

ABSTRACT
Actin and actin-binding proteins are incorporated into HIV-1 particles, and F-actin has been suggested to bind the NC domain in HIV-1 Gag. Furthermore, F-actin has been frequently observed in the vicinity of HIV-1 budding sites by cryo-electron tomography (cET). Filamentous structures emanating from viral buds and suggested to correspond to actin filaments have been observed by atomic force microscopy. To determine whether the NC domain of Gag is required for actin association with viral buds and for actin incorporation into HIV-1, we performed comparative analyses of virus-like particles (VLPs) obtained by expression of wild-type HIV-1 Gag or a Gag variant where the entire NC domain had been replaced by a dimerizing leucine zipper [Gag(LZ)]. The latter protein yielded efficient production of VLPs with near-wild-type assembly kinetics and size and exhibited a regular immature Gag lattice. Typical HIV-1 budding sites were detected by using cET in cells expressing either Gag or Gag(LZ), and no difference was observed regarding the association of buds with the F-actin network. Furthermore, actin was equally incorporated into wild-type HIV-1 and Gag- or Gag(LZ)-derived VLPs, with less actin per particle observed than had been reported previously. Incorporation appeared to correlate with the relative intracellular actin concentration, suggesting an uptake of cytosol rather than a specific recruitment of actin. Thus, the NC domain in HIV-1 Gag does not appear to have a role in actin recruitment or actin incorporation into HIV-1 particles.
IMPORTANCE HIV-1 particles bud from the plasma membrane, which is lined by a network of actin filaments. Actin was found to interact with the nucleocapsid domain of the viral structural protein Gag and is incorporated in significant amounts into HIV-1 particles, suggesting that it may play an active role in virus release. Using electron microscopy techniques, we previously observed bundles of actin filaments near HIV-1 buds, often seemingly in contact with the Gag layer. Here, we show that this spatial association is observed independently of the proposed actin-binding domain of HIV-1. The absence of this domain also did not affect actin incorporation and had a minor effect on the viral assembly rate. Furthermore, actin was not enriched in the virus compared to the average levels in the respective producing cell. Our data argue against a specific recruitment of actin to HIV-1 budding sites by the nucleocapsid domain of Gag.
INTRODUCTION
HIV-1 morphogenesis is driven by the viral Gag polyprotein, whose expression is sufficient for the production of extracellular virus-like particles (VLPs) with a size and protein lattice similar to those of the immature virion (1). Gag associates with and assembles at the plasma membrane, thereby inducing membrane curvature and bud formation. During assembly, the C-terminal p6 domain of Gag recruits the cellular endosomal sorting complex required for transport (ESCRT), which subsequently mediates abscission of the virus from the cell membrane. Gag is composed of the MA (matrix), CA (capsid), NC (nucleocapsid), and p6 domains, which are proteolytically separated in the nascent virus, leading to morphological maturation. In contrast, VLPs consisting of Gag alone remain immature, as they lack the viral protease (1). An immature HIV-1 particle or VLP with a diameter of 145 nm contains ∼2,400 Gag molecules arranged radially in a roughly spherical array with their N termini facing outward and their C termini facing inward (2, 3). MA is needed for membrane binding, CA mediates lateral protein interactions determining the immature lattice, and NC binds RNA, leading to the incorporation of the viral genome through its packaging signal Ψ (1, 4).
Many cellular proteins have been implicated in HIV-1 assembly, budding, and release. These include actin and several actin-binding proteins, but the precise role of actin in HIV-1 morphogenesis, if any, has not been determined so far. Based on its known properties, actin in principle could have positive or negative effects on HIV-1 particle production. First, polymerization of F-actin may aid in bud formation and thus drive virus release, potentially similar to the well-known role of filamentous actin (F-actin) in vaccinia virus spread (5). Alternatively, or in addition, dense cortical F-actin networks may present a barrier for bud formation, and depolymerization of cortical F-actin may be important for efficient virus formation. Early studies indicated that relatively large amounts of actin are incorporated into retroviral particles, with a 10:1 ratio for CA to actin in HIV-1 (6–8), corresponding to roughly 250 molecules of actin per virion. However, actin is one of the most abundant cytoplasmic proteins and is strongly enriched in the cortical region of the cell, and it is not known whether the amount of actin in the virus represents enrichment over its concentration in the vicinity of the budding site. Long-term treatment of HIV-1-infected cells with F-actin-disrupting agents (cytochalasin D or latrunculin B) or with an inhibitor of myosin light chain kinase (wortmannin) resulted in reduced retrovirus production (9–11), but it is unclear whether this was due to the blocking of a direct effect of actin on virus formation or to pleiotropic effects on the producer cell due to the prolonged action of the drug. Conversely, shorter treatment of virus-producing cells with actin-depolymerizing drugs appeared to enhance retroviral budding (9), which would be in line with cortical actin presenting an obstacle for virus formation.
Three reports provided evidence for a direct interaction of the NC domain of HIV-1 Gag with F-actin in vitro using cofractionation and coimmunoprecipitation experiments (8, 12, 13). Furthermore, Wilk and coauthors (8) performed immunogold labeling experiments, localizing actin to the position of the NC layer in the Gag lattice of immature HIV-1 VLPs. These results suggested a direct interaction of the NC domain of Gag with F-actin leading to actin recruitment and virion incorporation. Mutational analyses showed that F-actin binding to the NC domain did not require the interaction of this domain with the viral RNA (13, 14). A role of the NC domain in F-actin recruitment was supported by an investigation of viral budding sites at the surface of HIV-1 and murine leukemia virus (MLV) Gag-expressing HeLa and NIH 3T3 cells by atomic force microscopy (AFM) (15). That study reported prominent star-shaped structures (“asters”) with arm lengths of ∼0.5 to 2.5 μm centered at a subset of budding sites. They were interpreted as structured F-actin assemblies recruited by retroviral Gag. The presence of these asters was dependent on the presumed actin-interacting NC domain of Gag. No such structures were observed when the NC domain was replaced by a dimerizing leucine zipper from the Saccharomyces cerevisiae transcription factor GCN4 [Gag(LZ)] (15). Gag(LZ) expression was shown previously to yield the formation of extracellular VLPs with regular immature morphology, where the LZ domain substitutes for the role of RNA-NC interactions in concentrating and aligning Gag molecules during assembly (16–20).
A morphological connection between F-actin and nascent HIV-1 assembly sites was also suggested by our previous cryo-electron tomography (cET) analysis of budding sites at the membrane of HIV-1 Gag- or GagPol-expressing glioblastoma cells (21). F-actin structures were observed in the close vicinity of most budding sites analyzed and were pointing into the nascent particle in ∼50% of all cases. Furthermore, tips of actin filaments appeared to be connected with the inner rims of the immature Gag lattice in some cases, suggesting a direct interaction (21). We have now extended this cET analysis to budding sites of HIV-1 Gag(LZ), which lacks the putative actin interaction domain in NC. No difference regarding an association of budding sites with F-actin networks was observed for budding sites of wild-type Gag or Gag(LZ). Furthermore, actin concentrations were similar in both VLP preparations and were much lower than those in the respective producer cells. These results indicate that putative NC-actin interactions do not play a role in actin recruitment to HIV-1 budding sites, and the actin content in the virion may arise from the uptake of cytoplasmic material in general rather than from specific incorporation.
MATERIALS AND METHODS
Plasmids.
Adenoviral vectors for HIV-1 Gag (AdGag) and GagPol (AdGagPol) were constructed from Rev-independent expression plasmids (13), using BD Adeno-X Expression System 1 (BD Biosciences Clontech), as described previously (21). A Rev-independent Gag leucine zipper [Gag(LZ)] coding sequence was constructed by overlap PCR using an MA-CA-SP1-encoding fragment derived from pcDNA3.1synGagPol (22) and an LZ-p6-encoding fragment derived from a chimeric Gag variant containing codons 247 to 280 of the GCN4 leucine zipper in place of NC-SP2(Zwtp6) (16) (forward primer 5′-ACCAACACCGCCACCATCATGCTGCAGCGTATGAAGCAGCTCGAGG-3′ and reverse primer 5′-TATGGTACCTTATTGTGACGAGGGGTCGTTGCCAAAGAG-3′). The resulting PCR product was ligated into pShuttle2 for transfer into the Adeno-X viral DNA according to the manufacturer's protocol to obtain AdGag(LZ). Plasmids pCHIVunc(LZ) and pCHIVunc(LZ)eGFP are based on the non-replication-competent plasmid pCHIV (23). A synthetic BstEII-XmaI gene fragment encoding the GCN4 leucine zipper sequence (Geneart AG, Regensburg, Germany) was used to replace the corresponding BstEII-XmaI fragment (encoding the NC region) of the infectious proviral construct pNL43(unc) (24). From this, non-replication-competent derivatives were generated by cloning an SwaI fragment from pNL4-3unc(LZ) into pCHIV or pCHIVeGFP.
Cell culture, transfection, and recombinant adenovirus production.
HeLa (25), HEK293 (26), HEK293T (27), and U-373 MG (28) cells were grown in Dulbecco's modified Eagle's medium (DMEM). MT-4 cells (29) and primary CD4+ cells were cultured in RPMI 1640 medium. Both media were supplemented with 10% fetal calf serum (FCS; Biochrom), penicillin (100 IU/ml), streptomycin (100 μg/ml), and 4 mM glutamine. Peripheral blood mononuclear cells (PBMCs) were purified from buffy coats obtained from healthy blood donors by Ficoll gradient centrifugation according to standard procedures. CD4+ cells were isolated by using negative selection by a magnetically activated cell sorting (MACS) preparation (Miltenyi) according to the manufacturer's instructions. Recombinant adenoviruses were generated by transfection of 5 to 10 μg PacI-digested adenoviral plasmids into HEK293 cells plated onto 60-mm culture plates by using FugeneHD transfection reagent (Roche) according to the manufacturer's instructions. Cells were harvested at 10 to 14 days posttransfection. Approximately 50 to 100% of the harvested lysate (primary amplification lysate) was used for successive rounds of infections of HEK293 cells until cytopathic effects were evident within 5 to 6 days postinfection (p.i.). High-titer stocks were produced by infection of HEK293 target cells plated into a T75 flask with either primary amplification lysates or previously generated high-titer stocks. Titers of adenoviral vectors were determined by using the Adeno-X Rapid Titer kit (BD Biosciences Clontech). For all adenovirus transduction experiments, cells were infected with a multiplicity of infection (MOI) of 10 to 20.
Determination of cell volumes.
To achieve an accurate comparison of cell volumes between suspension and adherent cells, HEK293T cells were detached from the substrate by trypsinization. Cells were fixed in solution with 3% paraformaldehyde at room temperature, stained with 2 μg/ml phalloidin-Atto488 (Invitrogen) for 60 min to label the cell boundaries, washed in phosphate-buffered saline (PBS), and mounted onto coverslips. Spinning-disk confocal microscopy was performed at a magnification of ×100 (numerical aperture [NA] = 1.4) with a Z-spacing of 150 nm. Three-dimensional (3D) reconstructions using Volocity 6.0 software were performed for cells displaying a roundish or ellipsoid geometry. From this reconstruction, the ellipsoid volume was calculated.
HIV-1 and VLP purification.
HIV-1 was purified from infected MT-4 T cells or HEK293T cells transfected with the proviral plasmid pNL4-3 (30). Infection of MT-4 cells with HIV-1 strain NL4-3 by coculture and purification of the virus from cell culture supernatant by iodixanol gradient centrifugation were described previously (31). Immature HIV-1 particles were obtained by adding the HIV-1 protease inhibitor lopinavir (LPV) to a final concentration of 2 μM at 6 h after infection or transfection, followed by particle purification as described above. VLPs derived from adenovirus-transduced HEK293T cells were purified according to the same protocol as that used for complete HIV-1.
Immunoblotting and quantitation.
U-373 MG cells were seeded onto 6-well plates to a density of 2 × 105 cells/well and were incubated for 24 h at 37°C. Cells were transduced with adenoviral vectors as described above, followed by cell lysis and harvesting of cell culture supernatants at 48 h posttransduction (p.t.). Supernatants were clarified by centrifugation and filtered through 0.2-μm-pore-size filters, and particles were collected by ultracentrifugation through a 20% (wt/wt) sucrose cushion. Cell lysate and purified virion components were separated by SDS-PAGE (12.5% acrylamide) and transferred onto nitrocellulose membranes, and Gag-derived proteins were detected by using polyclonal sheep anti-CA antiserum. Secondary antibodies coupled to Alexa fluorescent dyes were used for detection with an Odyssey infrared imaging system as specified by the manufacturer (Li-Cor Biosciences, Lincoln, NE). For detection of actin, membranes were incubated with a rabbit anti-actin antibody (Sigma-Aldrich, St. Louis, MO). Amounts of Gag (or CA) antigen and actin were quantified by using standard curves generated from serial dilutions of purified HIV-1 CA (kindly provided by Vanda Lux), HIV-1 Gag (kindly provided by Johnson Mak, Melbourne, Australia), or human F-actin (kindly provided by Rasmus Schröder, Heidelberg, Germany). Standards and samples were always run on the same gel.
Sample preparation for cryo-electron tomography.
U-373 MG cells were grown directly on holey carbon film-coated electron microscopy (EM) gold grids (catalog number CF-2/1-2AU; Protochips Inc.). Prior to seeding of the cells, grids were glow discharged for 20 s in a Harrick PDC-002 plasma cleaner and washed in cell culture medium for 1 to 2 h at 37°C. The grids were subsequently placed into a well of a 6-well plate with the carbon film facing up. Cells were seeded typically at a low density (0.8 × 105 to 1 × 105 cells per well) and grown in low-serum medium (0.5% FCS) to increase cell adhesion and a flat-cell morphology. Transduction with AdGag, AdGagPol, and AdGag(LZ) was performed 4 h after seeding of the cells. At 2 days p.t., samples were plunge-frozen in liquid ethane or a mixture of liquid ethane and propane (67%:33% by volume) (32) after application of 10-nm bovine serum albumin (BSA)-coupled colloidal gold in PBS and blotting of the excess liquid with Whatman no. 1 filter paper.
Data acquisition for cET.
Automated ET was performed according to previously described basic principles including the predictive tracking method (33, 34). Tilt series of plunge-frozen specimens were acquired with an FEI G2 Polara transmission electron microscope operated at 300 keV and equipped with a Gatan GIF 2002 postcolumn energy filter and a 2,048-by-2,048 Multiscan charge-coupled-device (CCD) camera (Gatan, Pleasanton, CA). Data collection was performed with the energy filter operated in zero loss mode and with a 20-eV slit width. Tilt series were typically recorded from −60° to +60° with an angular increment of 1.5°, a total electron dose of 80 to 120 e−/Å2, and a defocus of −6 or −8 μm. In some cases, the tilt range could not be fully exploited due to proximity to a grid bar or a thick area on the specimen. Tilt series acquisition was performed by using SerialEM software (33). Image magnification was set to ×18,000, resulting in a pixel size at the specimen level of 7.7 Å for the G2 Polara microscope. The electron dose for a given tilt angle, α, was proportional to 1/cos(α), to compensate for the higher effective specimen thickness at higher tilt angles.
Image processing.
Three-dimensional reconstructions from tilt series were calculated by using the weighted back-projection method (35) and IMOD software (36). Reconstructed volumes were 1× binned and filtered by using the 3D low-pass-filter function of IMOD. Representative computational slices through 3D reconstructions presented here were generated by using Amira software (Visage Imaging).
Subtomogram averaging.
Subtomogram averaging analyses were performed as described previously (21). Subvolumes of 57.73 nm3 were extracted along the surface of the particles and preoriented normal to the surface. The subtomograms were then iteratively rotationally and translationally aligned against the average. Independent reconstructions were done for each viral particle, and each initial reference was the sum of the subtomograms prior to any alignment. Sixfold symmetry was applied to the average at each iteration. Alignment quality was assessed by using the cross-correlation between the reference and the subtomogram after the alignment. A threshold corresponding to the mean cross-correlation value was set, below which subtomograms were considered to be badly aligned, and these were not included in the final structure. All the analyses were done by using Matlab (Mathworks).
Radius-angle-frequency plots.
Radius-angle-frequency (RAF) plots were generated as described previously (37). The subtomograms extracted for subtomogram averaging were radially aligned and underwent an orthographic radial projection. The focus of the projection was determined as the geometric center of the particle from which the subtomogram was extracted. The resulting volumes corresponded to flattened subtomograms. Two-dimensional (2D) power spectra were calculated at each radius. The rotation autocorrelation function with a rotation range of between 0° and 180° was calculated for each power spectrum. For each subtomogram, this generates a 3D plot with radius on one axis and the other two axes representing those of the rotational autocorrelation function of the power spectrum, namely, angle and frequency. The analyses were done by using Matlab (Mathworks) and IMAGIC (Image Science Software GmbH).
Live-cell total internal reflection fluorescence microscopy and single-virus tracking.
HeLa cells were seeded at 1.5 × 104 cells/well into 8-well glass-bottom chamber slides (LabTek; Nunc). After 24 h of incubation at 37°C, cells were transfected with equimolar mixtures of either pCHIVunc and pCHIVunceGFP or pCHIVunc(LZ) and pCHIVunc(LZ)eGFP by using Fugene 6 (Roche) according to the manufacturer's instructions (500 ng DNA/well). At 20 h after transfection, the cells were transferred into imaging buffer (25 mM Na-HEPES [pH 7.4], 137 mM NaCl, 2.7 mM KCl, 1 mM CaCl2, 30 mM glucose), and live-cell total internal reflection fluorescence microscopy (TIR-FM) was performed. The microscope setup (Visitron Systems) was described previously (23). Briefly, TIR-FM was performed by using a Zeiss Axiovert 200M fluorescence microscope equipped with an Alpha Plan Fluar 100×/1.45 oil immersion objective. Image sequences were acquired by Metamorph (Visitron), using a sensitive EM-CCD camera (Cascade II, 512 by 512 pixels; Roper Scientific). Tracking of individual assembly sites was described previously (38, 39). Briefly, changes in enhanced green fluorescent protein-tagged Gag (Gag.eGFP) fluorescence intensity over time were analyzed as a signature for the HIV-1 Gag assembly process. Individual assembly sites were detected and localized by using an automated tracking algorithm based on particle localization using a Gaussian filter. A probabilistic approach was employed for the tracking of individual signals in consecutive frames of the movies (39). The background-corrected fluorescence intensity of detected particles was fitted to a saturating exponential function to obtain the rate of HIV-1 assembly (38). Only individual budding sites that remained discernible from neighboring sites throughout the exponential assembly phase were included in the analysis.
RESULTS
Biochemical and structural analysis of Gag(LZ) particles.
Analysis of vitrified cells by cET to investigate F-actin in spatial relation to assembling viral buds is restricted to areas where the cell is thinner than 500 nm, presenting a problem for data collection in many cell lines commonly used for EM analyses of HIV-1 budding sites (e.g., HeLa, COS-7, or T-cell lines). To overcome this problem, we took advantage of our previously established system based on the human glioblastoma cell line U-373 MG (21). This cell line displays an astrocytoid morphology characterized by long cellular protrusions and lamellipodium-like areas that are sufficiently thin for cET. In order to ensure efficient Gag expression in this cell line without cytopathic effects, we employed an adenoviral vector-based system that allowed transduction efficiencies near 100%. The suitability of this system for the expression of HIV-1 Gag (AdGag) and GagPol (AdGagPol) and analysis of viral structures by cET was demonstrated previously (21). In order to investigate whether actin filaments commonly found in the vicinity of HIV budding structures in the previous study might be recruited by the putative actin-interacting NC domain, we constructed an adenoviral vector expressing a Gag variant in which the NC-SP2 region was deleted and replaced by a dimerizing leucine zipper (LZ) domain from yeast GCN4 (Fig. 1A). This replacement has been shown to functionally compensate for the loss of the essential contributions of NC-RNA interactions to Gag multimerization and to allow efficient release of VLPs with immature morphology by thin-section EM (14, 16, 17, 19, 20).
FIG 1.
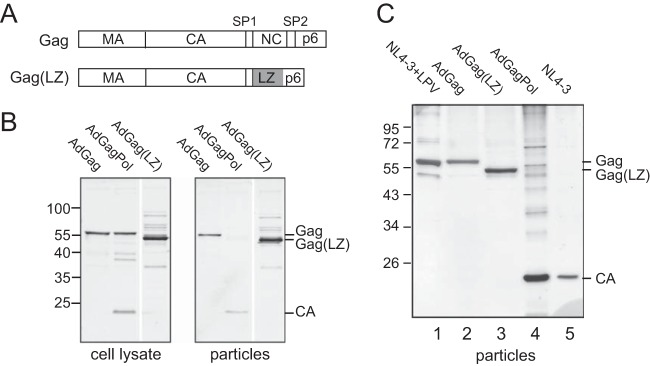
Expression of Gag, GagPol, and Gag(LZ) and particle production. (A) Schematic representation of the HIV-1 Gag and Gag(LZ) polyproteins. In Gag(LZ), the NC-SP2 region has been replaced by a leucine zipper (LZ) domain (gray) from the yeast transcription factor GCN4. (B) Gag expression and particle release. Cell lysates and tissue culture media of U-373 MG cells transduced with the indicated adenoviral vector variants were harvested at 48 h p.t. Particles were collected by ultracentrifugation of tissue culture media through a sucrose cushion. Samples were analyzed by SDS-PAGE and transferred onto a nitrocellulose membrane. Gag-derived proteins were detected by quantitative immunoblotting using polyclonal antiserum against HIV-1 CA. Positions of molecular mass standards are indicated to the left; positions of Gag-derived proteins are marked at the right. (C) Analysis of purified HIV-1 and VLPs released from HEK293T cells. Cells were either transduced with the indicated adenoviral vectors (lanes 2 to 4) or transfected with the proviral plasmid pNL4-3 (lanes 1 and 5), and particles were collected by ultracentrifugation through a sucrose cushion followed by velocity gradient centrifugation. For the generation of immature HIV-1 (lane 1), pNL4-3-transfected cells were grown in the presence of 2 μM of the protease inhibitor lopinavir (LPV). Samples were analyzed by SDS-PAGE, and proteins were visualized by silver staining according to standard procedures. Positions of molecular mass standards are indicated to the left; positions of Gag-derived proteins are marked at the right.
Transduction of U-373 MG cells with AdGag, AdGagPol, or AdGag(LZ) resulted in the expression of the respective protein and efficient VLP release in all cases (Fig. 1B). Extracellular particles released from AdGagPol-transduced cells displayed the expected mature Gag processing products, while AdGag- and AdGag(LZ)-derived particles remained unprocessed (Fig. 1B). Silver-stained gels of velocity gradient-purified particles produced from HEK293T cells that had been transduced with the different adenovirus vectors or transfected with a proviral HIV-1 plasmid in the absence or presence of the specific HIV-1 protease inhibitor lopinavir (LPV) showed the respective Gag or CA proteins as major particle constituents and revealed the purity of the preparations (Fig. 1C).
The structure of the assembled immature lattice was analyzed by cET of transduced U-373 MG cells and particles released from these cells (Fig. 2). VLPs produced from AdGag(LZ)-transduced cells were slightly larger than wild-type HIV-1 particles, with an average radius of 82 ± 8 nm (n = 25), compared to radii of 66 nm (21) and 73 nm (40) reported for different preparations of immature HIV-1. Visual inspection of computational slices from tomograms of extracellular VLPs (Fig. 2A) and HIV-specific budding sites (Fig. 2B) as well as analysis of the average radial density profile of Gag(LZ) particles (Fig. 2C) revealed two layers of density that were common to the immature wild-type and Gag(LZ) lattices: they could be attributed to the membrane/MA and CA layers, respectively. A third layer of density was clearly apparent in the immature wild-type lattice (Fig. 2A and B, red arrowheads) but appeared to be absent in immature Gag(LZ) shells. This layer was previously assigned to the complex of the NC domain and incorporated RNA (16, 17, 20, 41), which does not exist in the case of Gag(LZ).
FIG 2.

Structure of Gag(LZ) particles compared to immature HIV-1. (A and B) Immature VLPs (A) and budding sites (B) from U-373 MG cells transduced with AdGagPol (left) or AdGag(LZ) (right). In the case of AdGagPol-transduced cells, both mature and immature VLPs were detected in the sample, as described previously (21). Here, a representative immature particle is shown. Two-nanometer-thick computational slices through cryo-electron tomograms of representative structures are presented. White arrowheads indicate the electron-dense layer attributed to the CA lattice. Red arrowheads indicate density corresponding to the NC-RNA region, which is lacking in the case of AdGag(LZ) particles. Bar, 50 nm. (C) Radial density profile of immature AdGag(LZ) VLPs. The x axis indicates the radial distance relative to the middle of the viral membrane. Peaks corresponding to the viral membrane and MA domain (membrane/MA), the CA C-terminal domain (CTD), and the CA N-terminal domain (NTD) are marked. (D) Radius-angle-frequency plot for a 6-fold symmetric arrangement for AdGag(LZ)-derived VLPs indicating the presence of a regular lattice at the radial position of CA. The heat map is from blue to red. (E) Radial section with a thickness of 0.7 nm through a subtomogram average for AdGag(LZ)-derived VLPs. Density is colored black. Bar, 10 nm. (F) Surface rendering of the structure shown in panel E generated by subtomogram averaging. The color scheme follows an approximate radial division of the structure according to the different domains of Gag (membrane/MA in yellow, CA N-terminal domain in blue, CA C-terminal domain/SP1 in green, and LZ in gray).
In order to identify the regular hexameric arrangement characteristic of the CA layer of the immature assembled Gag lattice, we performed a radius-angle-frequency (RAF) analysis (37) (Fig. 2D). This analysis generates a three-dimensional density plot, within which a peak indicates the presence of a regular lattice. The position of the peak reveals the radius, rotational symmetry, and spacing of the lattice. The RAF plot for Gag(LZ) assemblies showed the presence of a regular signal at the radial position of CA with an angle of 60° and a frequency of 6.6 nm, corresponding to a hexagonal lattice with a unit-to-unit spacing of 7.5 nm. The same lattice parameters were previously reported for the immature HIV-1 CA lattice (37). Subtomogram averaging was performed on released VLPs to obtain a more detailed view of the 3D arrangement of the Gag lattice. A structure of Gag(LZ) VLPs generated by subtomogram averaging confirmed that the arrangement of the CA layer is very similar to that of wild-type immature HIV-1 particles (Fig. 2E and F) (37), indicating that replacement of the C-terminal NC-RNA region with a LZ does not significantly alter the formation of the regular CA lattice.
Assembly kinetics of Gag(LZ) determined by live-cell microscopy.
While Gag lattice formation per se was not affected by the absence of NC-RNA or putative NC–F-actin interactions, they could potentially be relevant for the kinetics of Gag assembly. We therefore determined the assembly kinetics of Gag(LZ) by live-cell microscopy. For this purpose, we took advantage of the fluorescently labeled HIV-1 derivative pCHIVeGFP (23), harboring an eGFP moiety between the MA and CA domains of Gag. In cells transfected with an equimolar mixture of pCHIVeGFP and its unlabeled counterpart, pCHIV, the formation of individual assembly sites can be traced based on total internal reflection fluorescence microscopy (TIR-FM) image series (38). Replacing the NC domain of Gag with LZ in the context of the viral genome (rather than in a vector expressing only Gag) would destroy the Gag-Pol translational frameshift signal and alter the overlapping pol reading frame, which could affect assembly kinetics as well. We therefore made use of pCHIVunceGFP (24), a variant of pCHIVeGFP in which the gag and pol reading frames are genetically uncoupled. This allows alteration of the 3′ region of gag without affecting frameshifting efficiency or the pol open reading frame (ORF) (24). Replacement of NC by LZ in this context yielded plasmids pCHIVunc(LZ) and pCHIVunc(LZ)eGFP, respectively. HeLa cells were transfected with equimolar mixtures of either pCHIVunc and pCHIVunceGFP or pCHIVunc(LZ) and pCHIVunc(LZ)eGFP. Individual cells in early stages of assembly site formation were subjected to live-cell TIR-FM at 20 h after transfection, and single-virus tracing of fluorescent punctae was performed as described previously (38) (Fig. 3B; see also Movies S1 and S2 in the supplemental material). Kinetic analysis of the early exponential phase of Gag.eGFP accumulation at individual assembly sites confirmed that the assembly rate for the uncoupled construct (k = 0.0057 ± 0.0035 s−1) was similar to that previously determined for wild-type HIV-1 (0.0043 ± 0.00054 s−1) (24).
FIG 3.

Assembly kinetics of a LZ-carrying HIV derivative. (A) Scheme of the Gag/Pol-encoding region of pCHIVunceGFP illustrating the insertion site for the leucine zipper. Arrowheads indicate cleavage sites for the HIV-1 protease. The leucine zipper sequence is flanked by short G/S-rich linker sequences. FS, translational frameshift signal; RT, reverse transcriptase. (B) Assembly of HIVunc(LZ)eGFP at the plasma membrane of a particle-producing cell. HeLa cells were transfected with an equimolar mixture of pCHIVunc(LZ) and pCHIVunc(LZ)eGFP. At 20 h after transfection, cells were analyzed by live-cell TIR-FM as described in Materials and Methods. Shown are individual frames from Movie S2 in the supplemental material recorded at the indicated times. Bar, 10 μm. (C) Rate of Gag assembly determined for HIVunc(LZ)eGFP. HeLa cells were transfected with equimolar mixtures of pCHIVunc(LZ) and pCHIVunc(LZ)eGFP. At 20 h after transfection, cells were analyzed by TIR-FM (see Movie S2 in the supplemental material), and individual assembly sites were tracked. Fluorescence intensities from the exponential assembly phase from 37 individual sites from 2 experiments were averaged as described previously (38). The graphs show mean values (black line) and standard deviations (gray bars); mean values were fitted to a saturating single exponential equation (white line), yielding the indicated rate constant. a.u., arbitrary units.
Analysis of cells expressing fluorescent wild-type Gag or Gag(LZ) revealed the appearance of very similar punctae following exponential kinetics in an early phase of expression in both cases. The assembly rate determined for Gag(LZ) (k = 0.0024 ± 0.002 s−1) was 2- to 3-fold lower than that for the corresponding wild-type construct (Fig. 3B and C). Based on visual inspection, numbers of nascent assembly sites per cell were in a similar range for cells expressing the wild-type and LZ constructs. However, Gag(LZ) assemblies gradually coalesced into larger fluorescent patches at later stages of observation (Fig. 3B; see also Movie S2 in the supplemental material), while distinct individual assemblies remained in the case of the wild-type constructs throughout the observation period (see Movie S1 in the supplemental material). Pronounced coalescence of individual buds was also not observed in our previous live-cell analyses of HIV-1 assembly (24, 38, 42). The formation of large fluorescent Gag patches was in agreement with extended electron-dense regions resembling the Gag lattice and found adjacent to or in direct connection with distinct virus budding sites upon cET of U-373 MG cells transduced with AdGag(LZ) (Fig. 4G and H, arrowheads).
FIG 4.

F-actin at Gag and Gag(LZ) budding sites detected by cET. (A to H) U-373 MG cells were transduced with AdGagPol (A and B) or AdGag(LZ) (C to H) and plunge-frozen at 48 h p.t. The figure shows computational slices with thicknesses of 2 nm (A, B, D to F, and H), 3 nm (G), and 4.5 nm (C) through representative cryo-electron tomograms exhibiting viral budding sites. The different panels illustrate different F-actin morphologies. Panels B1 and B2, D1 and D2, and G1 and G2, depict the same region at different z positions, as indicated in the figure. Bona fide structure designations are as follows: pm, plasma membrane; b, budding site; ip, immature particle; act, actin. In panel G, two cells (c1 [cell 1] and c2 [cell 2]) are separated by a filopodium (f). Adjacent plasma membranes are indicated (2xpm). White arrowheads in panels G and H indicate electron-dense patches attributed to an extended Gag(LZ) lattice. Bar, 100 nm. (I) F-actin phenotypes associated with Gag assembly sites. Budding sites in cells expressing either Gag/GagPol or Gag(LZ) were assigned to categories i to v, based on the presence and morphology of actin filaments in their vicinity, as previously reported (8). (i) At the side of actin-rich filopodia; (ii) at the tip of actin-rich filopodia; (iii) cortical F-actin parallel to the plasma membrane; (iv) cortical actin directed toward or protruding into the budding site; (v) not adjacent to filamentous actin. The top panel shows a schematic representation of each category, with the Gag lattice displayed in red, the membrane in blue, and F-actin in black. Numbers below correspond to the occurrence of budding sites for the respective categories. The Gag/GagPol data set includes data from our previously reported analysis (8).
F-actin morphologies at Gag(LZ) budding sites.
Ultrastructural characterization of Gag assembly sites at the membrane of U-373 MG cells by cET revealed clearly resolved cellular features such as ribosomes and actin filaments surrounding the viral buds (Fig. 4A and B). In our previous report (21), we commonly observed F-actin filaments in close proximity to HIV-1 budding sites, and this was confirmed in the present study (Fig. 4A and B). Quantitative analysis of F-actin morphologies at GagPol assembly sites manually distributed into five phenotypic classes revealed budding sites at the side (class i) and tip (class ii) (Fig. 4A) of F-actin-rich filopodia, together comprising around one-third of all structures analyzed (Fig. 4I). Most remaining assembly sites were near a dense cortical actin network that was either arranged parallel to the plasma membrane (class iii) or directed toward the bud (class iv) (Fig. 4B), with the latter class being more common (∼50% of all structures analyzed) (Fig. 4I); budding sites not adjacent to detectable F-actin (class v) were rare.
Analysis of U-373 MG cells transduced with AdGag(LZ) also revealed typical HIV-1 budding sites and clearly resolved cellular actin filaments (Fig. 4C to F and H). Again, the majority of buds were associated with densely packed F-actin, with <15% of all buds not being adjacent to detectable F-actin (Fig. 4G and I). HIV-1 buds were detected at the tip (Fig. 4C) or side (Fig. 4E) of F-actin-rich filopodia and were associated with a dense cortical actin network pointing toward the bud (Fig. 4D) in ∼50% of all Gag(LZ) structures analyzed (Fig. 4I), again similar to the wild-type construct. F-actin filaments extending into the viral bud and sometimes in close proximity to the innermost layer of the Gag shell were detected in the cases of both Gag and Gag(LZ) assemblies (Fig. 4A and C), indicating that this phenotype is not dependent on a direct association of F-actin filaments with the NC layer of the immature Gag lattice.
Actin incorporation into VLPs.
Actin and actin-interacting proteins have been detected in purified HIV-1 particles by biochemical methods, mass spectrometry, and immuno-EM (6–8). To determine whether actin incorporation is dependent on the presence of the NC domain in Gag, we performed quantitative analyses of actin incorporation for HIV-1 and VLPs derived from wild-type Gag or Gag(LZ). Particles produced in transfected or transduced HEK293T cells were purified by velocity gradient centrifugation (Fig. 1C). Wild-type HIV-1 was produced in the presence of an HIV-1 protease inhibitor to obtain immature particles. Particle preparations were subjected to quantitative immunoblotting using defined amounts of purified Gag and actin as standards (Fig. 5). Visual inspection of the immunoblots indicated no major differences in relative actin incorporation between wild-type HIV-1 and Gag VLPs and between Gag and Gag(LZ) VLPs. Comparison of the signals allowed quantification of the total amount of Gag and actin loaded in each case, from which we calculated the molar ratio of actin to Gag and the number of actin molecules per particle. These values were almost identical for Gag and wild-type HIV-1 and were also very similar for VLPs containing Gag(LZ) (Fig. 5). Clearly, replacement of the NC domain by a heterologous leucine zipper did not reduce actin incorporation, and NC is thus not required for actin incorporation into HIV-1.
FIG 5.
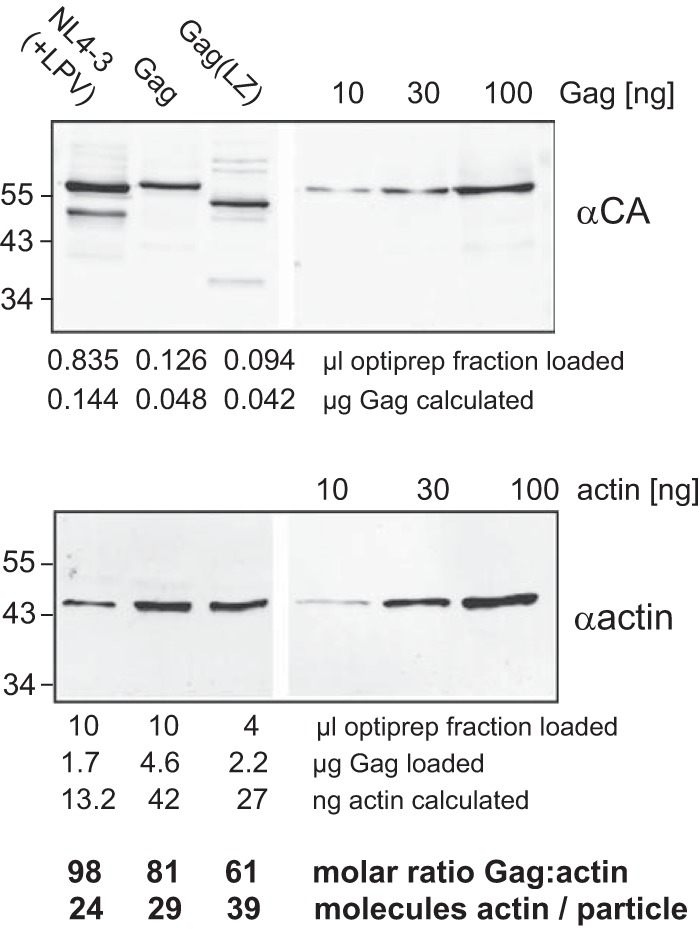
Quantification of actin in purified VLPs. Immature VLPs were produced in HEK293T cells transduced with AdGag or AdGag(LZ) or transfected with pNL4-3 in the presence of 2 μM LPV, as indicated. Particles were harvested by ultracentrifugation through a sucrose cushion and purified on velocity gradients. The indicated amounts of Optiprep-purified particles or quantified standards for Gag or actin were analyzed by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. Gag-derived proteins or actin was detected by immunoblotting using polyclonal rabbit antiserum against CA (top) or against human actin (bottom), respectively. Positions of molecular mass standards are indicated to the left. In order to compensate for different detection sensitivities, larger amounts of particle samples were loaded in the case of actin detection, as indicated below the blot. Amounts of Gag and actin were calculated based on signal intensities relative to the standards, yielding molar ratios for Gag and actin. Based on these data and assuming an average number of 2,400 Gag molecules per particle (52), the number of actin molecules per particle was calculated.
The observed molar ratio of actin to Gag of ∼1:100 differed substantially from the 1:10 ratio reported previously by others for HIV-1 particles purified from H9 and CEM cells (7). This discrepancy could not simply be explained by the use of Gag VLPs instead of HIV-1 virions, since we obtained a similarly low ratio for immature HIV-1 analyzed in parallel. The observed discrepancy may reflect differences in actin incorporation depending on the producer cell line, however. We therefore performed comparative experiments using MT-4 cells, HEK293T cells, and CD4-positive primary T cells prepared from human peripheral blood mononuclear cells (PBMCs) from four different donors. First, we determined cell volumes and total actin contents of the different cell types by confocal microscopy and immunoblotting, respectively (Table 1). In parallel, we purified HIV-1 particles from these cell types by velocity gradient centrifugation and determined their respective actin and CA contents. The calculated intracellular actin concentrations were roughly 3-fold higher in primary CD4+ T cells from each of the four donors than in the two cell lines analyzed. The higher actin concentration in primary T cells corresponded with a higher actin amount in HIV-1 particles produced from these cells. HIV-1 from primary CD4+ T cells exhibited an actin/Gag molar ratio of ∼1:20, in contrast to the 1:100 ratio determined for particles from HEK293T cells and the 1:200 ratio determined for particles from MT-4 cells (Table 1). These observations indicate that the producer cell type can influence actin incorporation into HIV-1. Importantly, the actin concentration determined for cells was substantially (3- to 10-fold) higher than that observed for the corresponding virus particles in all three cell types (Table 1). While we are aware that calculating “average” actin concentrations over the total cell volume, disregarding both the oligomeric status of the protein and its subcellular distribution, represents an oversimplification, these results do not suggest actin enrichment in HIV-1 particles.
TABLE 1.
Estimates of actin incorporation into HIV-1 particles derived from different cell typesa
| Parameter | Value for cell type |
||
|---|---|---|---|
| HEK293T | MT-4 | CD4+ T | |
| Cell vol (μm3) | 402 | 546 | 183 |
| Actin level/cell (pg) | 1.3 | 3.4 | 2.5 |
| Actin concn/cell (μM) | 77 | 117 | 327 |
| Molar ratio of CA/actin in virions | ∼110 | ∼220 | ∼20 |
| Actin concn/virion (μM) | 23 | 11 | 130 |
| Ratio of actin concn (virion/cell) | 0.3 | 0.09 | 0.4 |
| No. of actin molecules per virion | ∼22 | ∼11 | ∼125 |
For details, see Materials and Methods.
DISCUSSION
In this study, we analyzed the potential role of the NC domain in Gag in actin incorporation into HIV-1 particles and in the recruitment of F-actin to HIV-1 buds or reorganization of the cortical actin network. Our results suggest that these processes are independent of NC, and the reported NC-actin interaction does not appear to play a role in HIV-1 assembly. Furthermore, actin was not enriched in HIV-1 particles compared to producer cells, and results reported in the companion article (43) suggest that disruption of the F-actin network does not influence HIV-1 assembly rates. The combined results indicate that actin and its putative interaction with the NC domain do not have a major function in HIV-1 assembly.
Similar to previous studies (16–20), we observed the efficient release of Gag(LZ) particles with immature morphology from both HEK293T and U-373 MG cells. The Gag lattice in the CA region of the Gag(LZ) VLPs was indistinguishable from that observed in wild-type VLPs or immature HIV-1. Thus, NC-RNA interactions or the presence of the NC domain is not required for formation of the immature Gag lattice, and altering the interaction domain below CA does not have a significant effect on the CA arrangement. Furthermore, assembly rates were only moderately reduced for Gag(LZ) compared to wild-type Gag. Although HIV-1 preparations generally show considerable size heterogeneity, the observed average radius of 82 ± 8 nm for Gag(LZ) VLPs is larger than that previously observed for wild-type HIV-1 (21, 40). This larger size may be caused by a reduced tendency of the altered Gag protein to form a curved lattice (or an increased tendency to form a flat lattice). Aberrant large particles with a flat Gag lattice have previously been observed upon deletion or mutation of the SP1 region of Gag or N-terminally adjacent residues (44–46), indicating that this region controls lattice curvature. Changing the downstream NC domain may also influence lattice curvature. The most obvious difference between wild-type Gag and Gag(LZ) assembly was the appearance of large Gag patches in the latter case, where previously formed Gag assembly sites coalesced into these patches over time. It appears likely that the formation of confluent assembly sites is caused by the LZ domains, since this phenotype was not observed in our previous studies with wild-type Gag or other mutant Gag proteins. Large patches of an ordered lattice connected with apparently regular HIV-1 buds were detected by cET analyses of Gag(LZ)-expressing cells, and these structures are likely related to the coalescent assembly sites observed by light microscopy.
Our results confirmed the presence of actin in HIV-1 and VLPs, but the ratio of actin to Gag was lower than that previously reported. Quantitative immunoblotting indicated the presence of 10 to 40 actin molecules per virion produced from established cell lines. This number corresponds to a ratio of actin to Gag of 1:60 to 1:200, much lower than the previously reported ratio of 1:10 (7). The highest number of actin molecules (∼125 per particle) was observed for HIV-1 produced in primary T cells, where the ratio of actin to Gag was ∼1:20 and thus more similar to the previous report. However, these primary cells also exhibited a 3-fold-higher intracellular concentration of actin, and the amount of actin per virion correlated with the intracellular actin concentration for the different cell types. Given the presence of cortical actin in most cell types, one may assume that the local actin density at the plasma membrane is considerably higher than its average concentration in the cell. In contrast, actin concentrations in viral particles were much lower than the overall cellular concentration of actin, even for virus derived from primary T cells. These results do not exclude the possibility of active recruitment, but we consider it more likely that actin enters HIV-1 by passive incorporation as part of cytoplasmic material that is taken from the cell upon virus budding. This hypothesis is supported by the detection of many cytosolic proteins, including metabolic enzymes and heat shock proteins, in proteomic analyses of HIV-1 particles (see reference 47 and references therein; 48). While the incorporation of cellular proteins is generally discussed to be caused by their specific interaction with viral constituents and to be of functional importance (49), it is equally possible that HIV-1 incorporates cellular proteins, which are present in the membrane and in the confined cytosolic space at the assembly site and which are not excluded from the virion. Independent of the question of active or passive incorporation, NC or the presumed NC-actin interaction is clearly not important for actin incorporation into HIV-1 since Gag(LZ) particles contained similar amounts of actin as wild-type particles.
The observation of dense F-actin networks in the close vicinity of and often pointing toward viral buds in our cET study (21) as well as the AFM results showing asters at HIV-1 budding sites presumed to represent F-actin cables (15) suggested an active involvement of F-actin in HIV-1 assembly and/or release. Moreover, Gladnikoff et al. (15) reported that these asters were dependent on the presence of the NC domain in Gag and were not observed for Gag(LZ). Applying cET, we observed no difference in the presence or orientation of dense cortical F-actin in the vicinity of HIV-1 buds between wild-type Gag and Gag(LZ). Many buds were detected in regions with actin filaments pointing toward and sometimes extending into the viral bud. In some instances, these actin bundles were found in close proximity to the innermost density of the immature Gag lattice, but this was again not dependent on the NC domain (Fig. 4). The relative density of the F-actin network may have been biased by selecting flat cellular protrusions with dense cortical actin, which are accessible for cET and are thus overrepresented in the data sets. However, cells are also flat at the posterior end, where the actin meshwork is much less dense. The results of the companion paper (43) indicate that disruption of the F-actin network does not affect HIV-1 assembly rates, and the present study shows that NC-actin interactions are not needed for assembly or release and that actin is not enriched in the virion. The combined results argue against an important role for F-actin in HIV-1 assembly and show that the putative NC-actin interaction is not required for assembly. Our observations do not exclude a role of actin in processes prior or subsequent to particle assembly (i.e., intracellular trafficking or release), but the NC domain and its interaction with actin are clearly not needed for these processes either. The cortical actin network has been shown to be essential for organizing the virological synapse and for cell-to-cell transmission of HIV-1, however (50, 51), and it will be interesting to determine a potential contribution of the NC-actin interaction to synapse formation and cell-to-cell transmission in the future.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to R. Wagner (Regensburg, Germany) for Rev-independent Gag and GagPol expression vectors, G. Bernhardt (Regensburg, Germany) for providing the U-373 MG cell line, and H. Göttlinger (Boston, MA, USA) for the Zwtp6 construct. We thank V. Lux, J. Mak (Melbourne, Australia), and R. Schröder (Heidelberg, Germany) for purified CA, Gag, and F-actin, respectively.
This work was supported in part by DFG grants to H.-G.K., J.A.G.B., and K.G. within SPP1175 and to H.-G.K. within SFB638 (project A9). We also acknowledge a WTJIF award (060208/Z/00/Z) and a WT equipment grant (093305/Z/10/Z) to OPIC and WT core award 090532/Z/09/Z to the WTCHG. K.G. is supported by a Wellcome Trust SRF.
H.-G.K. and B.M. are investigators of the CellNetworks Cluster of Excellence (EXC81).
Footnotes
Published ahead of print 30 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00428-14.
REFERENCES
- 1.Sundquist WI, Kräusslich H-G. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2:a006924. 10.1101/cshperspect.a006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ganser-Pornillos B, Yeager M, Pornillos O. 2012. Assembly and architecture of HIV, p 441–465 In Rossmann MG, Rao VB. (ed), Viral molecular machines, vol 726 Springer, New York, NY: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Briggs JAG, Kräusslich H-G. 2011. The molecular architecture of HIV. J. Mol. Biol. 410:491–500. 10.1016/j.jmb.2011.04.021 [DOI] [PubMed] [Google Scholar]
- 4.D'Souza V, Summers MF. 2005. How retroviruses select their genomes. Nat. Rev. Microbiol. 3:643–655. 10.1038/nrmicro1210 [DOI] [PubMed] [Google Scholar]
- 5.Roberts KL, Smith GL. 2008. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 16:472–479. 10.1016/j.tim.2008.07.009 [DOI] [PubMed] [Google Scholar]
- 6.Ott DE, Coren LV, Johnson DG, Kane BP, Sowder RC, Kim YD, Fisher RJ, Zhou XZ, Lu KP, Henderson LE. 2000. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 266:42–51. 10.1006/viro.1999.0075 [DOI] [PubMed] [Google Scholar]
- 7.Ott DE, Coren LV, Kane BP, Busch LK, Johnson DG, Sowder RC, Chertova EN, Arthur LO, Henderson LE. 1996. Cytoskeletal proteins inside human immunodeficiency virus type 1 virions. J. Virol. 70:7734–7743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilk T, Gowen B, Fuller SD. 1999. Actin associates with the nucleocapsid domain of the human immunodeficiency virus Gag polyprotein. J. Virol. 73:1931–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Weisz OA, Stolz DB, Watkins SC, Montelaro RC. 2004. Differential effects of actin cytoskeleton dynamics on equine infectious anemia virus particle production. J. Virol. 78:882–891. 10.1128/JVI.78.2.882-891.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maldarelli F, King NW, Yagi MJ. 1987. Effects of cytoskeletal disrupting agents on mouse mammary tumor virus replication. Virus Res. 7:281–295. 10.1016/0168-1702(87)90043-8 [DOI] [PubMed] [Google Scholar]
- 11.Sasaki H, Nakamura M, Ohno T, Matsuda Y, Yuda Y, Nonomura Y. 1995. Myosin-actin interaction plays an important role in human immunodeficiency virus type 1 release from host cells. Proc. Natl. Acad. Sci. U. S. A. 92:2026–2030. 10.1073/pnas.92.6.2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibarrondo FJ, Choi R, Geng Y-Z, Canon J, Rey O, Baldwin GC, Krogstad P. 2001. HIV type 1 Gag and nucleocapsid proteins: cytoskeletal localization and effects on cell motility. AIDS Res. Hum. Retroviruses 17:1489–1500. 10.1089/08892220152644197 [DOI] [PubMed] [Google Scholar]
- 13.Liu B, Dai R, Tian CJ, Dawson L, Gorelick R, Yu XF. 1999. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J. Virol. 73:2901–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole E, Strappe P, Mok H-P, Hicks R, Lever AML. 2005. HIV-1 Gag-RNA interaction occurs at a perinuclear/centrosomal site; analysis by confocal microscopy and FRET. Traffic 6:741–755. 10.1111/j.1600-0854.2005.00312.x [DOI] [PubMed] [Google Scholar]
- 15.Gladnikoff M, Shimoni E, Gov NS, Rousso I. 2009. Retroviral assembly and budding occur through an actin-driven mechanism. Biophys. J. 97:2419–2428. 10.1016/j.bpj.2009.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Accola MA, Strack B, Göttlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74:5395–5402. 10.1128/JVI.74.12.5395-5402.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crist RM, Datta SA, Stephen AG, Soheilian F, Mirro J, Fisher RJ, Nagashima K, Rein A. 2009. Assembly properties of human immunodeficiency virus type 1 Gag-leucine zipper chimeras: implications for retrovirus assembly. J. Virol. 83:2216–2225. 10.1128/JVI.02031-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein KC, Reed JC, Tanaka M, Nguyen VT, Giri S, Lingappa JR. 2011. HIV Gag-leucine zipper chimeras form ABCE1-containing intermediates and RNase-resistant immature capsids similar to those formed by wild-type HIV-1 Gag. J. Virol. 85:7419–7435. 10.1128/JVI.00288-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popova E, Popov S, Göttlinger HG. 2010. Human immunodeficiency virus type 1 nucleocapsid p1 confers ESCRT pathway dependence. J. Virol. 84:6590–6597. 10.1128/JVI.00035-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Qian H, Love Z, Barklis E. 1998. Analysis of the assembly function of the human immunodeficiency virus type 1 Gag protein nucleocapsid domain. J. Virol. 72:1782–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson L-A, de Marco A, Oberwinkler H, Habermann A, Briggs JAG, Kräusslich H-G, Grünewald K. 2010. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 6:e1001173. 10.1371/journal.ppat.1001173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner R, Graf M, Bieler K, Wolf H, Grunwald T, Foley P, Überla K. 2000. Rev-independent expression of synthetic gag-pol genes of human immunodeficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum. Gene Ther. 11:2403–2413. 10.1089/104303400750038507 [DOI] [PubMed] [Google Scholar]
- 23.Lampe M, Briggs JAG, Endress T, Glass B, Riegelsberger S, Kräusslich H-G, Lamb DC, Bräuchle C, Müller B. 2007. Double-labelled HIV-1 particles for study of virus-cell interaction. Virology 360:92–104. 10.1016/j.virol.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 24.Radestock B, Morales I, Rahman SA, Radau S, Glass B, Zahedi RP, Müller B, Kräusslich H-G. 2013. Comprehensive mutational analysis reveals p6Gag phosphorylation to be dispensable for HIV-1 morphogenesis and replication. J. Virol. 87:724–734. 10.1128/JVI.02162-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scherer WF, Syverton JT, Gey GO. 1953. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 97:695–710. 10.1084/jem.97.5.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–72. 10.1099/0022-1317-36-1-59 [DOI] [PubMed] [Google Scholar]
- 27.Sena-Esteves M, Saeki Y, Camp SM, Chiocca EA, Breakefield XO. 1999. Single-step conversion of cells to retrovirus vector producers with herpes simplex virus/Epstein-Barr virus hybrid amplicons. J. Virol. 73:10426–10439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bigner DD, Bigner SH, Pontén J, Westermark B, Mahaley MS, Jr, Rouslathi E, Herschman H, Eng LF, Wikstrand CJ. 1981. Heterogeneity of genotypic and phenotypic characteristics of fifteen permanent cell lines derived from human gliomas. J. Neuropathol. Exp. Neurol. 40:201–229. 10.1097/00005072-198105000-00001 [DOI] [PubMed] [Google Scholar]
- 29.Miyoshi I, Taguchi H, Fujishita M, Niiya K, Kitagawa T, Ohtsuki Y, Akagi T. 1982. Asymptomatic type-C virus carriers in the family of an adult T-cell leukemia patient. Gann 73:339–340 [PubMed] [Google Scholar]
- 30.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welker R, Hohenberg H, Tessmer U, Huckhagel C, Kräusslich H-G. 2000. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 74:1168–1177. 10.1128/JVI.74.3.1168-1177.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tivol WF, Briegel A, Jensen GJ. 2008. An improved cryogen for plunge freezing. Microsc. Microanal. 14:375–379. 10.1017/S1431927608080781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastronarde DN. 2005. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152:36–51. 10.1016/j.jsb.2005.07.007 [DOI] [PubMed] [Google Scholar]
- 34.Ziese U, Janssen AH, Murk JL, Geerts WJ, Van der Krift T, Verkleij AJ, Koster AJ. 2002. Automated high-throughput electron tomography by pre-calibration of image shifts. J. Microsc. 205:187–200. 10.1046/j.0022-2720.2001.00987.x [DOI] [PubMed] [Google Scholar]
- 35.Frank J. 2006. Electron tomography: methods for three-dimensional visualization of structures in the cell, 2nd ed. Springer, Berlin, Germany [Google Scholar]
- 36.Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71–76. 10.1006/jsbi.1996.0013 [DOI] [PubMed] [Google Scholar]
- 37.de Marco A, Müller B, Glass B, Riches JD, Kräusslich H-G, Briggs JAG. 2010. Structural analysis of HIV-1 maturation using cryo-electron tomography. PLoS Pathog. 6:e1001215. 10.1371/journal.ppat.1001215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanchenko S, Godinez WJ, Lampe M, Kräusslich H-G, Eils R, Rohr K, Bräuchle C, Müller B, Lamb DC. 2009. Dynamics of HIV-1 assembly and release. PLoS Pathog. 5:e1000652. 10.1371/journal.ppat.1000652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Godinez WJ, Lampe M, Wörz S, Müller B, Eils R, Rohr K. 2009. Deterministic and probabilistic approaches for tracking virus particles in time-lapse fluorescence microscopy image sequences. Med. Image Anal. 13:325–342. 10.1016/j.media.2008.12.004 [DOI] [PubMed] [Google Scholar]
- 40.Briggs JAG, Simon MN, Gross I, Kräusslich HG, Fuller SD, Vogt VM, Johnson MC. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672–675. 10.1038/nsmb785 [DOI] [PubMed] [Google Scholar]
- 41.Popov S, Popova E, Inoue M, Göttlinger HG. 2008. Human immunodeficiency virus type 1 Gag engages the Bro1 domain of ALIX/AIP1 through the nucleocapsid. J. Virol. 82:1389–1398. 10.1128/JVI.01912-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baumgärtel V, Ivanchenko S, Dupont A, Sergeev M, Wiseman PW, Kräusslich H-G, Bräuchle C, Müller B, Lamb DC. 2011. Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 13:469–474. 10.1038/ncb2215 [DOI] [PubMed] [Google Scholar]
- 43.Rahman A, Koch P, Weichsel J, Godinez WJ, Schwarz U, Rohr K, Lamb DC, Kräusslich H-G, Müller B. 2014. Investigating the role of F-actin in human immunodeficiency virus assembly by live-cell microscopy. J. Virol. 88:7904–7914. 10.1128/JVI.00431-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Datta SA, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A. 2011. On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J. Virol. 85:4111–4121. 10.1128/JVI.00006-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gay B, Tournier J, Chazal N, Carriere C, Boulanger P. 1998. Morphopoietic determinants of HIV-1 Gag particles assembled in baculovirus-infected cells. Virology 247:160–169. 10.1006/viro.1998.9237 [DOI] [PubMed] [Google Scholar]
- 46.Kräusslich HG, Facke M, Heuser AM, Konvalinka J, Zentgraf H. 1995. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J. Virol. 69:3407–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Linde ME, Colquhoun DR, Ubaida Mohien C, Kole T, Aquino V, Cotter R, Edwards N, Hildreth JEK, Graham DR. 2013. The conserved set of host proteins incorporated into HIV-1 virions suggests a common egress pathway in multiple cell types. J. Proteome Res. 12:2045–2054. 10.1021/pr300918r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW, Sowder RC, Barsov E, Hood BL, Fisher RJ, Nagashima K, Conrads TP, Veenstra TD, Lifson JD, Ott DE. 2006. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 80:9039–9052. 10.1128/JVI.01013-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ott DE. 2002. Potential roles of cellular proteins in HIV-1. Rev. Med. Virol. 12:359–374. 10.1002/rmv.367 [DOI] [PubMed] [Google Scholar]
- 50.Jolly C, Mitar I, Sattentau QJ. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 81:5547–5560. 10.1128/JVI.01469-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vasiliver-Shamis G, Cho MW, Hioe CE, Dustin ML. 2009. Human immunodeficiency virus type 1 envelope gp120-induced partial T-cell receptor signaling creates an F-actin-depleted zone in the virological synapse. J. Virol. 83:11341–11355. 10.1128/JVI.01440-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlson L-A, Briggs JAG, Glass B, Riches JD, Simon MN, Johnson MC, Müller B, Grünewald K, Kräusslich H-G. 2008. Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host Microbe 4:592–599. 10.1016/j.chom.2008.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.