once with 1 IU, and a final three animals were each serially challenged up to three times with 0.2 IU until they became detectably viremic.
Viral loads.
Quantitative plasma SIV RNA loads were determined as previously described (23). Briefly, viral RNA (vRNA) was extracted from blood plasma by pelleting, followed by guanidine HCl-proteinase K lysis and isopropanol precipitation. Following cDNA synthesis, quantitative real-time PCR within the gag gene was performed with an overall assay sensitivity of 30 copies/ml.
SGA.
A 300-bp portion of the integrase gene surrounding the mutated site was amplified and sequenced by a limiting-dilution PCR where only a single genome is amplified (SGA) per reaction (6, 24, 25). VRNA was extracted with the QIAamp VRNA minikit (Qiagen) and then reverse transcribed into cDNA with SuperScript III reverse transcriptase according to manufacturer's recommendations (Invitrogen). In brief, a cDNA reaction mixture of 1× reverse transcription (RT) buffer, 0.5 mM each deoxynucleoside triphosphate, 5 mM dithiothreitol, 2 U/ml RNaseOUT (RNase inhibitor), 10 U/ml Superscript III reverse transcriptase, and 0.25 mM antisense primer SIVmacIntR1 (5′-AAG CAA GGG AAA TAA GTG CTA TGC AGT AA-3′) was incubated at 50°C for 60 min and at 55°C for 60 min, heat inactivated at 70°C for 15 min, and then treated with 1 U of RNase H at 37°C for 20 min. PCR was then performed with a reaction mixture of 1× PCR buffer, 2 mM MgCl2, 0.2 mM each deoxynucleoside triphosphate, 0.2 μM each primer, and 0.025 U/μl Platinum Taq polymerase (Invitrogen) in a 10-μl volume. Real-time PCR was performed with sense primer SIVmacIntF1 (5′-GAA GGG GAG GAA TAG GGG ATA TG-3′) and antisense primer SIVmacIntR3 ( 5′-CAC CTC TCT AGC CTC TCC GGT ATC C-3′) under the following conditions: 1 cycle of 94°C for 2 min and 40 cycles of 94°C for 15 s, 55°C for 30 s, 60°C for 1.5 min, and 72°C for 30 s. Template-positive reactions were determined by real-time PCR with SIVIntP 5′-TCC CTA CCT TTA AGA TGA CTG CTC CTT CCC CT-3′ with 6-carboxyfluorescein and ZEN/Iowa Black Hole Quencher (Integrated DNA Technologies) as a gene-specific probe and directly sequenced with SIVmacIntR3 by BigDye Terminator technology (Life Technologies). To confirm PCR amplification from a single template, chromatograms were manually examined for multiple peaks indicative of the presence of amplicons resulting from PCR-generated recombination events, Taq polymerase errors, or multiple variant templates. Minor variants found in any animal were confirmed by repeating the SGA analysis with a different aliquot of the same sample or a separate sample from a different time point or by reisolating vRNA from the original sample and repeating the SGA.
Statistical analyses.
Data in this study were subjected to standard contingency table analysis, log-linear modeling techniques, and related methods (26). Expansion/transfection by variant (clone) frequency tables was subjected to chi-square partitioning and decomposition methods; various hypotheses were tested, such that inferences regarding overall tables and relevant subtables were statistically independent. In cases where nonindependent tests were performed (e.g., percentage representation of particular variants to a specified proportion), probabilities were adjusted by the Benjamini-Hochberg strategy to control for compounding type I error rates.
RESULTS
Generation of molecularly tagged clones.
To generate multiple infectious molecular clones that are genetically distinguishable but have limited phenotypic differences, we introduced a series of synonymous mutations within the full-length SIVmac239 molecular clone. We identified a region of the integrase gene that was highly conserved at the amino acid level but was modestly polymorphic at the nucleotide level, including 11 sites within a 45-nucleotide stretch where synonymous mutations were found naturally in HIV-2 and in multiple SIV lineages, suggesting positions that may not adversely affect RNA structure or viral fitness. Nine unique SIVmac239 clones (identified as SIVmac239A through SIVmac239I) were generated by introducing two or three of these naturally occurring synonymous mutations within this region into WT SIVmac239 (Fig. 1A). Each tagged clone had at least one point mutation in common with other clones to control for potential fitness reductions associated with any one particular mutation. The entire genome of each clone was sequenced to confirm that only these mutations were introduced into the full-length clones.
In vitro characterization of transfection-derived SIVmac239X.
To evaluate the infectivity and replication capacity of the nine molecularly tagged clones relative to WT SIVmac239, infectious virus stocks of each individual clone were generated by transfection. With a single-cycle infectivity assay with TZM-bl reporter cells as targets, there was a <2-fold difference in infectivity between variants (range, 4.7 × 105 to 7.3 × 105 IU/ml), with mean titers of 5.4 × 105 IU/ml for the nine molecularly tagged clones and 4.9 × 105 IU/ml for WT SIVmac239 (Fig. 1B). Each variant was then compared to WT SIVmac239 in a virus growth curve assay with primary rhesus macaque PBMCs with input virus amounts normalized on the basis of these infectivity titers (Fig. 1C). Most of the variants replicated with kinetics nearly identical to those of WT SIVmac239; however, despite our attempts to limit phenotypic differences by introducing only synonymous mutations, variants D and I replicated slightly less efficiently than the WT and the other clones, with ∼3- to 4-fold lower measured p27 levels at days 6 and 10.
To generate an infectious synthetic swarm virus stock containing WT SIVmac239 and the nine molecularly tagged variants, we devised a cotransfection strategy that allowed the generation of a single virus stock with equal representation of each of the 10 viral variants. We opted for this strategy over proportional mixing of individual viral variant stocks at the time of infection to avoid inaccuracies in preparing each mixture prior to each challenge, which would diminish overall reproducibility, and to avoid virus stock waste. Small-scale transfections of 293T cells were used to iteratively determine the relative proportions of each plasmid that would yield a virus stock with equal representation of the 10 variants, as determined by sequence analysis of the vRNA from the transfection supernatant. We also determined the proportion of each variant in cellular DNA 24 h after infection of the SupT1-R5 human T lymphoblastoid line to confirm equivalent infectivity of each variant within the context of the synthetic swarm (data not shown). Once the optimal plasmid DNA ratio was determined, a large-scale transfection was performed with a total of 1 × 109 293T cells, yielding 2.5 liters of virus-containing supernatant, which was clarified and frozen in 0.5- and 1-ml aliquots. This stock contained 1 × 109 vRNA copies/ml with an infectious virus titer of 5 × 105 IU/ml on TZM-bl cells and a calculated specific infectivity of 1 × 10−3 IU/virion. We confirmed the relative proportion of each variant within this stock by extracting vRNA and performing SGA and direct sequencing of the region containing the molecular tags. We obtained 711 single-genome amplified sequences from the cotransfection-derived virus stock and found no statistically significant difference between each variant (range, 9 to 12%) and the expected 10% proportion (Fig. 2). Because this viral stock contained 10 unique variants of SIVmac239, we designated this pooled stock SIVmac239X.
FIG 2.

Stacked-bar graph representing the proportion of each variant following transfection (TNF) or from four independent infections (INF) produced virus harvested on day (d) 7, 8, or 9.
In vivo characterization of transfection-derived SIVmac239X.
We next sought to test the in vivo infectivity of this pooled, transfection-derived, synthetic-swarm SIVmac239X stock. Four Indian-origin rhesus macaques were challenged i.r. with 3 × 104 IU of SIVmac239X. However, only a single animal (4543) became viremic at this challenge dose, with viral loads greater than 107 RNA copies/ml at 14 days postinfection, while the remaining three animals showed no signs of infection (Fig. 3A). SGA analysis of day 9 and 14 plasma virus levels in animal 4543 indicated productive infection with two unique viral variants (B and F) detected at both time points (399 total sequences) (Table 1). We next evaluated infection rates with a 10-fold increase in the viral inoculum. Two animals previously challenged with 3 × 104 IU (4317 and 4324) and four naive animals were i.r. challenged with 3 × 105 IU of SIVmac239X. Following this challenge, all six animals were productively infected and the number of transmitted/founder (T/F) viruses was determined by SGA for each animal (Fig. 3B). We found the median number of T/F variants to be 2 with a range of 1 to 4 variants and a mean of 2.2 variants (Table 1), suggesting that this viral inoculum readily mediates mucosal infection and can be used in a manner that yields limited numbers of T/F variants, consistent with typical mucosal HIV-1 transmission (3, 5, 6, 8). The infection rate at 3 × 104 IU was 25%, and the mean number of T/F variants per challenge was 0.5, representing a useful repeated, specific-titer challenge model. At the 3 × 105-IU dose, the infection rate was 100% and the mean number of variants per challenge increased to just over two, representing a useful single, specific-titer challenge that infects all or nearly all naive animals with limited T/F variants detected systemically.
FIG 3.

Plasma viral load profiles of animals infected i.r. with transfection-derived SIVmac239X. (A) Animals challenged i.r. with 3 × 104 IU. (B) Animals challenged i.r. with 3 × 105 IU. Note that animals 4317 and 4324 were rechallenged with the larger dose (B) after showing no evidence of productive infection at the lower dose (A). The number of T/F variants was determined by sequence analysis of blood plasma and is displayed parenthetically for each animal.
TABLE 1.
Number of plasma sequences for each variant and percentage of each variant per sample
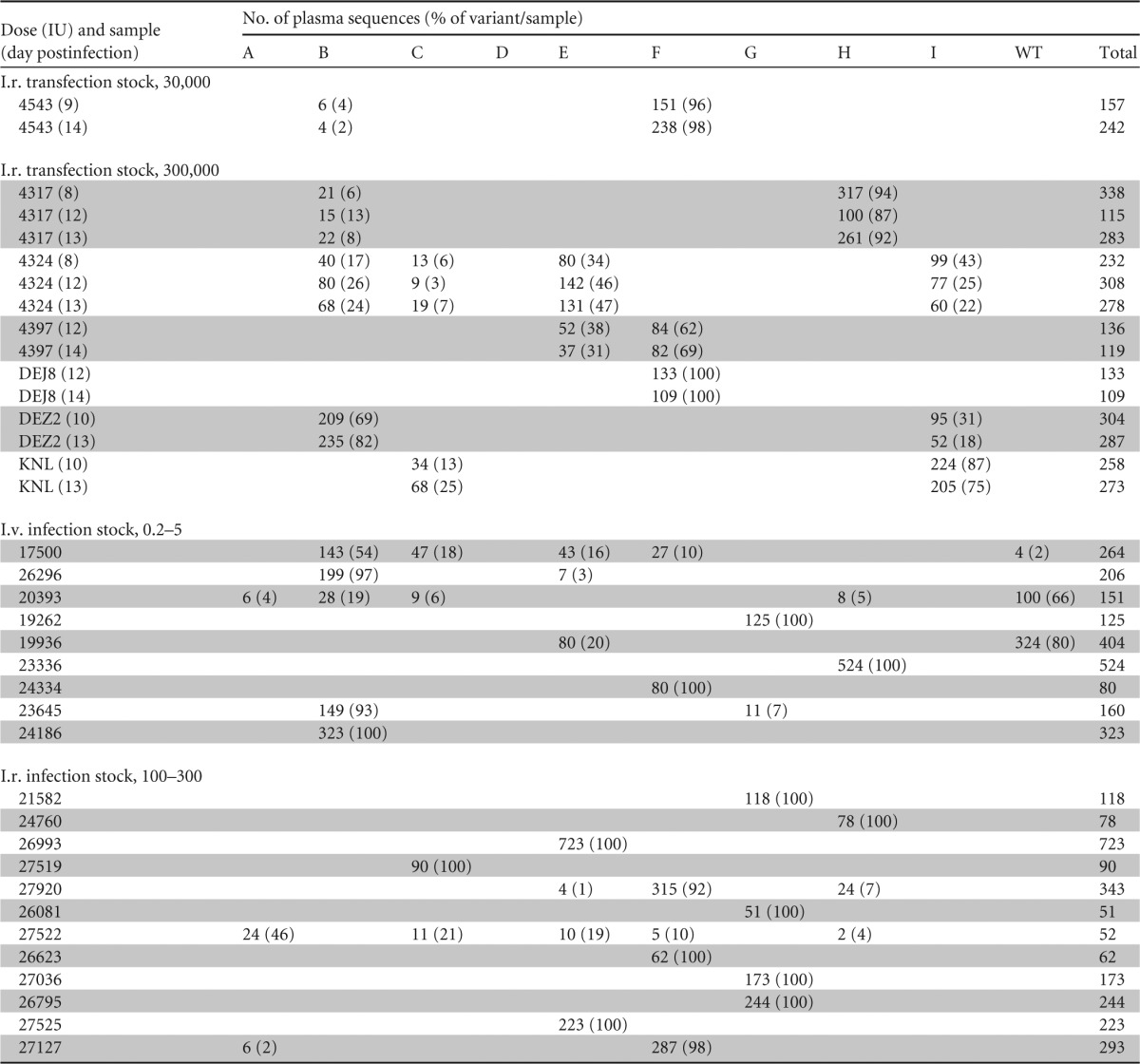
In vitro expansion of SIVmac239X.
While transfection-derived SIVmac239X is mucosally transmissible, a relatively large viral inoculum (3 × 105 IU) was required to provide high rates of infection with limited numbers of T/F variants. These data are consistent with accumulating but still incomplete data indicating that comparable in vitro titers of transfection-derived virus stocks are less infectious than infection-derived virus stocks for in vivo mucosal challenges. We have recently identified a number of differences between transfection- and infection-produced virus stocks that may account for the increased inoculum dose requirement for successful mucosal infection with transfection-derived virus (14). Among other differences, transfection-based production methods provide virus stocks with fewer and lower levels of cytokines and chemokines but with higher specific infectivity in vitro, while infection-derived stocks contain higher levels of cytokines and chemokines, which may promote inflammation and enhance in vivo infectivity. We therefore sought to generate and evaluate an infection-derived SIVmac239X stock. With the large volume transfection-derived stock described above as the in vitro seed inoculum, we tested triplicate independent small-scale expansions on pooled primary rhesus PBMCs. Culture supernatants were collected at days 7, 8, and 9 postinfection, and the proportion of each variant was assessed by analyzing 1,360 SGA-derived sequences from cell-free vRNA (Fig. 2). Consistent with our initial in vitro replication assay, for all three expansions, variants D and I were significantly underrepresented relative to the expected value of 10% for each clone (P < 0.001). Nearly all of the other variants were maintained at roughly equivalent proportions as in the parental transfection-derived stock, with within-culture fluctuations and small changes between independent cultures consistent with stochastic variability.
Despite the declines in variants D and I during in vitro small-scale test expansions, a large-scale infection-derived virus stock was generated with all 10 variants by infecting 1.4 × 108 PHA-stimulated, pooled rhesus PBMCs and collecting virus-containing supernatants at 7, 8, and 9 days postinfection. The proportion of each variant within the collected stocks was determined by analyzing 1,035 SGA-derived sequences. Although variant D was present at 3- to 6-fold lower levels than the other stock constituents following in vitro expansion, again suggesting a subtle replication defect for this clone relative to the other tagged clones, sequencing analysis of the large-scale stocks revealed only minor fluctuations in the relative proportions of the other nine viral variants, including variant I (Fig. 2). Only variant D was statistically significantly lower than WT SIVmac239 across all four experiments (P < 0.001). Importantly, for any given in vitro expansion, one or more variants (including WT SIVmac239) were over- or underrepresented, highlighting the variable nature of exponential-phase growth in culture to generate viral stocks, even for nearly identical viral variants. In contrast to transfection-derived stocks, which can be reproduced consistently between preparations and between laboratories, we note that infection-based expansion of the pooled stock in vitro yielded variability in the relative proportion of each viral variant for each independent culture, and thus, sequence analysis of each future expansion will be necessary to determine the relative proportion of each molecularly tagged clone. Virus stocks collected on days 7, 8, and 9 were comparable in vRNA contents (5.2 × 107, 3.9 ×107, and 1.45 ×107 copies/ml, respectively) and infectious titers (2.6 × 104, 2.9 × 104, and 2.0 × 104 IU/ml, respectively). All subsequent in vivo infections were performed with infection-derived stock harvested on day 8.
In vivo characterization of infection-derived SIVmac239X.
We first tested the in vivo infectivity and the relationship between infectious units and transmitted viral variants for the infection-derived stock by challenging naive, Indian-origin rhesus macaques i.v. Three out of three animals challenged with a single i.v. dose of 5 IU were productively infected with 2, 5, and 5 variants each based on SGA analysis of peak plasma viremia (Fig. 4A). We then reduced the inoculum by 5-fold and challenged three naive animals with a single i.v. dose of 1 IU. Again, all three animals were infected but the number of identifiable variants at peak plasma viremia was reduced to 1, 1, and 2 each (Fig. 4A). Finally, we reduced the inoculum by an additional 5-fold and performed repeated i.v. challenges of three additional naive macaques with 0.2 IU per challenge. With this dose, only one animal was infected by the first challenge, a second animal was infected by a second challenge, and the final animal was infected by a third challenge. Viral load data for these animals were plotted with the challenge time point responsible for productive infection set as day 0 in Fig. 4A. The number of variants at peak viremia was determined for each animal and was again limited to 1, 1, and 2 each. Interestingly, for this virus stock, the TZM-bl-determined infectious titer matched the in vivo infectivity when the virus was administered i.v.
FIG 4.

Plasma viral load profiles of animals infected i.v. (A) or i.r. (B) with infection-derived SIVmac239X. (A) Nine rhesus macaques were challenged i.v. with a single challenge with 5 or 1 IU or with repeated challenges with 0.2 IU. A single challenge with 5 IU (red) or 1 IU (yellow) led to productive infection of all six animals, but up to three challenges were required for productive infection of all three animals with the 0.2-IU dose (black). (B) Twelve rhesus macaques were challenged i.r. with repeated inoculations of infection-derived SIVmac239X. The eight animals indicated in black were infected with a 100-IU dose, while the four animals indicated in red remained uninfected after eight challenges with 100 IU and were subsequently challenge and infected with a 300-IU dose. All viral load time points were plotted so that the challenge date leading to productive infection was set as day 0. The number of T/F variants was determined by sequence analysis of plasma samples obtained at ramp-up and peak viremia and is displayed parenthetically for each animal. Additionally, light blue plot symbols indicate animals with MHC alleles known to be associated with an increased frequency of partial immune-mediated control of SIV infection. The specific genotypes are noted next to the animal names.
Next, we determined the titer of the infection-derived SIVmac239X stock i.r. by using a repeated, limiting-dose challenge. Twelve animals were challenged i.r. (atraumatically) with 100 IU of virus. At 1 week following each challenge, the animals were tested for plasma vRNA by quantitative RT-PCR and all aviremic animals were rechallenged 1 week thereafter. After 8 challenges, 4 of 12 animals remained uninfected and were challenged with an increased dose of 300 IU in the same repeated-challenge protocol (Fig. 4B). All four animals challenged at 300 IU became SIV positive within two or three challenges. Plasma viral load data for all of the infected animals were plotted with the challenge date responsible for productive infection set as day 0 and plotted in Fig. 4B. Of the eight animals infected with 100 IU, six were infected with a single variant and the remaining two were infected with three and five variants. Of the four animals infected with 300 IU, three were infected with a single variant and one was infected with two variants (Fig. 4B). In total, limiting-dose i.r. challenge initiated infection with a single T/F variant in 9 of 12 animals and a mean of 1.6 variants per animal.
We evaluated the rate of infection and any potential relationship between the number of challenges required for infection and the number of T/F variants. We found the rate of infection at a 100-IU dose to be 12% per exposure but with only 8 of 12 animals becoming infected within the first four challenges (Fig. 5). At the 300-IU dose, the rate of infection was higher at 58% per exposure, requiring three or fewer challenges to infect the remaining four animals. Interestingly, the mean and median numbers of T/F variants per animal infected by the 100-IU dose are similar to those of animals infected by the 300-IU dose, suggesting that this 3-fold increase in exposure overcame an apparent resistance to infection in the four animals uninfected after eight challenges with 100 IU while retaining only a few T/F variants.
FIG 5.

Kaplan-Meier plot indicating the portion of animals remaining uninfected after bimonthly i.r. challenges with infection-derived SIVmac239X. The number of variants detected by sequence analysis of plasma from each animal infected by each challenge is shown in parentheses.
The plasma viral load profiles of the i.v. and i.r. SIVmac239X-infected animals (Fig. 4) were indistinguishable from the viral load profiles previously reported for a repeated, low-dose i.r. challenge with WT SIVmac239 (27). Although several animals in both challenge groups controlled viral replication to levels below 105 RNA copies/ml plasma during the chronic phase of infection, all but one of these animals were positive for MHC class I alleles (Mamu-B*08 or -B*17) known to be associated with partial immune-mediated control of SIV infection (Fig. 4). After accounting for animals with controller MHC class I genotypes, there were no apparent differences in the viral load profiles of animals infected with limited T/F variants by the i.v. route compared with the viral load profiles of animals infected with limited T/F variants by the i.r. route.
Although these findings demonstrate the utility of SIVmac239X for identifying the number of T/F variants following in vivo inoculation, we sought to determine if any variants were systematically over- or underrepresented, which might suggest differential transmissibility or in vivo replication capacity. Although it is possible for the same tagged variant to initiate multiple independent infection events that could lead to systemic infection within a single animal, resampling of the same clone is unlikely in limited-dose inoculations such as those performed here. Therefore, for the purposes of these analyses, we defined each variant identified in plasma as representative of a single independent T/F infection event. Nine of the 10 SIVmac239X variants were each transmitted to at least three animals, with variant D as the only variant not found in any challenged animals, again suggestive of a replication defect in this particular clone (Table 1). Of the nine transmitted clones, three were each found in only three animals (A, I, and WT SIVmac239), three were each found in six animals (C, G, and H), and three were each found in nine animals (B, E, and F). Although the number of animals infected by each clone may reflect the intrinsic replication capacity of each clone, we noted that variant I (identified in three animals) predominated over variant C (identified in six animals) in animal KNL, which was infected with both clones (Table 1). Similarly, variant A (identified in three animals) predominated over variant F (identified in nine animals) in animal 27522, which was infected with both clones, whereas variant F predominated over variant A in animal 27127, which was also infected with both clones. In addition, WT SIVmac239 was a member of the group of clones transmitted to the fewest animals, suggesting that these differences in apparent clone transmissibility simply reflect the stochastic nature of limited-dose infections.
To examine this hypothesis further, we derived a simulation model to generate variant frequencies under the assumption that each of the nine clones being considered was equally represented, for a total of 54 infection events. Explicitly, for a given simulation experiment, we sampled (with replacement) nine distinct “variants” with a total of 54 infection events. We executed this procedure 10,000 times. Using this simulation model, we computed the range and variance of each simulated experiment. Thus, we obtained 10,000 ranges and 10,000 variances for the tabled counts in the simulations. The ranges (defined as the maximum count minus the minimum count in the table) ranged from 2 to 16, with first and third quartiles of 6 and 8, and with median and mean values of 7.0 and 7.2, respectively. Our observed infection results (Table 1) (A = 3, B = 9, C = 6, E = 9, F = 9, G = 6, H = 6, I = 3, and WT = 3) showed a median and mean value of 6 and a range of 6 (9 − 3). These results fall well within the central portions of the distributions of the tables generated from our simulated results. Furthermore, the variances of the simulation ranged from a minimum of 0.25 to a maximum of 23.5, with first and third quartiles of 3.75 and 7.5 and with median and mean values of 5.5 and 6.0, respectively, while our observed infection results revealed a variance of 6.75, which lies well within the first and third quartiles of the distributions of the tables generated from our 10,000 simulated results. In summary, the measures of central tendency (median and mean) and variability (range and variance) that we observed in our table of infection events are completely consistent with results obtained from thousands of simulated experiments in which nine distinct “variants” were sampled for 54 infection events. Therefore, the differences in the number of transmission events for each clone do not indicate inherent differential transmissibility but rather are consistent with the infection of nine phenotypically indistinguishable variants.
Proportion of each variant during acute and chronic infections.
Interestingly, in animals infected with more than one variant, the relative proportions of each variant in plasma were not equivalent (Fig. 6). A chi-square test was used to determine whether the observed proportions of each variant were significantly different from the expectation under the assumption that each variant was equally represented following transmission (e.g., 25% representation of each variant in an animal infected with four variants). In all of the animals infected with more than one variant, the observed proportion of each variant was significantly different from the expected value (P < 0.0001). In four of five animals infected with more than two variants (Fig. 6B), at least one variant in each animal was found at the expected proportion, but never all of the variants. As noted above, this phenomenon of systemic over- or underrepresentation of clones within animals infected with more than one T/F virus does not appear to be explained by particular variants being uniformly less fit than others, since we found examples of the same variant at both above and below the expected values in different animals. For example, variant B, identified in a total of eight animals, was the dominant lineage in four animals (DEZ2, 26296, 23645, and 17500), at the expected/predicted proportion in two animals (4324 and 20393), and a minor variant in two animals (4543 and 4317). Furthermore, in some animals sampled at multiple times during ramp-up viremia, we found a consistent proportion of each variant over time, with <2-fold changes in the relative proportion of any variant (Table 1), suggesting that differential levels of virus established during the earliest infection events are retained longitudinally. Lastly, we compared the relative proportion of each variant within the transfection-derived (Fig. 7A) and infection-derived SIVmac239X stocks (Fig. 7B) with the proportion of each variant within the SIVmac239X-infected animals (Fig. 7C). While variant D was undetected in any animal, all of the other variants were found as T/F lineages in 3 to 9 out of a total of 54 events. Taken together, these data suggest that very early initial events can alter the dynamics of viral replication, causing discrepancies between variants, which are preserved throughout the exponential phase of acute viremia within a system where all of the variants are nearly identical.
FIG 6.

Proportion of SGA sequences represented by each variant indicated in animals infected with two (A) or more than two (B) variants. For each animal, the proportion of each variant detected is compared to the theoretical expected value if it is equally represented (indicated by a dotted line). For each animal, the expected value was significantly different from that expected, except for the variants indicated by an asterisk.
FIG 7.

Pie charts indicating the proportion of each molecularly tagged variant as determined by sequence analysis within the transfection-produced SIVmac239X stock (A), the infection-derived SIVmac239X stock (B), or the total number of sequences derived from all of the infected animals as a fraction of the total number of sequences (C).
Finally, while we had specifically chosen mutation sites with known polymorphisms in naturally occurring SIV variants, we sought to determine the longitudinal in vivo stability of these mutations. To address this question, we performed SGA sequencing of 16 longitudinal plasma samples collected at 100 to 400 days postchallenge from 10 of the SIVmac239X-infected animals and compared the identified variants and sequence polymorphisms within the mutated domain with the originally identified T/F variants. In all 10 animals, only the originally identified T/F variants were found later in chronic infection (total of 535 sequences) (Table 2), with no gain or loss of variants, no evidence of reversion to WT SIVmac239, and no additional polymorphisms within the mutated sites.
TABLE 2.
Longitudinal retention of molecular tag up to 485 days postinfection

DISCUSSION
Limiting-dose SIV challenges, which involve a reduction of the experimental viral inoculum size such that systemic infection is initiated by either one or only a few transmitted viral variants, have become a common and preferred method of modeling mucosal HIV infection in NHPs (1, 10–13). This approach, which more closely recapitulates the inefficient nature of mucosal HIV transmission than traditional “high-dose” challenge models, requires the use of a virus stock containing sufficient sequence diversity to allow individual viral variants to be distinguished. Therefore, the use of a diverse challenge virus stock allows researchers to use infection rate and genetic sequencing approaches to discriminate and enumerate the T/F viruses to confirm the appropriateness of the challenge dose used and to evaluate the impact of a vaccine, microbicide, drug treatment, or other potential intervention strategies on the number of T/F viruses and, by extension, on the overall efficiency of virus transmission (28–36).
Although clonal virus stocks, such as those typically generated by transfection, can be used at specific-titer doses that provide reduced infection efficiencies, their genetic homogeneity precludes the ability to determine the number of T/F viruses initiating infection (14). Infection-derived virus stocks are generated via multiple rounds of error-prone viral replication and expansion in a permissive cell type and have been the favored stock type used for limiting-dose viral challenge models. There are, however, several potential drawbacks to using infection-derived viral swarms for limiting-dose challenge studies with NHPs. First, the genetic diversity contained within these stocks is generated by the selection pressures of an in vitro viral replication system, absent the relevant pressures that might be applied by an active immune system, as would be the case for an inoculum transmitted naturally in vivo. Second, recent work has shown that genetically diverse virus isolates can contain substantial phenotypic diversity as well (17). As a result, at limiting virus doses, the specific variant(s) that initiates infection within any given animal can be functionally quite different from the variant(s) initiating infection in a different animal, leading to confounding variability between study animals. These phenotypic differences can be profound for parameters such as susceptibility to neutralizing antibodies, cellular immunity, and even replicative capacity (14, 17). Lastly, we have previously shown that different infection-derived stocks of the same nominal virus isolate often comprise genetically distinct virus populations that do not overlap phylogenetically (14). Thus, there is ample potential for confounding variability, which may be amplified under limiting virus dose conditions, both within a given study with a single infection-derived virus stock and between studies with different uncloned isolate stocks.
To address these potential challenges while maintaining the ability to enumerate T/F variants following in vivo inoculation, we developed a novel approach to virus production that combines the consistency and predictability of clonal virus stocks with the diversity of an uncloned viral isolate. Using the SIVmac239 infectious molecular clone as a viral background, we developed a synthetic swarm of molecularly tagged viral clones that differ from one another by two or three synonymous changes per variant within a 41-nucleotide stretch of the viral integrase gene sequence. Although each of the noncoding changes chosen was identified in published SIV and HIV-2 sequences derived from in vivo infections, we first evaluated the function of each of the nine individual molecularly tagged variants in comparison with WT SIVmac239 (Fig. 1). In a single-cycle reporter cell line infectivity assay, all nine of the variants had infectious titers that were virtually identical to that of SIVmac239 (Fig. 1B). When evaluated for their replication capacity in primary rhesus macaque cells, again the molecularly tagged clones were virtually indistinguishable from WT SIVmac239 (Fig. 1C). The only exceptions to this finding were variants D and I, which both showed reduced peak p27 levels in infected culture supernatants. Surprisingly, this apparent subtle replication defect in variant D was associated with a substantial reduction in the representation of variant D in the pooled synthetic swarm following expansion in infected cells (Fig. 7A versus B), and variant D was not detected in any of the animals infected in this study (Fig. 7C and D). We note here that although variant D was not transmitted to any challenged animals, it was present at ∼3- to 6-fold lower proportions than the other variants in the infection-derived stock used to challenge 21 of the 29 animals in this study, making it less likely to be transmitted even in the absence of any in vivo replication defect. It is unclear if variant D can initiate systemic infection either by itself or as part of a multiple-variant infection. Because each of the molecular tags shared individual point mutations with at least one other molecular tag (see Fig. 1A), this reduced replication capacity of variant D could not be attributed to any individual point mutation within the molecular tag, suggesting that the specific combination of mutations is responsible. Importantly, the potential impact on replication and transmissibility imparted by several noncoding changes in variant D underscores the likelihood of extreme variability within uncloned viral isolate stocks, where each clone can contain a constellation of random coding and noncoding changes. Importantly to the field as a whole, for any given in vitro expansion, one or more variants (including WT SIVmac239) were over- or underrepresented, highlighting the variable nature of exponential growth in culture to generate viral stocks.
To generate a synthetic virus swarm containing all 9 molecularly tagged variants plus WT SIVmac239, we developed a cotransfection strategy in which all 10 viral variants would be generated in nearly equivalent proportions in a single transfected cell culture. We reasoned that generating the virus stock in this way would reduce the potential variability and complexities that would be introduced by generating each stock individually and creating an admixture of titers at each time of infection. We note here that although we opted to generate a pooled virus stock, there may be potential uses for individual stocks of each of the tagged clones, such as the use of different tagged variants in successive serial challenges to discern which challenge dose was responsible for systemic infection in serial-challenge studies. Individual variants from transfection-derived stocks, as well as the plasmid DNA itself, are available for use by the research community.
When used in i.r. challenges, the transfection-derived stock of SIVmac239X proved to be a useful tool for enumeration of the T/F variants that led to systemic infection, with each variant easily discernible by SGA sequencing of a small (300-bp) region of the integrase gene. We recently identified a number of key differences between transfection- and infection-derived virus stocks used for in vivo NHP models of HIV infection (14). While it is not clear which differential stock parameter is responsible for this difference or which challenge stock type represents a more accurate model of mucosal HIV infection, for a given in vitro infectious titer, infection-derived virus stocks appear to have greater in vivo mucosal infectivity than transfection-derived stocks. We therefore evaluated an expansion of SIVmac239X in an infected rhesus PBMC culture as an alternative SIVmac239X in vivo challenge stock. Although growing the tagged stock in infected cells skewed the relative proportions of the tags (likely because of a combination of stochastic occurrences and perhaps because of subtle differences in replicative capacity), which must be accounted for when evaluating the number of T/F variants and the specific variants transmitted, the infection-derived stock did appear to have greater relative mucosal infectivity while maintaining the ability to easily differentiate T/F variants by SGA sequencing of a small genome region. We are currently evaluating potential stock attributes that may be responsible for the apparent difference in in vivo infectivity in infection- and transfection-derived stocks. In future studies, the use of infection- or transfection-derived SIVmac239X stocks will require thoughtful consideration of which is most appropriate for the specific experimental questions being addressed; however, we show here that both provide a convenient way to enumerate T/F variants.
In addition to addressing potential experimental concerns about using uncloned viral isolate stocks for NHP mucosal challenges, SIVmac239X also provides technical advantages over traditional virus stocks. As shown by the data collected in this study, the ability to distinguish and enumerate T/F variants by sequencing only a small region of the viral genome allows far deeper SGA sequencing analysis without additional cost or time. This enables the detection of T/F variants that may be present in small proportions in the systemic virus population that might otherwise be missed when using a standard virus isolate stock. For example, animal 4543 was infected with two T/F variants (Fig. 3 and 6); however, one of these variants, B, was present at only 2 to 4% at two separate sequencing time points (Fig. 6 and Table 1). Detection of this variant was made possible by generating 150 and 240 sequences per time point (Table 1); however, generating this many 2.5- to 3.5-kb SGA sequences, as is typically required to definitively distinguish variants from an uncloned viral isolate, would be time and cost prohibitive. With a typical 30 sequences, variant B in this example would likely go undetected. One additional advantage of a small amplicon fragment is the ability to sequence from formalin-fixed tissues, including laser-captured microdissection sequencing (B. F. Keele, unpublished data).
In addition to improving upon existing models of HIV transmission, SIVmac239X will also allow the examination of phenomena that would be difficult or impossible to evaluate with conventional virus stocks. In our SIVmac239X animals that were infected with more than one T/F variant, we often observed unequal systemic proportions of the different variants. As described above, variant B provides an excellent example of a dominant lineage in several animals, an equivalent variant in other animals, and a minor variant in two other animals. Interestingly, although unequal systemic proportions have been noted previously following multivariant HIV infection, under those circumstances, the sizable genetic differences between variants were considered the likely explanation (6). Given the potential phenotypic differences inherent in an HIV quasispecies, replicative or fitness differences may well contribute to this unequal distribution. However, we observed this phenomenon in the setting of minimal genetic differences and limited phenotypic differences discernible in vitro, suggesting the involvement of factors other than viral replication. Given that there were no clear patterns to suggest that certain variants (other than D) were universally more or less fit than others, this observation suggests that this unequal proportionality reflects early replication dynamics, perhaps dictated by the local tissue environment, the initial cell type infected, or other nonviral parameters. The ability to track phenotypically similar but distinct and distinguishable variants over time provides additional insights into the dynamics of early replication in the absence of potentially confounding phenotypic differences among variants within a diverse challenge stock. To our knowledge, this is the first experiment to show that simultaneous inoculation leading to coinfection with two or more replicatively indistinguishable variants can lead to disparate proportions of each variant in the circulating plasma by the time of peak viremia, suggesting that there are differences in the dynamics of very early viral replication. The phenotypic equivalence of the variants indicates that the differences in proportional representation are not virologically driven but are dictated by either stochastic events (e.g., the replicative capacity of the first productively infected cell) or heterogeneous host effects that can impact very early viral replication (e.g., infection at sites of more or less robust innate responses). We suspect that these nonviral differences (whatever they may be) affect the virus very early, altering the first few days of replication. Once viral replication becomes established and disseminates from the site of entry, the impact of stochastic events on the entire viral progeny from one variant, but not on the other, vanishes. Further, we posit that once the virus has entered the lymphatic system, host responses would affect both lineages equally. Therefore, elucidating the nonviral mechanism(s) that can differentially modulate these earliest steps of viral infection at the portal of entry will be important for understanding viral replication at its most vulnerable time point—prior to systemic dissemination.
One limitation of the current molecularly tagged stock is that it is limited to only 10 distinct variants. For a repeated low-dose challenge, or a single limited-dose inoculation where the number of variants is limited to one or only a few variants, this stock is perfectly appropriate. If, however, the target number of T/F variants is greater than five, then this stock does not contain a sufficient total number of variants to accurately enumerate transmission events. When infection originates with more than five variants, the probability of a repeated infection resulting from one of the first five variants is greater than an infection resulting from an additional unique variant. If the number of variants that need to be discriminated is large, then additional variants will need to be generated to allow accurate enumeration and viral tracking. An alternative approach would be to generate viral stocks containing variants in different relative proportions to cover a much larger dynamic range. For example, one could generate a stock where variants A and B combined represented 60% of the total inoculum with variants C and E representing 30%, F and G representing 8%, and H and the WT representing the remaining 2%. After infection, one could estimate the number of T/F variants on the basis of the identification of variants at lower stock proportions. Alternative approaches to using these reagents will require further evaluation, but in limited-dose studies that most accurately mimic HIV-1 transmission, the 10 variants presented here can easily and accurately discriminate the few T/F variants.
While this initial characterization involved a SIVmac239-based synthetic swarm, this molecular-tagging approach should be more broadly applicable to other lentiviruses and models of HIV infection (e.g., other SIVs, SHIVs, minimally chimeric HIV, and viruses used in nonpathogenic hosts). Given the utility of the synthetic SIVmac239X swarm demonstrated here and the ability to combine various SIV clones to test specific hypotheses, we expect molecularly tagged synthetic virus swarms to be well suited for NHP models of AIDS focused on preclinical vaccine evaluations, microbicide or other intervention studies, basic transmission/early event studies, and even long-term pathogenesis or viral reservoir establishment studies. It is also possible to modify this approach for other virus-host models of disease beyond lentiviruses.
ACKNOWLEDGMENTS
This work was supported in part with federal funds from the National Cancer Institute; National Institutes of Health, under contract HHSN261200800001E and NIAID grant AI054292.
We acknowledge Julian Bess, Rodman Smith, and Terra Ireland at the Frederick National Laboratory for assistance in generating large-volume transfection stocks. We also thank Vicky Coalter, Adam Wiles, Rodney Wiles, and Donald Johnson at the Frederick National Laboratory for assistance with NHP study coordination and sample processing. The TZM-bl cell line was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
Footnotes
Published ahead of print 7 May 2014
REFERENCES
- 1.Fennessey CM, Keele BF. 2013. Using nonhuman primates to model HIV transmission. Curr. Opin. HIV AIDS 8:280–287. 10.1097/COH.0b013e328361cfff [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirsch VM, Lifson JD. 2000. Simian immunodeficiency virus infection of monkeys as a model system for the study of AIDS pathogenesis, treatment, and prevention. Adv. Pharmacol. 49:437–477. 10.1016/S1054-3589(00)49034-4 [DOI] [PubMed] [Google Scholar]
- 3.Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J. Virol. 83:3556–3567. 10.1128/JVI.02132-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baalwa J, Wang S, Parrish NF, Decker JM, Keele BF, Learn GH, Yue L, Ruzagira E, Ssemwanga D, Kamali A, Amornkul PN, Price MA, Kappes JC, Karita E, Kaleebu P, Sanders E, Gilmour J, Allen S, Hunter E, Montefiori DC, Haynes BF, Cormier E, Hahn BH, Shaw GM. 2013. Molecular identification, cloning and characterization of transmitted/founder HIV-1 subtype A, D and A/D infectious molecular clones. Virology 436:33–48. 10.1016/j.virol.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haaland RE, Hawkins PA, Salazar-Gonzalez J, Johnson A, Tichacek A, Karita E, Manigart O, Mulenga J, Keele BF, Shaw GM, Hahn BH, Allen SA, Derdeyn CA, Hunter E. 2009. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 5:e1000274. 10.1371/journal.ppat.1000274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557. 10.1073/pnas.0802203105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Bar KJ, Wang S, Decker JM, Chen Y, Sun C, Salazar-Gonzalez JF, Salazar MG, Learn GH, Morgan CJ, Schumacher JE, Hraber P, Giorgi EE, Bhattacharya T, Korber BT, Perelson AS, Eron JJ, Cohen MS, Hicks CB, Haynes BF, Markowitz M, Keele BF, Hahn BH, Shaw GM. 2010. High multiplicity Infection By HIV-1 in men who have sex with men. PLoS Pathog. 6:e1000890. 10.1371/journal.ppat.1000890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289. 10.1084/jem.20090378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novitsky V, Wang R, Margolin L, Baca J, Rossenkhan R, Moyo S, van Widenfelt E, Essex M. 2011. Transmission of single and multiple viral variants in primary HIV-1 subtype C infection. PLoS One 6:e16714. 10.1371/journal.pone.0016714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keele BF, Li H, Learn GH, Hraber P, Giorgi EE, Grayson T, Sun C, Chen Y, Yeh WW, Letvin NL, Mascola JR, Nabel GJ, Haynes BF, Bhattacharya T, Perelson AS, Korber BT, Hahn BH, Shaw GM. 2009. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J. Exp. Med. 206:1117–1134. 10.1084/jem.20082831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Keele BF, Li H, Keating S, Norris PJ, Carville A, Mansfield KG, Tomaras GD, Haynes BF, Kolodkin-Gal D, Letvin NL, Hahn BH, Shaw GM, Barouch DH. 2010. Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J. Virol. 84:10406–10412. 10.1128/JVI.01155-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma ZM, Keele BF, Qureshi H, Stone M, Desilva V, Fritts L, Lifson JD, Miller CJ. 2011. SIVmac251 is inefficiently transmitted to rhesus macaques by penile inoculation with a single SIVenv variant found in ramp-up phase plasma. AIDS Res. Hum. Retroviruses 27:1259–1269. 10.1089/aid.2011.0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stone M, Keele BF, Ma ZM, Bailes E, Dutra J, Hahn BH, Shaw GM, Miller CJ. 2010. A limited number of simian immunodeficiency virus (SIV) env variants are transmitted to rhesus macaques vaginally inoculated with SIVmac251. J. Virol. 84:7083–7095. 10.1128/JVI.00481-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Del Prete GQ, Scarlotta M, Newman L, Reid C, Parodi LM, Roser JD, Oswald K, Marx PA, Miller CJ, Desrosiers RC, Barouch DH, Pal R, Piatak M, Jr, Chertova E, Giavedoni LD, O'Connor DH, Lifson JD, Keele BF. 2013. Comparative characterization of transfection- and infection-derived simian immunodeficiency virus challenge stocks for in vivo nonhuman primate studies. J. Virol. 87:4584–4595. 10.1128/JVI.03507-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, Leitner T, Han CS, Gleasner CD, Green L, Lo CC, Nag A, Wallstrom TC, Wang S, McMichael AJ, Haynes BF, Hahn BH, Perelson AS, Borrow P, Shaw GM, Bhattacharya T, Korber BT. 2010. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 5:e12303. 10.1371/journal.pone.0012303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apetrei C, Kaur A, Lerche NW, Metzger M, Pandrea I, Hardcastle J, Falkenstein S, Bohm R, Koehler J, Traina-Dorge V, Williams T, Staprans S, Plauche G, Veazey RS, McClure H, Lackner AA, Gormus B, Robertson DL, Marx PA. 2005. Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J. Virol. 79:8991–9005. 10.1128/JVI.79.14.8991-9005.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopker M, Easlick J, Sterrett S, Decker JM, Barbian H, Learn G, Keele BF, Robinson JE, Li H, Hahn BH, Shaw GM, Bar KJ. 2013. Heterogeneity in neutralization sensitivities of viruses comprising the simian immunodeficiency virus SIVsmE660 isolate and vaccine challenge stock. J. Virol. 87:5477–5492. 10.1128/JVI.03419-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel MD, Letvin NL, King NW, Kannagi M, Sehgal PK, Hunt RD, Kanki PJ, Essex M, Desrosiers RC. 1985. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science 228:1201–1204. 10.1126/science.3159089 [DOI] [PubMed] [Google Scholar]
- 19.Regier DA, Desrosiers RC. 1990. The complete nucleotide sequence of a pathogenic molecular clone of simian immunodeficiency virus. AIDS Res. Hum. Retroviruses 6:1221–1231 [DOI] [PubMed] [Google Scholar]
- 20.Kestler HW, III, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. 1991. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65:651–662. 10.1016/0092-8674(91)90097-I [DOI] [PubMed] [Google Scholar]
- 21.Morcock DR, Thomas JA, Sowder RC, II, Henderson LE, Crise BJ, Gorelick RJ. 2008. HIV-1 inactivation by 4-vinylpyridine is enhanced by dissociating Zn(2+) from nucleocapsid protein. Virology 375:148–158. 10.1016/j.virol.2008.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81:8827–8832. 10.1128/JVI.00895-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cline AN, Bess JW, Piatak M, Jr, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 34:303–312. 10.1111/j.1600-0684.2005.00128.x [DOI] [PubMed] [Google Scholar]
- 24.Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Jr, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. 2005. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 43:406–413. 10.1128/JCM.43.1.406-413.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970. 10.1128/JVI.02660-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agresti A. 2013. Categorical data analysis, 3rd ed. Wiley, New York, NY [Google Scholar]
- 27.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolton DL, Song K, Wilson RL, Kozlowski PA, Tomaras GD, Keele BF, Lovingood RV, Rao S, Roederer M. 2012. Comparison of systemic and mucosal vaccination: impact on intravenous and rectal SIV challenge. Mucosal Immunol. 5:41–52. 10.1038/mi.2011.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenizia C, Keele BF, Nichols D, Cornara S, Binello N, Vaccari M, Pegu P, Robert-Guroff M, Ma ZM, Miller CJ, Venzon D, Hirsch V, Franchini G. 2011. TRIM5alpha does not affect simian immunodeficiency virus SIV(mac251) replication in vaccinated or unvaccinated Indian rhesus macaques following intrarectal challenge exposure. J. Virol. 85:12399–12409. 10.1128/JVI.05707-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pegu P, Vaccari M, Gordon S, Keele BF, Doster M, Guan Y, Ferrari G, Pal R, Ferrari MG, Whitney S, Hudacik L, Billings E, Rao M, Montefiori D, Tomaras G, Alam SM, Fenizia C, Lifson JD, Stablein D, Tartaglia J, Michael N, Kim J, Venzon D, Franchini G. 2013. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIVmac251 acquisition in an immunization regimen that mimics the RV-144 Thai trial. J. Virol. 87:1708–1719. 10.1128/JVI.02544-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qureshi H, Ma ZM, Huang Y, Hodge G, Thomas MA, DiPasquale J, DeSilva V, Fritts L, Bett AJ, Casimiro DR, Shiver JW, Robert-Guroff M, Robertson MN, McChesney MB, Gilbert PB, Miller CJ. 2012. Low-dose penile SIVmac251 exposure of rhesus macaques infected with adenovirus type 5 (Ad5) and then immunized with a replication-defective Ad5-based SIV gag/pol/nef vaccine recapitulates the results of the phase IIb step trial of a similar HIV-1 vaccine. J. Virol. 86:2239–2250. 10.1128/JVI.06175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds MR, Weiler AM, Piaskowski SM, Piatak M, Jr, Robertson HT, Allison DB, Bett AJ, Casimiro DR, Shiver JW, Wilson NA, Lifson JD, Koff WC, Watkins DI. 2012. A trivalent recombinant Ad5 gag/pol/nef vaccine fails to protect rhesus macaques from infection or control virus replication after a limiting-dose heterologous SIV challenge. Vaccine 30:4465–4475. 10.1016/j.vaccine.2012.04.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaccari M, Keele BF, Bosinger SE, Doster MN, Ma ZM, Pollara J, Hryniewicz A, Ferrari G, Guan Y, Forthal DN, Venzon D, Fenizia C, Morgan T, Montefiori D, Lifson JD, Miller CJ, Silvestri G, Rosati M, Felber BK, Pavlakis GN, Tartaglia J, Franchini G. 2013. Protection afforded by an HIV vaccine candidate in macaques depends on the dose of SIVmac251 at challenge exposure. J. Virol. 87:3538–3548. 10.1128/JVI.02863-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson NA, Keele BF, Reed JS, Piaskowski SM, MacNair CE, Bett AJ, Liang X, Wang F, Thoryk E, Heidecker GJ, Citron MP, Huang L, Lin J, Vitelli S, Ahn CD, Kaizu M, Maness NJ, Reynolds MR, Friedrich TC, Loffredo JT, Rakasz EG, Erickson S, Allison DB, Piatak M, Jr, Lifson JD, Shiver JW, Casimiro DR, Shaw GM, Hahn BH, Watkins DI. 2009. Vaccine-induced cellular responses control simian immunodeficiency virus replication after heterologous challenge. J. Virol. 83:6508–6521. 10.1128/JVI.00272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burton DR, Hessell AJ, Keele BF, Klasse PJ, Ketas TA, Moldt B, Dunlop DC, Poignard P, Doyle LA, Cavacini L, Veazey RS, Moore JP. 2011. Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc. Natl. Acad. Sci. U. S. A. 108:11181–11186. 10.1073/pnas.1103012108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roederer M, Keele BF, Schmidt SD, Mason RD, Welles HC, Fischer W, Labranche C, Foulds KE, Louder MK, Yang ZY, Todd JP, Buzby AP, Mach LV, Shen L, Seaton KE, Ward BM, Bailer RT, Gottardo R, Gu W, Ferrari G, Alam SM, Denny TN, Montefiori DC, Tomaras GD, Korber BT, Nason MC, Seder RA, Koup RA, Letvin NL, Rao SS, Nabel GJ, Mascola JR. 2014. Immunological and virological mechanisms of vaccine-mediated protection against SIV and HIV. Nature 505:502–508. 10.1038/nature12893 [DOI] [PMC free article] [PubMed] [Google Scholar]