

ABSTRACT
The lytic cycles of Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV) are induced in cell culture by sodium butyrate (NaB), a short-chain fatty acid (SCFA) histone deacetylase (HDAC) inhibitor. Valproic acid (VPA), another SCFA and an HDAC inhibitor, induces the lytic cycle of KSHV but blocks EBV lytic reactivation. To explore the hypothesis that structural differences between NaB and VPA account for their functional effects on the two related viruses, we investigated the capacity of 16 structurally related short- and medium-chain fatty acids to promote or prevent lytic cycle reactivation. SCFAs differentially affected EBV and KSHV reactivation. KSHV was reactivated by all SCFAs that are HDAC inhibitors, including phenylbutyrate. However, several fatty acid HDAC inhibitors, such as isobutyrate and phenylbutyrate, did not reactivate EBV. Reactivation of KSHV lytic transcripts could not be blocked completely by any fatty acid tested. In contrast, several medium-chain fatty acids inhibited lytic activation of EBV. Fatty acids that blocked EBV reactivation were more lipophilic than those that activated EBV. VPA blocked activation of the BZLF1 promoter by NaB but did not block the transcriptional function of ZEBRA. VPA also blocked activation of the DNA damage response that accompanies EBV lytic cycle activation. Properties of SCFAs in addition to their effects on chromatin are likely to explain activation or repression of EBV. We concluded that fatty acids stimulate the two related human gammaherpesviruses to enter the lytic cycle through different pathways.
IMPORTANCE Lytic reactivation of EBV and KSHV is needed for persistence of these viruses and plays a role in carcinogenesis. Our direct comparison highlights the mechanistic differences in lytic reactivation between related human oncogenic gammaherpesviruses. Our findings have therapeutic implications, as fatty acids are found in the diet and produced by the human microbiota. Small-molecule inducers of the lytic cycle are desired for oncolytic therapy. Inhibition of viral reactivation, alternatively, may prove useful in cancer treatment. Overall, our findings contribute to the understanding of pathways that control the latent-to-lytic switch and identify naturally occurring molecules that may regulate this process.
INTRODUCTION
Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), the two human gammaherpesviruses, both induce cancer. EBV is linked to lymphoid and epithelial cancers, such as Burkitt's lymphoma, Hodgkin's disease, diffuse large B cell lymphoma, lymphoproliferative disease in immunocompromised patients, nasopharyngeal carcinoma, and gastric carcinoma. KSHV causes Kaposi's sarcoma, primary effusion lymphoma (PEL), and multicentric Castleman's disease. EBV and KSHV persist in a latent stage where few viral genes are expressed. When the viruses enter the lytic stage, many viral genes are expressed, the viral DNA replicates, and new virions are produced. Lytic reactivation is necessary for transmission of the virus and may have direct oncogenic effects (1).
The switch between latency and lytic reactivation is highly regulated. Virally encoded transactivator genes are repressed during latency, but when they are expressed, the transactivator proteins drive the lytic cycle. The two EBV transactivator genes, BZLF1 and BRLF1, encode multifunctional proteins (ZEBRA and Rta) which, in concert, activate the viral lytic cascade (2–6). KSHV ORF50, the positional and functional homolog of EBV BRLF1, regulates early gene transcription (7–9) and DNA replication (10).
Endogenous stimuli that promote viral reactivation from latency are poorly characterized. However, a variety of chemical and biological agents induce the viral lytic cycle in cell culture. Phorbol esters such as tetradecanoyl phorbol acetate (TPA), which are protein kinase C (PKC) activators, 5-aza-2′-deoxycytidine, a DNA methyltransferase inhibitor, and the histone deacetylase (HDAC) inhibitors sodium butyrate (NaB) and trichostatin A (TSA) activate EBV (11–16) and KSHV (17–21). As a factor complicating the study of lytic cycle activation, the same inducing stimulus does not activate the lytic cycle in all cell backgrounds. For example, in HH-B2 PEL cells, KSHV is inducible by butyrate but not by phorbol esters or azacytidine (17, 18), whereas TPA induces the KSHV lytic cycle in several other cell lines. HDAC inhibitors activate EBV in a subclone of P3HR1 cells, HH514-16, but not in B95-8 cells; conversely, TPA activates EBV to enter the lytic cycle in B95-8 cells but not in HH514-16 cells (22, 23). EBV is activated in Akata cells following cross-linking of surface Ig (14, 24, 25). Some cell lines are notoriously resistant to lytic cycle activation by external stimuli. Even in cell lines that are responsive to lytic induction stimuli, a subpopulation of cells remains refractory to lytic cycle activation (26). Although some biochemical properties of the chemical inducing stimuli are known, the mechanisms by which various agents lead to expression of the lytic cycle activator genes, whether the pathways promoted by different agents converge, and whether the same pathways are involved with the two related viruses remain unknown.
Butyrate is an HDAC inhibitor of the short-chain fatty acid (SCFA) class. Reactivation of EBV by butyrate has been attributed to histone modifications and chromatin remodeling allowing access of transcription factors to the BZLF1 promoter. Treatment with butyrate, however, results in hyperacetylated histones in lytic cells and in cells refractory to lytic induction (27). Moreover, valproic acid (VPA; 2-propyl-pentanoic acid) (Fig. 1), a branched-chain SCFA and an HDAC inhibitor, does not induce the EBV lytic cycle, even though treatment with VPA results in hyperacetylated histones (28). In fact, VPA blocks viral lytic reactivation by butyrate and by all other inducing stimuli, including azacytidine, anti-IgG in Akata cells, and TPA in Raji cells (29). MS-275 and apicidin, representing two additional classes of HDAC inhibitors, and suberoylanilide hydroxamic acid (SAHA) reactivated EBV in HH514-16 cells; this activity was also inhibited by VPA (29). Conversely, VPA is a strong inducer of the lytic cycle of KSHV (29–31). Since butyrate is a 4-carbon straight-chain fatty acid and VPA is an 8-carbon branched-chain fatty acid, we evaluated the parameters of chain length and branching in lytic activation and repression.
FIG 1.

Structures of fatty acids. SCFAs have 2 to 5 carbon atoms, and MCFAs have 6 to 10 carbon atoms.
Here we study the responses of EBV and KSHV to butyrate, VPA, and 14 other SCFAs and medium-chain fatty acids (MCFAs) (Fig. 1) to provide a structure-function analysis of these fatty acids and their effects on the latent-to-lytic switch of the two viruses. SCFAs and MCFAs occur naturally in the diet, are produced by bacterial metabolism in the colon, and are products of amino acid catabolism (32). Fatty acids have known biological and medicinal properties. For example, 4-phenylbutyrate, an HDAC inhibitor, is used to induce fetal hemoglobin and to promote cancer cell differentiation (33). Isovalerate, found in the valerian plant, and VPA are anticonvulsants (34). We assessed a large panel of fatty acids for activation of the viral lytic cycles, a property of butyrate. By combining each fatty acid with an inducing agent appropriate for each cell line, we also tested the fatty acids for the capacity to block lytic reactivation, a property of VPA. We utilized EBV- and KSHV-infected cell lines in which the lytic cycle was activated by different inducing stimuli to determine if the ability of a fatty acid to prevent lytic gene expression was dependent on the cell background or activation pathway. BC-1 cells, which harbor both viruses, allowed a direct comparison of lytic reactivation of the two human gammaherpesviruses in the same cell background. We analyzed how the chemical and functional properties of the fatty acids correlated with their effects on EBV or KSHV lytic reactivation.
MATERIALS AND METHODS
Cell lines.
The EBV-infected Burkitt's lymphoma cell lines used were HH514-16, which is a subclone of the P3J-HR-1 cell line with low spontaneous lytic reactivation (35), and Raji (36). HH-B2 (9) and BC-3 are KSHV-infected PEL cell lines (37). These four cell lines were cultured in RPMI 1640 containing 8% fetal bovine serum, penicillin (50 U/ml), streptomycin (50 U/ml), and amphotericin B (1 μg/ml). BC-1 cells, a KSHV- and EBV-infected PEL cell line (38, 39), were cultured in RPMI 1640 containing 20% fetal bovine serum and gentamicin (50 μg/ml). All cells were grown at 37°C under 5% CO2.
Chemical treatment of cell lines.
Cells were subcultured at 3 × 105 ml−1; 48 h later, cells at 1 × 106 ml−1 were treated with chemical stimuli for the durations noted in the figure legends. All fatty acids were purchased from Sigma, unless noted otherwise. Sodium butyrate (NaB), sodium valproate (VPA), sodium acetate, sodium hexanoate, sodium octanoate, and sodium phenylbutyrate (Scandinavian Formulas) were prepared as 1 M solutions in water. Propanoic acid, valeric acid, heptanoic acid, 2-ethylbutyric acid, 2-methylbutyric acid, 2-ethylvaleric acid, isobutyric acid, 3-methylvaleric acid, and 4-methylvaleric acid were prepared as 1 M solutions in 1 M sodium hydroxide. A TPA (Calbiochem) stock made in dimethyl sulfoxide (DMSO) was used at 10 or 20 ng/ml. Cell viability was measured by counting cells that excluded staining with trypan blue.
Western blot analysis.
Cells that were left untreated or treated with chemical stimuli were harvested at the times indicated in the figure legends. Total cell extracts were electrophoresed in sodium dodecyl sulfate (SDS)–12% TGX polyacrylamide gels (Bio-Rad) and were transferred to nitrocellulose membranes (Bio-Rad). Antibodies were used to detect KSHV ORF50 (amino acids 351 to 691) (18), histone H3 acetylated at lysine 9 and lysine 14 (EMD Millipore), ZEBRA (BZ1) (40), phospho-H2AX(Ser139) and H2AX (Millipore), phospho-p53(Ser15) and p53 (Cell Signaling), and β-actin (Sigma). Protein levels were determined by densitometry. Immunoreactive protein levels were normalized to β-actin levels and expressed relative to the amounts in the untreated control cells. Data shown are representative of at least two biological replicates.
RT-qPCR.
Total RNA was isolated using an RNeasy kit with on-column DNase digestion (Qiagen). The cells that were the source of RNAs for reverse transcription-quantitative PCR (RT-qPCR) experiments were the same as those that were the source of proteins for the immunoblot experiments. The relative transcript levels were determined with gene-specific primers, using an iScript SYBR green RT-PCR kit (Bio-Rad). The primers used have been described previously (17, 29). Relative expression levels were calculated using the ΔΔCq method and normalized to 18S RNA. RNA samples were assayed in triplicate. Data shown in Fig. 2 to 7 are from representative experiments. Data averaged from biological replicates of all cell lines are presented in Fig. 8.
FIG 2.

Activation and blocking of the EBV lytic cycle by straight-chain fatty acids. EBV-infected HH514-16 cells were treated with a single straight-chain fatty acid alone (A and C) or combined with butyrate (3 mM) (B and D) for 18 h. The concentration of fatty acid was 10 mM unless noted as 3 mM. (A and B) Lytic induction was determined by the relative expression of BZLF1 and BGLF5 mRNAs, measured by RT-qPCR analysis, in triplicate, of RNAs extracted from untreated versus treated cells. Expression is presented relative to stimulation by butyrate (100%). The expression of BZLF1 was induced 11-fold by butyrate compared to that in untreated cells, and that of BGLF5 was induced 150-fold. (C and D) Lytic induction was measured by the level of ZEBRA protein detected on an immunoblot. HDAC inhibition was measured using an anti-acetyl (lysine 9 and lysine 14) H3 rabbit polyclonal antibody (AcH3). Immunoreactive protein levels were normalized to β-actin levels and expressed relative to the amount in the untreated control cells.
FIG 7.

Lytic reactivation of EBV and KSHV by branched-chain fatty acids in dually infected PEL cells. KSHV- and EBV-infected BC-1 cells were treated for 18 h with C-2-branched fatty acids (A and B) or C-3- or C-4-branched fatty acids (C and D) in the absence (A and C) or presence (B and D) of TPA (10 ng/ml). Fatty acids were used at 10 mM unless noted as 3 mM. The relative expression of EBV BRLF1 (top) and KSHV ORF50 (bottom) was measured by RT-qPCR analysis, in triplicate, of RNAs extracted from untreated versus treated cells. In panels B and D, expression is presented relative to stimulation by TPA (100%). TPA induced BRLF1 expression 19- to 78-fold and ORF50 expression 22- to 33-fold in biological triplicate experiments.
FIG 8.

Summary comparing activation or blocking of reactivation of the EBV and KSHV lytic cycles by fatty acids. The data from all EBV+ cell lines tested (HH514-16, Raji, and BC-1) (A and C) or all KSHV+ cell lines tested (HH-B2, BC-3, and BC-1) (B and D) were combined. Activation of EBV (A) or KSHV (B) in cells treated with each fatty acid was compared to that in untreated cells. Repression of EBV (C) or KSHV (D) in cells treated with a fatty acid combined with a known inducing agent (either butyrate or TPA) was compared to that with the known inducing agent alone, set at 100% lytic activation. The values are mean ± standard errors of the means (SEM). **, P < 0.0001; *, P < 0.05. (E) Summary comparing activation (red) or blocking (green) of reactivation of the EBV and KSHV lytic cycles by fatty acids. 1MCFAs block EBV reactivation in cells induced by butyrate.
Classification of the effects of fatty acids on activation and blocking of viral lytic cycles.
Lytic cycle activation was determined by the expression of lytic transactivator genes: BZLF1 or BRLF1 for EBV and ORF50 for KSHV. Activation by known inducing agents (butyrate for HH514-16, HH-B2, and BC-3 cells and TPA for BC-1 and Raji cells) was considered 100% activation. A fatty acid that activated the lytic cycle to >50% compared to induction by butyrate or TPA was classified as an activator. Fatty acids that induced the lytic cycle 20 to 50% were classified as moderate activators. Lytic induction of <20% by a fatty acid was classified as weak or no activation. Blocking of the EBV lytic cycle by each fatty acid was determined by combining the fatty acid with inducing agents that were active in each cell line. A fatty acid classified as a blocking agent decreased lytic induction to <20%. A decrease in lytic induction to 20 to 70% was classified as moderate blocking. A fatty acid was classified as weakly or not blocking when >70% lytic activation remained.
Plasmids and transfection.
Wild-type BZLF1 was expressed from a plasmid containing EBV genomic DNA driven by the cytomegalovirus (CMV) immediate early (IE) promoter in pHD1013 (gZ) (41). HH514-16 cells, subcultured to 3 × 106 cells/ml 2 days prior to the experiment, were transfected by nucleofection with 2 μg of DNA per 5 × 106 cells, using a Nucleofector V kit and program A-023 according to the manufacturer's protocol (Lonza). Cells were treated with fatty acid immediately after transfection and harvested 24 h later.
Luciferase reporter assays.
The fragment of the BZLF1 promoter (Zp) from positions −547 to +12 or −221 to +12 relative to the transcription start site was PCR amplified from the EBV genome of HH514-16 cells, inserted into the ZpCAT plasmid (42), and subcloned into the pGL2-Basic vector to create Zp−547/+12GL2 or Zp−221/+12GL2, respectively. Mutations in Zp reported to disrupt the activity of the ZIIIA/ZIIIB, ZIIR, and ZV/ZV′ response elements (43–45) were generated by site-directed mutagenesis using the following primers and their complements: 5′-CTTTTGGAAACTATGCAgaAttCACAGGatccGCTAATGTAC-3′, 5′-GCCTCAGAGACACACCTAAATTTAGCACggaattcACCATGACATCACAG-3′, and 5′-GGGAGATGTTAGACAGGccACTCACTAAACATTGCAttTTGCCAAGCTTGG-3′. HH514-16 cells were transfected by nucleofection with 1 μg pGL2-Basic or a Zp reporter construct and treated with fatty acids at 1 h posttransfection. Cells were harvested at 48 h posttransfection and lysed in cell culture lysis reagent (Promega). Luciferase assays were performed using a luciferase assay system (Promega). Relative luciferase units for each sample were normalized to the amount of total protein determined by bicinchoninic acid (BCA) assay (Pierce) with cell lysates pretreated with 2 volumes of iodoacetamide (100 mM) at 37°C for 15 min.
RESULTS
Straight-chain SCFAs activate the EBV lytic cycle, while MCFAs block EBV reactivation.
In addition to the well-known property of butyrate as an inducer of the EBV lytic cycle (13), pentanoate (valerate) and propionate also induce expression of viral early antigen and viral capsid antigen (46, 47). However, the effects of MCFAs on EBV reactivation have not been described previously. Here we tested straight-chain fatty acids with 2 to 8 carbon atoms (C2 to C8) as activators and blockers of the EBV lytic cycle. EBV-infected Burkitt's lymphoma cells (HH514-16) were treated with straight-chain fatty acids for 18 h. Lytic reactivation was measured by expression of two viral lytic mRNAs, BZLF1 and BGLF5, and by the amount of ZEBRA protein expressed from the BZLF1 gene. In all experiments, cell viability was >85%, as measured by counting cells that excluded staining with trypan blue (data not shown). Increases in viral lytic gene expression were observed in cells treated with the SCFAs propionate, butyrate, and valerate (C3, C4, and C5), but not acetate (C2), and the MCFAs hexanoate, heptanoate, and octanoate (C6, C7, and C8) (Fig. 2A and C).
VPA (valproate), a branched-chain fatty acid, blocks EBV reactivation by butyrate (28, 29). To determine if any straight-chain fatty acids block viral reactivation induced by butyrate, HH514-16 cells were treated with butyrate plus another straight-chain fatty acid. The lytic cycle was induced by butyrate in the presence of acetate or by butyrate together with the SCFAs that activated it alone, i.e., propionate and valerate (Fig. 2B and D). However, the addition of hexanoate in combination with butyrate led to 60 to 80% decreases in the amounts of BZLF1 and BGLF5 mRNAs and ZEBRA protein compared with those in cells treated with butyrate alone. No EBV lytic gene expression was detected when heptanoate or octanoate was added in addition to butyrate.
We also examined the effects of straight-chain fatty acids on EBV lytic reactivation in the Burkitt's lymphoma cell line Raji. The EBV lytic gene BRLF1 in Raji cells was not activated by butyrate or any other fatty acid. The protein kinase C agonist TPA activates the lytic cycle in Raji cells, and butyrate synergizes with TPA (28, 48). Valerate, which activated EBV in HH514-16 cells, also synergized with TPA in Raji cells (see Fig. 8). Hexanoate, heptanoate, and octanoate blocked EBV reactivation induced by TPA. The same medium-chain fatty acids blocked lytic reactivation induced by butyrate in HH514-16 cells and by TPA in Raji cells. We concluded that blocking of EBV lytic reactivation is related to the length of the fatty acid carbon chain.
Straight-chain SCFAs that activate the EBV lytic cycle are HDAC inhibitors; however, MCFAs that block EBV reactivation do not prevent HDAC inhibition.
SCFAs are general inhibitors of class I and class II histone deacetylases in many cell types (49, 50). In HH514-16 Burkitt's lymphoma cells, propionate, butyrate, and valerate behaved as HDAC inhibitors, increasing the levels of acetylated histone H3 (AcH3) (Fig. 2C, lanes 3 to 5). These SCFAs also activated EBV (Fig. 2A). When each of the other SCFAs was combined with butyrate, the level of AcH3 increased to levels comparable to those seen in cells treated with butyrate alone (Fig. 2D). Hexanoate, heptanoate, and octanoate blocked EBV reactivation (Fig. 2B) but did not prevent increased histone H3 acetylation by butyrate (Fig. 2D, lanes 6 to 10).
Straight-chain SCFAs also activate the KSHV lytic cycle, but MCFAs do not block KSHV lytic transcript reactivation.
The lytic cycle of KSHV can be activated by butyrate (9, 19, 51). VPA also promotes lytic activation of KSHV, unlike its blocking effect in EBV (29, 30). The effects of fatty acids other than butyrate and VPA on KSHV lytic reactivation had not previously been examined. KSHV+ HH-B2 PEL cells were treated with fatty acids for 12 h, and lytic activation was measured by expression of ORF50, encoding the KSHV lytic transactivator Rta protein (7, 9). Compared to that in untreated or acetate-treated HH-B2 cells, ORF50 mRNA was expressed to 240-fold higher levels in cells treated with propionate, butyrate, or valerate (C3, C4, and C5) (Fig. 3A); ORF50 protein expression was also stimulated by these SCFAs (Fig. 3B). Treatment of HH-B2 cells with hexanoate or heptanoate induced ORF50 mRNA to about 50% the level induced by butyrate; octanoate induced ORF50 mRNA to only ∼20% the level induced by butyrate. Moreover, MCFAs did not reduce the abundance of ORF50 mRNA after treatment of cells with butyrate (Fig. 3A). We concluded that MCFAs stimulate and do not block expression of KSHV ORF50 mRNA (Fig. 3A). The same MCFAs fail to stimulate and do block expression of EBV lytic mRNAs. However, the ORF50 protein was not detected in HH-B2 cells treated with heptanoate or octanoate. These two MCFAs at 10 mM, but not 3 mM, blocked ORF50 protein expression (Fig. 3C). There may be separate effects of MCFAs on gene expression and protein translation or protein stability.
FIG 3.

Activation and blocking of the KSHV lytic cycle by straight-chain fatty acids. KSHV-infected HH-B2 cells were treated with a single straight-chain fatty acid or with butyrate (3 mM) combined with another straight-chain fatty acid (10 mM) for 12 h. (A) Lytic induction was determined by the relative expression of ORF50 mRNA, measured by RT-qPCR analysis, in triplicate, of RNAs extracted from untreated versus treated cells. Expression is presented relative to stimulation by butyrate (100%). ORF50 mRNA was induced 240-fold by butyrate compared to that in untreated cells. (B and C) Lytic induction was measured by the ORF50 protein levels on an immunoblot probed with anti-ORF50 antibody.
The effects of straight-chain fatty acids on KSHV lytic reactivation were measured in another PEL cell line, BC-3, where butyrate was the strongest inducer of the lytic cycle. Propionate, valerate, and hexanoate also induced ORF50 expression (see Fig. 8). Heptanoate and octanoate did not induce KSHV ORF50 expression in BC-3 cells. When combined with butyrate, none of the straight-chain fatty acids reduced ORF50 mRNA expression by more than 50%. The results for two KSHV+ PEL cell lines, HH-B2 and BC-3, were similar in that medium-chain fatty acids (C6 and C8) did not reduce initiation of KSHV lytic reactivation as measured by ORF50 mRNA. We concluded that MCFAs block lytic reactivation of EBV more efficiently than that of KSHV.
Activation and blocking of EBV and KSHV lytic cycles in coinfected cells.
The effects of fatty acids on EBV and KSHV reactivation were compared directly in BC-1 primary effusion lymphoma cells, which are infected with both viruses. BC-1 cells were left untreated or treated with each fatty acid alone for 18 h; total RNA was extracted, and viral mRNAs encoding lytic cycle activators of both viruses were detected simultaneously. EBV lytic activation was measured by expression of BRLF1, which encodes the EBV Rta transactivator protein; KSHV activation was measured by expression of ORF50, encoding KSHV Rta. Both EBV and KSHV were induced by propionate, butyrate, and valerate (Fig. 4A). KSHV was activated moderately by hexanoate and weakly by heptanoate or octanoate. EBV was not activated by these MCFAs. These data were consistent with results for cell lines infected by either virus alone, showing that MCFAs activate lytic gene expression of KSHV but not EBV.
FIG 4.

Lytic reactivation of EBV and KSHV by straight-chain fatty acids in dually infected cells. KSHV- and EBV-infected BC-1 cells were treated with a single fatty acid (10 mM unless noted as 3 mM) (A) or with TPA (20 ng/ml) combined with a straight-chain fatty acid (B) for 18 h. The relative expression of EBV BRLF1 (top) and KSHV ORF50 (bottom) was measured by RT-qPCR analysis, in triplicate, of RNAs extracted from untreated versus treated cells.
In BC-1 cells, KSHV was strongly induced by propionate, butyrate, and valerate, while EBV was activated to a lower level (Fig. 4A). However, EBV and KSHV were induced to comparable levels by TPA in this cell background. Therefore, to test for blocking of lytic activation by straight-chain fatty acids, we compared lytic induction by TPA in the absence and presence of fatty acids. Acetate did not inhibit lytic induction of either virus by TPA. Propionate, butyrate, and valerate increased expression of lytic mRNAs of both EBV and KSHV in the presence of TPA (Fig. 4B). Hexanoate (C6), which blocked EBV lytic reactivation in HH514-16 and Raji cells, did not block EBV BRLF1 expression induced by TPA in BC-1 cells, possibly due to the presence of KSHV in these cells. Hexanoate increased the expression of KSHV ORF50 by TPA compared to that with TPA alone, consistent with the low-level activation of KSHV by hexanoate alone. Expression of EBV BRLF1 was blocked when cells were treated with octanoate (C8) plus TPA compared to TPA alone. Blocking of the EBV lytic cycle by octanoate was observed in all cell lines. Octanoate did not block KSHV ORF50 mRNA reactivation by TPA in BC-1 cells. We concluded that octanoate selectively blocks EBV lytic gene expression.
Isovalerate is the only branched-chain fatty acid that activates the EBV lytic cycle.
The chemical structures of butyrate and VPA differ in the total number of carbon atoms, but also in the absence and presence of branching; butyrate is a straight chain, whereas VPA (2-propylvalerate) is branched at the second carbon (C-2) (Fig. 1). To test whether branching helps to explain the difference between the effects of butyrate and VPA on EBV, we tested other branched-chain fatty acids as inducers or repressors of viral reactivation. We first examined fatty acids branched at the second carbon, like VPA, which has 8 carbon atoms. The other C-2-branched fatty acids were composed of 4, 5, or 6 carbon atoms. For example, isobutyrate has 4 carbon atoms, the same number as butyrate. The EBV lytic cycle in HH514-16 cells was not reactivated by any C-2-branched fatty acid, including VPA, isobutyrate (2-methylpropionate), 2-methylbutyrate, 2-ethylbutyrate, and 2-methylvalerate (Fig. 5A and C).
FIG 5.

Activation and blocking of the EBV lytic cycle by branched-chain fatty acids. EBV-infected HH514-16 cells were treated with fatty acids branched at C-2 (A and C), C-3, or C-4 (B and D) in the absence or presence of butyrate for 18 h. The concentration of fatty acid was 10 mM unless noted as 3 mM. (A and B) Lytic induction was determined by the relative expression of BZLF1 and BGLF5, measured by RT-qPCR analysis, in triplicate, of RNAs extracted from untreated versus treated cells. Expression is presented relative to stimulation by butyrate (100%). The expression of BZLF1 was induced 23-fold by butyrate compared to that in untreated cells, and that of BGLF5 was induced 150-fold. (C and D) Lytic induction was measured by an immunoblot probed with anti-ZEBRA antibody. HDAC inhibition was measured using an anti-acetyl (lysine 9 and lysine 14) H3 rabbit polyclonal antibody (AcH3).
To gain further insight about the structural properties of fatty acids that affect EBV activation, the C-3 branch location in the fatty acid was examined. We found that isovalerate (3-methylbutyrate)-treated EBV+ HH514-16 cells expressed BZLF1 and BGLF5 mRNAs and ZEBRA protein to levels comparable to those stimulated by butyrate (Fig. 5B and D). Interestingly, there was no comparable increase in EBV lytic gene expression in HH514-16 cells treated with 3-methylvalerate, 4-methylvalerate, or phenylbutyrate. Thus, only a single C-3-branched fatty acid, isovalerate, activated EBV lytic gene expression.
Several branched-chain fatty acids are repressors of EBV.
To determine if C-2-branched fatty acids other than VPA blocked EBV reactivation, HH514-16 cells were treated with the combination of butyrate and a branched-chain fatty acid (Fig. 5A and C). Compared to butyrate alone, isobutyrate plus butyrate did not decrease ZEBRA protein expression. The addition of isobutyrate decreased BZLF1 and BGLF5 mRNA levels, but this was not statistically significant among biological replicates (see Fig. 8). 2-Methylbutyrate did not decrease levels of EBV lytic mRNAs or ZEBRA protein compared to those with butyrate alone. However, 2-ethylbutyrate and 2-methylvalerate decreased both the levels of lytic mRNAs and ZEBRA protein by half. Of the C-2-branched fatty acids, VPA was unique in completely blocking the expression of BZLF1 and BGLF5 induced by butyrate. VPA does not globally block transcription, as VPA induces the expression of many cellular genes (29). Also, the same number of cells remained viable after VPA treatment and butyrate treatment for 18 h in this experiment, and for 48 h as well (data not shown).
Fatty acids branched at C-3 and C-4 were also examined as blockers of the EBV lytic cycle (Fig. 5B and D). The combination of isovalerate and butyrate stimulated the same level of BZLF1 and BGLF5 gene expression as that with butyrate alone. 3-Methylvalerate, 4-methylvalerate, and phenylbutyrate, however, decreased BGLF5 and BZLF1 RNA and protein expression induced by butyrate. 4-Methylvalerate was a slightly more potent blocker than 3-methylvalerate. Phenylbutyrate was the most potent blocking agent; no lytic RNA or ZEBRA protein was detected in the presence of phenylbutyrate. The blocking effects of phenylbutyrate that we observed were comparable to those of VPA.
The effects of branched-chain fatty acids were also tested in the EBV+ Raji cell line, in which the lytic cycle is activated by TPA and butyrate synergizes with TPA. BRLF1 mRNA was induced to the same level in Raji cells treated with TPA or a combination of TPA and the C-2-branched fatty acid isobutyrate or 2-methylbutyrate; 2-ethylbutyrate partially blocked BRLF1 mRNA expression compared to TPA alone (see Fig. 8). VPA, however, completely blocked BRLF1 mRNA expression in Raji cells. The activity of fatty acids branched at C-3 or C-4 in Raji cells was similar to their properties in HH514-16 cells. Isovalerate, the only branched-chain fatty acid to activate EBV in HH514-16 cells, also synergized with TPA in Raji cells. The addition of 3-methylvalerate or 4-methylvalerate to TPA decreased BRLF1 expression versus that with TPA alone. Phenylbutyrate, a strong blocker of butyrate induction of EBV in HH514-16 cells, completely blocked TPA-induced EBV BRLF1 expression in Raji cells.
The capacity of branched-chain fatty acids to reactivate or block EBV does not correlate with their behavior as HDAC inhibitors.
Butyrate and other HDAC inhibitors, such as TSA and MS-275, induce the EBV lytic cycle. VPA, which is also an HDAC inhibitor (52), fails to activate the EBV lytic cycle and blocks all inducing stimuli (Fig. 5; also see Fig. 7) (28, 29). Among the branched-chain fatty acids, we identified examples of HDAC inhibitors that reactivate EBV, that block EBV reactivation, and that have no effect on induction of the lytic cycle. Isovalerate, a documented HDAC inhibitor (53) that caused a 7-fold induction of AcH3 in our experiments, was the only branched-chain fatty acid to activate EBV (Fig. 5D). The EBV lytic cycle was blocked by phenylbutyrate. Phenylbutyrate, also a known HDAC inhibitor, produced a 3-fold increase in AcH3 (Fig. 5D). Other branched-chain fatty acids, i.e., 2-ethylbutyrate, 2-methylvalerate, 3-methylvalerate, and 4-methylvalerate, increased the level of AcH3 above background, though not to the same extent as that with butyrate; yet each decreased EBV lytic reactivation. Isobutyrate, an HDAC inhibitor (49), caused a 15-fold increase in the level of AcH3 in treated cells compared to untreated cells (Fig. 5C). However, isobutyrate did not activate EBV in any cell line tested. Isobutyrate also did not block expression of ZEBRA protein induced by butyrate in HH514-16 cells or BRLF1 mRNA expression induced by TPA in Raji or BC-1 cells (Fig. 5; also see Fig. 7). 2-Methylbutyrate also did not activate or block EBV reactivation, but it produced an increase in AcH3. No fatty acid prevented the increase in AcH3 in cells treated with butyrate. Therefore, neither activation nor blocking of the EBV lytic cycle can be explained by global effects of the HDAC inhibitor activity of any fatty acid.
Branched-chain fatty acids can activate, and do not block, KSHV lytic reactivation.
One major difference between lytic induction of EBV and KSHV is that only KSHV is activated by VPA (29, 30). We identified isobutyrate as another branched-chain fatty acid that activated KSHV but not EBV (Fig. 6A). Among the other C-2-branched fatty acids, low levels of ORF50 mRNA and protein were stimulated by 2-ethylbutyrate; 2-methylbutyrate and 2-methylvalerate did not cause an increase in ORF50 expression. None of the C-2-branched fatty acids blocked ORF50 expression induced by butyrate. With the C-3- and C-4-branched fatty acids, lytic gene expression increased in isovalerate-treated HH-B2 cells (Fig. 6B), as it did in EBV+ cells. Though 4-phenylbutyrate blocked EBV lytic reactivation, in HH-B2 cells it potently induced KSHV ORF50 expression. 3-Methylvalerate and 4-methylvalerate also increased ORF50 expression. None of the C-3- or C-4-branched fatty acids blocked ORF50 expression induced by butyrate (Fig. 6B). In BC-3 cells, another PEL cell line, the KSHV lytic cycle was activated by the branched-chain fatty acids isobutyrate, valproate, isovalerate, and phenylbutyrate (see Fig. 8), which also promoted the KSHV lytic cycle in HH-B2 cells.
FIG 6.
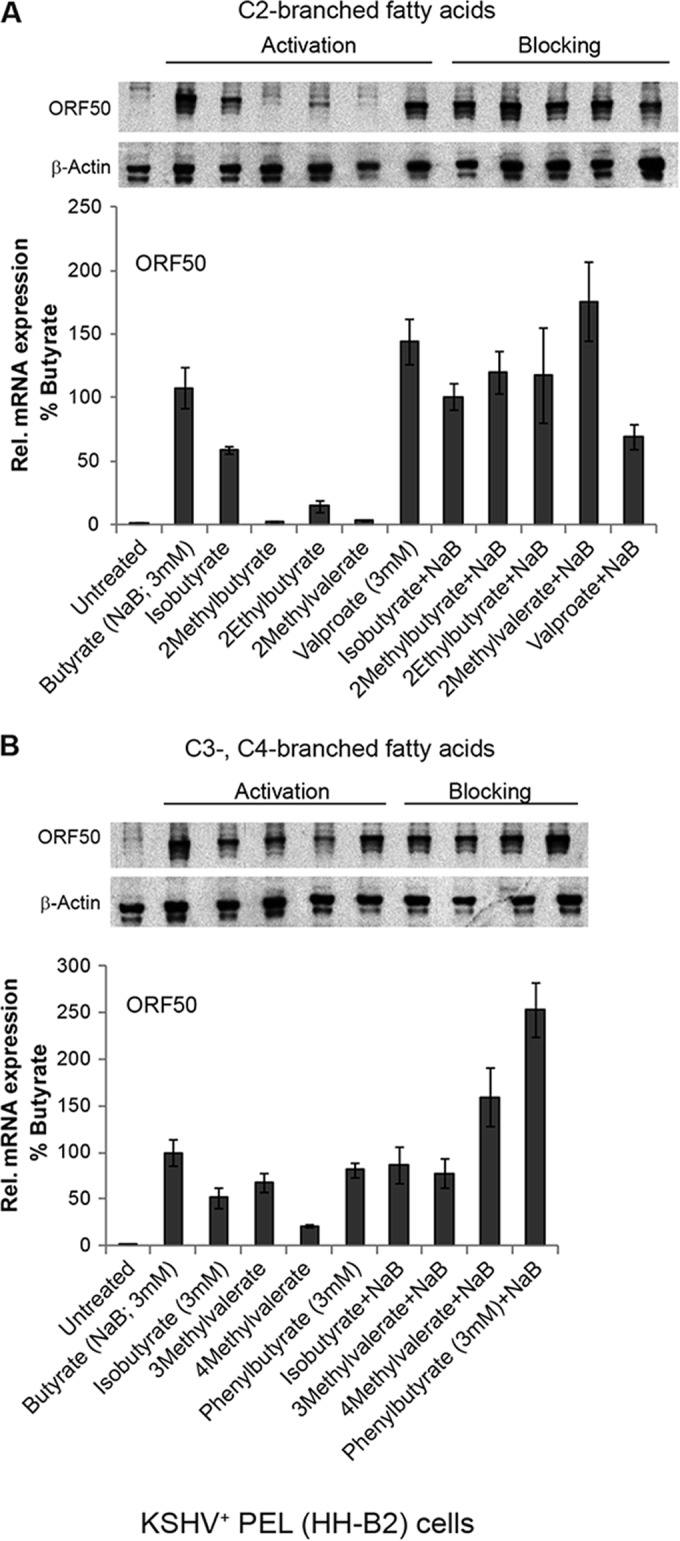
Activation and blocking of the KSHV lytic cycle by branched-chain fatty acids. KSHV-infected HH-B2 cells were treated with C-2-branched fatty acids (A) or C-3- or C-4-branched fatty acids (10 mM unless noted as 3 mM) (B) in the absence or presence of butyrate (3 mM) for 12 h. Lytic induction was determined by the relative expression of ORF50, measured by RT-qPCR analysis, in triplicate, of RNAs extracted from untreated versus treated cells. Expression is presented relative to stimulation by butyrate (100%). ORF50 mRNA was induced 108-fold by butyrate compared to that in untreated cells. Lytic induction was also measured by an immunoblot probed with anti-ORF50 antibody.
EBV and KSHV lytic cycle activation and blocking by branched-chain fatty acids in coinfected cells.
The effects of branched-chain fatty acids on lytic reactivation were compared in BC-1 PEL cells coinfected with EBV and KSHV. None of the C-2-branched fatty acids induced EBV in BC-1 cells (Fig. 7A, top panel). The EBV lytic cycle was induced by isovalerate and weakly induced by 3-methylvalerate and 4-methylvalerate, but not by phenylbutyrate (Fig. 7C, top panel). KSHV ORF50 mRNA, however, was activated strongly by isobutyrate, valproate (Fig. 7A, bottom panel), isovalerate, and phenylbutyrate and moderately by 3-methylvalerate and 4-methylvalerate (Fig. 7C, bottom panel). The branched-chain fatty acids were tested for prevention of lytic cycle reactivation induced by TPA, which induces both viruses. Again, no fatty acid blocked the KSHV lytic cycle (Fig. 7B, bottom panel, and D, bottom panel). EBV reactivation was blocked most effectively by valproate (Fig. 7B, top panel). The results for the dually infected BC-1 cells confirm that isobutyrate, VPA, and 4-phenylbutyrate are potent activators of KSHV but not EBV. The activating and blocking effects of short- and medium-chain fatty acids on the EBV and KSHV lytic cycles in all cell lines tested are compared in Fig. 8.
Effects of fatty acids on the EBV, but not KSHV, lytic cycle correlate with fatty acid lipophilicity.
We sought a chemical property that predicts the effects of fatty acids on viral lytic activation. The acid dissociation constants (pKa) of all fatty acids were similar (4.76 to 4.85) and did not correlate with effects on either virus (data not shown). Intriguingly, the lipophilicity values of fatty acids predicted the blocking of EBV. Lipophilicity, or hydrophobicity, is quantified by calculating the octanol-water partition coefficient, or distribution coefficient, at the biologically relevant pH 7.4. Fatty acids that activated EBV had lipophilicity values (LogD) between −2.4 and −1.4 (Fig. 9A). Fatty acids with LogD values in this range, but with branching at the second carbon (isobutyrate and 2-methylbutyrate), did not activate EBV. This structural feature prevents activation. Fatty acids with LogD values of −1.0 to −0.3 decreased EBV lytic reactivation, while the fatty acids with LogD values of >0 completely blocked activation of the EBV lytic cycle. For KSHV, this correlation of fatty acid lipophilicity with viral reactivation was not observed. Fatty acids that either had no effect or caused reactivation of KSHV had LogD values that spanned the entire range (Fig. 9B).
FIG 9.
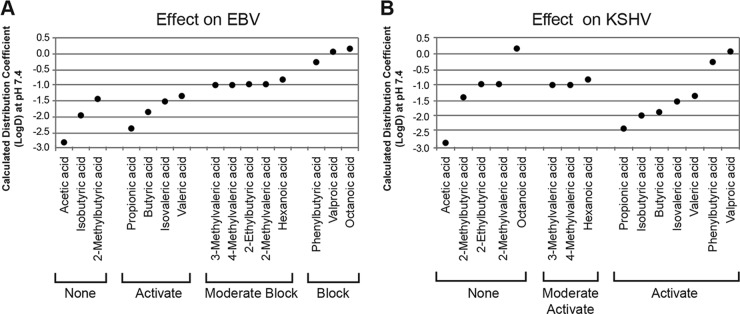
Correlation between lipophilicity and activation or blocking of EBV and KSHV lytic cycles. The calculated octanol-water distribution coefficients at pH 7.4 (LogD) (ChEMBL database [90]) of the straight- and branched-chain fatty acids, an assessment of lipophilicity, were plotted against their effects on EBV (A) and KSHV (B) lytic reactivation.
VPA inhibits activation of the EBV BZLF1 promoter (Zp).
We performed luciferase reporter assays to determine if fatty acids activate or block expression from the BZLF1 promoter (Zp) (Fig. 10A). Fatty acids that induced the EBV lytic cycle in HH514-16 cells also induced expression of a Zp luciferase reporter (Fig. 10B). Similarly, fatty acids that failed to induce the lytic cycle also failed to activate Zp-GL2. Furthermore, VPA blocked expression of Zp−221/+12GL2 induced by butyrate (Fig. 10C). This experiment supports the hypothesis that VPA acts at the level of Zp to decrease transcription. Since the reporter assays were conducted in EBV-positive cells and Zp is autostimulated by the ZEBRA protein, Zp reporter activation might occur through induction of ZEBRA protein produced from the viral genome. A reporter containing mutations within the ZIIIA and ZIIIB sites, where autostimulation occurs (43), was still induced by butyrate but not by VPA; butyrate induction was blocked by VPA (Fig. 10C).
FIG 10.

Fatty acids alter activation of the BZLF1 promoter (Zp). (A) Schematic diagram of the EBV BZLF1 promoter (Zp) from positions −221 to +12 relative to the transcription start site, with known response elements labeled. (B) Effects of short-chain fatty acids (10 mM, except for butyrate [3 mM]) on the expression of luciferase regulated by a fragment of Zp from positions −547 to +12. HH514-16 cells were transfected with Zp−547/+12-GL2 by nucleofection and incubated at 37°C for 1 h. Fatty acids were then added to cells from one transfection divided into a 96-well plate for 48 h. (C and D) Effects of butyrate, VPA, or the fatty acids combined on the expression of Zp−221/+12-GL2, either wild type or with inactivating mutations in the ZIIIA/ZIIIB, ZIIR, ZV/ZV′, or ZIIR/ZV/ZV′ elements. The luciferase activities were normalized to the total protein level. The data are averages for at least three separate transfections. RLU, relative light units.
Negative regulatory elements, namely, ZIIR and ZV/ZV′, have been identified in Zp (44, 54, 55). To test the hypothesis that VPA blocks expression of BZLF1 via these elements, we measured expression of a luciferase reporter controlled by Zp with mutations known to disrupt the function of either or both of these repressor elements. We found, as expected, that induction of the Zp luciferase reporter by butyrate was increased ∼6- to 8-fold in the ZIIR and ZIIR/ZV/ZV′ combined mutants. VPA blocked induction by butyrate of Zp reporters with mutations in ZIIR and ZV/ZV′ (Fig. 10D). We concluded that the inhibitory effects of VPA at Zp are not mediated by those sites in Zp that are known to be involved in autostimulation or constitutive repression.
VPA does not block the transcriptional activation function of ZEBRA protein.
Our results demonstrating that VPA blocks activity at Zp do not exclude the possibility that VPA also inhibits the function of ZEBRA protein. To examine this possibility, we scored for EBV lytic proteins when ZEBRA was overexpressed in the absence or presence of VPA (Fig. 11). EBV-positive HH514-16 cells were nucleofected with empty vector or a plasmid that expresses the ZEBRA protein under the control of the CMV IE promoter. The cells were left untreated or treated with butyrate (NaB), VPA, or NaB plus VPA for 24 h. Unlike its effect on Zp, VPA did not block expression of ZEBRA driven by the CMV IE promoter in the plasmid. In fact, both butyrate and VPA enhanced ZEBRA expression from the plasmid. VPA did not inhibit the ability of ZEBRA protein to activate expression of the EBV early lytic proteins Rta and EA-D. We concluded that VPA does not exert an inhibitory effect on the capacity of ZEBRA to stimulate expression of early lytic genes.
FIG 11.
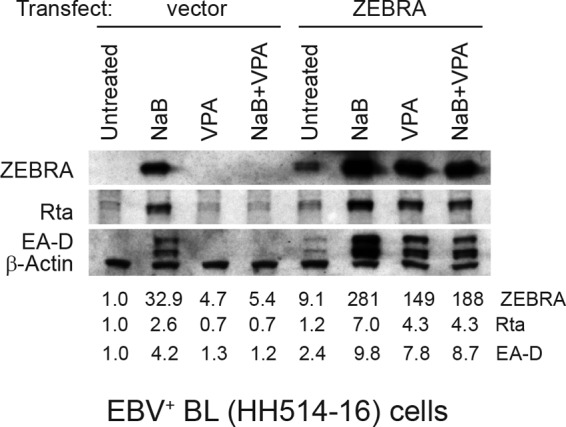
EBV early proteins are expressed in the presence of VPA when ZEBRA is overexpressed. HH514-16 cells were transfected with empty vector or a plasmid that expresses the ZEBRA protein under the control of the CMV IE promoter. Immediately after transfection, the cells were treated with butyrate (NaB; 3 mM), VPA (10 mM), or NaB plus VPA for 24 h. Expression of Rta, EA-D, ZEBRA, and β-actin was measured by immunoblotting. Data are representative of biological triplicate experiments.
VPA does not block activation of the ATM/p53 pathway prior to EBV lytic induction.
Induction of the EBV lytic cycle by HDAC inhibitors has been shown to require p53 and ATM kinase (56–59). To test the hypothesis that VPA blocks EBV lytic induction by blocking the ATM pathway, we compared the effects of butyrate and VPA treatment of HH514-16 cells on phosphorylation of H2AX (γH2AX), a measure of ATM activation during the DNA damage response. A time course experiment showed that the ZEBRA protein was expressed after 15 h of butyrate treatment (Fig. 12A), and BZLF1 mRNA was detected by 8 h in butyrate-induced HH514-16 cells (23). We compared times upstream of lytic reactivation (2 and 4 h) with 24 h, when ZEBRA was strongly expressed. Total H2AX levels were constant in untreated cells and in those treated with either butyrate or VPA when monitored at 2, 4, and 24 h (Fig. 12B). We did not detect any change in the levels of γH2AX in cells treated with butyrate or VPA or a combination of the two fatty acids for 2 or 4 h. Increased γH2AX was detected at 24 h only in butyrate-treated cells (Fig. 12B, lane 7), not in cells treated with VPA or both fatty acids. Therefore, induction of γH2AX was coincident with EBV reactivation. This experiment shows that induction of the DNA damage response that accompanies lytic activation is blocked by VPA but that VPA neither stimulates nor inhibits the DNA damage response preceding lytic activation.
FIG 12.
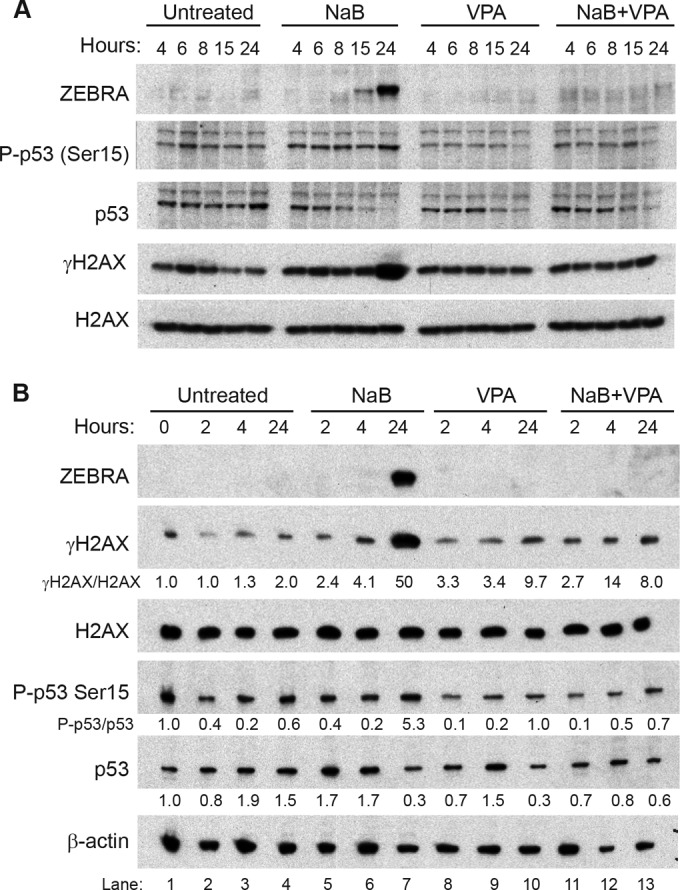
Effects of butyrate and VPA on activation of H2AX and p53 at early and late times. EBV-infected HH514-16 cells were treated with butyrate (3 mM) and/or VPA (10 mM) for 4, 6, 8, 15, or 24 h (A) or 0, 2, 4, or 24 h (B). H2AX, γH2AX, p53, and phospho-p53(Ser15) levels in cell lysates were measured by immunoblotting. The ratios of γH2AX to total H2AX and phospho-p53(Ser15) to total p53 are presented relative to those in untreated cells at time zero. Lytic induction was measured by the level of ZEBRA protein. Data are representative of at least triplicate biological experiments.
We tested the hypothesis that VPA prevents expression of ZEBRA by altering the p53 protein. We investigated the effects of butyrate and VPA on levels of total p53 protein and activation of p53, as detected by phosphorylation at serine 15 at times before and after induction of the EBV lytic cycle. After 24 h of butyrate treatment in HH514-16 cells, when the lytic cycle was reactivated, we observed a 5-fold decrease in total p53 (Fig. 12B, compare lanes 4 and 7). Since ZEBRA was induced by butyrate at this time, this result is consistent with reports of degradation of p53 by ZEBRA protein (60). However, VPA also decreased total p53 protein levels at 24 h, without activating the lytic cycle. Even though total p53 was decreased in both butyrate- and VPA-treated cells, phospho-p53(Ser15) increased only in butyrate-treated cells. Cells treated with either VPA alone or both butyrate and VPA did not demonstrate increased levels of phospho-p53(Ser15) at 24 h. Induction of the EBV lytic cycle has been shown to induce DNA damage response pathways (61). Therefore, at 24 h, the inhibition of induction of the DNA damage response by VPA correlates with its capacity to inhibit the lytic cycle. Treatment with either butyrate or VPA alone, or the two fatty acids combined, resulted in no robust changes in total p53 or phospho-p53(Ser15) levels at 2 or 4 h (Fig. 12B). We obtained no evidence that VPA alters the ATM/p53 pathway in order to block initiation of EBV lytic activation.
DISCUSSION
Reactivation of herpesviruses from latency is required for the viruses to persist and to spread among cells and hosts. Environmental conditions induce herpesviruses to replicate (62, 63), but the mechanisms by which these stimuli function in vivo have not been elucidated. The EBV and KSHV lytic cycles can be induced in cell culture by stimuli including butyrate, an SCFA and an HDAC inhibitor. However, VPA, which is also an SCFA and an HDAC inhibitor, induces the KSHV lytic cycle, but VPA does not reactivate EBV and blocks EBV reactivation. Why do butyrate and VPA have opposing effects on EBV? Are fatty acids endogenous chemical promoters and suppressors of viral lytic reactivation? The following conclusions from our work address these questions. (i) Fatty acids that are HDAC inhibitors induce KSHV, but HDAC inhibition by fatty acids does not predict their activity toward EBV reactivation. (ii) No fatty acid tested blocks KSHV lytic gene expression. (iii) The EBV lytic cycle is blocked by the more lipophilic MCFAs at the BZLF1 promoter.
Structure-function analysis of short- and medium-chain fatty acids and viral lytic reactivation.
To investigate different effects of butyrate and VPA on EBV and KSHV lytic reactivation, we performed a structure-function analysis of a panel of SCFAs and MCFAs with straight and branched carbon chains. The data summary shows that individual fatty acids may activate, block, or have no effect on lytic reactivation of EBV and KSHV (Fig. 8). The results demonstrate that the length of the carbon chain in straight-chain fatty acids, which relates to lipophilicity (Fig. 9), greatly influences the effect on EBV lytic activation.
VPA is branched at the second carbon. Therefore, we investigated whether any other C-2-branched fatty acid could reactivate EBV or whether C-2 branching promotes blocking of EBV reactivation. EBV was not activated by any C-2-branched fatty acid, including isobutyrate, which has the same number of carbon atoms as butyrate. Therefore, the lipophilicity of a fatty acid, which is related to the total number of carbon atoms, does not solely determine EBV activation. Rather, the results show that C-2 branching prevents a fatty acid from activating the EBV lytic cycle. This structural feature may prevent binding of the SCFA to a specific receptor that leads to lytic activation. Branching at the third carbon, however, does not prevent activation, since EBV was reactivated by isovalerate (Fig. 5B and D and 7C) (46).
C-2 branching by itself is not sufficient for blocking, since some C-2-branched fatty acids, such as isobutyrate and 2-methylbutyrate, did not block EBV reactivation. In addition, fatty acids branched at C-3 or C-4, such as 3-methylvalerate and 4-phenylbutyrate, blocked EBV reactivation induced by butyrate. Our observation that phenylbutyrate blocks EBV lytic activation differs from other reports where phenylbutyrate was found to upregulate EBV lytic genes in BL cells or in nasopharyngeal carcinoma cells after 3 days of exposure (64, 65). Regardless of branch location, the more lipophilic fatty acids blocked EBV lytic reactivation (Fig. 9). Our findings are consistent with studies showing that the EBV lytic cycle is inhibited by saturated and unsaturated long-chain fatty acids (66), as well as the lipophilic small molecules moronic acid and resveratrol (67, 68). The blocking of EBV reactivation by lipophilic molecules may be mediated by a receptor that recognizes lipophilic molecules or by interactions between the fatty acids and lipids in the cell membrane generating a cellular response that prevents triggering of the EBV lytic cycle.
The KSHV lytic cycle is induced by the smallest and largest C-2-branched fatty acids tested, isobutyrate and VPA, but not other C-2-branched fatty acids. There is no correlation between lipophilicity and KSHV reactivation. Similarly, fatty acids branched at C-3 or C-4, with low or high lipophilicity, i.e., isovalerate and phenylbutyrate, were more potent activators of KSHV than fatty acids with intermediate lipophilicity, such as 4-methylvalerate. Unlike EBV, no fatty acid blocked KSHV reactivation, regardless of lipophilicity or branch structure.
EBV and KSHV reactivate from latency by different pathways.
Our work shows that branched-chain MCFAs produce different outcomes on EBV and KSHV lytic reactivation. These findings add to accumulating evidence that despite many similarities, the two genetically related gammaherpesviruses behave differently upon entering the lytic cycle. The latent-to-lytic switch of both viruses can be activated in cell culture by similar chemical stimuli, such as HDAC inhibitors and PKC agonists. However, new protein synthesis is required before EBV, but not KSHV, lytic activation (17). A second difference is in the responses of the two viruses to HDAC inhibitors. There is an invariant correlation between HDAC inhibition and lytic activation of KSHV. Changes in histone acetylation, chromatin structure, and nucleosome positioning near the ORF50 promoter have been observed during KSHV reactivation by butyrate (69). However, HDAC inhibitors may not only target histones in KSHV; they could also alter acetylation of nonhistone proteins involved in ORF50 transcription, such as Sp1 (18, 69), latency-associated nuclear antigen (70), or cohesin (71).
In EBV, histone-modifying enzymes and markers of open chromatin, such as acetylation, are detected at the BZLF1 and BRLF1 promoters in cells where the lytic cycle has been activated by HDAC inhibitors (72, 73). However, not all HDAC inhibitors activated EBV (Fig. 2 and 5). Additionally, histone H3 acetylation is increased at EBV transactivator promoters by VPA, which does not induce the lytic cycle, and by butyrate in the refractory subpopulation of cells and in cell lines in which the EBV lytic cycle is not induced by butyrate (27, 28). Therefore, control of the EBV latent-to-lytic switch may involve a more complicated model of chromatin remodeling including other histone modifications and acetylation of nonhistone proteins (74, 75).
Possible mechanisms of inhibition of EBV lytic reactivation by VPA.
We investigated three different but not mutually exclusive mechanisms to explain how VPA blocks EBV reactivation. We found that VPA did not inhibit the transcriptional activation function of exogenously expressed ZEBRA protein. However, VPA blocked butyrate-induced activation of the BZLF1 promoter (Zp). VPA blocking was not attributed to autostimulation or the ZIIR or ZV/ZV′ negative-response elements. We investigated the effects of butyrate and VPA on the ATM/p53 pathways, which have been implicated in EBV lytic reactivation. VPA blocked activation of the ATM and p53 pathways that accompany lytic reactivation by butyrate, but VPA did not alter γH2AX or p53 temporally upstream of lytic activation. The effects of fatty acids on cellular signaling pathways may be transitory and not identified at the times examined. Another possibility is that only a small percentage of cells respond to the fatty acids, which would not be detectable in biochemical assays. Numerous cellular transcription factors bind to Zp, and new protein synthesis is required to activate Zp. Further work is required to learn how fatty acids activate or repress Zp by modulating the expression or function of cellular gene products.
Short- and medium-chain fatty acids are naturally occurring metabolites.
Exposure of virally infected cells to fatty acids in vivo may impinge on the process of viral reactivation. The SCFAs acetate, propionate, and butyrate are produced in large amounts by bacteria in the large intestine; butyrate reaches a concentration of 20 mM in the colon. Lymphocytes latently infected with EBV or KSHV may be exposed to a high concentration of SCFAs during immune surveillance of the gut. SCFAs are produced by bacteria present in the mouth and pharynx, sites of reactivation of EBV-infected tonsillar B cells. The culture medium of oral bacterial species induces the lytic cycle of EBV (76) and KSHV (77). Millimolar concentrations of propionate and butyrate (30 mM and 8 mM, respectively) and trace amounts of isobutyrate and isovalerate are present in dental plaque (78, 79). The herpesviruses EBV, human cytomegalovirus, human herpesvirus 6 (HHV-6), HHV-7, and KSHV can be recovered from gums of patients with periodontal disease (80, 81). SCFAs such as isobutyrate, which we found to activate only KSHV, and propionate, butyrate, and isovalerate, which induce both EBV and KSHV, can be detected in the bloodstream (32).
SCFAs and MCFAs are also naturally found in foods. In children on a diet high in medium-chain triglycerides for the treatment of obesity, hypertension (82), and epilepsy (83), the average plasma concentrations of octanoate and decanoate were 0.3 mM and 0.16 mM, respectively (84). Our studies on the roles of SCFAs and MCFAs in viral reactivation address a more general problem of the connection between diet and carcinogenesis and provide clues to how SCFAs and MCFAs could be applied for clinical use.
Viral lytic activation by fatty acids as therapy for virus-associated cancers.
The presence of latent virus in EBV- and KSHV-associated cancers provides a potential target for therapy. Since currently available antiviral drugs target lytic replication but are ineffective against the virus in the latent state, drugs that reactivate the virus have been investigated as therapies for virus-positive cancers. Our studies of the differential activities of SCFAs and MCFAs as inducers or inhibitors of viral reactivation have implications for oncolytic strategies. The HDAC inhibitors butyrate, phenylbutyrate, and VPA have been investigated as lytic activators in cells, mice, and patients (31, 85–89). One risk of applying lytic induction therapy is that incomplete inhibition of viral replication by antiviral drugs could allow secondary infection and disease progression. Another challenge is the low efficiency of lytic reactivation. Hence, it is likely that a reservoir of latently infected cells remains. Our results show that VPA and phenylbutyrate do not induce EBV reactivation in B cell lymphoma, but block it. While HDAC inhibitors may activate cellular gene expression that is beneficial in treating cancer, they seem to have limited usefulness for inducing EBV lytic genes. In fact, our data suggest that MCFAs, such as VPA and phenylbutyrate, may be used to block EBV lytic reactivation. One population that merits clinical investigation with these agents includes patients with posttransplant lymphoproliferative disorders (PTLD). This type of therapy could also be applicable to immunocompromised patients with oral hairy leukoplakia (OHL), where EBV is lytically active. Given the effects we observed on EBV and KSHV lytic reactivation, dietary sources of fatty acids may also be considered for treating virus-associated cancers.
ACKNOWLEDGMENTS
This work was supported by grants from the National Cancer Institute (grants CA012055 and CA016038 to G.M. and F32CA165705 to K.L.G.).
We thank Ethel Cesarman for the gift of BC-1 cells. We thank Ayman El-Guindy, Lyn Gradoville, Danielle Lyons, Grace McInerney, and Ruth Wang'ondu for helpful discussions.
Footnotes
Published ahead of print 7 May 2014
REFERENCES
- 1.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE, Kenney SC. 2011. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 85:165–177. 10.1128/JVI.01512-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Countryman J, Jenson H, Seibl R, Wolf H, Miller G. 1987. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J. Virol. 61:3672–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Countryman J, Miller G. 1985. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. U. S. A. 82:4085–4089. 10.1073/pnas.82.12.4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardwick JM, Lieberman PM, Hayward SD. 1988. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J. Virol. 62:2274–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ragoczy T, Heston L, Miller G. 1998. The Epstein-Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J. Virol. 72:7978–7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zalani S, Holley-Guthrie E, Kenney S. 1996. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc. Natl. Acad. Sci. U. S. A. 93:9194–9199. 10.1073/pnas.93.17.9194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- 9.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207–6212. 10.1128/JVI.74.13.6207-6212.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Tang Q, Maul GG, Yuan Y. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J. Virol. 80:12171–12186. 10.1128/JVI.00990-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Sasson SA, Klein G. 1981. Activation of the Epstein-Barr virus genome by 5-aza-cytidine in latently infected human lymphoid lines. Int. J. Cancer 28:131–135. 10.1002/ijc.2910280204 [DOI] [PubMed] [Google Scholar]
- 12.Davies AH, Grand RJ, Evans FJ, Rickinson AB. 1991. Induction of Epstein-Barr virus lytic cycle by tumor-promoting and non-tumor-promoting phorbol esters requires active protein kinase C. J. Virol. 65:6838–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luka J, Kallin B, Klein G. 1979. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94:228–231. 10.1016/0042-6822(79)90455-0 [DOI] [PubMed] [Google Scholar]
- 14.Takada K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27–32. 10.1002/ijc.2910330106 [DOI] [PubMed] [Google Scholar]
- 15.zur Hausen H, O'Neill FJ, Freese UK, Hecker E. 1978. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 272:373–375. 10.1038/272373a0 [DOI] [PubMed] [Google Scholar]
- 16.Saemundsen AK, Kallin B, Klein G. 1980. Effect of n-butyrate on cellular and viral DNA synthesis in cells latently infected with Epstein-Barr virus. Virology 107:557–561. 10.1016/0042-6822(80)90326-8 [DOI] [PubMed] [Google Scholar]
- 17.Ye J, Gradoville L, Daigle D, Miller G. 2007. De novo protein synthesis is required for lytic cycle reactivation of Epstein-Barr virus, but not Kaposi's sarcoma-associated herpesvirus, in response to histone deacetylase inhibitors and protein kinase C agonists. J. Virol. 81:9279–9291. 10.1128/JVI.00982-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye J, Shedd D, Miller G. 2005. An Sp1 response element in the Kaposi's sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 79:1397–1408. 10.1128/JVI.79.3.1397-1408.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 21.Wang SE, Wu FY, Chen H, Shamay M, Zheng Q, Hayward GS. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248–4267. 10.1128/JVI.78.8.4248-4267.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gradoville L, Kwa D, El-Guindy A, Miller G. 2002. Protein kinase C-independent activation of the Epstein-Barr virus lytic cycle. J. Virol. 76:5612–5626. 10.1128/JVI.76.11.5612-5626.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Countryman J, Gradoville L, Bhaduri-McIntosh S, Ye J, Heston L, Himmelfarb S, Shedd D, Miller G. 2009. Stimulus duration and response time independently influence the kinetics of lytic cycle reactivation of Epstein-Barr virus. J. Virol. 83:10694–10709. 10.1128/JVI.01172-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mellinghoff I, Daibata M, Humphreys RE, Mulder C, Takada K, Sairenji T. 1991. Early events in Epstein-Barr virus genome expression after activation: regulation by second messengers of B cell activation. Virology 185:922–928. 10.1016/0042-6822(91)90574-U [DOI] [PubMed] [Google Scholar]
- 25.Takada K, Horinouchi K, Ono Y, Aya T, Osato T, Takahashi M, Hayasaka S. 1991. An Epstein-Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes 5:147–156. 10.1007/BF00571929 [DOI] [PubMed] [Google Scholar]
- 26.Bhaduri-McIntosh S, Miller G. 2006. Cells lytically infected with Epstein-Barr virus are detected and separable by immunoglobulins from EBV-seropositive individuals. J. Virol. Methods 137:103–114. 10.1016/j.jviromet.2006.06.006 [DOI] [PubMed] [Google Scholar]
- 27.Daigle D, Megyola C, El-Guindy A, Gradoville L, Tuck D, Miller G, Bhaduri-McIntosh S. 2010. Upregulation of STAT3 marks Burkitt lymphoma cells refractory to Epstein-Barr virus lytic cycle induction by HDAC inhibitors. J. Virol. 84:993–1004. 10.1128/JVI.01745-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Countryman JK, Gradoville L, Miller G. 2008. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol. 82:4706–4719. 10.1128/JVI.00116-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daigle D, Gradoville L, Tuck D, Schultz V, Wong'ondu R, Ye J, Gorres K, Miller G. 2011. Valproic acid antagonizes the capacity of other HDAC inhibitors to activate the Epstein-Barr virus lytic cycle. J. Virol. 85:5628–5643. 10.1128/JVI.02659-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw RN, Arbiser JL, Offermann MK. 2000. Valproic acid induces human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS 14:899–902. 10.1097/00002030-200005050-00021 [DOI] [PubMed] [Google Scholar]
- 31.Klass CM, Offermann MK. 2005. Targeting human herpesvirus-8 for treatment of Kaposi's sarcoma and primary effusion lymphoma. Curr. Opin. Oncol. 17:447–455. 10.1097/01.cco.0000172823.01190.6c [DOI] [PubMed] [Google Scholar]
- 32.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. 1987. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28:1221–1227. 10.1136/gut.28.10.1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iannitti T, Palmieri B. 2011. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 11:227–249. 10.2165/11591280-000000000-00000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eadie MJ. 2004. Could valerian have been the first anticonvulsant? Epilepsia 45:1338–1343. 10.1111/j.0013-9580.2004.27904.x [DOI] [PubMed] [Google Scholar]
- 35.Heston L, Rabson M, Brown N, Miller G. 1982. New Epstein-Barr virus variants from cellular subclones of P3J-HR-1 Burkitt lymphoma. Nature 295:160–163. 10.1038/295160a0 [DOI] [PubMed] [Google Scholar]
- 36.Epstein MA, Achong BG, Barr YM, Zajac B, Henle G, Henle W. 1966. Morphological and virological investigations on cultured Burkitt tumor lymphoblasts (strain Raji). J. Natl. Cancer Inst. 37:547–559 [PubMed] [Google Scholar]
- 37.Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM, Cesarman E. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88:2648–2654 [PubMed] [Google Scholar]
- 38.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714 [PubMed] [Google Scholar]
- 39.Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, Garber R, Pellett PE, McGeoch DJ, Chang Y. 1996. Primary characterization of a herpesvirus agent associated with Kaposi's sarcomae. J. Virol. 70:549–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young LS, Lau R, Rowe M, Niedobitek G, Packham G, Shanahan F, Rowe DT, Greenspan D, Greenspan JS, Rickinson AB, Farrell PJ. 1991. Differentiation-associated expression of the Epstein-Barr-virus Bzlf1 transactivator protein in oral hairy leukoplakia. J. Virol. 65:2868–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Francis AL, Gradoville L, Miller G. 1997. Alteration of a single serine in the basic domain of the Epstein-Barr virus ZEBRA protein separates its functions of transcriptional activation and disruption of latency. J. Virol. 71:3054–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ragoczy T, Miller G. 1999. Role of the Epstein-Barr virus RTA protein in activation of distinct classes of viral lytic cycle genes. J. Virol. 73:9858–9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flemington E, Speck SH. 1990. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64:1227–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu P, Liu S, Speck SH. 1998. Identification of a negative cis element within the ZII domain of the Epstein-Barr virus lytic switch BZLF1 gene promoter. J. Virol. 72:8230–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, McCarthy PJ, Wang Z, Gorlen DA, Mertz JE. 2012. Shutoff of BZLF1 gene expression is necessary for immortalization of primary B cells by Epstein-Barr virus. J. Virol. 86:8086–8096. 10.1128/JVI.00234-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawanishi M, Ito Y. 1980. Effect of short-chain fatty acids on Epstein-Barr virus early and viral capsid antigen induction in P3HR-1 cells. Cancer Lett. 11:129–132. 10.1016/0304-3835(80)90103-2 [DOI] [PubMed] [Google Scholar]
- 47.Kishishita M, Yanase S, Ito Y. 1982. Activation of Epstein-Barr virus expression in human-lymphoblastoid P3hr-1 and Raji cells with propionic-acid and with culture fluids of propionic acid-producing anaerobes. Cancer Lett. 16:117–120. 10.1016/0304-3835(82)90051-9 [DOI] [PubMed] [Google Scholar]
- 48.Anisimova E, Prachova K, Roubal J, Vonka V. 1984. Effects of n-butyrate and phorbol ester (TPA) on induction of Epstein-Barr virus antigens and cell differentiation. Arch. Virol. 81:223–237. 10.1007/BF01309995 [DOI] [PubMed] [Google Scholar]
- 49.Sealy L, Chalkley R. 1978. The effect of sodium butyrate on histone modification. Cell 14:115–121. 10.1016/0092-8674(78)90306-9 [DOI] [PubMed] [Google Scholar]
- 50.Cousens LS, Gallwitz D, Alberts BM. 1979. Different accessibilities in chromatin to histone acetylase. J. Biol. Chem. 254:1716–1723 [PubMed] [Google Scholar]
- 51.Miller G, Rigsby MO, Heston L, Grogan E, Sun R, Metroka C, Levy JA, Gao SJ, Chang Y, Moore P. 1996. Antibodies to butyrate-inducible antigens of Kaposi's sarcoma-associated herpesvirus in patients with HIV-1 infection. N. Engl. J. Med. 334:1292–1297. 10.1056/NEJM199605163342003 [DOI] [PubMed] [Google Scholar]
- 52.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. 2001. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276:36734–36741. 10.1074/jbc.M101287200 [DOI] [PubMed] [Google Scholar]
- 53.McBain JA, Eastman A, Nobel CS, Mueller GC. 1997. Apoptotic death in adenocarcinoma cell lines induced by butyrate and other histone deacetylase inhibitors. Biochem. Pharmacol. 53:1357–1368. 10.1016/S0006-2952(96)00904-5 [DOI] [PubMed] [Google Scholar]
- 54.Kraus RJ, Mirocha SJ, Stephany HM, Puchalski JR, Mertz JE. 2001. Identification of a novel element involved in regulation of the lytic switch BZLF1 gene promoter of Epstein-Barr virus. J. Virol. 75:867–877. 10.1128/JVI.75.2.867-877.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu X, Wang Z, Mertz JE. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 3:e194. 10.1371/journal.ppat.0030194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chua HH, Chiu HY, Lin SJ, Weng PL, Lin JH, Wu SW, Tsai SC, Tsai CH. 2012. p53 and Sp1 cooperate to regulate the expression of Epstein-Barr viral Zta protein. J. Med. Virol. 84:1279–1288. 10.1002/jmv.23316 [DOI] [PubMed] [Google Scholar]
- 57.Hagemeier SR, Barlow EA, Kleman AA, Kenney SC. 2011. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. J. Virol. 85:4318–4329. 10.1128/JVI.01856-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hagemeier SR, Barlow EA, Meng Q, Kenney SC. 2012. The cellular ataxia telangiectasia-mutated kinase promotes Epstein-Barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J. Virol. 86:13360–13370. 10.1128/JVI.01850-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang SS, Lo YC, Chua HH, Chiu HY, Tsai SC, Chen JY, Lo KW, Tsai CH. 2008. Critical role of p53 in histone deacetylase inhibitor-induced Epstein-Barr virus Zta expression. J. Virol. 82:7745–7751. 10.1128/JVI.02717-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato Y, Shirata N, Kudoh A, Iwahori S, Nakayama S, Murata T, Isomura H, Nishiyama Y, Tsurumi T. 2009. Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 388:204–211. 10.1016/j.virol.2009.03.017 [DOI] [PubMed] [Google Scholar]
- 61.Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 280:8156–8163. 10.1074/jbc.M411405200 [DOI] [PubMed] [Google Scholar]
- 62.Chang P-J, Ye J, Miller G. 2009. Regulation of expression, mode of action and downstream targets of ORF50 protein in KSHV lytic cycle activation, p 521–553 In Damania B, Pipas JM. (ed), DNA tumor viruses. Springer-Verlag, New York, NY [Google Scholar]
- 63.Amon W, Farrell PJ. 2005. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 15:149–156. 10.1002/rmv.456 [DOI] [PubMed] [Google Scholar]
- 64.Bar-Ner M, Thibault A, Tsokos M, Magrath IT, Samid D. 1999. Phenylbutyrate induces cell differentiation and modulates Epstein-Barr virus gene expression in Burkitt's lymphoma cells. Clin. Cancer Res. 5:1509–1516 [PubMed] [Google Scholar]
- 65.Chung YL, Lee YH, Yen SH, Chi KH. 2000. A novel approach for nasopharyngeal carcinoma treatment uses phenylbutyrate as a protein kinase C modulator: implications for radiosensitization and EBV-targeted therapy. Clin. Cancer Res. 6:1452–1458 [PubMed] [Google Scholar]
- 66.Akihisa T, Tokuda H, Ogata M, Ukiya M, Iizuka M, Suzuki T, Metori K, Shimizu N, Nishino H. 2004. Cancer chemopreventive effects of polyunsaturated fatty acids. Cancer Lett. 205:9–13. 10.1016/S0304-3835(03)00284-2 [DOI] [PubMed] [Google Scholar]
- 67.Chang FR, Hsieh YC, Chang YF, Lee KH, Wu YC, Chang LK. 2010. Inhibition of the Epstein-Barr virus lytic cycle by moronic acid. Antiviral Res. 85:490–495. 10.1016/j.antiviral.2009.12.002 [DOI] [PubMed] [Google Scholar]
- 68.Yiu CY, Chen SY, Chang LK, Chiu YF, Lin TP. 2010. Inhibitory effects of resveratrol on the Epstein-Barr virus lytic cycle. Molecules 15:7115–7124. 10.3390/molecules15107115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. 2003. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 77:11425–11435. 10.1128/JVI.77.21.11425-11435.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282. 10.1128/JVI.02541-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen HS, Wikramasinghe P, Showe L, Lieberman PM. 2012. Cohesins repress Kaposi's sarcoma-associated herpesvirus immediate early gene transcription during latency. J. Virol. 86:9454–9464. 10.1128/JVI.00787-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang LK, Liu ST. 2000. Activation of the BRLF1 promoter and lytic cycle of Epstein-Barr virus by histone acetylation. Nucleic Acids Res. 28:3918–3925. 10.1093/nar/28.20.3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jenkins PJ, Binne UK, Farrell PJ. 2000. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 74:710–720. 10.1128/JVI.74.2.710-720.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, Kanda T, Tsurumi T. 2012. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J. Virol. 86:4752–4761. 10.1128/JVI.06768-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramasubramanyan S, Osborn K, Flower K, Sinclair AJ. 2012. Dynamic chromatin environment of key lytic cycle regulatory regions of the Epstein-Barr virus genome. J. Virol. 86:1809–1819. 10.1128/JVI.06334-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Asai S, Nakamura Y, Yamamura M, Ikezawa H, Namikawa I. 1991. Quantitative analysis of the Epstein-Barr virus-inducing properties of short-chain fatty acids present in the culture fluids of oral bacteria. Arch. Virol. 119:291–296. 10.1007/BF01310678 [DOI] [PubMed] [Google Scholar]
- 77.Morris TL, Arnold RR, Webster-Cyriaque J. 2007. Signaling cascades triggered by bacterial metabolic end products during reactivation of Kaposi's sarcoma-associated herpesvirus. J. Virol. 81:6032–6042. 10.1128/JVI.02504-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singer RE, Buckner BA. 1981. Butyrate and propionate—important components of toxic dental plaque extracts. Infect. Immun. 32:458–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Margolis HC, Duckworth JH, Moreno EC. 1988. Composition of pooled resting plaque fluid from caries-free and caries-susceptible individuals. J. Dent. Res. 67:1468–1475. 10.1177/00220345880670120601 [DOI] [PubMed] [Google Scholar]
- 80.Contreras A, Umeda M, Chen C, Bakker I, Morrison JL, Slots J. 1999. Relationship between herpesviruses and adult periodontitis and periodontopathic bacteria. J. Periodontol. 70:478–484. 10.1902/jop.1999.70.5.478 [DOI] [PubMed] [Google Scholar]
- 81.Mardirossian A, Contreras A, Navazesh M, Nowzari H, Slots J. 2000. Herpesviruses 6, 7 and 8 in HIV- and non-HIV-associated periodontitis. J. Periodontal Res. 35:278–284. 10.1034/j.1600-0765.2000.035005278.x [DOI] [PubMed] [Google Scholar]
- 82.Nagao K, Yanagita T. 2010. Medium-chain fatty acids: functional lipids for the prevention and treatment of the metabolic syndrome. Pharmacol. Res. 61:208–212. 10.1016/j.phrs.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 83.Kessler SK, Neal EG, Camfield CS, Kossoff EH. 2011. Dietary therapies for epilepsy: future research. Epilepsy Behav. 22:17–22. 10.1016/j.yebeh.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haidukewych D, Forsythe WI, Sills M. 1982. Monitoring octanoic and decanoic acids in plasma from children with intractable epilepsy treated with medium-chain triglyceride diet. Clin. Chem. 28:642–645 [PubMed] [Google Scholar]
- 85.Feng WH, Kenney SC. 2006. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res. 66:8762–8769. 10.1158/0008-5472.CAN-06-1006 [DOI] [PubMed] [Google Scholar]
- 86.Perrine SP, Hermine O, Small T, Suarez F, O'Reilly R, Boulad F, Fingeroth J, Askin M, Levy A, Mentzer SJ, Di Nicola M, Gianni AM, Klein C, Horwitz S, Faller DV. 2007. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 109:2571–2578. 10.1182/blood-2006-01-024703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Phillips JA, Griffin BE. 2007. Pilot study of sodium phenylbutyrate as adjuvant in cyclophosphamide-resistant endemic Burkitt's lymphoma. Trans. R. Soc. Trop. Med. Hyg. 101:1265–1269. 10.1016/j.trstmh.2007.06.020 [DOI] [PubMed] [Google Scholar]
- 88.Jones K, Nourse J, Corbett G, Gandhi MK. 2010. Sodium valproate in combination with ganciclovir induces lysis of EBV-infected lymphoma cells without impairing EBV-specific T-cell immunity. Int. J. Lab. Hematol. 32:e169–e174. 10.1111/j.1751-553X.2008.01130.x [DOI] [PubMed] [Google Scholar]
- 89.Lechowicz M, Dittmer DP, Lee JY, Krown SE, Wachsman W, Aboulafia D, Dezube BJ, Ratner L, Said J, Ambinder RF. 2009. Molecular and clinical assessment in the treatment of AIDS Kaposi sarcoma with valproic acid. Clin. Infect. Dis. 49:1946–1949. 10.1086/648447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. 2012. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40:D1100–D1107. 10.1093/nar/gkr777 [DOI] [PMC free article] [PubMed] [Google Scholar]