

Abstract
(E)-4-[3-(1-Adamantyl)-4′-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC) induces the cell-cycle arrest and apoptosis of leukemia and cancer cells. Studies demonstrated that 3-Cl-AHPC bound to the atypical orphan nuclear receptor small heterodimer partner (SHP). Although missing a DNA-binding domain, SHP heterodimerizes with the ligand-binding domains of other nuclear receptors to repress their abilities to induce or inhibit gene expression. 3-Cl-AHPC analogues having the 1-adamantyl and phenolic hydroxyl pharmacophoric elements replaced with isosteric groups were designed, synthesized, and evaluated for their inhibition of proliferation and induction of human cancer cell apoptosis. Structure–anticancer activity relationship studies indicated the importance of both groups to apoptotic activity. Docking of 3-Cl-AHPC and its analogues to an SHP computational model that was based on the crystal structure of ultraspiracle complexed with 1-stearoyl-2-palmitoylglycero-3-phosphoethanolamine suggested why these 3-Cl-AHPC groups could influence SHP activity. Inhibitory activity against Src homology 2 domain-containing protein tyrosine phosphatase 2 (Shp-2) was also assessed. The most active Shp-2 inhibitor was found to be the 3′-(3,3-dimethylbutynyl) analogue of 3-Cl-AHPC.
Introduction
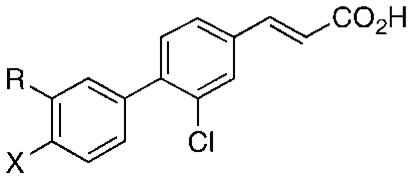
Recently, we identified (E)-4-[3′-(1-adamantyl)-4′-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC,a 1 in Figure 1) as a potential candidate for treating acute myeloid leukemia (AML) on the basis of its induction of AML cell apoptosis (programmed cell death) in blood samples obtained from patients1–3 and its extension of survival in an AML animal model.1 Prior to identifying 1,1 we focused our initial structure–activity studies on modifying substituents on the 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthoic acid (AHPN, 13) and (E)-4-[3′-(1-adamantyl)-4′-hydroxyphenyl]cinnamic acid (AHPC, 15) scaffolds to improve their induction of apoptosis induction and/or their antiproliferative activity in retinoid-resistant human HL-60R myeloid leukemia and MDA-MB-231 breast cancer cells or in human microvascular endothelial (HMVE) cells.2,4,5 Their inhibition of HMEV cell proliferation was used to assess anti-angiogenic activity against solid tumor growth.4
Figure 1.
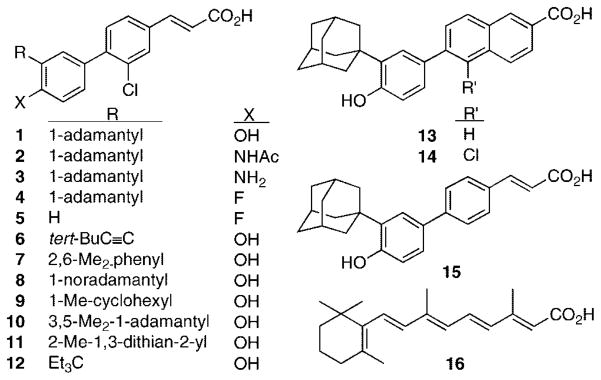
Structures of 3-Cl-AHPC (1), analogues 2–12, AHPN (13), 5-Cl-AHPN (14), AHPC (15), and all-trans-retinoic acid (16).
Although AHPN (13) was originally reported as a retinoic acid receptor (RAR) γ-selective retinoid,6 we first observed that it possessed unique apoptotic activity.7 We subsequently observed that, when administered to mice at doses found to induce cancer cell apoptosis in vitro, 13 and its related (E)-cinnamic acid AHPC (15), which we first synthesized in 1997, produced adverse systemic effects.1 Because the direct target(s) of 13 and 15 that led to apoptosis had not been identified, indirect design methods were used to generate a pharmacophoric model for apoptosis-inducing activity based on the AHPN and AHPC scaffolds.2 At the same time, we sought to reduce their retinoid activity on the premise that such a reduction would be accompanied by a similar reduction in retinoid-like adverse effects. These efforts led to the introduction of a halogen atom ortho to the diaryl ring junction on the AHPN and AHPC scaffolds and produced 5-Cl-AHPN (14) and 3-Cl-AHPC (1).1–3,8–10 Thus, the CO2H group of 1 is representative of the two-component hydrogen bond acceptor in the pharmacophoric model, its 3′-(1-adamantyl) group, 3′(1-Ad), is the large hydrophobic group, and its 4′-phenolic OH group is the hydrogen-bond donor or acceptor. Next, we established that, of 10 isosteric and hydrogen-bond acceptor or donor groups, the CO2H group was the most potent contributor to cancer cell apoptosis.4
Here, we report the impact of replacing the 4′-OH and 3′-(1-Ad) groups of 3-Cl-AHPC (1) with other polar and hydrophobic groups, respectively. Preliminary studies on substitutional variations using the AHPN (13) and AHPC (15) scaffolds produced the heretofore unreported structure–activity data listed in Tables 1–3. The various substitutions suggested that the binding pocket of the putative target required interaction with both pharmacophoric elements for maximal apoptosis induction. Removal of the 4′-OH group from 13 was accompanied by loss of HL-60R and MDA-MB-231 cell apoptosis-inducing activity in 18 (Table 1), although moderate HMEV cell growth inhibition occurred at 0.5 μM 18. Methylation of the 4′-OH group of 13 produced methyl ether 19 accompanied by a reduction in the level of HL-60R apoptosis from 73 to 3% at 0.1 μM, although no reduction occurred at 1.0 μM. 4′-(2-Methoxyethoxy)methyl ether 20 at 1.0 μM was far less active (21% HL-60R apoptosis) than 19. Shifting the 4′-OH group to the 5′- or 6′-position eliminated the ability of 1.0 μM 32 or 34, respectively, to induce MDA-MB-231 cell apoptosis (Table 2). 5′-Methyl ether 33 induced weak (20%) apoptosis of MDA-MB-231 cells, whereas 6′-methyl ether 35 had no HL-60R apoptosis-inducing activity. Introducing a 5′-OMe group onto 13 removed MDA-MB-231 apoptosis induction from 37. The unstable 4′,5′-catechol 36 was observed to be inactive, most likely by being degraded in cell culture. Switching the 3′- and 4′-group positions of 19 eliminated HL-60R apoptosis by 38 at 1.0 μM. Analogue 39 having a 6′-CH2OH-3′-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)phenyl ring rather than the 3′-(1-Ad)-4′-OH-phenyl ring was also inactive. These results suggested that the 4′-position for the OH group was optimal for apoptotic and antiproliferative activities. Because some phenolic ethers can be metabolically or otherwise cleaved in cell culture, these results did not permit us to fully assess hydrogen-bond donor or acceptor properties at the 4′-position. Therefore, the impact of other polar or electronegative groups as shown in 2–4 in Figure 1 was examined.
Table 1.
Effects on Human Cancer Cell Apoptosis and Microvascular Endothelial Cell Proliferation Caused by Replacing the 3′-(1-Adamantyl), 4′-Hydroxyl, or 5-Hydrogen of AHPN (13) with Other Substituents in AHPN Analogues 14 and 17–31
|
apoptosis (%)
|
HMVE cell growth inhibition at 72 ha
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| HL-60R at 24 h
|
MDA-MB-231 at 96 h
|
||||||||
| compd | R | X | R′ | 0.1 μM | 1.0 μM | 1.0 μM | 2.0 μM | IC50 (μM) | 0.5 μM (%) |
| 13 (AHPN) | 1-Adb | OH | H | 73 | 98 | 26 | 46 | 0.33 | 70 |
| 14 (5-Cl-AHPN) | 1-Ad | OH | Cl | 62 | 94 | 21 | 41 | >0.5d | 45 |
| 17 (5-Me-AHPN) | 1-Ad | OH | Me | 29 | 70 | 28 | 43 | 0.34 | 65 |
| 18 | 1-Ad | H | H | 2c | 2c | 2 | 7 | >0.5 | 36 |
| 19 | 1-Ad | OMe | H | 3 | 95 | ndd | ndd | ndd | ndd |
| 20 | 1-Ad | OCH2O(CH2)2OMe | H | ndd | 21 | ndd | ndd | ndd | ndd |
| 21 | tert-Bu | OH | H | ndd | ndd | ndd | ndd | >0.5 | 10 |
| 22 | MeO | OH | Me | −4 | 7 | 5 | 7 | ndd | ndd |
| 23 | 2-Ad | OH | H | 0 | 0 | ndd | ndd | >0.5 | 35 |
| 24 | 2,4,6-Me3-Ph | OH | Me | 0 | 0 | ndd | ndd | >0.5 | 17 |
| 25 | 3,3,5,5-Me4-cyclohexyl | OH | H | 26 | 60 | 10 | 49 | 0.5 | 50 |
| 26 | Me2C=CH(CH2)2CMe=CHCH2 | OH | H | ndd | 0 | ndd | ndd | ndd | ndd |
| 27 | 1-(1-Ad)Et | OH | H | ndd | 0 | ndd | ndd | >0.5 | 30 |
| 28 | 5,6,7,8-H4-5,5,8,8-Me4-2-naphthyl | OH | Me | 0 | 0 | ndd | ndd | 0.6e | 60 |
| 29 | 4aβ,8aα-decalin-2α-yl | OH | H | ndd | 0 | ndd | ndd | >0.5 | 0 |
| 30 | 4a,8aα-decalin-2β-yl | OH | H | ndd | 0 | ndd | ndd | ndd | ndd |
| 31 | 4aα,8a-decalin-1α-yl | OH | H | ndd | 0 | ndd | ndd | >0.5 | 10 |
| 16 (trans-RA) | 0 | 0 | 0 | 0 | >0.5 | 10 | |||
At 0.5 μM, the highest concentration tested.
1-Ad, 1-adamantyl.
Growth inhibition by 18 of 4% at 0.1 μM and 8% at 1.0 μM.
Not determined.
Extrapolated value.
Table 3.
Effects on Human Cancer Cell Apoptosis and Microvascular Endothelial Cell Proliferation Caused by Replacing the 3′-(1-Adamantyl) Group of AHPC (15) with Other Cyclic Ring Systems in Analogues 40–43
|
apoptosis (%)
|
HMVE cell growth inhibition at 72 ha
|
||||||
|---|---|---|---|---|---|---|---|---|
| HL-60R at 24 h
|
MDA-MB-231 at 96 h
|
|||||||
| compd | R | R′ | 0.1 μM | 1.0 μM | 1.0 μM | 2.0 μM | IC50 (μM) | 0.5 μM (%) |
| 1 (3-Cl-AHPC) | 1-Adb | Cl | 51 | 94 | 31 | 43 | ndc | ndc |
| 15 (AHPC) | 1-Ad | H | 90 | 98 | 53 | 58 | 0.1 | 90 |
| 40 | 2-Ad | H | 2 | 90 | 20 | 47 | ndc | ndc |
| 41 | 2,3,3-Me3-norbornyl | H | 2 | 2 | ndc | ndc | >0.5 | 40 |
| 42 | 3α-pentylbicyclo[2.2.2]octyl | H | 0 | 0 | ndc | ndc | ndc | ndc |
| 43 | 5,6,7,8-H4-5,5,8,8-Me4-2-naphthyl | H | ndc | 0 | ndc | ndc | ndc | ndc |
At 0.5 μM, the highest concentration tested.
1-Ad, 1-adamantyl.
Not determined.
Table 2.
Effects on Human Cancer Cell Apoptosis Caused by Shifting the Positions of Phenyl Ring Substituents and/or Introducing Additional Substituents on This Ring in AHPN (13) Analogues 32–38 and by Replacing the AHPN Phenyl Ring with a Tetrahydronaphthalene Ring in Analogue 39
|
apoptosis (%) at 1.0 μM
|
|||||
|---|---|---|---|---|---|---|
| compd | R | R1 | R2 | R3 | HL-60R at 24 h | MDA-MB-231 at 96 h |
| 32 | 1-Ada | H | OH | H | ndb | 0 |
| 33 | 1-Ad | H | OMe | H | ndb | 20 |
| 34 | 1-Ad | H | H | OH | ndb | 0 |
| 35 | 1-Ad | H | H | OMe | 0 | ndb |
| 36 | 1-Ad | OH | OH | H | ndb | 0c |
| 37 | 1-Ad | OH | OMe | H | ndb | 0 |
| 38 | OMe | 1-Ad | H | H | 0 | ndb |
| 39 | Me2C(CH2)2 | CMe2 | H | CH2OH | ndb | 0 |
1-Ad, 1-adamantyl.
Not determined.
Unstable under assay conditions.
Replacing the 3′-(1-Ad) group of AHPN (13) with the smaller tert-butyl group reduced HMVE cell growth inhibition by 7-fold (to 10%) in 21 at 0.5 μM, while replacing it with OMe removed apoptotic activity in 22. As these results suggested that a small hydrophobic alkyl or hydrogen-bond acceptor group at the 3′-position did not promote apoptosis, we probed chemical space constraints using AHPN analogues 27–31 (Table 1) and AHPC analogues 41–43 (Table 3) having the following 3′-R groups: 1-(1-Ad)ethyl, 5,6,7,8-tetrahydro-5,5,8,8-tetramethylnaphth-2-yl, 4aβ,8aα-decalin-2α-yl, 4a,8aα-decalin-2β-yl, 4a,8aα-decalin-1α-yl, 2,3,3-trimethylnorbornyl, and 3a-pentylbicyclo[2.2.2]octyl. Although the calculated molecular surface areas of the resulting 3′-R-4′-OH-phenyl groups were 5–31% larger than that of the 3′-(1-Ad)-4′-OH-phenyl group (210 Å2) (see Table S1 of the Supporting Information), none of these analogues at 1.0 μM induced HL-60R cell apoptosis; however, three (27, 28, and 41) at 0.5 μM inhibited HMVE proliferation by 30–60%. These results suggested that hydrophobic groups more similar to 3′-(1-Ad) such as the 3,3,5,5-tetramethylcyclohexyl and 2-Ad analogues of 13 and 15 (25 and 40) might display higher apoptotic or antiproliferative activity. At 1.0 μM, both were more apoptotic (60 and 90%, respectively) than 27–31. However, the absence of any correlation between apoptosis and the 3′-R-4′-hydroxyphenyl calculated molecular surface areas, which ranged from 29% larger to 2% smaller than that of the 3′-(1-Ad)-4′-OH-phenyl group of 13, suggested that hydrophobic properties such as symmetry and length might also play roles and led to the analogues in Figure 1 in which the 3′-R substituents were selected on the basis of structural similarity to the 3′-(1-Ad) or the comparable distance between the 3′-carbon atom of the phenyl ring and the most distant R-carbon atom. Correlations between apoptotic activity and 3′-substituents on the AHPN and AHPC scaffolds have not been reported by other groups, although Charpentier et al. introduced tert-butyl and 1-methylcyclohexyl at this position to improve RARγ subtype binding selectivity.6 The results of our modifications at the 3′-position on the 3-Cl-AHPC scaffold to give analogues 6–12 in Figure 1 are reported.
Recently, we determined that [5,5′-3H2]AHPN (13) bound with low nanomolar affinity to recombinant human small or short heterodimer partner protein (SHP or NR0B2, respectively)5,10 and, therefore, first experimentally demonstrated that SHP possessed a ligand-binding site, a result that supported the pocket found in a SHP computational model11 derived from the crystal structure of ultraspiracle (USP) complexed to a phospholipid.12,13 SHP is a member of the class I orphan nuclear receptor (NR) subfamily of steroid/thyroid hormone NR transcription factors. The orphan NRs earned this classification because, when discovered, no natural ligand had been identified.14
SHP is one of two unique orphan NRs in lacking characteristic N-terminal domains A–D.15 In typical NRs, these domains contain the ligand-independent activation function 1 (AF-1) (AB), DNA-binding domain (C), and the hinge region (D), which links these domains to the ligand-binding domain (LBD or E), and the C-terminal domain (F).16 Although SHP has yet to be crystallized, two structural models based on its homology to crystallized NR ligand-binding domains (LBDs) have been constructed. In the first derived from the structures of the hepatic nuclear factor (HNF)-4α and estrogen-related receptor (ERR)γ LBDs by Choi and co-workers,17 helix H12 of SHP occupies the classical transcriptional agonist or AF-2 site conformation, such as that found in the crystal structures of liganded nonpermissive NR LBDs, and its L6–7 loop between helices H6 and H7 exists as a random coil. The second model constructed by Pellicciari and co-workers11 was derived from the crystal structure of the retinoid X receptor orthologue ultraspiracle (USP) LBD complexed with 1-stearoyl-2-palmitoyl-sn-glycero-3-phosphoethanolamine (Protein Data Bank entry 1G2N).12 The second SHP model displayed 11 helices (H1 and H3–H12) and the L1–3 loop between helices H1 and H3 characteristic of NRs.11 SHP helix H12 adopted the transcriptional antagonist conformation shown by the USP LBD H12 that allowed it to stabilize the L1–3 loop between H1 and H3, and its L6–7 loop exhibited a defined, rather than random, structure.
SHP has several atypical or unique features. First, SHP can function as a homodimer18 or a heterodimer with a variety of NRs, including those that modulate transcription in a manner independent of ligand, those requiring ligand binding to function, and those whose function is modulated by ligand binding. Thus far, heterodimeric partners include the androgen receptor, aryl hydrocarbon receptor nuclear translocator, constitutive androstane receptor (CAR), estrogen receptors, ERRs, fetoprotein transcription factor/liver receptor homologue-1 (LRH-1), glucocorticoid receptor, HNF-4, liver X receptor, nur77/TR3/NGFI-B, peroxisome proliferator-activated receptors (PPARs), RARs, and RXRs.14,19 Second, SHP has three Lxx(L/I/V)L motifs (NR boxes 1–3, respectively) (Figure 2A), which in the model reside in helices H1, H6, and H10, respectively,11 through which it can prevent (i) NR AF-2 and AF-1 sites from interacting with a coactivator to induce or enhance gene transcription, (ii) NR dimerization, or (iii) NR binding to its response element to induce gene transcription. Thus, SHP does not function as a classical transcription factor but as a NR transcriptional core-pressor or a bridging protein between a NR and a repressor protein, such as the E1A-like inhibitor of differentiation 1 (EID-1)20 or the Sin3A multiprotein repressor complex, which contains histone deacetylases with which SHP also interacts.21 Third, the SHP NR boxes exhibit selectivity for particular NRs. Crystallographic analysis demonstrated that the AF-2 site in LRH-1 H12 interacted with a peptide corresponding to NR box 1 in SHP H1.22 Deletion analysis indicated that the RXR interaction domain of SHP resided between residues 92 and 148,15 which in the SHP model encompasses H6 NR box 2.11 Fourth, a large insertion loop (residues 128–139) occurs between H6 and H7. Fifth, the SHP model does not display the β-sheet structure found in the L5–6 loop of RXRα23,24 and other NRs.
Figure 2.
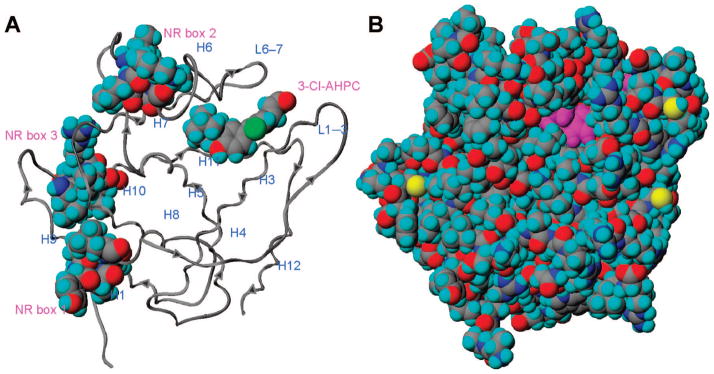
Small heterodimer partner (SHP) model. (A) Displayed in ribbon format with the 11 helices and three nuclear receptor (NR) boxes labeled. The docked structure of 3-Cl-AHPC (1) and the NR boxes are shown in space-filling format with atoms colored as follows: carbon, gray; chlorine, green; hydrogen, cyan; nitrogen, blue; oxygen, red. Arrows depict the N-terminal to C-terminal direction of the backbone. (B) Displayed in space-filling format with SHP atoms colored as in panel A and sulfur colored yellow; atoms of 1 are colored magenta except for the 3-chlorine atom which is colored green. The model suggests that a portion of the 3′-(1-adamantyl)-4′-hydroxyphenyl ring is exposed to the environment.
Pellicciari and co-workers hypothesized that, like USP, SHP should have a ligand-binding pocket comprised of residues from helices H3, H5, H7, and H11 and then showed that the phospholipid found complexed to the USP LBD in the crystal structure could be computationally docked into this pocket.11 The recent identification of a high-affinity endogenous ligand for USP, methyl farnesoate,25 provides evidence that a functional ligand-binding pocket exists in USP and supports the USP-based SHP model. Here, we report docking of 3-Cl-AHPC (1) and several new analogues to this SHP model.
Chemistry
Design
The reduction in adverse effects found on oral administration to mice led to shifts in the scaffold used for structure–activity studies from AHPN (13) to AHPC (15) and then to 3-Cl-AHPC (1). The impact on apoptotic activity of replacing the 4′-OH group of 1 was explored using 2–4 (Figure 1), while 3′-des(1-Ad)-4′-Fl analogue 5 was to be used for comparison to 4. Next, the impact of replacing 3′-(1-Ad) with similar hydrophobic groups was explored in 6–12 (see the Supporting Information for a discussion on analogue design and Table S1 for physical parameters calculated for analogues). Although 9 was previously reported to have retinoid activity lower than that of 13 in RAR transcriptional activation and F9 teratocarcinoma cell differentiation induction assays,6 its anti-proliferative activity was not described.
Synthesis
The syntheses of 2–12 were based on that used for constructing 3-Cl-AHPC2,4 (1), namely, convergent sequences involving a Suzuki–Miyaura palladium-catalyzed coupling26 between an arylboronic acid and an aryl bromide29 or triflate to introduce the 4–1′ diaryl bond. A full description of the synthetic routes and schemes is found in the synthesis section and in Schemes S1 and S2 of the Supporting Information. Synthetic highlights include a selective para bromination27,28 and application of the Pettus procedure for introducing a tert-alkyl group without rearrangement.32 NMR spectral assignments for the aromatic protons in 4 and 5 (see Method A and Method B) were based on reported shift and H–19F coupling constant values.30,31
Biological Results
Apoptotic and Antiproliferative Activities
4-Substituted cinnamic acids 1–12 were evaluated for their abilities to induce the apoptosis of human KG-1 AML cells and to inhibit the proliferation of these cells and human DU-145 prostate, MDA-MB-231 breast, and H292 non-small cell lung cancer cell lines (Table 4). Although KG-1 cells have an inducible tumor suppressor p53, in contrast to HL-60 AML cells in which p53 is deleted, KG-1 cells are resistant to such chemotherapy drugs as etoposide and daunorubicin33 and the antibody–drug conjugate gemtuzumab ozogamicin.34 Like KG-1 and HL-60R AML cells, the three solid tumor cell lines are resistant to the antiproliferative effects of all-trans-retinoic acid (16). DU-145 prostate and MDA-MB-231 cells have a mutated nonfunctional p53 tumor suppressor gene, whereas H292 cells are reported to overexpress p53, which is also indicative of poor survival.35 3-Cl-AHPC (1) continued to be the most potent KG-1 apoptosis inducer with an IC50 value of 4.2 μM at 48 h. Thus, the apoptosis induction assay results further confirm the importance of both 3′-(1-Ad) and 4′-OH groups to apoptotic activity. Replacing the 4′-OH group of 1 with a 4′-NH2 group weakened the ability of aniline 3 to induce apoptosis by 67% at 1.0 μM and by 27% at 5.0 μM and increased the IC50 value for KG-1 proliferation inhibition by almost 3.6-fold compared to that for 1 (IC50 = 1.4 μM). Acetylation of the amino group of 3 eliminated KG-1 apoptotic activity in acetanilide 2 at 5.0 μM and further reduced antiproliferative activity at 5.0 μM by 50%. The 4′-fluoro group rendered 4 inactive in both KG-1 cell assays.
Table 4.
Effects on Apoptosis Induction and Proliferation in Leukemia and Cancer Cell Lines and on Inhibition of Shp-2 Protein Tyrosine Phosphatase Activity Caused by Replacing the 3′-(1-Adamantyl) or 4′-Hydroxyl Group of 3-Cl-AHPC (1) in Analogues 2–12
|
KG-1 apoptosis at 48 h (%)
|
IC50 (μM) for proliferation inhibition (% inhibition at highest concentration evaluated)
|
Shp-2 enzyme activity inhibition IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R | X | 1.0 μM | 5.0 μM | KG-1 AMLa,f | DU-145 prostate cancerg | MDA-MB-231 breast cancerg | H-292 lung cancerg | |
| 1h | 1-Adi | OH | 30 | 55j | 1.4 (67) | 0.48 (85)b | 0.95 (63)b | 0.4 (92)b | 2.10 |
| 2 | 1-Ad | NHAc | 3 | 7 | >5 (25) | 5 (8)a | >5 (−5)a | >5 (13)a | 4.55 |
| 3 | 1-Ad | NH2 | 10 | 40j | 5.0 (50) | 1.3 (79)a | 4.0 (61)a | 6.0l (45)a | 1.83 |
| 4 | 1-Ad | F | 0 | 0 | >5 (0) | 16 (84)c | 67 (83)c | 6.0 (95)c | 2.11 |
| 5 | H | F | 0 | 0 | >5 (7) | >100 (23)c,l | >100 (9)c,l | 47 (60)c,l | 500m |
| 6 | t-BuC≡C | OH | 0 | 0 | >5 (22) | >10 (13)d | >10 (4)d | >10 (14)d | 0.45 |
| 7 | 2,6-Me2-Ph | OH | 0 | 0 | >5 (0) | >10 (4)d | >10 (0)d | >10 (0)d | 19.9 |
| 8 | 1-noradamantyl | OH | 2 | 16j | 0.92 (94) | 7.9 (59)d | 9.8 (51)d | 6.8 (69)d | 3.91 |
| 9 | 1-Me-cyclohexyl | OH | 0 | 0 | >5 (0) | 8.0l (32)a | >5 (9)a | 3.9 (63)a | 6.40 |
| 10 | 3,5-Me2-1-Ad | OH | 0 | 0j,k,k | >5 (0) | >5 (23)a | >5 (7)a | 3.6 (66)a | 2.27 |
| 11 | 2-Me-1,3-dithian-2-yl | OH | 5 | 5 | >5 (0) | >20 (8)e | >20 (13)e | >20 (6)e | 10.2 |
| 12 | 1,1-Et2-propyl | OH | 3 | 4k | >5 (0) | >5 (2)a | >5 (2)a | >5 (5)a | 5.37 |
Highest concentration evaluated, 5 μM.
Highest concentration evaluated, 2.5 μM.
Highest concentration evaluated, 100 μM.
Highest concentration evaluated, 10 μM.
Highest concentration evaluated, 20 μM.
At 48 h.
At 72 h.
3-Cl-AHPC.
1-Ad, 1-adamantyl.
IC50 values: 1, 4.2 μM; 3, 9.3 μM; 8, 230 μM; 10, 13.5 μM.
Apoptosis at 25 μM: 10, 83%; 12, 14%.
Percent inhibition at 10 μM 5: DU-145, 15%; MDA-MB-231, 0%; H292, 18%.
Extrapolated value.
Of the 3′-modifications, only 3′-(1-noradamantyl) analogue 8, in which one 2-CH2 group of the 1-Ad ring system is absent, retained appreciable apoptotic activity at 5.0 μM with 31% of that exhibited by 1. However, apoptosis induced by 1.0 μM 8 dropped to the vehicle alone control value of 2%. At 5.0 μM, analogues 6, 7, and 9–12 having 3,3-dimethylbutynyl, 2,6-dimethylphenyl, 1-methylcyclohexyl, 3,5-dimethyl-1-Ad, 2-methyl-1,3-dithian-2-yl, and 1,1-diethylpropyl, respectively, as 3′-R groups were essentially inactive. The calculated surface areas for these groups bridged that of the 1-Ad. Interestingly, 8 at 5.0 μM inhibited KG-1 proliferation more robustly (1.2-fold) than 1 and had a lower IC50 value (0.92 μM), whereas 6 at 5.0 μM had only approximately one-third of the antiproliferative activity of 1. The other analogues were unable to inhibit KG-1 cell growth.
The 3′-(1-Ad)-4′-NH2-phenyl analogue 3 and 3′-(1-noradamantyl)-4′-OH-phenyl analogue 8 inhibited the proliferation of the three solid tumor cell lines, although their calculated IC50 values were at least 3–4- and 10–20-fold higher, respectively, than those of 3-Cl-AHPC (1). In these lines, 3 was more potent than 8. The 3′-(1-Ad)-4′-Fl-phenyl and 3′-(1-methylcyclohexyl)-4′-OH-phenyl analogues (4 and 9, respectively) exhibited modest antiproliferative activity against the prostate and lung cancer cell lines, with 9 being more potent. The 3′-(3,5-dimethyl-1-Ad) analogue 10 had modest antiproliferative activity against the lung cancer cell line. Other analogues in Table 4 exhibited minimal or no antiproliferative activity.
SHP Binding Studies
Of the analogues in Table 4, three (4, 10, and 12) at 50 μM specifically displaced [5,5′-3H2]AHPN from recombinant human SHP with a robustness (100 ± 10%) similar to that of 3-Cl-AHPC (1) at 50 μM (Figure 3A). Dose–response curves were then generated (Figure 3B) to demonstrate that displacement of label occurred in the concentration range at which apoptosis induction was observed (Figure 3B). On the basis of displacements at 1.0, 5.0, and 50 μM, their IC50 values of 4, 10, and 12 were estimated to be 1.0, 0.5, and 8.0 μM, respectively, compared to an IC50 of 2.5 μM for 1. Displacement of label by 8 and 9 at 50 μM was less robust but still within 25% of that achieved by 1.
Figure 3.

Binding of 3-Cl-AHPC (1) and its analogues to small heterodimer partner (SHP). (A) Relative displacement of 5 nM [5,5′-3H2]AHPN from recombinant human GST–SHP protein by 50 μM 2–12 compared to displacement by 50 μM 1 as 100%. (B) Concentration-dependent displacement of 5 nM [5,5′-3H2]AHPN from SHP by 1.0, 5.0, and 50 μM 1, 4, 9, 10, and 12 after incubation in the presence of 5 nM radiolabeled AHPN for 2 h. Ligand-bound protein was isolated by being bound to glutathione–Sepharose 4B beads, which were then washed twice and counted. Specific binding was calculated by subtracting nonspecific binding from total binding.
Although we examined whether a correlation existed between the binding affinities of the 12 analogues for SHP and their abilities to induce KG-1 cell apoptosis or to inhibit KG-1 proliferation (Table 4), correlations were not evident. The 4′-F, 3′-(3,5-dimethyl-1-Ad), and 3′-(1,1-diethylpropyl) analogues (4, 10, and 12, respectively) did not induce KG-1 apoptosis at 5.0 μM; however, their relative displacements of labeled AHPN at 50 μM rivaled that of 3-Cl-AHPC (1), being 90, 103, and 90%, respectively, whereas the 4′-NH2 analogue 3 and 3′-noradamantyl analogue 8 with relative label displacements of 61 and 85% induced 40 and 16% apoptosis, respectively. However, if only the activities of the KG-1 apoptosis inducers (1–3 and 8) or proliferation inhibitors (1–3, 6, and 8) at 5.0 μM were plotted versus their relative abilities at 50 μM to displace the radiolabel from SHP, the resulting graphs would suggest that a linear relationship would exist if the number of active compounds was increased. Earlier, we found a correlation between MDA-MB-231 breast cancer cell apoptosis induction and SHP binding for apoptotic analogues having modifications at the CO2H position.5
The binding data underscore the importance of the 4′-OH group for interaction of the analogue with SHP. Replacement with 4′-NH2 and 4′-AcNH groups weakened binding of 3 and 2 by 40 and 53%, respectively. The electronegative 4′-Fl did not have such a deleterious effect on binding by apoptotically inactive 4, which at 50 μM had 90% of the ability of 1 to displace the radiolabel. The weakest binders among the 3′-R variants were the 3′-H-4′-Fl, 3′-(2,6-dimethylphenyl), and 3′-(2-methyl-1,3-dithian-2-yl) analogues (5, 7, and 11, respectively).
Inhibition of Shp-2 Protein Tyrosine Phosphatase Activity
Because KG-1 cells were reported to overexpress Src homology 2 domain-containing protein tyrosine phosphatase (Shp-2), which was considered to have a role in leukemia development,36 the inhibitory effects of 2–12 on Shp-2 activity were compared to that of 3-Cl-AHPC (1) (Table 4). In this series, 3′-(3,3-dimethylbutynyl) analogue 6 was the most potent inhibitor with an IC50 value of 0.45 μM, followed in descending potency by the 3′-(1-Ad)-4′-NH2 analogue 3, 1, and the 3′-(1-Ad)-4′-Fl and 3′-(3,5-dimethyl-1-Ad) analogues (4 and 10, respectively). The 4′-Fl analogue 5 lacking the 3′-(1-Ad) group was essentially inactive.
Computational Studies
Docking to the SHP Model
The phospholipid-binding pocket of the holo-USP-derived SHP model11 is comprised of residues from helices H3, H5, H7, and H11 and has an interior surface that resembles a boat. The polar Arg238 guanidinium group resides against the bow; the hydrophobic groups of Ala59 in H3, Leu97 and Leu100 in H5, Ala145 and Trp148 in H7, Leu231 and Leu235 in H11, and Ile240 at the beginning of the L11–12 loop, which line the pocket, rest against the sides of the hull, and the indole ring of Trp148 in H7 and the phenyl ring of Phe96 in H5 reside against the top and bottom of the stern, respectively (see Figures 2, 4, and 5). Initial computational studies were performed using Glide, and then BioMed Cache software was used to support the results. Docking 3-Cl-AHPC (1) to the SHP model using Glide and BioMed Cache produced the poses shown in Figures 4 and 5, respectively. Both docking poses complement the pocket residues in that the carboxylate group of 1 in the bow of the boat electrostatically interacts or forms a salt bridge with the positively charged Arg238 guanidinium group at the C-terminus of H11, while the two hydrophobic aromatic rings of 1 contact hydrophobic groups covering the hull. Docking with Glide suggested that the CO2H group of 1 interacts through one hydrogen bond with the side chain amino group of Gln43 in the L1–3 loop and electrostatically interacts with the positively charged Arg238 in H11 (Figure 4B). Although the measured distance between the carbon of the CO2H group of 1 and the guanidinium carbon of Arg238 is 7.1 Å, it could be argued that the highly flexible Arg238 side chain could undergo a ligand-induced conformational change to shorten the interaction distance. While no hydrogen-bond formation was observed with Gln43 in the BioMed Cache docking pose for 1, the 4.2 Å distance between the carbon of the CO2H group of 1 and the guanidinium carbon of Arg238 indicates the presence of a stronger electrostatic interaction. In both docking poses, the 3′-(1-Ad) group and the adjacent 4′-OH group contact the stern.
Figure 4.
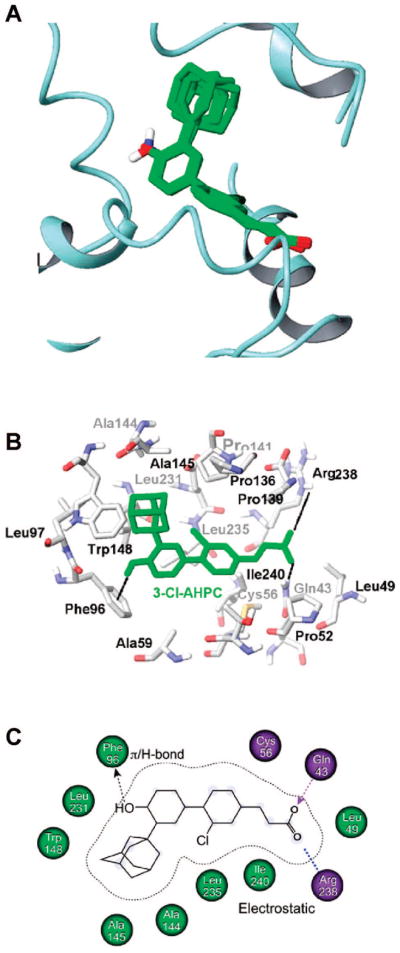
Docking of 3-Cl-AHPC (1) to the small heterodimer partner (SHP) model using Glide. (A) Common pose of docking of 1, 3, and 8 to SHP. SHP helices are shown in ribbon format; in overlaid analogues, atoms are colored as follows: carbon, green; hydrogen, white; nitrogen, blue; oxygen, red. (B) 1 docked into the SHP binding-pocket represented in stick format. The numbering of pocket residues corresponds to that in the SHP protein sequence. SHP atoms are colored as follows: carbon, white; nitrogen, blue; oxygen, red; sulfur, yellow. The docked pose of 1 is colored green. The π–H interaction between the 4′-hydroxyl hydrogen of 1 and the phenyl ring of Phe96 and the electrostatic interactions between its carboxylic acid oxygens and the Arg238 and Gln43 side chains are depicted as dotted lines. (C) Cartoon representation of 1 docked in the SHP-binding pocket with hydrophobic residues labeled in light green and polar residues in lavender.
Figure 5.

Docking of 3-Cl-AHPC (1) to the small heterodimer partner (SHP) model using BioMed Cache or SYBYL. (A) 1 docked into the SHP-binding pocket represented in ball-and-stick format. Atoms of 1 are colored magenta except for the 3-chlorine atom which is colored green. SHP pocket atoms are colored as follows: carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow. SHP pocket residues are labeled with numbers corresponding to those of the SHP protein sequence. Note the proximity of the 3-chlorine atom of 1 to the Cys56 mercapto hydrogen and the 4′-hydroxyl hydrogen to the Phe96 ring. The C6′–C1′–C4–C5 dihedral angle for 1 is 115°. (B) Space-filling representation of 1 docked in the SHP-binding pocket with 1 and SHP atoms colored as in panel A. In panels A and B, note potential interactions between the 3′-(1-adamantyl) group of 1 and the Try148 indole ring and its 4′-phenolic hydroxyl hydrogen and the Phe96 phenyl ring, and the proximity of its carboxylate oxygens to the Arg 238 side chain nitrogen atoms and of its 3-chlorine atom to the mercapto group of Cys56 and/or the hydroxyl group of Thr55. (C) Connolly solvent-accessible surface of the ligand-binding cavity of SHP. Pocket surface atom coloration: carbon, white indicating a hydrophobic region; nitrogen, blue indicating an electropositive region; oxygen, red indicating an electronegative region; and sulfur, yellow. Color-coding of atoms in the stick representation of 1 is as follows: carbon, magenta; chlorine, green; hydrogen, cyan; oxygen, red. This figure was generated using the MOLCAD module in SYBYL version 7.1. The C6′–C1′-C4–C5 dihedral angle for the diaryl bond of 1 is 116°. In panels A and C, nonpolar hydrogens of 1 have been omitted for clarity.
Docking using Glide and BioMed Cache also unveiled a noteworthy edge-to-face interaction between the hydroxyl-substituted phenyl ring of 3-Cl-AHPC (1) and the side chain ring of Phe96 (Figures 4B and 5A). This interaction was enforced by the nonconventional hydrogen bond between the hydrogen of the 4′-OH group of 1 and the π-electron cloud of the Ph96 phenyl ring. Such nontraditional types of hydrogen bonds, which have been described to occur naturally, can provide roughly half of the energetic stabilization of conventional hydrogen bonds.37
A major difference in the docking poses for 3-Cl-AHPC (1) is the orientation of the cinnamyl ring that causes the 3-Cl atom to point in opposite directions (Figures 4B and 5A,C). Glide docking indicated that the Cl atom interacts with Leu235 in H11, whereas BioMed Cache docking suggested that the H atom of the OH group of Thr55 or the SH group of Cys56 in helix H3 could form a stabilizing hydrogen–halogen bond38 with the 3-Cl atom of 1 if a shift in the position of a side chain or the H3 backbone occurs during binding. In the latter pose, distances measured between the 3-Cl atom of 1 and the Thr55 O and Cys56 S atoms were less than 5 and 4 Å, respectively. Interestingly, the binding pose identified by Glide was conserved among the KG-1 apoptosis inducers 1, 3, and 8 (rmsd < 0.8 Å) (Figure 4A) but absent in the remaining poorly active or inactive compounds (rmsd > 1.0 Å). Moreover, in both poses, the binding site for 1 is located close to the insertion loop between H6 and H7 (residues 128–139), which was demonstrated to be involved in transcriptional inhibitor recruitment by SHP, and close to H6 NR box 2 (residues 118–122) (Figure 2A).11 Unlike NR boxes 1 and 3, SHP NR box 2 selectively interacts with the AF-2 site in several NRs. Thus, binding of 1, 3, and 8 to SHP could directly impact the stabilization of both the insertion loop and NR box 2. The similar docking poses assumed by 1, 3, and 8 (Figure 4A) suggest that large and sterically stringent hydrophobic contacts between their hydrophobic 3′-(1-Ad) and 3′-(noradamantyl) groups and SHP are required to promote conformational shifts by the SHP insertion loop and NR box 2.
Because part of the ligand-binding site in the SHP model11 is constituted of helix H3 (Figure 2A), which also stabilizes the antagonistic conformation of H12 and the N-terminal L1–3 loop, binding by 1, 3, and 8 may also affect the conformation of NR box 1 in H1. Conversely, we could speculate that the binding poses of poorly active or inactive compounds identified by Glide, though presenting relatively good scores, may partly or insufficiently involve active conformational rearrangements of the SHP insertion loop and NR boxes.
The Glide and BioMed Cache energetical scores obtained for docking 1–12 to the SHP model did not correlate with their binding affinities as represented by their abilities to displace radiolabeled AHPN from recombinant human SHP (Figure 3A). The lack of correlation could be due to the small sample size or clustering of values for radiolabel displacement (29–103% of that exhibited by 1) or for docking scores (84 kcal/mol range) or could suggest that the SHP structural model could be improved once the X-ray crystallographic coordinates of liganded SHP are available or that SHP, as in the case of NR LBDs,23,24 could undergo appreciable conformational changes upon binding a functional ligand. A correlation between binding affinities and docking scores was not observed for the three most robust KG-1 apoptosis inducers (1, 3, and 8); however, a linear relationship between their docking scores and levels of apoptosis induction was observed. Replacement of the 4′-OH group of 3-Cl-AHPC (1) with 4′-NHAc and 4′-NH2 groups reduced the binding affinity of 2 and 3, respectively, to suggest that neither group could provide the hydrogen–π bond stabilizing interaction with the Phe96 aromatic ring that the OH group could. The docked pose for 3 differed from those of other analogues in that its 4′-NH2 group was much farther from the phenyl ring of Phe96 (7.1 Å). Our observation that removal of the 4′-OH group eliminated the apoptotic activity of 18 (see Table 1) further supports the importance of the hydrogen–π bond interaction. The more potent binding affinity of 4′-Fl analogue 4 compared to 3 suggests that withdrawal of an electron by fluorine may stabilize the edge-to-face interaction of its substituted phenyl ring with the aromatic ring of Phe96. The BioMed Cache docking poses may explain why some analogues have lower KG-1 cell apoptosis-inducing activity than 3-Cl-AHPC (1). The greater distances (>3.8 Å) between the O atoms of the CO2H groups of 4, 7, 8, and 11 and the side chain N atoms of Arg238 compared to those in 1 (2.9 and 3.3 Å) suggest that their binding would have weakened electrostatic interactions. The docked pose for aniline 3 suggests that its 4′-NH2 group would be so far (7.1 Å) from the center of the Phe96 ring that a stabilizing hydrogen–π bond interaction could not occur. The docked pose for 3′-(2,6-dimethylphenyl) analogue 7 suggests that one of its methyl groups would clash with the Leu100 side chain while the other would clash with Leu231 so that its 4′-OH hydrogen would be forced away from the phenyl ring of Phe96.
Construction of an Shp-2 Protein Tyrosine Phosphatase Pharmacophore
The structures of 1–12 were examined for use in generating pharmacophoric models or a hypothesis of Shp-2 enzyme activity inhibition. As a result, 4′-Fl analogue 5 was considered inactive, whereas the others were classified as active. The five possible models generated had similar phase scores but slightly different energetics scores. Selection of the most probable model was done on the basis of its highest phase score, quantitative structure–activity relationship (QSAR) r2 value, and lowest conformational gap energy. This model is characterized by two hydrophobic groups at the 3- and 3′-positions (H2 and H1, respectively), a negatively charged group (N), and a central aromatic ring (R) and for 6 would be represented by the 3-Cl and 3′-(3,3-dimethylbutynyl) groups, the carboxylate, and the cinnamyl ring as shown in Figure 6A. To investigate the influence of the molecular shape of H1 on phosphatase inhibition, a QSAR study was conducted using linear regression analysis and the molecular surface descriptors of common overlapping steric volume (COSV) and noncommon overlapping steric volume (NCOSV) (Figure 6B). The molecular shapes of active inhibitors 1–4 and 7–12 were aligned with the conformation of the most active inhibitor (6) to give the model (r2 = 0.95). In Figure 6C, conformers of the most potent and least potent inhibitors (6 and 7, respectively) were overlaid to demonstrate differences.
Figure 6.

QSAR analysis of Shp-2 protein tyrosine phosphatase activity inhibition by 3-Cl-AHPC (1) and active inhibitors 2–4 and 6–12. (A) Pharmacophore overlaid with the most potent inhibitor, 3′-(3,3-dimethylbutynyl) analogue 6, which is shown in stick format. In the pharmacophore, hydrophobic components H1 and H2 are represented with green spheres, the negative group N atom is represented with an orange sphere, and the aromatic ring R is represented with the orange ring. Atoms are colored as follows: carbon, gray; chlorine, green; hydrogen, white; oxygen, red. (B) Plot of the correlation between observed inhibitory activity (Experimental pIC50) and the predicted inhibitory activity (Predicted pIC50) of active inhibitors. The QSAR equation is shown below. COSV, common surface volume; NCOSV, noncommon surface volume. (C) Superposition of the most potent analogue, 6, in gray stick format and the least potent, 3′-(2,6-dimethylphenyl) analogue 7, in orange stick format, showing the excluded volumes between 6 (white dots) and 7 (red dots) for the hydrophobic site H1. Atoms other than carbon are colored as in panel A.
Discussion and Conclusions
Small Heterodimer Partner
3-Cl-AHPC (1) is one of the first ligands identified for the orphan nuclear receptor (NR) small heterodimer partner (SHP).5 Thus, SHP can now join CAR, FXR, ERR, LHR-1, LXR, PPARs, and RXRs as an adopted orphan NR. Our studies indicate that 1 has a functional role in modulating SHP activity that subsequently leads to enhanced interaction between SHP and a multiprotein repressor complex containing the repressor mSin3A, the NR corepressor NCoR, histone deacetylase 4, and heat shock protein 90. These binding events are then followed by attenuation of proto-oncogene c-Myc mRNA and protein expression5 and ultimately by induction of apoptosis.1 Interestingly, AHPN (13) treatment of MDA-MB-468 breast cancer cells was found to induce the expression of cell-cycle and apoptosis regulatory protein-1 (CARP-1),39 followed by a reduction in the level of c-Myc expression.5 Because 1 and 13–15 enhance the corepressor activity of SHP in terms of its ability to interact with the mSin3a repressor complex,5 they could be classified as SHP functional agonists.
Previously, we established that induction of apoptosis by SHP required the presence of the terminal CO2H group on the AHPN (13) or AHPC (15) scaffold.4 Here, we demonstrate that optimal apoptotic activity also requires both the 3′-(1-Ad) and 4′-OH groups (Table 4). This finding is supported by our previous work on these scaffolds as shown in Tables 1–3. After our initial report on the apoptosis-inducing activity of 13,7 other groups initiated analogue development that also supports our results with respect to the 4′-OH group. The methyl ether (19)6 of 13 was reported to be less potent at inducing DU-145 prostate cancer cell apoptosis than 13.40 In terms of caspase activation as a measure of apoptosis-inducing activity in Jurkat leukemia cells, the 4′-CH2OH and 4′,5′-dioxomethylene analogues of 13 were also less active.41 The 4′-CH2OH analogue40 was the more effective inducer of caspase activity41 and reduced the extent of DU-145 cell proliferation in culture.40 While inactive in the latter assay, the 4′,5′-dioxomethylene was reported to inhibit H292 lung cancer xenograft growth in an athymic mouse model.40 Although both analogues, as observed for 13, transactivated RARγ in reporter construct assays, they also inhibited retinoid-insensitive cancer cell growth to support a retinoid-independent pathway.40
The anticancer activity of AHPC (15) was also reported by other groups beginning in 2003.42–46 The almost two log reduction in antiproliferative activity against ovarian cancer cells caused by removal of the 4′-OH group from 15 also supports the importance of this group. Recently, modifications at the 5′-position on the AHPC scaffold were reported.45 On the basis of IC50 values for IGROV ovarian cancer and NB4 leukemia cell growth inhibition, analogues having an additional 5′-OH or 5′-OMe substituent had lower activity than 15, in agreement with the results listed in Table 1. Growth inhibition by the 4′-OH-5′-CH2NH2, 4′,5′-dioxomethylene, and 4′,5′-dioxoethylene analogues approached that by 15. As indicated by propidium iodide staining, apoptosis induced in the ovarian cancer cell line by the first and second analogues at their antiproliferative IC80 values was comparable to that induced by 15 (50%).
In addition to our discovery of its role in inducing apoptosis, SHP is reported to play important roles in negatively regulating the conversion of cholesterol to bile acids and in interfering with drug metabolism by interacting with LRH-1, PXR, HNF-4, and CAR to repress the expression of several cytochrome P450 catabolic enzymes.20 SHP also regulates the expression of genes with roles in bile acid transport, lipid metabolism, and gluconeogenesis.20 Thus, regulation of SHP function by ligands may have roles in treatment of cancer, diabetes, and non-alcohol-related fatty liver disease.
Protein Tyrosine Phosphatase Shp-2
This enzyme is thought to play important roles in cell proliferation, differentiation, and migration47 and is known to relay signals from growth factor receptors to p21(ras).48 Constitutively active Shp-2 mutants can cause Noonan syndrome, characterized by cognitive impairment, short stature, and congenital heart defects,49 and induce leukomyogenesis.50 Shp-2 also has an important role in insulin-induced adipogenesis.51 Recently, overexpression of Shp-2 was found to protect immortalized mouse embryonic fibroblasts from epigallocatechin 3-gallate-mediated apoptosis.47 The overexpression and upregulation of phosphorylated, and hence activated, Shp-2 by the bcr/abl oncoprotein was found in 23 of 25 chronic myeloid leukemia cell samples obtained from patients.36 Shp-2 overexpression was associated with cell proliferation and resistance to apoptosis. These authors also reported that Shp-2 was expressed in the KG-1 myeloid leukemia cell line, which was then used as a positive control.36 Earlier, we reported that 2-Cl-AHPC (1) inhibited Shp-2 protein tyrosine phosphatase (PTP) activity (IC50 = 2.10 μM).4 This result led us to hypothesize that inhibition of Shp-2 PTP activity by 1 could have a role in apoptosis induction in KG-1 cells or in inhibition of their proliferation. However, the results of our Shp-2 activity inhibition assays (Table 4) clearly show that Shp-2 did not act as a major player in KG-1 cell apoptosis induced by 1 or its analogues. In fact, despite having an IC50 value of 0.45 μM for inhibiting Shp-2 activity, 3′-(3,3-dimethylbutynyl) analogue 6, which was the most active enzyme inhibitor, was unable to induce KG-1 cell apoptosis (0%) and inhibited KG-1 cell proliferation by only 22% after treatment at 5.0 μM for 48 h. Moreover, 4′-Fl analogue 5 with an IC50 value of 2.11 μM for Shp-2 activity inhibition was impotent at 5.0 μM as either a KG-1 apoptosis inducer (0%) or growth inhibitor (0%). Nevertheless, replacement of the 3′-(1-Ad) group of 1 with the 3′-(3,3-dimethylbutynyl) group enhanced the Shp-2 inhibitory potency of 6 more than 4-fold and also removed SHP functional activity. Thus, 6 may serve as a lead for generating a selective Shp-2 PTP inhibitor. Finding such a selective Shp-2 inhibitor may have therapeutic potential in treating diseases in which Shp-2 is overexpressed or constitutively activated by mutations in its inhibitory loop.
Computational Studies
Molecular Docking Experiments
(i) Method A
Compound structures were prepared using the LIGPREP interface as implemented in MAESTRO (Schrödinger, Mannheim, Germany) and the OPLS-2005 force field. The model of SHP11 was prepared using the protein preparation wizard script in MAESTRO. Docking experiments were conducted using GLIDE 4.5 (Schrödinger) and a flexible docking approach with the extra precision method as implemented in the software. Docking results were ranked by calculating the energy score E-model as implemented in the Schrödinger suite. This scoring function combined an energy grid score, binding affinity predicted by GlideScore, and the internal strain energy of the ligand resulting from its binding conformation.
(ii) Method B
The three-dimensional model used for docking was that derived by homology modeling of the holo-ultraspiracle receptor (USP) crystal structure.11 Docking of 3-Cl-AHPC and analogues into the putative binding pocket was performed using BioMed (Beaverton, OR) Cache 6.2. The diaryl dihedral (torsion) angle (C6′–C1′–C4–C5) of the starting conformer was set to 50° on the basis of molecular dynamics studies on 1.2 The putative binding pocket site was derived by selecting all neighboring residues within 4 Å (radius) of the phosphatidylethanolamine that was docked to the SHP model.11 This distance was large enough to encompass the putative binding pocket. Each analogue was docked into a 15 Å × 15 Å × 15 Å box located at the center of the active site using a united atom potential of mean force with a genetic algorithm that ran for 20000 generations. Other parameters were left at their respective default values. Both the ligand and side chains of the pocket residues were allowed to be flexible during the docking process. The final docking poses were analyzed by measurement of interatom distances after overlaying analogue structures on the basis of superposition of their 3′-, 4-, and 5-carbon atoms.
Pharmacophoric Model Generation
Compounds were built using the program MAESTRO (Schrödinger) and energetically minimized using the OPLS-2001 force field implemented in the program.52 Conformational analysis for each compound was performed using PHASE (Schrödinger), with conformational space sampled using a combination of the Monte Carlo Multiple Minimum approach53 and the Low Mode conformational search algorithm.54 Conformations within 10 kcal/mol of the global minima were retained and used to generate the pharmacophoric model. Structurally redundant replicates were omitted. The following chemical features for each conformer were defined: H-bond acceptor (A), H-bond donor (D), general hydrophobic site (H), negatively charged group (N), positively charged group (P), and aromatic ring (R).
PHASE generated and scored the models using a weighted combination of three independent scoring functions: vector, site, and volume. The vector score measured how well vectors for hydrogen-bond acceptors and donors and aromatic rings were aligned to the structures used to generate the pharmacophoric model. The site score measured how closely chemical features were superimposed on structure alignment. The volume score measured the average value of how much the volumes of contributing compounds overlapped on alignment. An energetic score (kilocalories per mole) was used to evaluate the conformational gap energy between the aligned conformation of each compound and the global energy minimum conformation. Scoring produced five models, each having four chemical features (see the Supporting Information). The models were used by PHASE to build a QSAR equation endowed with a correlation coefficient (r2) indicating fitness between experimental and predicted compound activities. Descriptors of molecular volumes of the hydrodrophobic site H1 were calculated using Molecular Shape analysis as implemented in Cerius 2 (Accelrys, San Diego, CA). Common overlapping steric volumes (COSV) and noncommon overlapping steric volumes (NCOSV) were calculated by aligning the shape of compounds 1–4 and 7–12 with the most active compound, 6. The QSAR study was conducted using linear regression analysis as implemented in Cerius 2.
Biology
Cell Growth
Tumor cell lines (ATCC, Manassas, VA) were grown in medium containing 10% fetal bovine serum (FBS): KG-1 leukemia cells (H. P. Koeffler, UCLA, Cedars-Sinai Medical Center, Los Angeles, CA) and retinoid-resistant HL-60R leukemia cells (S. Collins, University of Washington, Seattle, WA) in medium containing 5% heat-inactivated FBS and HMVE cells (Clonetics, San Diego, CA) in MEV growth medium containing 5% fetal calf serum and the growth factors and antimicrobials in the accompanying kit (Clonetics) under conditions we have described previously.4
Growth Inhibition
MDA-MB-231, DU-145, and H292 cells were allowed to attach for 24 h and then treated for 72 h with each analogue dissolved in Me2SO or with Me2SO alone (final Me2SO concentration of 0.1%). Media and compounds were replaced after 48 h, as described previously.4 Viable cell numbers at each concentration were determined on three replicatesusinga3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt reduction assay (Cell Titer 96 AQueous Nonradioactive Cell Proliferation Assay, Promega, Madison, WI).
KG-1, HL-60R, and MDA-MB-231 cells in medium containing 5% FBS were allowed to attach overnight, treated for the indicated times with analogues at the specified concentrations in Me2SO or Me2SO alone (final Me2SO concentration of 0.1%), and then harvested.4 Cell numbers were counted in triplicate wells using a hemocytometer.
HMVE cells in medium were allowed to attach for 24 h before being treated. Compounds dissolved in Me2SO were added to give final concentrations of 100, 125, 250, and 500 nM (final Me2SO concentration of ≤1%). The medium and test compound were replaced every other day. Cell growth after treatment for 7 days was assessed by the Alamar blue method (BioSource International, Camarillo, CA) at 590 nm after excitation at 530 nm using a Cytoflor fluorometer as previously described.4 Results in Tables 1 and 2 are averages of five replicates.
Cell Apoptosis
KG-1 and HL-60R leukemia and MDA-MB-231 breast cancer cells were seeded and incubated as described for the growth inhibition experiments. Apoptotic cells were identified using acridine orange staining on at least three replicates as described previously.4
Receptor Binding Assay
Expression of recombinant glutathione S-transferase–human SHP chimeric protein (GST–SHP), cell lysis, isolation of GST–SHP using glutathione–Sepharose beads, and incubation of beads with [5,5′-3H2]AHPN10 in the presence or absence of 3-Cl-AHPC (1) or an analogue followed by separation of bound from nonbound label were performed as we described previously.4
Inhibition of SHP-2 PTP Activity
The PTP-catalyzed hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (Invitrogen, Carlsbad, CA) in the presence of each analogue in Me2O or Me2O alone (final concentration of 5%) was assayed as we described previously.4
Supplementary Material
Scheme 1a.

a (a) 1,1′-dioctadecyl-4,4′-bipyridinium dibromide, NaHSO3, K2CO3, CH2Cl2/H2O, room temperature (rt) to 30 °C. (b) CuCl2, t-BuNO2, MeCN, 0 °C to rt. (c) TsOH·H2O, (CH2OH)2, MePh, reflux. (d) n-BuLi, THF, −78 °C; (MeO)3B, −78 °C to rt; aqueous HCl. (e) 1-Adamantanol, (PhNH)3Al, o,m,p-Et2-benzene, 195 °C. (f) NH4Br, H2O2, HOAc. (g) Ac2O, pyridine, CH2Cl2. (h) Pd(PPh3)4, LiCl, K3PO4, DME, reflux. (i) [(EtO)2P(O)CH2CO2Et, KN(SiMe3)2, THF, −78 °C], −78 °C to rt; aqueous HCl. (j) LiOH ·H2O, THF/MeOH/H2O; H3O+. (k) Concentrated HCl, EtOH, reflux. (l) Aqueous HCl, NaNO2; HPF6; 150 °C. (m) n-BuLi, THF, −78 °C; (i-PrO)3B, −78 °C to rt; H3O+. (n) BnBr, K2CO3, Me2CO, reflux. (o) BBr3, CH2Cl2, −78 °C; H2O. (p) Tf2O, pyridine, CH2Cl2, 0 °C to rt. (q) Ph3P=CHCO2Et, PhMe, reflux. (r) Pd(PPh3)4, LiCl, aqueous Na2CO3, DME, reflux. (s) Aqueous NaOH, MeOH, reflux; H3O+. (t) NBS, FSO3H, MeCN, 0 °C to rt. (u) 3,3-Dimethyl-1-butyne, (Ph3P)2PdCl2, PPh3, CuI, Et2NH, reflux. (v) SnCl2 ·2H2O, EtOH, 80 °C (72) or 92 °C (81); aqueous NaOH. (w) CuBr2, t-BuNO2, MeCN, 0 °C to rt. (x) NaI, NaOCl, KOH, H2O, MeOH, 0 °C to rt. (y) 3,3-Dimethyl-l-butyne, (Ph3P)2PdCl2, CuI, Et3N, THF, 0 °C to rt. (z) 2,6-Me2C6H4B(OH)2, Pd(PPh3)4, aqueous Na2CO3, DME, 95 °C. (aa) n-Hexyl nitrite, CHBr3, 93 °C.
Scheme 2a.

a (a) NaOCl, CCl4, HOAc, 0 °C. (b) KOH, MeOH, reflux, 6 h. (c) Tf2O, 50% H2O2, Na2HPO4, CH2Cl2. (d) LiAlH4, Et2O, −78 °C to rt; H2O. (e) 1-Noradamantanol, MeSO3H, from 80 to 140 °C. (f) BnBr, K2CO3, Me2CO, reflux. (g) n-BuLi, THF, −78 °C; (i-PrO)3B, −78 °C to rt; aqueous HCl. (h) 64, Pd(PPh3)4, LiCl, aqueous Na2CO3, DME, reflux. (i) BBr3, CH2Cl2, −78 °C; H2O. (j) LiOH ·H2O, THF/MeOH/H2O; H3O+. (k) 1-Methylcyclohexanol, MeSO3H, CH2Cl2, reflux. (l) 3,5-Dimethyl-1-adamantanol, MeSO3H, CH2Cl2, reflux. (m) 1,3-Propanedithiol, BF3 · Et2O, CH2Cl2, 0 °C to rt. (n) t-BuMe2SiCl, DMAP, Et3N, DMF. (o) (t-Boc)2O, NaH, THF, 0 °C to rt. (p) EtMgBr, Et2O, −20 to 0 °C; H2O. (q) (n-Bu)4NBr3, CHCl3.
Acknowledgments
We are grateful for support of this research by National Institutes of Health Grants P01 CA51993 (M.I.D., J.A.F., and X.-K.Z.) and R01 CA109370 (M.I.D. and J.A.F.) and California Tobacco-Related Diseases Research Grant 11RT-0081 (M.I.D. and X.-K.Z.).
Footnotes
Abbreviations: 3-Cl-AHPC, (E)-4-[3′-(1-adamantyl)-4′-hydroxyphenyl]-3-chlorocinnamic acid; AHPC, (E)-4-[3′-(1-adamantyl)-4′-hydroxyphenyl-]cinnamic acid; AHPN, 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthalenecarboxylic acid; AML, acute myelocytic leukemia; HMVE, human microvascular endothelial; RAR, retinoic acid receptor; RXR, retinoid X receptor; SHP, small or short heterodimer partner; Shp-2, Src homology 2 domain-containing protein tyrosine phosphatase 2.
Supporting Information Available: Table S1 giving surface and volume parameters, Table S2 showing data and calculations illustrated by models in Figure S1, and Tables S3 and S4 listing HPLC analyses of target compounds using two different solvent systems. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zhang Y, Dawson MI, Mohammad R, Rishi AK, Farhana L, Feng KC, Leid M, Peterson V, Zhang XK, Edelstein M, Eilander D, Biggar S, Wall N, Reichert U, Fontana JA. Induction of apoptosis of human B-CLL and ALL cells by a novel retinoid and its nonretinoidal analog. Blood. 2002;100:2917–2925. doi: 10.1182/blood.V100.8.2917. [DOI] [PubMed] [Google Scholar]
- 2.Dawson MI, Harris DL, Liu G, Hobbs PD, Lange CW, Jong L, Bruey-Sedano N, James SY, Zhang XK, Peterson VJ, Leid M, Farhana L, Rishi AK, Fontana JA. Antagonist analogue of 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN) family of apoptosis inducers that effectively blocks AHPN-induced apoptosis but not cell-cycle arrest. J Med Chem. 2004;47:3518–3536. doi: 10.1021/jm030524k. [DOI] [PubMed] [Google Scholar]
- 3.Farhana L, Dawson MI, Fontana JA. Apoptosis induction by a novel retinoid-related molecule requires nuclear factor-κB activation. Cancer Res. 2005;65:4909–4917. doi: 10.1158/0008-5472.CAN-04-4124. [DOI] [PubMed] [Google Scholar]
- 4.Dawson MI, Xia Z, Liu G, Fontana JA, Farhana L, Patel BB, Arumugarajah S, Bhuiyan M, Zhang XK, Han YH, Stallcup WB, Fukushi J, Mustelin T, Tautz L, Su Y, Harris DL, Waleh N, Hobbs PD, Jong L, Chao W, Schiff LJ, Sani BP. An adamantyl-substituted retinoid-derived molecule that inhibits cancer cell growth and angiogenesis by inducing apoptosis and binds to small heterodimer partner nuclear receptor: Effects of modifying its carboxylate group on apoptosis, proliferation, and protein-tyrosine phosphatase activity. J Med Chem. 2007;50:2622–2639. doi: 10.1021/jm0613323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farhana L, Dawson MI, Leid M, Wang L, Moore DD, Liu G, Xia Z, Fontana JA. Adamantyl-substituted retinoid-related molecules bind to small heterodimer partner and modulate the Sin3A repressor. Cancer Res. 2007;67:318–325. doi: 10.1158/0008-5472.CAN-06-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charpentier B, Bernardon JM, Eustache J, Millois C, Martin B, Michel S, Shroot B. Synthesis, structure-affinity relationships, and biological activities of ligands binding to retinoic acid receptor subtypes. J Med Chem. 1995;38:4993–5006. doi: 10.1021/jm00026a006. [DOI] [PubMed] [Google Scholar]
- 7.Shao ZM, Dawson MI, Li XS, Rishi AK, Sheikh MS, Han QX, Ordonez JV, Shroot B, Fontana JA. p53 independent G0/G1 arrest and apoptosis induced by a novel retinoid in human breast cancer cells. Oncogene. 1995;11:493–504. [PubMed] [Google Scholar]
- 8.Zhang Y, Dawson MI, Ning Y, Polin L, Parchment RE, Corbett T, Mohamed AN, Feng KC, Farhana L, Rishi AK, Hogge D, Leid M, Peterson VJ, Zhang XK, Mohammad R, Lu JS, Willman C, VanBuren E, Biggar S, Edelstein M, Eilender D, Fontana JA. Induction of apoptosis in retinoid-refractory acute myelogenous leukemia by a novel AHPN analog. Blood. 2003;102:3743–3752. doi: 10.1182/blood-2003-01-0108. [DOI] [PubMed] [Google Scholar]
- 9.Farhana L, Dawson MI, Huang Y, Zhang Y, Rishi AK, Reddy KB, Freeman RS, Fontana JA. Apoptosis signaling by the novel compound 3-Cl-AHPC involves increased EGFR proteolysis and accompanying decreased phosphatidylinositol 3-kinase and AKT kinase activities. Oncogene. 2004;23:1874–1884. doi: 10.1038/sj.onc.1207311. [DOI] [PubMed] [Google Scholar]
- 10.Fontana JA, Dawson MI, Leid M, Rishi AK, Zhang Y, Hsu CA, Lu JS, Peterson VJ, Jong L, Hobbs P, Chao WR, Shroot B, Reichert U. Identification of a unique binding protein specific for a novel retinoid inducing cellular apoptosis. Int J Cancer. 2000;86:474–479. doi: 10.1002/(sici)1097-0215(20000515)86:4<474::aid-ijc5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 11.Macchiarulo A, Rizzo G, Costantino G, Fiorucci S, Pellicciari R. Unveiling hidden features of orphan nuclear receptors: The case of the small heterodimer partner (SHP) J Mol Graphics Modell. 2006;24:362–372. doi: 10.1016/j.jmgm.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 12.Billas IM, Moulinier L, Rochel N, Moras D. Crystal structure of the ligand-binding domain of the ultraspiracle protein USP, the ortholog of retinoid X receptors in insects. J Biol Chem. 2001;276:7465–7474. doi: 10.1074/jbc.M008926200. [DOI] [PubMed] [Google Scholar]
- 13.Clayton GM, Peak-Chew SY, Evans RM, Schwabe JW. The structure of the ultraspiracle ligand-binding domain reveals a nuclear receptor locked in an inactive conformation. Proc Natl Acad Sci US A. 2001;98:1549–1554. doi: 10.1073/pnas.041611298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benoit G, Cooney A, Giquere V, Ingraham H, Lazar M, Mucat G, Perlmann T, Renaud JP, Schwabe J, Sladek F, Tsai MJ, Laudet V International union of pharmacology. LXVI. Orphan nuclear receptors. Pharmacol Rev. 2006;58:798–836. doi: 10.1124/pr.58.4.10. [DOI] [PubMed] [Google Scholar]
- 15.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336–1339. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 16.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 17.Park YY, Kim HJ, Kim JY, Kim MY, Song KH, Park KC, Yu KY, Shong M, Kim KH, Choi S. Differential role of the loop region between helices H6 and H7 within the orphan nuclear receptors small heterodimer partner and DAX-1. Mol Endocrinol. 2004;18:1082–1095. doi: 10.1210/me.2003-0339. [DOI] [PubMed] [Google Scholar]
- 18.Iyer AK, Zhang YH, McCabe ERB. Dosage-sensitive sex reversal adrenal hypoplasia congenital critical region on the X chromosome, gene 1 (DAX1) (NR0B1) and small heterodimer partner (SHP) (NR0B2) form homodimers individually, as well as DAX1-SHP heterodimers. Mol Endocrinol. 2006;20:2326–2342. doi: 10.1210/me.2005-0383. [DOI] [PubMed] [Google Scholar]
- 19.Lee YS, Chanda D, Sim J, Park YY, Choi HS. Structure and function of the atypical orphan nuclear receptor small heterodimer partner. Int Rev Cytol. 2007;261:117–158. doi: 10.1016/S0074-7696(07)61003-1. [DOI] [PubMed] [Google Scholar]
- 20.Båvner A, Johansson L, Toresson G, Gustafsson J-Å, Treuter E. A transcriptional inhibitor targeted by the atypical orphan nuclear receptor SHP. EMBO Rep. 2002;3:478–484. doi: 10.1093/embo-reports/kvf087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gobinet J, Carascossa S, Cavailles V, Vignon F, Nicolas JC, Jalaguier S. SHP represses transcriptional activity via recruitment of histone deacetylases. Biochemistry. 2005;44:6312–6320. doi: 10.1021/bi047308d. [DOI] [PubMed] [Google Scholar]
- 22.Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, Guan Z, Tripathy A, Raetz CR, McDonnell DP, Moore DD. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat Struct Mol Biol. 2005;12:357–363. doi: 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- 23.Egea PF, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRα ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19:2592–2601. doi: 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egea PF, Mitschler A, Moras D. Molecular recognition of agonist ligands by RXRs. Mol Endocrinol. 2002;16:987–997. doi: 10.1210/mend.16.5.0823. [DOI] [PubMed] [Google Scholar]
- 25.Jones G, Jones D, Teal P, Sapa A, Wozniak M. The retinoid-X receptor ortholog, ultraspiracle, binds with nanomolar affinity to an endogenous morphogenetic ligand. FEBS J. 2006;273:4983–4996. doi: 10.1111/j.1742-4658.2006.05498.x. [DOI] [PubMed] [Google Scholar]
- 26.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 27.Olifirov DI, Koshchii VA, Kozlikovskii Ya B. Reaction of 1-hydroxyadamantane with aromatic amines in the presence of orthoalkylation catalysts. Zh Org Khim. 1992;28:182–187. [Google Scholar]
- 28.Krishna Mohan KVV, Narender N, Srinivasu P, Kulkarni SJ, Raghavan KV. Novel bromination method for anilines and anisoles using NH4Br/H2O2 in CH3COOH. Synth Commun. 2004;34:2143–2152. [Google Scholar]
- 29.Holzapfel CW, Dwyer C. Still and Suzuki cross-coupling reactions of o-nitrophenyl triflates: A versatile route to a variety of heterocycles. Heterocycles. 1998;48:1513–1518. [Google Scholar]
- 30.Ailprandi B, Cacace F, Fornarini S. Gas phase alkylation of halobenzenes by free isopropyl cations. Tetrahedron. 1987;43:2831–2841. [Google Scholar]
- 31.Bertha F, Fetter J, Kajtár-Peredy M, Lempert K, Czira G. Simple and condensed β-lactams, Part 31. Acid catalyzed ring closures and ring transformations of some 3-aryloxy-4-oxoazetidine-2-carbaldehydes. Tetrahedron. 1998;54:15227–15242. [Google Scholar]
- 32.Van De Water RW, Magdziak DJ, Chau JN, Pettus TRR. New construction of ortho ring-alkylated phenols via generation and reaction of assorted o-quinone methides. J Am Chem Soc. 2000;122:6502–6503. [Google Scholar]
- 33.Gieseler F, Bauer E, Nuessler V, Clark V, Valsamas S. Molecular effect of topoisomerase II inhibitors in AML cell lines: Correlation of apoptosis with topoisomerase II activity but not with DNA damage. Leukemia. 1999;13:1859–1863. doi: 10.1038/sj.leu.2401570. [DOI] [PubMed] [Google Scholar]
- 34.Amico D, Barbui AM, Erba E, Rambaldi A, Introna M, Golay J. Differential response of human acute myeloid leukemia cell to gemtuzumab ozogamicin in vitro: Role of Chk1 and Chk2 phosphoprylation and caspase 3. Blood. 2003;101:4589–4597. doi: 10.1182/blood-2002-07-2311. [DOI] [PubMed] [Google Scholar]
- 35.Ju MZ, Kapoor K, Newton K, Cheon RL, Strong LC, Koo JS. Global detection of molecular changes reveals concurrent alteration of several biological pathways in nonsmall cell lung cancer cells. Mol Genet Genomics. 2005;274:141–154. doi: 10.1007/s00438-005-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu XZ, Yu YZ, Fang YM, Liang Y, Lu QH, Xu RZ. Overexpression of Shp-2 is associated with the unlimited growth and apoptosis resistance of p210 bcr-al-mediated chronic myeloid leukemia. Zhonghua Yi Xue Za Zhi. 2005;85:1903–1906. [PubMed] [Google Scholar]
- 37.Scheiner S, Kar T, Pattanayak J. Comparison of various types of hydrogen bonds involving aromatic amino acids. J Am Chem Soc. 2002;124:13257–13264. doi: 10.1021/ja027200q. [DOI] [PubMed] [Google Scholar]
- 38.Kovács A, Varga Z. Halogen acceptors in hydrogen bonding. Coord Chem Rev. 2006;250:710–727. [Google Scholar]
- 39.Rishi AK, Zhang L, Boyanapalli M, Wali A, Mohammad RM, Yu Y, Fontana JA, Hatfield JS, Dawson MI, Majumdar AP, Reichert U. Identification and characterization of a cell cycle and apoptosis regulatory protein-1 as a novel mediator of apoptosis signaling by retinoid CD437. J Biol Chem. 2003;278:33422–33435. doi: 10.1074/jbc.M303173200. [DOI] [PubMed] [Google Scholar]
- 40.Lu XP, Fanjul A, Picard N, Shroot B, Pfahl M. A selective retinoid with high activity against an androgen-resistant prostate cancer cell type. Int J Cancer. 1999;80:272–278. doi: 10.1002/(sici)1097-0215(19990118)80:2<272::aid-ijc17>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 41.Ortiz MA, Lopez-Hernandez FJ, Bayon Y, Pfahl M, Piedrafita FJ. Retinoid-related molecules induce cytochrome c release and apoptosis through activation of c-Jun NH2-terminal kinase/p38 mitogen-activated protein kinases. Cancer Res. 2001;61:8504–8512. [PubMed] [Google Scholar]
- 42.Pisano C, Merlini L, Penco S, Carminati P, Zunino F. Cellular and pharmacological bases of the antitumor activity of a novel adamantyl retinoid, ST1926. J Chemother. 2004;(Suppl 4):74–76. doi: 10.1179/joc.2004.16.Supplement-1.74. [DOI] [PubMed] [Google Scholar]
- 43.Cincinelli R, Dallavalle S, Nannei R, Carella S, De Zani D, Merlini L, Penco S, Garattini E, Giannini G, Pisano C, Vesci L, Carminati P, Zuco V, Zanchi C, Zunino F. Synthesis and structure-activity relationships of a new series of retinoid-related biphenyl-4-ylacrylic acids endowed with antiproliferative and proapoptotic activity. J Med Chem. 2005;48:4931–4946. doi: 10.1021/jm049440h. [DOI] [PubMed] [Google Scholar]
- 44.Cincinelli R, Dallavalle S, Merlini L, Penco S, Pisano C, Carminati P, Giannini G, Vesci L, Gaetano C, Illy B, Zuco V, Supino R, Zunino F. A novel atypical retinoid endowed with proapoptotic and antitumor activity. J Med Chem. 2003;46:909– 912. doi: 10.1021/jm025593y. [DOI] [PubMed] [Google Scholar]
- 45.Cincinelli R, Dallavalle S, Nannei R, Merlini L, Penco S, Giannini G, Pisano C, Vesci L, Ferrara FF, Zuco V, Zanchi C, Zunino F. Synthesis and structure-activity relationships of new antiproliferative and proapoptotic retinoid-related biphenyl-4-yl-acrylic acids. Bioorg Med Chem. 2007;15:4863–4875. doi: 10.1016/j.bmc.2007.04.057. [DOI] [PubMed] [Google Scholar]
- 46.Pisano C, Vesci L, Foderà R, Ferrara FF, Rossi C, De Cesare M, Zuco V, Pratesi G, Supino R, Zunino F. Antitumor activity of the retinoid-related molecules (E)-3-(4′-hydroxy-3′-adamantylbiphenyl-4-yl)acrylic acid (ST1926) and 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (CD437) in F9 teratocarcinoma: Role of retinoic acid receptor γ and retinoid-independent pathways. Mol Pharmacol. 2006;70:909–924. doi: 10.1124/mol.106.023614. [DOI] [PubMed] [Google Scholar]
- 47.Amin ARM, Thakur VS, Paul RK, Feng GS, Qu CK, Mukhtar H, Agarwal ML. SHP-2 tyrosine phosphatase inhibits p73-dependent apoptosis and expression of a subset of p53 target genes induced by EGCG. Proc Natl Acad Sci US A. 2007;104:5419–5424. doi: 10.1073/pnas.0700642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yart A, Mayeux P, Raynal P. Gab1, SHP-2 and other novel regulators of Ras: Targets for anticancer drug discovery. Curr Cancer Drug Targets. 2003;3:177–192. doi: 10.2174/1568009033481976. [DOI] [PubMed] [Google Scholar]
- 49.Van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007;2:4. doi: 10.1186/1750-1172-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bocchinfuso G, Stella L, Martinelli S, Flex E, Carta C, Pantaleoni F, Pispisa B, Venanzi M, Tartaglia M, Palleschi A. Structural and functional effects of disease-causing amino acid substitutions affecting residues Ala72 and Glu76 of the protein tyrosine phosphatase SHP-2. Proteins. 2007;66:963–974. doi: 10.1002/prot.21050. [DOI] [PubMed] [Google Scholar]
- 51.Uehara T, Suzuki K, Yamanaka H, Kizaki T, Sakurai T, Ishibashi Y, Ishida H, Ohno H. SHP-2 positively regulates adipogenic differentiation in 3T3-L1 cells. Int J Mol Med. 2007;19:895–900. [PubMed] [Google Scholar]
- 52.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and proprieties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 53.Chang G, Guida WC, Still WC. An internal coordinate Monte-Carlo method for searching conformational space. J Am Chem Soc. 1989;111:4379–4386. [Google Scholar]
- 54.Kolossváry I, Guida WC. Low Model Search. An efficient, automated computational method for conformational analysis: Application to cyclic and acyclic alkanes and cyclic peptides. J Am Chem Soc. 1996;118:5011–5019. JM800456K. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.