

ABSTRACT
The varicella-zoster virus (VZV) open reading frame 54 (ORF54) gene encodes an 87-kDa monomer that oligomerizes to form the VZV portal protein, pORF54. pORF54 was hypothesized to perform a function similar to that of a previously described herpes simplex virus 1 (HSV-1) homolog, pUL6. pUL6 and the associated viral terminase are required for processing of concatemeric viral DNA and packaging of individual viral genomes into preformed capsids. In this report, we describe two VZV bacterial artificial chromosome (BAC) constructs with ORF54 gene deletions, Δ54L (full ORF deletion) and Δ54S (partial internal deletion). The full deletion of ORF54 likely disrupted essential adjacent genes (ORF53 and ORF55) and therefore could not be complemented on an ORF54-expressing cell line (ARPE54). In contrast, Δ54S was successfully propagated in ARPE54 cells but failed to replicate in parental, noncomplementing ARPE19 cells. Transmission electron microscopy confirmed the presence of only empty VZV capsids in Δ54S-infected ARPE19 cell nuclei. Similar to the HSV-1 genome, the VZV genome is composed of a unique long region (UL) and a unique short region (US) flanked by inverted repeats. DNA from cells infected with parental VZV (VZVLUC strain) contained the predicted UL and US termini, whereas cells infected with Δ54S contained neither. This result demonstrates that Δ54S is not able to process and package viral DNA, thus making pORF54 an excellent chemotherapeutic target. In addition, the utility of BAC constructs Δ54L and Δ54S as tools for the isolation of site-directed ORF54 mutants was demonstrated by recombineering single-nucleotide changes within ORF54 that conferred resistance to VZV-specific portal protein inhibitors.
IMPORTANCE Antivirals with novel mechanisms of action would provide additional therapeutic options to treat human herpesvirus infections. Proteins involved in the herpesviral DNA encapsidation process have become promising antiviral targets. Previously, we described a series of N-α-methylbenzyl-N′-aryl thiourea analogs that target the VZV portal protein (pORF54) and prevent viral replication in vitro. To better understand the mechanism of action of these compounds, it is important to define the structural and functional characteristics of the VZV portal protein. In contrast to HSV, no VZV mutants have been described for any of the seven essential DNA encapsidation genes. The VZV ORF54 deletion mutant described in this study represents the first VZV encapsidation mutant reported to date. We demonstrate that the deletion mutant can serve as a platform for the isolation of portal mutants via recombineering and provide a strategy for more in-depth studies of VZV portal structure and function.
INTRODUCTION
Herpesvirus DNA encapsidation is understood only in general terms. The mechanistic events and the function(s) of the proteins involved are still under investigation. During viral replication, progeny DNA genomes are synthesized in the nucleus as long, branched head-to-tail concatemers. The capsid proteins are synthesized in the cytoplasm, transported to the nucleus, and assembled around a protein scaffold. During the encapsidation process, concatemeric DNA is processed and packaged into the procapsid. Successful DNA packaging includes removal of the scaffold proteins and the tight packing of one unit length of herpesvirus genomic DNA into the capsid.
Studies of the proteins that form the molecular complexes involved in DNA cleavage and packaging are important to understand the intricacies of these small but powerful molecular motors, and these proteins are of increasing importance as novel antiviral targets. DNA packaging motors have been described in detail for a number of the tailed bacteriophages (1–3). The essential components include the viral terminase, viral procapsid, viral genomic DNA, and portal protein. Portals are a multimeric structure (12- or 13-mer) embedded at the 5-fold vertex of the capsid shell. As originally proposed by Hendrix, these circular structures include a channel or tunnel for the translocation of viral DNA during particle assembly (4). The portal serves as an interface between the unfilled procapsid and uncleaved viral DNA and is the docking site for the viral terminase complex. Together, these components generate the significant force necessary to load viral DNA into the procapsid (5). A comparison of different phage and herpesvirus portal proteins yields little amino acid sequence similarity. There are also significant differences in the molecular masses of the monomeric subunits; some phage portal proteins are <40 kDa, and the largest herpesvirus portal proteins are >85 kDa. Despite these differences, all portals share a similar ring-like morphology (6–8) and all have a common core structure consisting of three nonparallel alpha helices, two of which are separated by an amino acid stretch comprising the tunnel loop. Delivery of DNA into the procapsid involves sequential conformational changes at the portal-viral DNA interface (3, 10–13). Studies by Lebedev et al. on the SPP1 bacteriophage portal protein revealed that translocation of viral DNA through the portal multimer is a dynamic event (9). Jing et al. recently described a one-way valve mechanism for DNA translocation through the bacteriphage phi29 connector (80). Whereas the analysis of crystal structures and structure-function studies of bacteriophage portals have begun to reveal the details of phage encapsidation, similar studies involving mammalian herpesvirus portals are not as advanced.
The portal proteins for herpes simplex virus (HSV) and varicella-zoster virus (VZV) are encoded by the UL6 and open reading frame 54 (ORF54) genes, respectively. Previous studies provided evidence that HSV-1 pUL6 forms the portal protein through which DNA can enter the viral procapsid (8, 14–17). Subsequently, we showed that ORF54 encodes the homologous portal protein for VZV, pORF54 (18, 19). Small molecules that target the HSV (20) and VZV (21) portal proteins have been described. The HSV and VZV portal inhibitor series are thiourea compounds with in vitro 50% inhibitory concentrations (IC50s) in the nanomolar range. Each series is highly specific for its respective virus, but only minor chemical changes are required to switch its specificity. Viral infection in the presence of portal inhibitors results in the accumulation of empty capsids in the nucleus. Isolates resistant to the portal compounds contain mutations that map to the portal gene, but the exact mechanism of inhibition has not been determined. To date, no deletion mutants have been isolated for any of the VZV DNA encapsidation genes. Isolation of an ORF54 null mutant and a companion complementing cell line are critical to future studies of VZV encapsidation, the VZV portal, and the portal inhibitor series.
Seven genes have been shown to be essential in the HSV DNA encapsidation process: UL6, -15, -17, -25, -28, -32, and -33 (14, 17, 22–33). When any of the seven genes were deleted from the viral genome, empty capsids accumulated in the nucleus. Few studies have been done on the VZV homologs—ORF54, -45/42, -43, -34, -30, -26, and -25 (19, 21, 34–36). Studies of VZV encapsidation have lagged behind those of other alphaherpesviruses in part due to the highly cell-associated nature of VZV. Recently, new tools have emerged to more readily manipulate herpesvirus genomes, including that of VZV. The advent of recombineering using VZV bacterial artificial chromosome (BAC) constructs allows for the efficient and precise construction of VZV mutants (37, 38).
In this report, VZV ORF54 was targeted for deletion to define its role in viral replication. Considering its homology to pUL6, pORF54 is predicted to be essential for DNA encapsidation. Therefore, a human retinal pigmented epithelial cell line stably expressing pORF54 (ARPE54) was isolated and used to complement a recombineered VZV ORF54 deletion construct. The parental virus was a previously engineered VZV strain (VZVLUC) that contains both the green fluorescent protein (GFP) and firefly luciferase genes (39). The VZVLUC BAC was manipulated in Escherichia coli to replace either the entire 2,310-bp ORF54 gene (Δ54L) or a 1,223-bp internal region of ORF54 (Δ54S) with a selectable marker, galK. A Δ54S repaired virus (54R) was isolated by replacing galK with the parental ORF54 gene. pORF54 was shown to be essential for viral replication and specifically for viral DNA cleavage and packaging. In addition, the BAC constructs Δ54S and Δ54L proved useful in the isolation of specific ORF54 point mutants that conferred resistance to portal inhibitors.
MATERIALS AND METHODS
Cells and viruses.
ARPE19 cells (human retinal pigmented epithelial cells; ATCC CRL-2302), ARPE54 cells, and MeWo cells (human melanoma cells; ATCC HTB-65) were maintained at 37°C and 5% CO2 in minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. ARPE19 cells were used for propagation of VZV strains and construction of the ORF54 stable cell line, ARPE54. Infected cell stocks were prepared by resuspending trypsinized monolayers in 90% FBS with 10% dimethyl sulfoxide (DMSO) and subjecting them to a slow freeze at −80°C overnight. Frozen cells were moved to liquid nitrogen for long-term storage.
Cell-free VZV was prepared by scraping ∼1 × 107 infected ARPE19 or ARPE54 cell monolayers (∼70% cytopathic effect [CPE]) into 10 ml of phosphate-buffered saline (PBS)-sucrose-glutamate-serum buffer (PSGC) followed by brief probe sonication three times for 15 s each time using a Sonics Vibra-Cell ultrasonic disintegrator (21 kHz and 9 μM amplitude) fitted with a 0.5-in solid, tapered probe (40). Sonicates were checked microscopically for efficient cell disruption and clarified at 1,000 × g for 15 min, and supernatants were stored at −80°C. Cell-free titers were determined with the appropriate cell lines and ranged from 103 to 104 PFU/ml.
DNA sequencing.
All DNA sequencing was performed by Eurofins WMG Operon (Louisville, KY). PCR products generated with single-nucleotide changes were cloned and sequenced to confirm the desired change prior to recombineering. The entire ORF54 gene from all VZV BAC genomic DNAs (whether a single nucleotide change, galK insertion, or ORF54 repair) was sequenced to confirm that only the intended change was introduced.
BAC DNA isolation and agarose gel electrophoresis.
BAC DNAs were prepared using a NucleoBond BAC 100 kit (Clontech Laboratories). Briefly, E. coli grown overnight in LB with chloramphenicol (25 mg/ml) was pelleted at 4,500 × g for 15 min at 4°C. Cells were resuspended in S1/RNase A buffer and lysed in S2 buffer. Clarified lysates were applied to NucleoBond columns, washed, and eluted in prewarmed N5 buffer. DNAs precipitated with isopropanol were centrifuged at 15,000 × g for 30 min at 4°C, washed with 70% ethyl alcohol (EtOH), dried, and resuspended in 10 mM Tris (pH 8.5). BAC DNAs were digested with SapI and analyzed using 0.65% agarose gels stained with ethidium bromide (EtBr). Virus was reconstituted by transfecting 1 × 106 ARPE19 or ARPE54 cells with 2 μg of BAC DNA complexed with 6 μl of Fugene 6 transfection reagent (Promega). Plates were incubated for 3 to 5 days and observed for GFP-positive plaques.
Recombineering Δ54L and Δ54S.
The VZV ORF54 mutant viruses were constructed by recombineering (41) using the VZVLUC BAC system described by Zhang et al. (39) with the following modifications. Two ORF54 mutants were constructed: a null mutant with an exact deletion of the entire ORF54 coding region (Δ54L) and an internal deletion mutant missing 1,223 bp (Δ54S). A galK cassette flanked by 50-bp homology arms was generated via PCR using primer combinations Δ54S-galK forward (F) and reverse (R) for Δ54L and Δ54L-galK F and R for Δ54S (Table 1). Amplicons were digested with DpnI and purified using a QIAquick PCR purification kit (Qiagen). An overnight culture of SW102 E. coli (NCI) harboring the VZVLUC BAC was diluted 1:50 in 25 ml of LB with chloramphenicol (25 μg/ml) in a 50-ml baffled conical flask and incubated at 32°C in a shaking incubator to an optical density at 600 nm (OD600) of 0.4. Cells were heat shocked at 42°C for exactly 15 min in a shaking water bath. The culture was cooled for 2 min in an ice water bath and transferred to a 50-ml conical centrifuge tube. Cells were pelleted at 4,500 rpm at 0°C for 5 min. The supernatant was removed and the pellet resuspended in 20 ml ice-cold double-distilled water (ddH2O) by gently swirling the tubes in an ice water bath. Cells were pelleted at 4,500 rpm at 0°C for 5 min and washed a second time. Pelleted cells were resuspended in 100 μl ice-cold ddH2O. Electrocompetent SW102 cells (30 μl) were electroporated in a 0.1-cm cuvette using a Bio-Rad Gene Pulser X Cell (Bio-Rad Laboratories) with 50 ng of the purified galK homology cassette (25 mF, 1.80 kV, and 200 Ω). Cells were allowed to recover with shaking (200 rpm) at 32°C in 1 ml of LB for 1 h, washed in 1 ml of M9 buffer 4 times, and resuspended in 1 ml of M9 buffer. Serial dilutions of 1:10, 1:100, and 1:1,000 were plated on M63 plates containing 25 μg/ml chloramphenicol and 0.2% galactose. Plates were incubated at 32°C for 3 to 5 days. E. coli containing VZVLUC BAC with the galK cassette was positively selected on minimal medium with galactose as the sole carbon source. Colonies were picked on MacConkey agar plates supplemented with 25 μg/ml chloramphenicol and 0.2% galactose. Pink colonies were streaked for isolation, and a single pink colony was selected for characterization.
TABLE 1.
BACs, plasmids, primers, and strains used in this study
| Reagent | Description | Source, reference, or sequencea |
|---|---|---|
| BACs | ||
| VZVLUC | VZV pOKA containing firefly luciferase and green fluorescent protein | 79 |
| Δ54L | galK cassette in place of entire ORF54 in VZVLUC | This study |
| Δ54S | galK cassette in place of ORF54 bp 301 to 1574 in VZVLUC | This study |
| 54R | Wild-type ORF54 replacing galK cassette in Δ54S BAC | This study |
| VZV5448r | Mutation at ORF54 amino acid 48 conferring resistance to thiourea compounds | This study |
| VZV54407r | Mutation at ORF54 amino acid 407 conferring resistance to thiourea compounds | This study |
| Plasmid | ||
| pgalK | Used to make galK cassette with flanking ORF54 homology arms | 41 |
| Bacterial strain | ||
| SW102 | Used to propagate/manipulate VZV BAC clones | 41 |
| Primersb | ||
| Δ54L-galK F | Used to generate homology arms for entire ORF54 deletion | AACCGGTTACCTCCCGCGCCTCGCATACGAATCTTGGTATTGCTTGTATTcctgttgacaattaatcatcggca |
| Δ54L-galK R | Used to generate homology arms for entire ORF54 deletion | TTTGGGTGAAGAACTGTCTACAGAAATTGATCTTTTCATCGTGGTTTTCAtcagcactgtcctgctcctt |
| Δ54S-galK F | Used to generate homology arms for internal ORF54 deletion | ATTGAAAACTATTGGCAATATATGCATCATCCCCTATTATGCCGGTAAGAcctgttgacaattaatcatcggca |
| Δ54S-galK R | Used to generate homology arms for internal ORF54 deletion | ACTGTAATTCGCCAAGTTCAGGCAACCGTTTTGATGAACGCGTTGGATGCtcagcactgtcctgctcctt |
| 54R F | Used to generate ORF54 to repair Δ54S | ATTGAAAACTATTGGCAATATATGCATCATCCCCTATTATGCCGGTAAGA |
| 54R R | Used to generate ORF54 to repair Δ54S | ACTGTAATTCGCCAAGTTCAGGCAACCGTTTTGATGAACGCGTTGGATGC |
| 54 iF | Internal ORF54 primer to confirm reconstitution of ORF54 in ORF54R | TGTTTCTTCTGCCATTGTAATA |
| 54 iR | Internal ORF54 primer to confirm reconstitution of ORF54 in ORF54R | GGATCCGTATGCAAAATGTTGCTATT |
| ARPE54 F | Used to validate the presence or ORF54 genomic integration | ATGGCCGAAATAACGTCTCT |
| ARPE54 R | Used to validate the presence or ORF54 genomic integration | CTAAGATCTTCGATCACGTC |
| TRS F | Terminal repeat short probe for Southern analysis | AGATCCCAGACCCGGAGGAT |
| TRS R | Terminal repeat short probe for Southern analysis | CAGGTTGGCAAACGCAGTCT |
| TRL F | Terminal repeat long probe for Southern analysis | TCGGATGGCGACCGTGCACTACT |
| TRL R | Terminal repeat long probe for Southern analysis | GATGGCGACGTTGGCTTTGGTGA |
| 48 F-1 | Mutation at ORF54 nucleotide 142 conferring resistance | CTATATCCGGGGCGTACTtATTCCAGTAT |
| 48 R-1 | Used to generate homology arms outside ORF54 coding region (5′ ORF55) | CTCATTCCTCTGCATTTCAGGAGGCGCTT |
| 48 F-2 | Used to generate homology arms outside ORF54 coding region (5′ ORF53) | CCGTATACACCCTATCTTCAACCGCAGTT |
| 48 R-2 | Mutation at ORF54 nucleotide 142 conferring resistance | ATACTGGAATaAGTACGCCCCGGATATAG |
| 407 F-1 | Mutation at ORF54 nucleotide 1220 conferring resistance | CAGAATGTGATTGtCCGTTTCTTCTAGATAGGA |
| 407 R-1 | Used to generate homology arms within 5′ ORF54 | ATGGCCGAAATAACGTCTCTTTTTAATAACAGTT |
| 407 F-2 | Used to generate homology arms within 3′ ORF54 | CTAAGATCTTCGATCACGTCGCTCACATCCAAC |
| 407 R-2 | Mutation at ORF54 nucleotide 1220 conferring resistance | TCCTATCTAGAAGAAACGGaCAATCACATTCTG |
VZV sequences are in uppercase, VZV point mutations in lowercase boldface, and galK sequences in lowercase roman type.
Primers are forward (F) and reverse (R) with respect to the genome map.
A repaired VZV isolate was generated for Δ54S by following the procedure described above with the following modifications. Electrocompetent SW102 cells containing VZVLUC-Δ54S BAC were transformed with 500 ng of a full-length ORF54 DNA amplicon generated via PCR with primer pair 54R F and 54R R (Table 1). Cells were allowed to recover with shaking (200 rpm) at 32°C in 10 ml of LB for 4 h, washed, and plated as described above except that M63 plates were supplemented with 25 mg/ml chloramphenicol, 0.2% 2-deoxygalactose (DOG), and 0.2% glycerol. Plates were incubated at 32°C for 5 to 7 days. E. coli containing VZVLUC BAC that lost the galK cassette was negatively selected by resistance to DOG on minimal plates with glycerol as the carbon source. (When DOG is phosphorylated by galK, a toxic intermediate, 2-deoxy-galactose-1-phosphate, is formed.) Colonies from DOG plates were screened by colony lift hybridization using an internal ORF54 probe (generated with primer pair 54i F/54i R; Table 1) labeled with a DIG High Prime DNA labeling kit (Roche). Positive colonies were further verified by PCR using the same set of internal ORF54 PCR primers.
The ORF54 gene sequence of all VZV BAC DNA constructs was confirmed by DNA sequencing prior to transfection into ARPE19 or ARPE54 cells.
VZV replication kinetics.
Viral replication was determined via a previously described luciferase based assay (39) with the following modifications. Twelve-well plates containing 3 × 105 ARPE19, APRE54, or MeWo cells/well were infected in triplicate with 300 PFU of cell-associated VZVLUC, Δ54S, or 54R. Infected monolayers were washed with PBS, and 250 μl of luciferase assay lysis buffer was added per well at designated times postinfection (from 6 h up to 7 days). Plates were rocked gently at room temperature for 15 min, and samples were stored at −80°C. On the day of the assay, 20-μl portions of each sample were placed into individual wells of Costar flat-bottom white polystyrene 96-well assay plates (Corning). Next, 25 μl of freshly reconstituted luciferin (0.6 mg/ml) was added to each well, and the 96-well plate was incubated for 10 min at room temperature. Relative luminescence units (RLU) were recorded using a Veritas microplate luminometer (Turner Biosystems). Data were normalized based on 6-h RLU readings to account for variations in initial inocula. The data were graphed as the averages from triplicate samples ± standard deviations.
Preparation of a pORF54 C-terminal-specific antibody.
A peptide corresponding to amino acids 726 to 739 of the C terminus of pORF54 was synthesized and conjugated to KLH (GenScript). Conjugated peptide was used to immunize rabbits for the production of polyclonal pORF54 C terminus-specific antiserum. Antibody was affinity purified using the peptide antigen and tested for reactivity against the peptide by enzyme-linked immunosorbent assay (ELISA). The ELISA titer of preimmune serum was <1:1,000 compared to >1:512,000 for the purified C terminus antibody.
Immunoprecipitation and immunoblotting.
ARPE19 or ARPE54 cells (5 × 106) were mock infected or infected with ∼104 PFU of cell-associated VZVLUC, Δ54S, or 54R for 72 h. Plates were washed with phosphate-buffered saline (PBS) and resuspended in 1 ml immunoprecipitation assay buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, and protease inhibitor cocktail [0.5 mM phenylmethylsulfonyl fluoride {PMSF}, 150 nM aprotinin, 1 μM leupeptin, 1 μM pepstatin A]). After incubation on ice for 30 min, the lysates were briefly sonicated and clarified at 14,000 rpm for 15 min at 4°C in a microcentrifuge. Supernatants were incubated with 10 μl of rabbit antiserum directed against the pORF54 C terminus for 1.5 h at 4°C. Thirty microliters of a 50% slurry of protein A/G-Sepharose beads (Amersham Pharmacia Biotech) was added, and the mixture was incubated for 1 h at 4°C with constant rocking. The beads were washed five times with 1 ml cold immunoprecipitation assay buffer, and immune complexes were boiled in loading buffer (62.5 mM Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 5% β-mercaptoethanol, 12.5% glycerol). The immunoprecipitates were separated on 10% SDS-PAGE, and proteins were transferred to polyvinylidene difluoride (PVDF) membranes for immunoblot analysis. pORF54 was detected using a previously described anti-pORF54 guinea pig (1:500) serum (19) and protein A-horseradish peroxidase (HRP; 1:15,000) conjugate (BD Biosciences). Chemiluminescent detection was performed using a SuperSignal West Pico chemiluminescent substrate system (Pierce).
For immunoblotting only, aliquots of the cell extracts described above were fractionated via SDS-PAGE, transferred to PVDF membranes, and analyzed for VZV protein expression using a monoclonal antibody specific for VZV gE (Millipore; catalog no. MAB8612).
DNA cleavage analysis.
Approximately 107 ARPE19 cells were infected with 1 × 104 PFU 54R- or Δ54S-infected cells in MEM containing 2% FBS for 48 h. Infected monolayers were washed twice with ice-cold PBS and solubilized in 0.8 ml of N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES) with sodium dodecyl sulfate (10 mM Tris [pH 7.5], 0.6% SDS, 50 mM NaCl, 10 mM EDTA). The lysate was incubated in the presence of 100 μg/ml proteinase K (Sigma) overnight at 37°C. RNase A (Sigma) was added to a concentration of 25 μg/ml, and the lysates were incubated for an additional 45 min at 37°C. Sodium acetate was added to a concentration of 0.15 M and extracted with phenol-chloroform. DNA was precipitated with two volumes of ice-cold ethanol and gently resuspended in 10 mM Tris, pH 8.5. Total infected-cell DNA (7 μg) was digested with BamHI, separated on a 0.8% agarose gel, and transferred to Amersham Hybond N+ membranes (GE Healthcare Life Sciences). DNA amplicons specific for the short (TRS) and long (TRL) terminal repeats were generated using primer pairs TRS F/TRS R and TRL F/TRL R, respectively (Table 1). Probes were labeled using a Roche DIG High Prime labeling and detection kit II (Roche; catalog no. 11585614910). Membranes were hybridized overnight at 40°C and washed, blocked, and developed per the manufacturer's instructions.
Transmission electron microscopy.
ARPE19 cell monolayers (∼106) were infected with 104 PFU of VZV pOKA, Δ54S, or 54R and harvested at 72 h postinfection. Cells were washed twice with PBS and fixed in 4% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate (NaCac) buffer, pH 7.4. Fixed cells were washed in 0.1 M NaCac buffer, osmicated with 2% osmium tetroxide, stained en bloc with 2% uranyl acetate, and dehydrated with a graded ethanol series prior to embedding in Epon-Araldite resin. Thin sections (70 nm) were cut with a diamond knife using a Leica EM UC6 ultramicrotome (Leica Microsystems), collected on 200-mesh copper grids, and stained with 5% methanolic uranyl acetate and Reynolds lead citrate. Cells were observed with a JEM 1230 transmission electron microscope (JEOL USA) at 110 kV and imaged with an UltraScan 4000 charge-coupled-device (CCD) camera (Gatan Inc.).
Isolation and characterization of VZV5448r and VZV54407r.
Previously, a thiourea inhibitor-resistant isolate (VZV EllenrB) with a mutation at nucleotide 1220 of ORF54 was shown to have a single amino acid change, from glycine to aspartic acid, at residue 407 (21). This mutation was engineered into Δ54S via recombineering. Two overlapping DNA amplicons containing the desired nucleotide change were generated via PCR with Phusion Hi Fidelity polymerase (New England BioLabs) with primer pairs 407 F-1/407 R-1 and 407 F-2/407 R-2 (Table 1). Equal amounts of purified PCR products were used in a primer extension reaction with Q5 Hi Fidelity DNA polymerase (New England BioLabs). The fragment containing the point mutation was subcloned using a CloneJet PCR cloning kit (Thermo Scientific) and transformed into SmartCells (Genlantis). Plasmid DNA was prepared using a QIAprep Spin miniprep kit (Qiagen) and sequenced to confirm the G-to-A change at bp 1220. An ORF54 PCR product was then generated using primer pair 407 F-2/407 R-1 (Table 1) and electroporated into SW102 cells containing Δ54S. Colonies selected on DOG plates were screened for recombinant BAC containing the mutated ORF54 gene.
A new thiourea inhibitor-resistant isolate (VZV Ellenr750) was generated as described previously (21). A mutation at nucleotide 142 of ORF54 was identified by sequence analysis and suggested that a single amino acid change, from glutamic acid to lysine, at residue 48 conferred resistance to the inhibitor. This mutation was engineered into Δ54L via recombineering as described above with the following modifications. Two overlapping DNA fragments containing the desired nucleotide change were generated via PCR with primer pairs 48 F-1/48 R-1 and 48 F-2/48 R-2 (Table 1). Plasmid DNA was prepared and sequenced to confirm the G-to-A change at bp 142. An ORF54 PCR product was then generated using primer pair 48 F-2/48 R-1 (Table 1) and electroporated into SW102 cells containing Δ54L. Colonies selected on DOG plates were screened for recombinant BAC containing the mutated ORF54 gene.
VZV plaque reduction assay.
ARPE19 cells were infected with approximately 100 PFU of VZV-infected cell stock (VZVLUC, VZV5448r, or VZV54407r) per well. Sensitivity of isolates was determined by diluting the compound to the desired concentrations (0.03, 0.06, 0.125, 0.250, 0.500, 1.00, or 2.00 μg/ml) in MEM containing 0.3% dimethyl sulfoxide (DMSO) at the time of infection. Positive-control wells were VZV-infected cells in MEM containing 0.3% DMSO without the compound. Monolayers were incubated for 5 days at 37°C, washed with PBS, and harvested in lysis buffer. Relative luminescence units (RLU) were determined for each sample using a firefly luciferase glow assay kit (Pierce) as described above. The signal observed for infected cells without compound was set as 100% replication. The data were graphed as the averages from triplicate samples ± standard errors.
RESULTS
Isolation of an ORF54-expressing cell line, ARPE54.
The viral portal protein of HSV-1 was shown to be essential for viral replication (14, 17). Therefore, it was necessary to isolate a stable cell line capable of supporting the replication of VZV ORF54 mutants. The 2,310-bp ORF54 gene was cloned downstream of the EF-1alpha promoter in a lentivirus expression construct containing a puromycin selection marker (Fig. 1A). A stable cell line (ARPE54) was generated after transduction of ARPE19 cells and puromycin selection. No gross morphological changes were observed for ARPE54 cells (Fig. 1B). Genomic integration of ORF54 was validated by performing ORF54-specific PCR with total genomic DNA isolated from ARPE19 and ARPE54 cells. A single PCR product of 2.3 kb was observed in the reaction mixture containing ARPE54 genomic DNA (Fig. 1C, lane 3) but not in that with ARPE19 DNA (Fig. 1C, lane 2).
FIG 1.
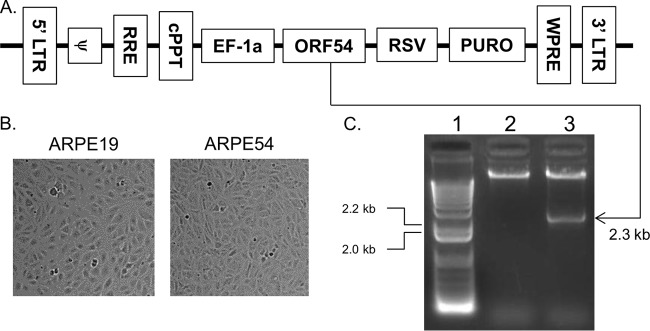
Isolation of an ORF54 stable cell line. (A) The full-length ORF54 gene was cloned into the Gentarget lentivirus vector pLenti-EF-1a-Rsv-Puro. Control regions include the 5′ long terminal repeat (LTR) and 3′ LTR, packaging sequence (ψ), Rev-responsive element (RRE), central polypurine tract (cPPT), human elongation factor 1 alpha promoter (EF1a), Rous sarcoma virus promoter (RSV), and woodchuck hepatitis posttranscriptional regulatory element (WPRE). Open reading frames include the VZV open reading frame 54 (ORF54) and puromycin resistance gene (PURO). (B) ARPE-19 cells were transduced with ORF54 lentivirus, and selection was performed with puromycin. (C) Genomic integration of ORF54 was validated by PCR using genomic DNA from the stable cell clone. Genomic integration of ORF54 into human retinal epithelial cells (ARPE19 cells) was validated by performing PCR with ORF54-specific primers and analyzing the products on a 1% agarose gel. Lane 1, lambda HindIII DNA molecular weight markers. Lane 2, ARPE19 genomic DNA. Lane 3, ORF54-specific PCR of ARPE54 genomic DNA.
Construction of BAC ORF54 deletions Δ54S and Δ54L via recombineering.
Two different null mutations were constructed via recombineering using the parental virus, VZVLUC (Fig. 2A). Initially, homology arms located precisely outside the 5′ and 3′ ends of the ORF54 coding region were used to delete 2,310 bp encoding all 770 amino acids of pORF54, resulting in the isolation of BAC Δ54L (Fig. 2B). Analysis of the genomic map in the ORF54 region revealed that (i) the 5′ coding region and likely promoter of VZV ORF53, an essential gene of unknown function, overlaps the 3′ coding region of ORF54 and (ii) the likely promoter of VZV ORF55, encoding the essential VZV helicase, overlaps the 5′ coding region of ORF54 (42). It was possible that deletion of the entire ORF54 coding region would affect one or both of these other VZV genes and that the cell line would not complement the deletion mutation. Therefore, homology arms internal to ORF54 were used to create a 1,223-bp deletion corresponding to amino acids 117 to 525, resulting in the isolation of BAC Δ54S (Fig. 2C). BAC Δ54S was designed to remove a significant portion of ORF54 yet not affect adjacent genes. A repaired BAC, BAC 54R, was isolated by recombineering the wild-type ORF54 gene into BAC Δ54S.
FIG 2.
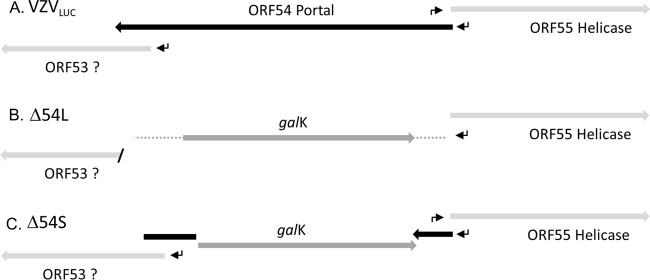
Predicted genome structures and features of parental and mutant viruses in the ORF54 region. (A) VZVLUC parental virus indicating shared coding regions of ORF53 and ORF54. The ORF55 promoter is predicted to fall within the 3′ coding sequence of ORF54. (B) Δ54L, or large 54 deletion, indicates a complete (2,310-bp) deletion and replacement of ORF54 sequences with galK sequences, resulting in deletion of the ORF53 promoter and 3′ coding sequences. The predicted ORF55 promoter is also deleted. (C) Δ54S, or small 54 deletion, indicates an internal deletion of 1,223 bp that conserves ORF53 and ORF55 coding regions and promoters. Arrows, promoters; galK, E. coli galactokinase gene.
The genomic structures of BACs VZVLUC, Δ54L, Δ54S, and 54R were analyzed by restriction enzyme digestion (Fig. 3). BAC DNAs were shown to contain the expected changes in the ORF54 region with HindIII (data not shown) and SapI (Fig. 3). New restriction fragments of 13.1 and 1.8 kb for BAC Δ54L, and 13.4 and 2.5 kb for BAC Δ54S, were predicted based on insertion of the galK gene with an internal SapI site. The overall digestion pattern also indicated that there were no gross genomic alternations in the constructs. The genomic pattern of BAC 54R was identical to that of the parental virus VZVLUC.
FIG 3.

Confirmation of VZV BAC genome structures. SapI restriction sites flanking the ORF54 gene and the resulting predicted fragment sizes are shown for VZVLUC, Δ54L, and Δ54S. Insertion of galK results in an additional SapI site. SapI-digested BAC DNAs were visualized after electrophoresis on a 0.65% agarose gel and staining with EtBr.
Plaque phenotype of BAC Δ54S and BAC Δ54L on noncomplementing (ARPE19) and complementing (ARPE54) cell lines.
Amino acid homology between pORF54 and HSV-1 pUL6 suggests that pORF54 is essential for viral replication (77). To test this hypothesis, recombinant BAC DNAs were transfected into ARPE54 cells. Seven days posttransfection, cell-free virus was harvested and used to infect new cell monolayers. Infected ARPE19 and ARPE54 cells were observed for plaque formation and GFP expression (Fig. 4A). Transfection of BAC Δ54L did not yield viable plaques on either cell line, indicating that disruption of one or both of the adjacent ORFs was lethal (data not shown). However, BAC Δ54S formed plaques on ARPE54 (Fig. 4A, left panels) but not ARPE19 (Fig. 4A, right panels) cell monolayers. Individual intense GFP-positive cells could be observed on ARPE19 monolayers, suggesting that virus could infect but not spread in ARPE19 cells. Virus harvested from BAC 54R DNA-transfected AREP54 cells was able to form plaques identical to those seen with VZVLUC on either cell line (data not shown).
FIG 4.

Growth kinetics. (A) ARPE54 or ARPE19 cells were infected with cell-free Δ54S, and a representative field was photographed at low and high magnification 5 days postinfection. Monolayers of (B) ARPE19, (C) ARPE54, or (D) MeWo cells were infected in triplicate with VZVLUC, Δ54S, or 54R. Cells were harvested in luciferase lysis buffer at the indicated time points. Firefly luciferase activity is shown in relative luminescent units (RLU). Each point is the average of results for three independent samples (the standard deviation is provided).
Δ54S can replicate in ARPE54 cells but not in ARPE19 cells.
Using a previously described luciferase-based assay, replication of VZVLUC, Δ54S and 54R was analyzed in ARPE19 and ARPE54 cells. Monolayers were infected with 300 PFU of cell-associated virus stock and harvested at the time points indicated in Fig. 4. Only VZVLUC and 54R replicated efficiently in ARPE19 cells (Fig. 4B). Δ54S could neither form plaques nor replicate efficiently (Fig. 4B) in ARPE19 cells. All three viruses replicated similarly in ARPE54 cells (Fig. 4C). Δ54S replicated efficiently (Fig. 4C) and formed plaques similar to those formed by the parental virus in ARPE19 cells. The results suggested that ORF54 is required for viral replication and that the ORF54 complementing cell line could effectively provide pORF54 in trans to support replication of ORF54 mutant viruses.
Δ54S cannot replicate in MeWo cells.
ARPE19 are highly permissive for VZV but are not representative of virus replication and spread in skin. VZV-infected ARPE19 cells typically do not undergo extensive fusion. In contrast, VZV-infected human melanoma cells (MeWo cells) exhibit multinucleated giant cells and large syncytia similar to the pathology observed for human skin infection (43–46). It was possible that the Δ54S mutant might spread cell to cell via fusion without the requirement for DNA-containing capsids. To address this possibility, MeWo cell monolayers were infected with approximately 300 PFU of cell-associated virus stock and harvested at the time points indicated in Fig. 4. VZVLUC and 54R replicated efficiently in MeWo cells (Fig. 4D). Δ54S did not replicate (Fig. 4D) in MeWo cells, and there was little evidence of syncytium formation (data not shown). The results suggested that ORF54 is required for viral replication in MeWo cells.
pORF54 expression is absent from Δ54S-infected ARPE19 cells.
Immunoprecipitation followed by Western blot analysis was used to confirm the absence of pORF54 in Δ54S-infected ARPE19 cells. Virus-infected or mock-infected ARPE19 cell extracts were immunoprecipitated using a pORF54-specific rabbit antiserum. Precipitates were fractionated by SDS-PAGE and analyzed by immunoblotting with a pORF54-specific guinea pig serum. An 87-kDa protein was immunoprecipitated from VZVLUC- and 54R-infected but not Δ54S- or mock-infected ARPE19-infected cell extracts (Fig. 5A). The 87-kDa protein is consistent with the size previously reported for the putative portal monomer, pORF54, encoded by the VZV ORF54 gene (19, 35). The VZVLUC, Δ54S, and 54R samples contained VZV gE (Fig. 5B), confirming virus infection. The results showed that pORF54 was not expressed in ARPE19 cells infected with Δ54S.
FIG 5.
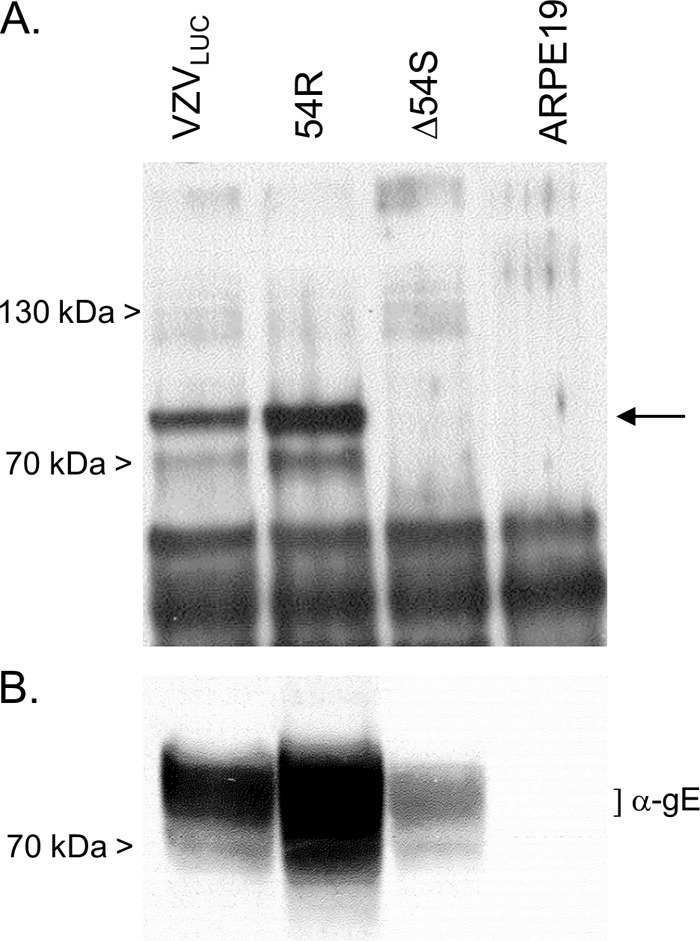
Expression of pORF54 in Δ54S-infected cells. (A) Mock-infected or virus-infected ARPE19 cell extracts were immunoprecipitated with anti-pORF54 rabbit serum. Precipitated proteins were fractionated by SDS-PAGE and analyzed by Western blotting with an anti-pORF54 guinea pig serum. (B) Aliquots of cell extracts precipitated in panel A were immunoblotted with VZV anti-gE (α-gE) monoclonal antibody. The arrow indicates the position of the pORF54 monomer.
Viral DNA is not processed in Δ54S-infected ARPE19 cells.
The absence of pORF54 was hypothesized to result in a defect in viral DNA processing. To examine the encapsidation phenotype of Δ54S, ARPE19 cells were infected with Δ54S and 54R for 48 h. Total infected cell DNA was isolated, digested with BamHI, and analyzed by Southern blotting with probes specific for the terminal repeat regions of the VZV genome (Fig. 6). The repaired virus, 54R, showed normal DNA processing. The TRL- and TRS-specific probes detected fragments of 1.9 and 0.9 kb, respectively. The 2.8-kb fragment detected by both probes represents uncleaved viral DNA. Δ54S DNA was inefficiently cleaved, and as predicted, little to no 1.9- or 0.9-kb DNA fragments were observed. The 2.8-bp fragment representing uncleaved DNA was present. The results suggest that pORF54 is necessary for the proper cleavage of viral genomic DNA. These results are consistent with previous studies showing that the HSV homolog, pUL6, was required for proper viral DNA processing (14, 17).
FIG 6.

DNA cleavage assay. Monolayers of ARPE19 cells were infected with Δ54S or 54R. Cells were harvested at 24 h postinfection. Total infected cell DNA was digested with BamHI, fractionated by agarose gel electrophoresis, and analyzed by Southern blotting with probes that specifically recognize the long (TRL) and short (TRS) terminal repeats. DNA fragments representing uncleaved (2.8 kb), TRL (1.9 kb), or TRS (0.9 kb) are indicated.
Δ54S-infected ARPE19 cells do not contain DNA-filled capsids.
Since viral DNA cleavage is tightly linked to genome packaging, we hypothesized that mutant-infected ARPE19 cells would contain large numbers of empty (unfilled) viral capsids. Thin sections of VZVLUC-, Δ54S-, and 54R-infected ARPE19 cells were examined by transmission electron microscopy. VZVLUC-infected (Fig. 7A and Fig. 8A and B) and 54R-infected (Fig. 7C) ARPE19 cells showed typical features of wild-type-infected cells. Both electron-dense, DNA-filled and empty capsids were observed in the nucleus. In Δ54S-infected cells (Fig. 7B and Fig. 8A and B), only empty capsids could be observed. Examination of numerous Δ54S sections revealed the accumulation of empty capsids in groups or clusters which represent crystalline packing of large numbers of capsids, as reported by others (47–51).
FIG 7.

Electron microscopy of VZVLUC-, Δ54S-, and 54R-infected AREP19 cells. Monolayers of ARPE19 cells were infected with (A) VZVLUC-, (B) Δ54S-, or (C) 54R-infected cell stocks (multiplicity of infection [MOI], ∼0.001). Cells were harvested at 72 h postinfection, fixed, and examined via transmission electron microscopy. Boxed images in the right column represent the boxed regions in the corresponding left column. The solid black arrows in panels A and C indicate intranuclear DNA-filled capsid. All other capsids are unfilled and represent either A- or B-type capsids. No DNA-filled capsids were observed in the nuclei of Δ54S-infected ARPE19 cells (B). Budding virions containing an envelope, capsid, and DNA could be observed at the plasma membrane of VZVLUC- and 54R-infected ARPE19 cells (star).
FIG 8.

Electron microscopy of VZVLUC- and Δ54S-infected ARPE19 cells. Monolayers of ARPE19 cells were infected with Δ54S-infected cell stocks (MOI, ∼0.001). Cells were harvested at 72 h postinfection, fixed, and examined via transmission electron microscopy. (A) DNA-filled and unfilled empty capsids in the nucleus of a VZVLUC-infected ARPE19 cell. Δ54S cells contained aggregated and individual empty capsids but no DNA-filled capsids. (B) Reverse images of the exact same fields as those represented in panel A. DNA-filled capsids contain bright white centers (white arrows), while empty capsids have gray or dark interiors. The star represents an empty capsid between the inner and outer nuclear membranes of a Δ54S-infected ARPE19 cell.
Previously, we reported that VZV infection in the presence of encapsidation inhibitors that target VZV pORF54 resulted in only empty viral capsids (21). As in the previous study, capsids with various phenotypes were observed. Some appeared completely empty (presumably A-type capsids), while others had an obvious inner ring which may represent the scaffold protein of B-type capsids (Fig. 7B).
In addition, virus particles in various states of envelopment, including DNA-containing enveloped particles, could be observed for VZVLUC (Fig. 7A) and 54R (Fig. 7C). Although light particles could be observed at the plasma membrane in some Δ54S sections, DNA-containing, enveloped particles were not observed (data not shown).
Δ54S and Δ54L as platforms for the isolation of specific ORF54 point mutants.
The development of a complementing cell line and the two ORF54 BAC deletion constructs should allow for easy isolation of ORF54 point and deletion mutants. As proof of principle, single point mutations conferring resistance to the previously described N-α-methylbenzyl-N′-arylthiourea VZV-specific, encapsidation inhibitor series were recombineered into Δ54S or Δ54L. Δ54S was reconstituted with an ORF54 gene containing a single base change at nucleotide 1220, resulting in the substitution of aspartic acid (D) for glycine (G) at amino acid 407. Δ54L was reconstituted with an ORF54 gene containing a single base change at nucleotide 142, resulting in the substitution of lysine (K) for glutamic acid (E) at amino acid 48. All changes were confirmed via sequence analysis. A luciferase based plaque reduction assay was performed to determine the IC90 for the parent virus (VZVLUC), and the recombineered isolates were grown in the presence of encapsidation inhibitor (Fig. 9). As reported for the original VZV Ellen resistant isolate (EllenrB), substitution at amino acid 407 conferred resistance and resulted in a 4.5-fold increase in the IC90 compared to that for VZVLUC. Substitution at amino acid 48 was also sufficient to confer resistance, resulting in a 6.4-fold increase in the IC90 compared to that for VZVLUC. The results suggest that both Δ54L and Δ54S will be valuable in constructing mutations throughout ORF54 to generate a fine structure-function map of pORF54.
FIG 9.
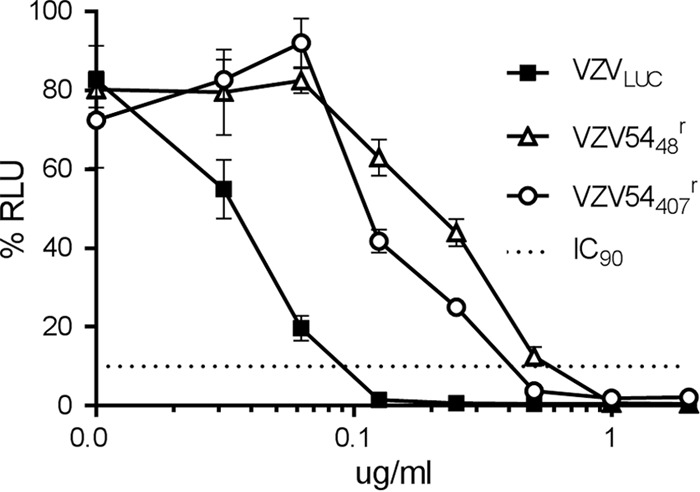
Plaque reduction assay. Monolayers of ARPE19 cells were infected in triplicate with VZVLUC, VZV5448r, or VZV54407r in the presence of various concentrations of thiourea inhibitor (0, 0.03, 0.06, 0.125, 0.250, 0.500, 1.00, or 2.00 μg/ml). After incubation for 5 days at 37°C, cells were harvested in luciferase lysis buffer and assayed for luciferase activity. Each point is the average of results from three independent samples (the standard error is provided), and the data are presented as percent activity of the maximum RLU recorded for each virus. The dotted line is provided to compare IC90 values between viruses.
DISCUSSION
Varicella-zoster virus (VZV) is a human herpesvirus that causes an acute disseminated primary infection, chickenpox, and latent virus may reactivate from within dorsal root ganglia to cause herpes zoster, commonly known as shingles (52–54). Primary varicella presents as a rash with vesicular skin lesions and in most healthy children is a self-limiting disease. In adults, primary varicella infection can result in substantially more severe disease with secondary complications, including extensive rash and viral pneumonia. Congenital varicella syndrome is a concern through at least the first half of pregnancy, with risk of malformation to the fetus (55, 56). Infants born to mothers with active primary disease are at increased risk for severe and sometimes fatal varicella (57, 58). Reactivation of latent VZV can occur in individuals previously infected with the virus and generally presents as radicular pain with vesicular rash in a unilateral dermatomal distribution, most often affecting thoracic or trigeminal ophthalmic nerves (59–61). The appearance of new lesions involving adjacent dermatomes is not uncommon. Although reactivation can occur in any age group, most reactivations occur in individuals greater than 60 years of age or in immunosuppressed populations. Neurological involvement resulting in pain lasting over 1 month from the onset of zoster is termed postherpetic neuralgia (PHN). PHN is often a debilitating condition characterized by burning, itching, and tingling sensations that are disruptive to an individual's daily routine (59, 60, 62).
A live, attenuated (Oka strain) varicella vaccine (Varivax; Merck) is available for immunization of healthy children, adolescents, and adults. Vaccination has proven effective at reducing the occurrence and severity of primary varicella in millions of individuals worldwide (63, 64). The recent introduction of a second live, attenuated vaccine (Zostavax; Merck) to reduce the occurrence of zoster in the elderly has proven modestly effective (63, 65).
Of some concern are reports that the live vaccine can cause disease in healthy (66–71) and immunocompromised (72–74) individuals. A case of zoster that later disseminated due to an acyclovir-resistant Oka strain has been reported (75).
Recently, Doerr (76) described the need for an efficacious, inactivated vaccine for selected patient populations. We propose that VZV portal mutants could represent the next-generation varicella-zoster vaccine. The current vaccine is safe for immunocompetent individuals, although local and/or systemic side effects, including mild zoster reactivations, do occur. A replication-defective, ORF54 mutant virus (i.e., Δ54S) can replicate only in a complementing cell line expressing ORF54. Virus obtained after infection of the complementing cell line could be used for human vaccination. Although Δ54S could infect host cells and express viral antigens, no infectious virions would be produced; thus, spread beyond the injection site cannot occur. Additionally, a replication-defective vaccine would not be capable of reactivating and therefore cannot cause recurrent disease in patient populations. Because of the potentially enhanced safety profile, new patient populations, such as cancer patients, transplant patients, and pregnant women, could be targeted for vaccination.
Beyond the utility of portal mutants as single-cycle vaccines, our laboratory is interested in the structure and function of herpesvirus portal proteins. The phenotypic effect of inhibiting pORF54 function was consistent with the genetic evidence provided by previous studies with HSV UL6 deletion mutants (14, 17). Cells infected with HSV mutants containing loss-of-function mutations in the UL6 gene failed to properly process viral DNA. UL6 deletion mutants are defective in both DNA cleavage and packaging, and electron microscopy confirmed large numbers of empty capsids in nuclei of mutant-infected cells. Studies by Newcomb et al. showed that pUL6 assembled into ring-like structures in vitro that form a portal of entry for viral DNA into the capsid (15). VZV pORF54 shows 44% amino acid identity with its HSV-1 pUL6 homolog (18, 77), and it recently was shown to form portal-like structures when expressed in insect cells (18, 19). The results reported in this paper suggest that pORF54 performs a functional role similar to that of pUL6 during VZV replication.
Small-molecule inhibitors of herpesvirus portal protein function have significant potential in antiviral therapy. High-throughput screening resulted in the identification of such inhibitors for HSV and VZV (20, 21, 77). Mutations in the UL6 and ORF54 genes of inhibitor-resistant isolates strongly suggest that the portal protein is the target of the inhibitors. Structural and functional studies of portal proteins are necessary to understand how and where the inhibitors act to prevent encapsidation. Newcomb et al. provided evidence suggesting that the HSV-specific inhibitors reduced the incorporation of portal and terminase proteins into HSV capsids (78). It is possible that the inhibitors prevent (i) portal oligomerization, (ii) association with the capsid vertex, (iii) association with one of more terminase subunits, and/or (iv) important conformational changes required during the movement of DNA into preformed capsids. It is not known if the inhibitors bind a specific pocket within the portal multimer or in the individual portal monomers. The ORF54-expressing cell line and ORF54 mutant constructs reported here will be used to generate additional portal mutants that should prove valuable in elucidating portal functions and inhibitor mechanisms.
ACKNOWLEDGMENTS
These studies were supported by a grant from the National Institutes of Health (7R15AI062713-03) and seed funding from Mercer University School of Medicine.
We gratefully acknowledge the staff at Georgia Regents University for assistance with transmission electron microscopy.
Footnotes
Published ahead of print 7 May 2014
REFERENCES
- 1.Oliveira L, Tavares P, Alonso JC. 2013. Headful DNA packaging: bacteriophage SPP1 as a model system. Virus Res. 173:247–259. 10.1016/j.virusres.2013.01.021 [DOI] [PubMed] [Google Scholar]
- 2.Morais MC. 2012. The dsDNA packaging motor in bacteriophage o29. Adv. Exp. Med. Biol. 726:511–547. 10.1007/978-1-4614-0980-9_23 [DOI] [PubMed] [Google Scholar]
- 3.Rao VB, Feiss M. 2008. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42:647–681. 10.1146/annurev.genet.42.110807.091545 [DOI] [PubMed] [Google Scholar]
- 4.Hendrix RW. 1978. Symmetry mismatch and DNA packaging in large bacteriophages. Proc. Natl. Acad. Sci. U. S. A. 75:4779–4783. 10.1073/pnas.75.10.4779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. 2001. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature 413:748–752. 10.1038/35099581 [DOI] [PubMed] [Google Scholar]
- 6.Valpuesta JM, Carrascosa JL. 1994. Structure of viral connectors and their function in bacteriophage assembly and DNA packaging. Q. Rev. Biophys. 27:107–155. 10.1017/S0033583500004510 [DOI] [PubMed] [Google Scholar]
- 7.Orlova EV, Dube P, Beckmann E, Zemlin F, Lurz R, Trautner TA, Tavares P, van Heel M. 1999. Structure of the 13-fold symmetric portal protein of bacteriophage SPP1. Nat. Struct. Biol. 6:842–846. 10.1038/12303 [DOI] [PubMed] [Google Scholar]
- 8.Trus BL, Cheng N, Newcomb WW, Homa FL, Brown JC, Steven AC. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78:12668–12671. 10.1128/JVI.78.22.12668-12671.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. 2007. Structural framework for DNA translocation via the viral portal protein. EMBO J. 26:1984–1994. 10.1038/sj.emboj.7601643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Padilla-Sanchez V, Gao S, Kim HR, Kihara D, Sun L, Rossmann MG, Rao VB. 2014. Structure-function analysis of the DNA translocating portal of the bacteriophage T4 packaging machine. J. Mol. Biol. 426:1019–1038. 10.1016/j.jmb.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alam TI, Draper B, Kondabagil K, Rentas FJ, Ghosh-Kumar M, Sun S, Rossmann MG, Rao VB. 2008. The headful packaging nuclease of bacteriophage T4. Mol. Microbiol. 69:1180–1190. 10.1111/j.1365-2958.2008.06344.x [DOI] [PubMed] [Google Scholar]
- 12.Ghosh-Kumar M, Alam TI, Draper B, Stack JD, Rao VB. 2011. Regulation by interdomain communication of a headful packaging nuclease from bacteriophage T4. Nucleic Acids Res. 39:2742–2755. 10.1093/nar/gkq1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao S, Rao VB. 2011. Specificity of interactions among the DNA-packaging machine components of T4-related bacteriophages. J. Biol. Chem. 286:3944–3956. 10.1074/jbc.M110.196907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamberti C, Weller SK. 1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 226:403–407. 10.1006/viro.1996.0668 [DOI] [PubMed] [Google Scholar]
- 15.Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, Brown JC. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75:10923–10932. 10.1128/JVI.75.22.10923-10932.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel AH, MacLean JB. 1995. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206:465–478. 10.1016/S0042-6822(95)80062-X [DOI] [PubMed] [Google Scholar]
- 17.Patel AH, Rixon FJ, Cunningham C, Davison AJ. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217:111–123. 10.1006/viro.1996.0098 [DOI] [PubMed] [Google Scholar]
- 18.Visalli RJ, Howard AJ. 2014. Non-axial view of the varicella-zoster virus portal protein reveals conserved crown, wing and clip architecture. Intervirology 57:121–125. 10.1159/000360225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howard AJ, Sherman DM, Visalli MA, Burnside DM, Visalli RJ. 2012. The Varicella-zoster virus ORF54 gene product encodes the capsid portal protein, pORF54. Virus Res. 167:102–105. 10.1016/j.virusres.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Zeijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O'Hara B, Bloom JD, Johann SV. 2000. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J. Virol. 74:9054–9061. 10.1128/JVI.74.19.9054-9061.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Visalli RJ, Fairhurst J, Srinivas S, Hu W, Feld B, DiGrandi M, Curran K, Ross A, Bloom JD, van Zeijl M, Jones TR, O'Connell J, Cohen JI. 2003. Identification of small molecule compounds that selectively inhibit varicella-zoster virus replication. J. Virol. 77:2349–2358. 10.1128/JVI.77.4.2349-2358.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNab AR, Desai P, Person S, Roof LL, Thomsen DR, Newcomb WW, Brown JC, Homa FL. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 72:1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavalcoli JD, Baghian A, Homa FL, Kousoulas KG. 1993. Resolution of genotypic and phenotypic properties of herpes simplex virus type 1 temperature-sensitive mutant (KOS) tsZ47: evidence for allelic complementation in the UL28 gene. Virology 197:23–34. 10.1006/viro.1993.1563 [DOI] [PubMed] [Google Scholar]
- 24.Addison C, Rixon FJ, Preston VG. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J. Gen. Virol. 71(Part 10):2377–2384. 10.1099/0022-1317-71-10-2377 [DOI] [PubMed] [Google Scholar]
- 25.Tengelsen LA, Pederson NE, Shaver PR, Wathen MW, Homa FL. 1993. Herpes simplex virus type 1 DNA cleavage and encapsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J. Virol. 67:3470–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu D, Sheaffer AK, Tenney DJ, Weller SK. 1997. Characterization of ICP6::lacZ insertion mutants of the UL15 gene of herpes simplex virus type 1 reveals the translation of two proteins. J. Virol. 71:2656–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baines JD, Poon AP, Rovnak J, Roizman B. 1994. The herpes simplex virus 1 UL15 gene encodes two proteins and is required for cleavage of genomic viral DNA. J. Virol. 68:8118–8124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poon AP, Roizman B. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67:4497–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cunningham C, Davison AJ. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116–124. 10.1006/viro.1993.1572 [DOI] [PubMed] [Google Scholar]
- 30.Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn J, Leege T, Granzow H, Fuchs W, Mettenleiter TC, Klupp BG. 2010. Analysis of pseudorabies and herpes simplex virus recombinants simultaneously lacking the pUL17 and pUL25 components of the C-capsid specific component. Virus Res. 153:20–28. 10.1016/j.virusres.2010.06.022 [DOI] [PubMed] [Google Scholar]
- 32.Taus NS, Salmon B, Baines JD. 1998. The herpes simplex virus 1 UL 17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115–125. 10.1006/viro.1998.9439 [DOI] [PubMed] [Google Scholar]
- 33.al-Kobaisi MF, Rixon FJ, McDougall I, Preston VG. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388. 10.1016/0042-6822(91)90043-B [DOI] [PubMed] [Google Scholar]
- 34.Visalli RJ, Knepper J, Goshorn B, Vanover K, Burnside DM, Irven K, McGauley R, Visalli M. 2009. Characterization of the Varicella-zoster virus ORF25 gene product: pORF25 interacts with multiple DNA encapsidation proteins. Virus Res. 144:58–64. 10.1016/j.virusres.2009.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visalli RJ, Nicolosi DM, Irven KL, Goshorn B, Khan T, Visalli MA. 2007. The Varicella-zoster virus DNA encapsidation genes: Identification and characterization of the putative terminase subunits. Virus Res. 129:200–211. 10.1016/j.virusres.2007.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vizoso Pinto MG, Pothineni VR, Haase R, Woidy M, Lotz-Havla AS, Gersting SW, Muntau AC, Haas J, Sommer M, Arvin AM, Baiker A. 2011. Varicella zoster virus ORF25 gene product: an essential hub protein linking encapsidation proteins and the nuclear egress complex. J. Proteome Res. 10:5374–5382. 10.1021/pr200628s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Huang Y, Zhu H. 2010. An efficient protocol for VZV BAC-based mutagenesis. Methods Mol. Biol. 634:75–86. 10.1007/978-1-60761-652-8_5 [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, Huang Y, Zhu H. 2008. A highly efficient protocol of generating and analyzing VZV ORF deletion mutants based on a newly developed luciferase VZV BAC system. J. Virol. Methods 148:197–204. 10.1016/j.jviromet.2007.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Selariu A, Warden C, Huang G, Huang Y, Zaccheus O, Cheng T, Xia N, Zhu H. 2010. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog. 6:e1000971. 10.1371/journal.ppat.1000971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harper DR, Mathieu N, Mullarkey J. 1998. High-titre, cryostable cell-free varicella zoster virus. Arch. Virol. 143:1163–1170. 10.1007/s007050050364 [DOI] [PubMed] [Google Scholar]
- 41.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davison AJ, Scott JE. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67(Pt 9):1759–1816 [DOI] [PubMed] [Google Scholar]
- 43.Grose C, Brunel PA. 1978. Varicella-zoster virus: isolation and propagation in human melanoma cells at 36 and 32 degrees C. Infect. Immun. 19:199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole NL, Grose C. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 13:207–222. 10.1002/rmv.377 [DOI] [PubMed] [Google Scholar]
- 45.Muraki R, Baba T, Iwasaki T, Sata T, Kurata T. 1992. Immunohistochemical study of skin lesions in herpes zoster. Virchows Arch. A Pathol. Anat Histopathol. 420:71–76. 10.1007/BF01605987 [DOI] [PubMed] [Google Scholar]
- 46.Moffat JF, Stein MD, Kaneshima H, Arvin AM. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69:5236–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliver SL, Brady JJ, Sommer MH, Reichelt M, Sung P, Blau HM, Arvin AM. 2013. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 110:1911–1916. 10.1073/pnas.1216985110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoover SE, Cohrs RJ, Rangel ZG, Gilden DH, Munson P, Cohen JI. 2006. Downregulation of varicella-zoster virus (VZV) immediate-early ORF62 transcription by VZV ORF63 correlates with virus replication in vitro and with latency. J. Virol. 80:3459–3468. 10.1128/JVI.80.7.3459-3468.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reichelt M, Joubert L, Perrino J, Koh AL, Phanwar I, Arvin AM. 2012. 3D reconstruction of VZV infected cell nuclei and PML nuclear cages by serial section array scanning electron microscopy and electron tomography. PLoS Pathog. 8:e1002740. 10.1371/journal.ppat.1002740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harson R, Grose C. 1995. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J. Virol. 69:4994–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grose C, Yu X, Cohrs RJ, Carpenter JE, Bowlin JL, Gilden D. 2013. Aberrant virion assembly and limited glycoprotein C production in varicella-zoster virus-infected neurons. J. Virol. 87:9643–9648. 10.1128/JVI.01506-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen JI, Brunell PA, Straus SE, Krause PR. 1999. Recent advances in varicella-zoster virus infection. Ann. Intern. Med. 130:922–932. 10.7326/0003-4819-130-11-199906010-00017 [DOI] [PubMed] [Google Scholar]
- 53.Arvin AM. 1996. Varicella-zoster virus. Clin. Microbiol. Rev. 9:361–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gershon AA, Gershon MD. 2013. Pathogenesis and current approaches to control of varicella-zoster virus infections. Clin. Microbiol. Rev. 26:728–743. 10.1128/CMR.00052-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gnann JW., Jr 2002. Varicella-zoster virus: atypical presentations and unusual complications. J. Infect. Dis. 186(Suppl 1):S91–S98. 10.1086/342963 [DOI] [PubMed] [Google Scholar]
- 56.Enders G, Miller E, Cradock-Watson J, Bolley I, Ridehalgh M. 1994. Consequences of varicella and herpes zoster in pregnancy: prospective study of 1739 cases. Lancet 343:1548–1551. 10.1016/S0140-6736(94)92943-2 [DOI] [PubMed] [Google Scholar]
- 57.Prober CG, Gershon AA, Grose C, McCracken GH, Jr, Nelson JD. 1990. Consensus: varicella-zoster infections in pregnancy and the perinatal period. Pediatr. Infect. Dis. J. 9:865–869. 10.1097/00006454-199012000-00001 [DOI] [PubMed] [Google Scholar]
- 58.Snoeck R, Andrei G, De Clercq E. 2000. Novel agents for the therapy of varicella-zoster virus infections. Expert Opin. Investig. Drugs 9:1743–1751. 10.1517/13543784.9.8.1743 [DOI] [PubMed] [Google Scholar]
- 59.Tenser RB. 2001. Herpes zoster infection and postherpetic neuralgia. Curr. Neurol. Neurosci. Rep. 1:526–532. 10.1007/s11910-001-0057-z [DOI] [PubMed] [Google Scholar]
- 60.Wood MJ. 1996. How to measure and reduce the burden of zoster-associated pain. Scand. J. Infect. Dis. Suppl. 100:55–58 [PubMed] [Google Scholar]
- 61.Kost RG, Straus SE. 1996. Postherpetic neuralgia–pathogenesis, treatment, and prevention. N. Engl. J. Med. 335:32–42. 10.1056/NEJM199607043350107 [DOI] [PubMed] [Google Scholar]
- 62.Arvin AM. 1996. Varicella-zoster virus: overview and clinical manifestations. Semin. Dermatol. 15:4–7 [PubMed] [Google Scholar]
- 63.Andrei G, Snoeck R. 2013. Advances in the treatment of varicella-zoster virus infections. Adv. Pharmacol. 67:107–168. 10.1016/B978-0-12-405880-4.00004-4 [DOI] [PubMed] [Google Scholar]
- 64.Kim SR, Khan F, Tyring SK. 2014. Varicella zoster: an update on current treatment options and future perspectives. Expert Opin. Pharmacother 15:61–71. 10.1517/14656566.2014.860443 [DOI] [PubMed] [Google Scholar]
- 65.Doan HQ, Ung B, Ramirez-Fort MK, Khan F, Tyring SK. 2013. Zostavax: a subcutaneous vaccine for the prevention of herpes zoster. Expert Opin. Biol. Ther. 13:1467–1477. 10.1517/14712598.2013.830101 [DOI] [PubMed] [Google Scholar]
- 66.Brunell PA, Argaw T. 2000. Chickenpox attributable to a vaccine virus contracted from a vaccinee with zoster. Pediatrics 106:E28. 10.1542/peds.106.2.e28 [DOI] [PubMed] [Google Scholar]
- 67.LaRussa P, Steinberg S, Meurice F, Gershon A. 1997. Transmission of vaccine strain varicella-zoster virus from a healthy adult with vaccine-associated rash to susceptible household contacts. J. Infect. Dis. 176:1072–1075. 10.1086/516514 [DOI] [PubMed] [Google Scholar]
- 68.Uebe B, Sauerbrei A, Burdach S, Horneff G. 2002. Herpes zoster by reactivated vaccine varicella zoster virus in a healthy child. Eur. J. Pediatr. 161:442–444. 10.1007/s00431-002-0981-1 [DOI] [PubMed] [Google Scholar]
- 69.Chouliaras G, Spoulou V, Quinlivan M, Breuer J, Theodoridou M. 2010. Vaccine-associated herpes zoster ophthalmicus [correction of opthalmicus] and encephalitis in an immunocompetent child. Pediatrics 125:e969–972. 10.1542/peds.2009-2633 [DOI] [PubMed] [Google Scholar]
- 70.Goulleret N, Mauvisseau E, Essevaz-Roulet M, Quinlivan M, Breuer J. 2010. Safety profile of live varicella virus vaccine (Oka/Merck): five-year results of the European Varicella Zoster Virus Identification Program (EU VZVIP). Vaccine 28:5878–5882. 10.1016/j.vaccine.2010.06.056 [DOI] [PubMed] [Google Scholar]
- 71.Tseng HF, Schmid DS, Harpaz R, Larussa P, Jensen NJ, Rivailler P, Radford K, Folster J, Jacobsen SJ. 2014. Herpes zoster caused by vaccine-strain varicella zoster virus in an immunocompetent recipient of zoster vaccine. Clin. Infect. Dis. 58:1125–1128. 10.1093/cid/ciu058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levy O, Orange JS, Hibberd P, Steinberg S, LaRussa P, Weinberg A, Wilson SB, Shaulov A, Fleisher G, Geha RS, Bonilla FA, Exley M. 2003. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J. Infect. Dis. 188:948–953. 10.1086/378503 [DOI] [PubMed] [Google Scholar]
- 73.Bryan CJ, Prichard MN, Daily S, Jefferson G, Hartline C, Cassady KA, Hilliard L, Shimamura M. 2008. Acyclovir-resistant chronic verrucous vaccine strain varicella in a patient with neuroblastoma. Pediatr. Infect. Dis. J. 27:946–948. 10.1097/INF.0b013e318175d85c [DOI] [PubMed] [Google Scholar]
- 74.Leung J, Siegel S, Jones JF, Schulte C, Blog D, Scott Schmid D, Bialek SR, Marin M. 2014. Fatal varicella due to the vaccine-strain varicella-zoster virus. Hum. Vaccin. Immunother. 10:146–149. 10.4161/hv.26200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levin MJ, Dahl KM, Weinberg A, Giller R, Patel A, Krause PR. 2003. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus, in an immunosuppressed child. J. Infect. Dis. 188:954–959. 10.1086/378502 [DOI] [PubMed] [Google Scholar]
- 76.Doerr HW. 2013. Progress in VZV vaccination? Some concerns. Med. Microbiol. Immunol. 202:257–258. 10.1007/s00430-013-0298-x [DOI] [PubMed] [Google Scholar]
- 77.Visalli RJ, van Zeijl M. 2003. DNA encapsidation as a target for anti-herpesvirus drug therapy. Antiviral Res. 59:73–87. 10.1016/S0166-3542(03)00108-6 [DOI] [PubMed] [Google Scholar]
- 78.Newcomb WW, Brown JC. 2002. Inhibition of herpes simplex virus replication by WAY-150138: assembly of capsids depleted of the portal and terminase proteins involved in DNA encapsidation. J. Virol. 76:10084–10088. 10.1128/JVI.76.19.10084-10088.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Z, Rowe J, Wang W, Sommer M, Arvin A, Moffat J, Zhu H. 2007. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J. Virol. 81:9024–9033. 10.1128/JVI.02666-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jing P, Haque F, Shu D, Montemagno C, Guo P. 2010. One-way traffic of a viral motor channel for double-stranded DNA translocation. Nano Lett. 10:3620–3627. 10.1021/nl101939e [DOI] [PMC free article] [PubMed] [Google Scholar]