

Abstract
Site-specific recombinases are tremendously valuable tools for basic research and genetic engineering. By promoting high-fidelity DNA modifications, site-specific recombination systems have empowered researchers with unprecedented control over diverse biological functions, enabling countless insights into cellular structure and function. The rigid target specificities of many sites-specific recombinases, however, have limited their adoption in fields that require highly flexible recognition abilities. As a result, intense effort has been directed toward altering the properties of site-specific recombination systems by protein engineering. Here, we review key developments in the rational design and directed molecular evolution of site-specific recombinases, highlighting the numerous applications of these enzymes across diverse fields of study.
Keywords: protein engineering, recombinase, genome engineering
Introduction
Site-specific recombinases are highly specialized enzymes that promote DNA rearrangements between specific target sites (Grindley et al., 2006; Fig. 1). In nature, these enzymes control and coordinate a number of diverse eukaryotic and prokaryotic functions, including the integration and excision of viral genomes, the activation of developmentally relevant genes and the transposition of mobile genetic elements. Most known site-specific recombinases exhibit distinct and strict sequence specificities, an evolutionary result of the tightly regulated role that DNA re-organization plays in key biological pathways. The elucidation of the minimal nucleotide sequence recognized by several site-specific recombinases has allowed researchers to take advantage of these systems for genetic (Branda and Dymecki, 2004) and metabolic (Jantama et al., 2008; Yuan et al., 2006) engineering, as well as synthetic biology (Cheng and Lu, 2012; Table I). Long-standing applications of this technology include site-specific integration (Fukushige and Sauer, 1992; O'Gorman et al., 1991; Sauer and Henderson, 1990) and excision of transgenic elements and selectable markers (Dale and Ow, 1991), tissue-specific (Kuhn et al., 1995) and conditional knockouts (Feil et al., 1996; Logie and Stewart, 1995), and the induction of chromosomal deletions (Tsien et al., 1996; Wagner et al., 1997) and translocations (Ramirez-Solis et al., 1995; Smith et al., 1995). These technologies have also enabled investigators to manipulate chromosome structure across dozens of organisms and achieve previously unattainable forms of control over numerous biological functions (Branda and Dymecki, 2004).
Figure 1.
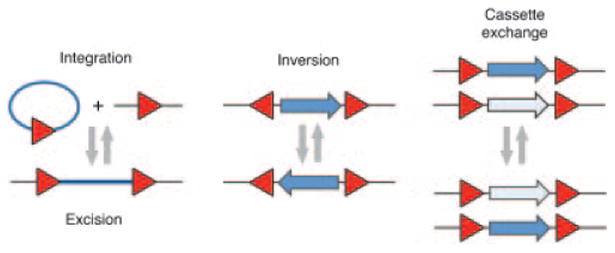
Possible outcomes of site-specific recombination. Red triangles indicate recombination sites.
Table I.
Site-specific DNA recombination systems used for genome engineering in mammalian cells.
| Recombinase | Origin | Classification | Target site | Target sequence |
|---|---|---|---|---|
| Flp | S. cerevisiae | Tyrosine | FRT | 5′-GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC-3′ |
| KD | K. drosophilarum | Tyrosine | KDRT | 5′-AAACGATATCAGACATTTGTCTGATAATGCTTCATTATCAGACAAATGTCTGATATCGTTT-3′ |
| B2 | Z. bailii | Tyrosine | B2RT | 5′-GAGTTTCATTAAGGAATAACTAATTCCCTAATGAAACTC-3′ |
| B3 | Z. bisporus | Tyrosine | B3RT | 5′-GGTTGCTTAAGAATAAGTAATTCTTAAGCAACC-3′ |
| R | Z. rouxii | Tyrosine | RSRT | 5′-TTGATGAAAGAATAACGTATTCTTTCATCAA-3′ |
| Cre | Phage P1 | Tyrosine | loxP | 5′-ATAACTTCGTATAGCATACATTATACGAAGTTAT-3′ |
| VCre | Vibriosp. | Tyrosine | VloxP | 5′-TCAATTTCTGAGAACTGTCATTCTCGGAAATTGA-3′ |
| SCre | Shewanellasp. | Tyrosine | SloxP | 5′-CTCGTGTCCGATAACTGTAATTATCGGACATGAT-3′ |
| Vika | V. coralliilyticus | Tyrosine | vox | 5′-AATAGGTCTGAGAACGCCCATTCTCAGACGTATT-3′ |
| Dre | Bacteriophage D6 | Tyrosine | rox | 5′-TAACTTTAAATAATGCCAATTATTTAAAGTTA-3′ |
| λ-Int | Phage λ | Tyrosine | attP | 5′-CAGCTTTTTTATACTAAGTTG-3′ |
| attB | 5′-CTGCTTTTTTATACTAACTTG-3′ | |||
| HK022 | Phage HK022 | Tyrosine | attP | 5′-ATCCTTTAGGTGAATAAGTTG-3′ |
| attB | 5′-GCACTTTAGGTGAAAAAGGTT-3′ | |||
| φC31 | Phage φC31 | Serine | attP | 5′-CCCCAACTGGGGTAACCTTTGAGTTCTCTCAGTTGGGG-3′ |
| attB | 5′-GTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCG-3′ | |||
| Bxb1 | Phage Bxb1 | Serine | attP | 5′-GGTTTGTCTGGTCAACCACCGCGGTCTCAGTGGTGTACGGTACAAACC-3′ |
| attB | 5′-GGCTTGTCGACGACGGCGGTCTCCGTCGTCAGGATCAT-3′ | |||
| Gin | Phage Mu | Serine | gix | 5′-TTATCCAAAACCTCGGTTTACAGGAA-3′ |
| Tn3 | E. coli | Serine | res site I | 5′-CGTTCGAAATATTATAAATTATCAGACA-3′ |
For the prototypical site-specific recombinases Cre, Flp, λ-Int, φC31, Gin and Tn3, crossover regions are underlined.
Virtually all site-specific recombinases can be categorized within one of two structurally and mechanistically distinct groups: the tyrosine (e.g., Cre, Flp, and the λ integrase) (Grainge and Jayaram, 1999) or serine (e.g., φC31 integrase, γδ resolvase, and Gin invertase) recombinases (Smith and Thorpe, 2002). Both recombinase families recognize target sites composed of two inversely repeated binding elements that flank a spacer sequence where DNA breakage and religation occur. In most cases, sequence identity within this central crossover region is critical for the recombination reaction. This highly coordinated, co-factor independent process requires concomitant binding of two recombinase monomers to each target site: two DNA-bound dimers then join to form a synaptic complex, leading to crossover and strand exchange. These two classes of enzymes vary in a number of ways, however, including the reliance on different nucleophilic amino acid residues (i.e., tyrosine or serine) that attack DNA to form transient covalent protein-DNA linkages. The recombination mechanisms of these two groups are also distinct. The tyrosine recombinases break and rejoin pairs of single DNA strands to generate Holliday junction intermediates, while the serine recombinases cleave all four DNA strands before promoting strand exchange and religation. The key steps for these processes are illustrated in Figure 2. The mechanisms of site-specific recombination have also been reviewed at length elsewhere (Chen and Rice, 2003; Grindley et al., 2006).
Figure 2.
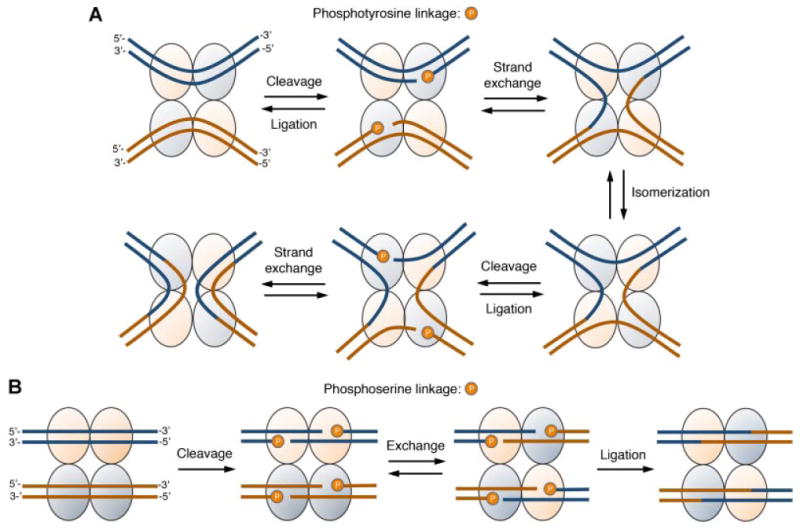
Mechanisms of site-specific recombination. A: Cartoon of tyrosine recombinase-mediated DNA recombination. Two DNA duplexes are bound by four recombinases assembled in a head-to-tail orientation. Nucleophilic attack of the scissile phosphate bonds by two catalytically active monomers (blue circles) leads to the formation of a covalent 3′ phosphotyrosine intermediate. Attack of the opposite phosphotyrosine linkage by the 5′ hydroxyl leads to the generation of a Holliday junction intermediate. Isomerization of the complex activates the second set of recombinase monomers and leads to an additional round of DNA cleavage and strand exchange, and ultimately, release of the recombined DNA product. Sequence identity between the central crossover regions is required for strand exchange. B: Cartoon of serine recombinase-catalyzed DNA recombination. The recombinase binds its cognate target site cooperatively as a head-to-head dimer (orange and blue circles). Two DNA-bound recombinase dimers then assemble to form a tetrameric synaptic complex, enabling nucleophilic attack and covalent attachment of each recombinase subunit to the 5′ phosphate at the cleavage site. This allows for exchange between two recombinase subunits and their covalently attached DNA, followed by reversal of the serine-phosphodiester linkage and ligation of the cleaved DNA. Proper base pairing between the central core dinucleotide residues is necessary for recombination.
While site-specific recombination systems are diverse, the property that makes these enzymes so enticing—the ability to specifically and autonomously integrate, excise, or invert defined sequences of DNA—also limits their practical utility. Because site-specific recombinases have evolved to perform essential biological functions, they demonstrate remarkably strict specificity toward their natural target. Indeed, application of these enzymes in mammalian cells requires either the presence of rare pre-existing pseudo-recognition sites or the pre-introduction of specific target sites within the host genome by homologous recombination, an inefficient and time-consuming process. Thus, while site-specific recombinases represent potentially transformative tools for targeted genetic engineering, their application has been impeded by technical constraints. In order for this technology to reach its full potential, methods for the evolution and design of custom recombinases capable of modifying investigator-defined DNA sequences are needed.
Over the past two decades, protein engineering has emerged as a versatile and powerful approach for tailoring the properties of biomolecules for diverse and complex tasks (Jackel et al., 2008). Amongst the techniques employed by researchers for altering the properties of enzymes, two of the most common are directed evolution and rational design. Based on the principles that guide natural evolution; namely, diversification (e.g., gene mutagenesis, DNA shuffling, etc.), selection (e.g., identifying altered variants by phage, yeast or ribosome display, or fluorescence-activated cell sorting) and amplification, directed evolution enables rapid evaluation of large (>107) gene libraries in the absence of prior knowledge of protein structure (Yuan et al., 2005). Rational or computational design-based methods, which rely on structural, functional or mechanistic information, offer an alternative approach for modifying biomolecular properties by enabling the introduction of mutations at defined sites (Cedrone et al., 2000). Significantly, protein engineering has been employed to alter the properties of a variety of site-specific recombination systems with great success. Such features include recognition specificity, enzyme thermostability, and recombination efficiency. Indeed, these strategies are now poised to provide researchers with a means of generating novel site-specific recombinases capable of reengineering complex biological systems. Here, we review the history and recent developments in the directed evolution and rational design of site-specific recombinases. We examine how these various methods have been applied to generate site-specific recombinases with new, exceptional properties, and discuss many of the challenges facing their maturation. We emphasize unique insights into the evolution of site-specific recombination and highlight applications of these designer enzymes for genetic and metabolic engineering.
Improving Site-Specific DNA Recombination Systems by Directed Evolution
A contemporary goal of researchers in protein and genetic engineering has been the establishment of methods that allow for complete re-programming of site-specific recombinase specificity. Due primarily to the lack of structural information available, however, many early efforts within this field instead focused on improving recombinase activity for biotechnological applications. One such example is the evolution of Flp recombinase thermostability. Isolated from Saccharomyces cerevisiae, Flp recombinase is a prototypical tyrosine recombinase that catalyzes intramolecular recombination between two inverted 599-bp repeats within the yeast 2-μM DNA plasmid (Broach and Hicks, 1980; Broach et al., 1982; Hartley and Donelson, 1980; Fig. 3). The minimal 34-bp Flp recognition target (FRT) site consists of two 13-bp inverted repeat binding elements that flank a central 8-bp asymmetric spacer sequence (Andrews et al., 1985), which serves as the crossover site for Flp-mediated recombination (Table I). To date, the Flp-FRTrecombination system has been used for gene (i.e., cassette) integration, excision and exchange in numerous cell lines and organisms, including mice, Drosophila, C. elegans, plants, fungi and bacteria (Branda and Dymecki, 2004). However, because Flp is derived from S. cerevisiae, which has an optimal growth temperature of 25–30°C, it has not evolved to support high-activity at human physiological temperatures. Indeed, the reduction in Flp recombinase activity observed at temperatures above 35°C initially indicated that it might not be an optimal choice for applications in mammalian cells (Buchholz et al., 1996).
Figure 3.
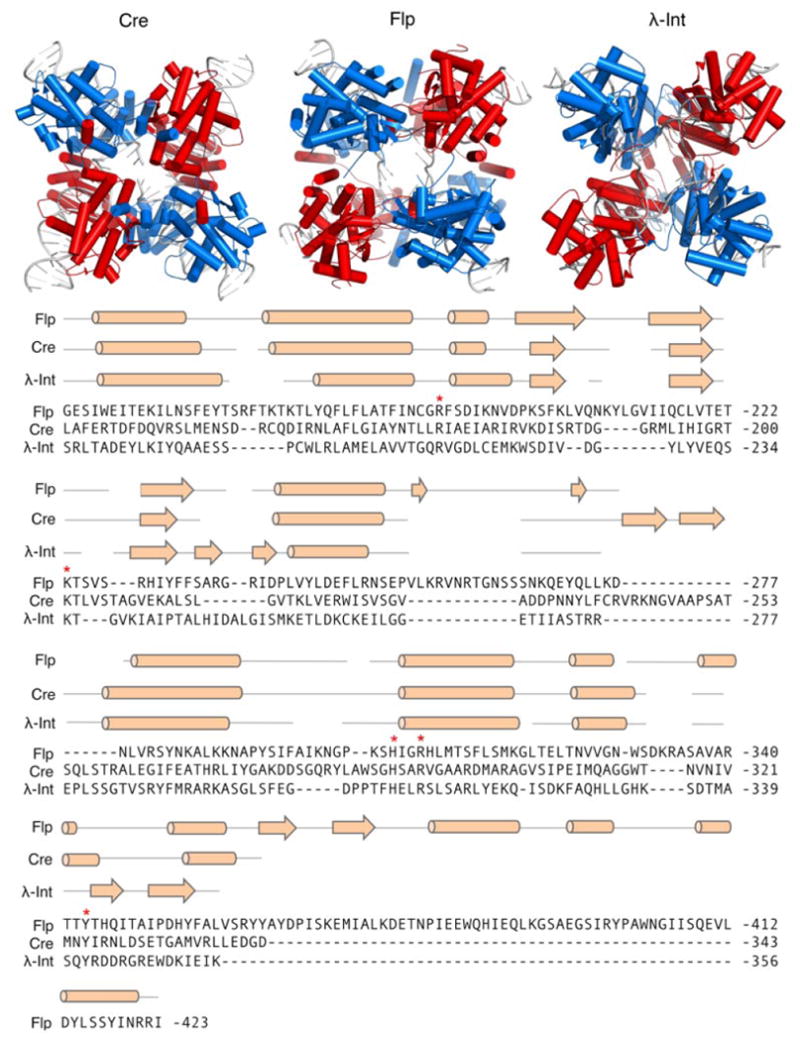
Overview of the tyrosine recombinases. Top: Structures of the Cre, Flp, and λ-Int tetramers complexed with their respective DNA targets. Pairs of recombinase dimers are colored red and blue. DNA depicted as gray cartoon. PDB IDs are Cre: 1CRX (Guo et al., 1997), Flp: 1FLO (Chen et al., 2000), and λ-Int: 1Z1G (Biswas et al., 2005). Bottom: Sequence alignment of Cre, Flp, and λ-Int C-terminal domains. Secondary structural elements for each enzyme are indicated above alignment. Cylinders and arrows indicate α-helix and β-sheet secondary structures, respectively. Red asterisks indicate conserved amino acid residues critical for catalysis.
To address this limitation, Buchholz and Stewart utilized random mutagenesis by error-prone PCR, DNA shuffling, and a lacZ-based blue/white recombination screen to generate Flp variants with enhanced thermostability (Buchholz et al., 1998). Error-prone PCR relies on the mis-incorporation of nucleotides by DNA polymerases to generate point mutations within a gene sequence (Cadwell and Joyce, 1992; Guo et al., 2010), whereas DNA shuffling recombines genetic diversity from parental genes to create new genetic variants (Stemmer, 1994a,b), a process that requires digesting the parental gene variants into random fragments and reassembling those fragments into full-length genes by PCR. In this approach, the digested fragments serve as both template and primer, annealing to other digested fragments based on sequence homology, resulting in full-length, recombined genes. To select for thermostable Flp variants, Buchholz and Stewart devised a genetic screen that links the blue/white colony readout to Flp-mediated recombination at elevated temperatures. In this system, two FRTsites flank the lacZ gene, such that, in the absence of recombination, β-galactosidase is expressed and blue colonies form, while recombination leads to excision of the lacZ gene and formation of white colonies. Mutations that support increased activity were therefore identified by white colony formation. The most improved variant, FLPe (P2S, L33S, Y108N, and S294P), showed a 4- and 10-fold increase in recombination at 37°C and 40°C, respectively, in comparison to wild-type enzyme in vitro (Buchholz et al., 1998). FLPe also showed increased recombination efficiency in human embryonic kidney (HEK) 293 and mouse embryonic stem (ES) cells. Crystallographic studies of the DNA-bound FLPe tetramer have revealed that all four mutations reside near the surface of the enzyme and do not influence packing of the protein core (Conway et al., 2003). In particular, P2S and S294P are located at the N-termini of two separate α-helices, L33S is present within an a-helical kink, and Y108N lies within a turn connecting the N- and C-terminal domains. To date, FLPe has proven useful for a wide range of applications, including the generation of transgenic “deleter” mice (Rodriguez et al., 2000), the large-scale production of helper-dependent adenoviral particles (Umana et al., 2001), and the cloning of artificial bacterial chromosomes (Liu et al., 2003). More broadly, the findings by Buchholz and Stewart indicate that directed evolution is a viable method for enhancing the efficiency of site-specific recombination.
Understanding the Determinants of DNA Recognition Specificity: Swapping λ and HK022 Integrase Specificity
Although directed evolution is commonly viewed as a means to identify protein variants with new properties, it also affords the opportunity to study the contribution of individual amino acids to the function of a protein (Yuen and Liu, 2007). The bacteriophage integrases catalyze recombination between attachment sites in the phage (attP) and bacterial (attB) genomes (Groth and Calos, 2004). For the tyrosine integrases, these attachment sites are typically composed of two inverted binding sites separated by a 6- to 8-bp spacer sequence where strand exchange occurs. Interestingly, while the integrases (Int) of bacteriophage λ and HK022 share nearly 70% sequence identity and catalyze very similar reactions, they recognize sites that share only 40% identity (Yagil et al., 1989; Table I). Toward the goal of understanding λ-Int target specificity, Yagil et al. (1995) investigated the possibility of switching λ-Int specificity to that of the Int-HK022 by shuffling the genes of these two enzymes to generate a library of chimeric integrases (Table II). By evaluating the activity of individual chimeras, Weisberg et al. discovered a mutant of λ-Int origin, containing 13 substitutions derived from Int-HK022, capable of specifically recognizing the HK022 target (Yagil et al., 1995). While analysis of individual point mutants revealed that no single mutation was capable of shifting λ-Int specificity toward the HK022 attachment site, a detailed examination of the combined effects of these mutations led Weisberg et al. to discover a network of five substitutions (N99D, S282P, G283K, R287K, and E319R) that mediate the conversion of λ-Int specificity (Yagil et al., 1995). Subsequent mutational analyses revealed that chimeras harboring only isolated subsets of the original 13 mutations displayed relaxed specificity, demonstrating activity on both λ-Int and Int-HK022 sites (Dorgai et al., 1995). These findings suggest the unique possibility that artificially evolved recombinase specificity may emerge by multi-step changes that first relax and then restrict target recognition.
Table II.
Directed evolution of recombinase specificity.
| Recombinase | Mutant target sitea | Mutagenesis method(s) | Selection method | Specificity | Refs. |
|---|---|---|---|---|---|
| λ-Int | 5′-GCACTTTAGGTGAAAAAGGTT-3′ | Family shuffling | Blue/white screen | Switched | Yagil et al. (1995) |
| 5′-GCACTTTAGGTGAAAAAGGTT-3′ | Error-prone PCR | Blue/white screen | Switched | Dorgai et al. (1995) | |
| 5′-CTGCTTTCTTATACCAAGTGG-3′ | Error-prone PCR | SLiPEb by IVCc | Relaxed | Tay et al. (2010) | |
| Flp | 5′-GAAGTTCCTATAGTCTAGAAACTATAGGAACTTC-3′ | Error-prone PCR | Blue/red screen | Relaxed | Voziyanov et al. (2002) |
| 5′-GAAGTTCCTATAGTCTAGAAACTArAGGAACTTC-3′ | Error-prone PCR/DNA shuffling | Blue/red/white screen | Switched | Voziyanov et al. (2003) | |
| 5′-CTAATTCCTTTACTCATGTAAGTATCAAATCACT-3′ | Site-saturation/DNA shuffling | Blue/red/white screen | Relaxed | Bolusani et al. (2006) | |
| Cre | 5′-ATATATACGTATATAGACATATATACGTATATAT-3′ | Error-prone PCR/DNA shuffling | SLiPE | Relaxed | Buchholz and Stewart (2001) |
| 5′-ATAACTCTATATAGCATACATTATATAGAGTTAT-3′ | Site-saturation | FACS | Switched | Santoro and Schultz (2002) | |
| 5′-ACAACATCCTATTACACCCTATATGCCAACATGG-3′ | Error-prone PCR/DNA shuffling | SLiPE | Switched | Sarkar et al. (2007) | |
| Gind | 5′-TCCAAAACCATAAATTATCA-3′ | Error-prone PCR/DNA shuffling | SLiPE | Relaxed | Gordley et al. (2007) |
| 5′-CGAAATATTATAAATTATCA-3′ | Site-saturation | SLiPE/split gene reassembly | Switched | Gaj et al. (2011) | |
| 5′-NNNNAAABNWWNVTTTNNNN-3′ | Site-saturation | SLiPE/split gene reassembly | Switched | Gaj et al. (2013b) | |
| Tn3d | 5′-CTGACTACTATACTTTGAGC-3′ | Error-prone PCR | Red/white screen | Relaxed | Proudfoot et al. (2011) |
| φC31 | 5′-TAAGTACTTGGGTTTCCCTTGGTGTCCCCATGGAGATTT-3′ | Error-prone PCR/DNA shuffling | Blue/white screen | Switched | Sclimenti et al. (2001) |
N indicates A, T, C or G; B indicates T, C or G; V indicates A, C or G; and W indicates A or T.
Target site mutations are underlined.
Substrate-linked protein evolution.
In vitro compartmentalization.
Central 20-bp core sequence shown only.
Since these studies, the λ-Int has also been adapted for molecular cloning, enabling sophisticated tasks such as the generation of biosynthetic gene clusters in several Streptomyces strains (Eustaquio et al., 2005) and the production of minicircle DNA in E. coli (Kay et al., 2010). However, unlike other prototypical tyrosine recombinases, such as Cre and Flp, the λ-Int naturally performs site-specific recombination with assistance from accessory factor proteins, which help coordinate the directionality (i.e., integration and excision) of the recombination reaction (Grindley et al., 2006). In order to overcome this constraint, accessory-factor independent variants of the λ-Int have been identified by selection in the presence of defective integration host factor activity (Miller et al., 1980) and reversion analysis (Wu et al., 1997). Dröge and colleagues have subsequently shown that rationally designed λ-Int variants based on two mutations identified by these studies, Int-h (E174K) and Int-h/218 (E174K, E218K), catalyze both integrative and excisive recombination in human cells (Lorbach et al., 2000). These mutants have also allowed significant effort to be devoted to characterizing (Christ et al., 2002) and altering (Rutkai et al., 2003) the recognition specificity of λ-Int. More recent studies have revealed that λ-Int can be coaxed to recognize unnatural attachment sites by in vitro compartmentalization (IVC; Tay et al., 2010), an emulsion-based technology for the generation of cell-like compartments (Tawfik and Griffiths, 1998) that has enabled the directed evolution of several types of DNA-modifying and processing enzymes, including DNA methyltransferases (Cohen et al., 2004) and polymerases (Ghadessy et al., 2001), restriction endonucleases (Doi et al., 2004) and transcription factors (Fen et al., 2007). While the λ-Int variants generated by these selections showed only modest shifts in specificity, this study nevertheless demonstrates the potential of in vitro evolution systems for generation of recombinase variants with unnatural qualities. Additionally, Yagil et al. have demonstrated that Int-HK022 promotes bi-directional site-specific recombination in mammalian cells (Kolot et al., 2003), yet variants with altered recognition abilities have not been reported.
The Evolution of Flp Recombinase Variants with Unnatural Target Specificity
Previously the subject of activity enhancement, Flp recombinase is also a convenient platform for altering target specificity. As noted above, Flp catalyzes recombination between FRT sites that contain two 13-bp inverted repeat binding elements that flank a central 8-bp asymmetric spacer sequence. To test whether directed evolution could be used to alter the specificity of the Flp recombinase, Voziyanov et al. (2002) developed a dual-reporter screen that enabled direct readout of the effects of individual mutations on Flp specificity. In this system, one reporter contained the lacZ gene flanked by one of several mutant FRT target sites, while the other contained the red fluorescent protein (RFP) gene flanked by native FRT target sites, enabling blue/red color-based determination of the recombination specificity of the Flp variant carried by each colony. Using this approach, Voziyanov et al. identified Flp variants that tolerated several mutant FRT sites with substitutions at “position 1,” the centermost base within each FRT binding element. Notably, the study revealed that the residue at Flp position 82 is an important determinant in target specificity, as each of the most active variants contained one of three different substitutions at this site (K82M, K82H, and K82Y; Voziyanov et al., 2002). Voziyanov et al. (2003) next showed that Flp could be progressively adapted to recombine mutant FRT sites that contained individual or combined mutations within each FRT binding element (Table II). Similar to the study mentioned above, random mutagenesis and bacterial screening led to the identification of variants with relaxed specificity, however, subsequent cycles of DNA shuffling resulted in the generation of Flp variants with selective DNA binding properties. Impressively, one of the selected mutants (K82Y, V226A, and N264I) demonstrated a >50-fold shift in specificity toward a mutant FRT target with substitutions at “position 1” (Voziyanov et al., 2003; Fig. 4).
Figure 4.
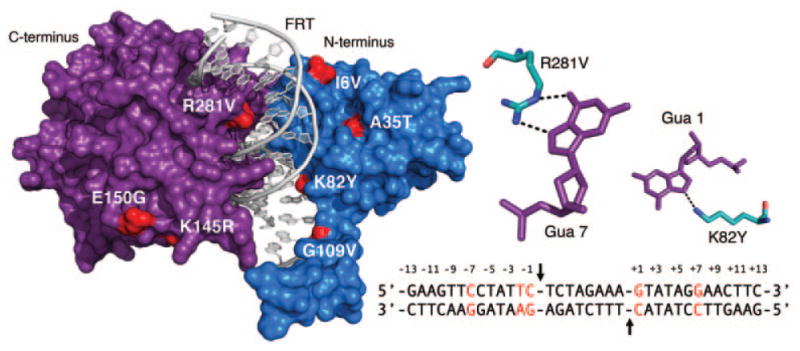
Distribution of mutations that contribute to evolved Flp specificity. Left: Surface illustration of the Flp monomer bound to FRT DNA target. Mutations that contribute to the interactions between evolved Flp variants and unnatural FRT targets (Voziyanov et al., 2003) are highlighted red. DNA depicted as a gray cartoon. Flp N-terminal domain colored blue, C-terminal domain colored purple. Not visible are the substitutions V266A and N264I. PDB ID is 1FLO (Chen et al., 2000). Right upper: The interactions between Arg 281 of wild-type Flp and Gua 7 of wild-type FRT, and Lys 82 and Gua 1 are shown. Gua 7 and Gua 1 were among five positions substituted in the mutant FRT site by Voziyanov et al. (Right lower) FRT target. Positions substituted for evolution studies highlighted red. Black arrows indicate the location of the scissile phosphates.
Mutagenic and crystallographic studies of the Flp recombinase-DNA complex have revealed that Flp recognition of the central spacer sequence is based on interactions with the phosphate backbone and is therefore largely nonspecific (Bruckner and Cox, 1986; Chen and Rice, 2003; Chen et al., 2000). Enzyme specificity is determined almost entirely by a series of direct side-chain-to-base contacts and indirect water-mediated interactions between the major groove of the FRT inverted repeat binding elements and the C-terminal domain of Flp. As a result, increasingly sophisticated stepwise regimens have been used to identify Flp mutants capable of recognizing unnatural target sites in clinically relevant genes, such as human interleukin 10 (IL10; Bolusani et al., 2006; Table II). To achieve this, the proposed FRT binding elements within the IL10 gene were compared to the parental FRT inverted repeats; for each mismatched FRT base pair, the corresponding Flp residue that contacted it was targeted for random mutagenesis. Selection was performed using a hybrid, asymmetric target site consisting of one native FRT binding element and one IL10-derived pseudo-FRT binding element. The most active Flp mutant (FV8: M44V, A55S, M58V, S59N, S130P, E166K, K285H, and A349T) successfully recombined the IL10 target sequence, but retained the ability to recombine wild-type FRT (Bolusani et al., 2006). This relaxation in target specificity could be due to the lack of base symmetry within each pseudo-FRT site used over the course of the evolutions. Combinatorial selection against symmetrical, hybrid recombination sites that contain elements from native FRT and mutant target sites may enable the identification of variants that overcome relaxation effects. These efforts have inspired the development of computational tools that enable the identification of pseudo-FRT sites in the human genome as a means to expand the utility of Flp for genome engineering (Shultz et al., 2011). Similar tools have also been developed for several other recombinases (Surendranath et al., 2010).
Random and Targeted Approaches for Altering Cre Specificity
The protein engineering efforts described thus far relied primarily on the manual screening of individual library clones using colorimetric colony screens. In order improve the throughput of the selection process and to generate recombinase variants with well-defined target specificities, Buchholz and Stewart developed a selection method that physically links individual recombinase variants to their DNA substrate (Buchholz and Stewart, 2001). This technique—termed Substrate-Linked Protein Evolution (SLiPE)—enables the isolation of rare recombinase variants in liquid culture, even in high-background settings, by PCR. As proof-of-principle of SLiPE, Buchholz and Stewart sought to evolve variants of the Cre recombinase (Fig. 3), which is widely used throughout molecular biology for conditional gene inactivation and recombinase-mediated cassette exchange (Nagy, 2000). Cre is derived from bacteriophage P1 and recognizes a 34-bp DNA sequence termed loxP that consists of two 13-bp palindromic binding-site elements, which flank a central asymmetric 8-bp spacer sequence that contains the 6-bp crossover region where recombination occurs (Hoess and Abremski, 1985; Hoess et al., 1982; Table I). As with Flp and λ-Int, Cre is a prototypical member of the tyrosine recombinases, and as such, nearly two decades of effort has led to a comprehensive structural and mechanistic understanding of Cre-mediated recombination (Guo et al., 1997; Van Duyne, 2001). Buchholz and Stewart initiated their studies by using SLiPE to evolve Cre variants capable of recombining a 34-bp pseudo-loxP site present on human chromosome 22 (Table II). The selected mutant loxP site contained three mutations within each loxP inverted binding element, as well as substitutions at each position within the central 8-bp spacer sequence. After 35 cycles of SLiPE and 11 rounds of DNA shuffling, a highly active population of Cre variants was isolated, with an average mutation rate of ∼11 amino acid substitutions per variant. Mapping of these amino acid mutations onto the Cre structure revealed clustering within two regions: the active site and an area of the enzyme postulated to position the loxP spacer sequence for cleavage in the pre-cleaved protein-DNA complex (Fig. 5A). Importantly, Buchholz and Stewart found that several Cre variants displayed activity in mammalian cells, albeit with reduced recombination efficiency and relaxed specificity compared to wild-type Cre. Nevertheless, these results indicated that substrate-linked directed evolution represents an innovative and streamlined approach for selecting recombinase variants. More recently, our laboratory has developed a SLiPE system based on recombinase-mediated reassembly of the geneencoding TEM-1 β-lactamase that enables quantitative and high-throughput recovery of rare (<10−6) site-specific recombinase variants (Gersbach et al., 2010).
Figure 5.
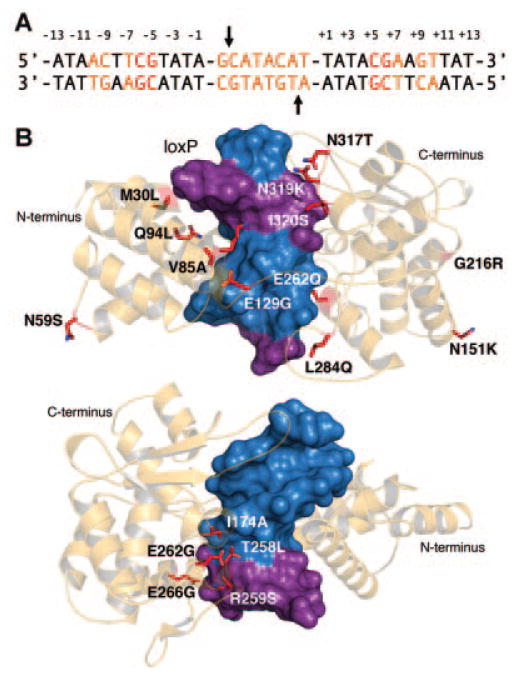
Evolution of Cre specificity. A: loxP target. Base positions altered for selection with randomly mutated Cre variants highlighted orange (Buchholz and Stewart, 2001), those positions altered for selection with Cre variants that contain targeted substitutions highlighted red (Santoro and Schultz, 2002). Black arrows indicate the location of the scissile phosphates (B). Top and bottom: Cre monomer (light orange cartoon) in complex with loxP (blue and purple surface). Mutations identified by (top) substrate-linked protein evolution (SLiPE) and (bottom) those positions targeted for randomization for FACS selection shown as red sticks. Top and bottom: Substituted positions within each loxP target denoted purple. PDB ID is 1CRX (Guo et al., 1997).
One potential explanation for the emergence of relaxed Cre variants within pools selected by SLiPE is that positive selection alone is not sufficient to ensure the dominance of recombinase variants with strict specificity for a new target. To test this hypothesis, Santoro and Schultz (2002) attempted to identify Cre recombinase variants that could recognize a series of minimally mutated loxP sites by developing a fluorescence-activated cell sorting (FACS) screen that utilizes both positive and negative selection. In this bacterial system, Cre variants capable of recombining mutant loxP sites drive expression of GFPuv, while Cre variants that maintain the ability to recognize wild-type loxP drive enhanced yellow fluorescent protein (EYFP) expression, allowing simple FACS selection of mutants. This strategy was used to select Cre variants that could recombine a mutant loxP site with substitutions at positions 7, 6, and 5 within the inverted loxP binding elements (Table II). To achieve this, the Cre residues (Ile 174, Thr 258, Arg 259, Glu 262, and Glu 266) known to contact these positions were targeted for randomization. After only five rounds of positive and negative selection, a fairly conserved population of Cre variants with converted specificity was identified (Santoro and Schultz, 2002; Fig. 5B). Positive selection alone, however, led to the selection of Cre mutants with relaxed specificity. Interestingly, when this approach was attempted with an alternate mutant loxP site that contained substitutions at positions 3 and 2, only relaxed variants were identified, indicating that no individual Cre variants within the library satisfied both positive and negative selection requirements and that indirect factors may contribute to target recognition. In a follow-up study, Baldwin, Santoro, and Schultz determined the co-crystal structures of two evolved Cre-DNA complexes and found that recognition specificity was indeed the product of unexpected macromolecular plasticity and a unique network of water-mediated protein-DNA contacts (Baldwin et al., 2003), indicating that these enzymes can be artificially evolved to utilize indirect mechanisms for sequence discrimination.
Chimeric Recombinases With Designer Specificity
Thus far, attempts to alter recombinase specificity have typically required iterative rounds of mutagenesis and complex selection strategies. However, these approaches have met with limited success. The use of chimeric recombinases with designer specificity presents an alternative strategy that could overcome many of the technical limitations associated with these previously described methods. In particular, the resolvase/invertase family of serine recombinases (Smith and Thorpe, 2002) may represent an effective platform for such types of programming (Fig. 6 and Table I). These enzymes are modular in both form and function (Abdel-Meguid et al., 1984; Sanderson et al., 1990; Yang and Steitz, 1995): a C-terminal DNA-binding domain directs sequence-specific association with DNA, while an N-terminal catalytic domain recognizes a central core sequence and promotes sequence-specific recombination (Fig. 7A). In nature, these recombinases rely on accessory factors or multiple binding sites for activation and regulation of catalysis (Grindley et al., 2006); however, mutants of several resolvase/invertase variants have been identified that function without these additional factors (Arnold et al., 1999; Klippel et al., 1988). Together, these findings prompted Stark and colleagues to investigate the fusion of a hyperactivated, accessory-factor independent mutant of the catalytic domain of the Tn3 resolvase to the Zif268 zinc-finger protein (Akopian et al., 2003). Remarkably, Stark and co-workers found that these engineered zinc-finger recombinase (ZFR) fusion proteins possessed the anticipated chimeric target specificity and catalyzed unrestricted recombination in bacterial cells (Akopian et al., 2003) and subsequently in vitro (Prorocic et al., 2011).
Figure 6.
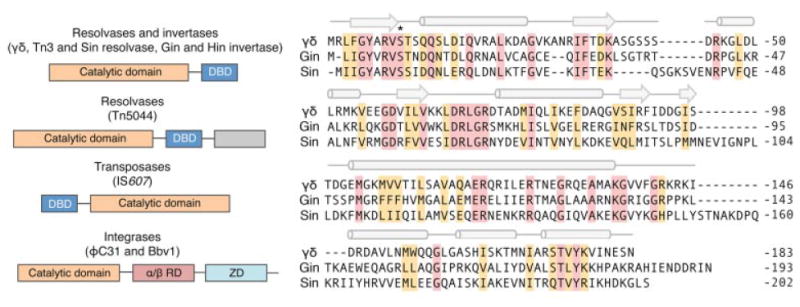
Overview of the serine recombinases. Left: Domain organization of the serine recombinases. α/β RD and ZD indicate C-terminal α/β recombinase and zinc-nucleated integrase domains, respectively (Rutherford et al., 2013). Gray indicates domains of unknown function. Right: Comparison of representative members of the resolvase/invertase family of serine recombinases. Conserved residues are shaded pink. Secondary structural elements within the γδ resolvase are indicated above alignment. Cylinders and arrows indicate α-helix and β-sheet structures, respectively. Asterisk indicates the conserved serine residue critical for catalysis.
Figure 7.
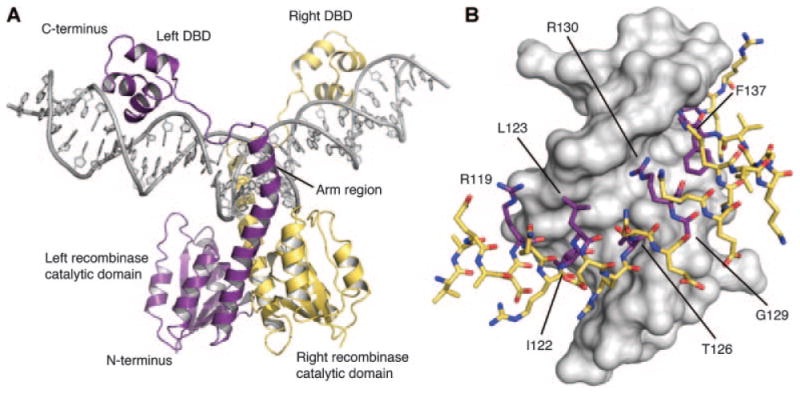
DNA recognition by the resolvase/invertase family of serine recombinases. A: The γδ resolvase dimer (purple and yellow) in complex with target DNA (gray). PDB ID: 1GDT (Yang and Steitz, 1995). DBD indicates DNA-binding domain. B: Specific recognition of DNA (gray) by the serine recombinase arm region (sticks). Residues that confer catalytic domain specificity and have been subject to reprogramming are highlighted purple (Gaj et al., 2011, 2013b).
The modular structure of zinc-finger proteins makes them an attractive building block for the design of custom DNA-binding proteins (Beerli and Barbas, 2002). Indeed, the versatility of ZFRs arises from the ability to customize the DNA-binding domain to recognize a wide variety of DNA sequences. To this end, advances in the design and selection of zinc-finger domains by our laboratory (Beerli et al., 1998; Dreier et al., 2001, 2005; Segal et al., 1999) and others (Doyon et al., 2008; Isalan et al., 2001; Maeder et al., 2008; Sander et al., 2011) have enabled the generation of ZFRs with expanded target specificities (Gordley et al., 2007; Nomura et al., 2012). We have shown that rationally designed ZFRs utilizing the Gin and Tn3 catalytic domains are capable of catalyzing targeted integration with exceptional specificity at pre-introduced sites in HEK293 cells (Gordley et al., 2009) and that ZFRs can be used in tandem with the PiggyBac transposase for highly efficient, two-step gene transfer in mouse and human cells (Gersbach et al., 2011). Our laboratory has also employed directed evolution to generate catalytic domains with expanded specificities by using SLiPE to evolve for “generalist” recombinases that recognize abroad spectrum of target sites (Gersbach et al., 2010; Gordley et al., 2007; Table II). These selected catalytic domains are capable of excising transgenes flanked by their chimeric ZFR target sites with efficiencies of > 15% in human cells (Gordley et al., 2007). Similarly, Proudfoot and Stark have evolved a series of catalytic domain variants capable of recombining a panel of asymmetric core sequences derived from the bovine β-casein gene (Proudfoot et al., 2011), a locus that could potentially enable high-level expression of protein into milk from integrated gene sequences (Table II).
Although “generalist” catalytic domains may allow for recombination between an extended range of DNA sequences, the relaxed specificity profiles that these enzymes exhibit may not be desirable for applications that require precise targeting. As such, our laboratory has sought to develop a comprehensive design platform that enables the generation of ZFRs with exact investigator-defined specificity. As a first step toward achieving this goal, we have shown that the target specificities of two distinct serine recombinases—Gin and Tn3—can be directly interconverted by targeted mutagenesis of the C-terminal arm, a region of the recombinase that connects the catalytic and DNA-binding domains, as well as contacts substrate DNA (Gaj et al., 2011; Fig. 7B and Table II). These redesigned catalytic domains demonstrated a >10,000-fold shift in specificity, and targeted integration into pre-introduced target sites in the human genome with high specificity. Expanding on this proof-of-principle work, we have more recently shown that directed evolution also allows for the generation of a diverse array of Gin catalytic domains that are capable of recombining a broad range of user-defined DNA targets with high specificity in the context of ZFRs (Gaj et al., 2013b; Table II). This customization strategy has led to the design of ZFRs capable of achieving targeted modification of endogenous genetic elements in human cells. These findings indicate that ZFRs may be suitable for a number of diverse applications, including targeted integration of single or multiple transgenes for metabolic pathway engineering. We have also recently shown that the Gin recombinase catalytic domain can be fused to transcription activator-like effector (TALE) proteins to generate chimeric TALE recombinases (TALERs; Mercer et al., 2012). Derived from the plant pathogenic bacterial genus Xanthomonas, TALEs are naturally occurring proteins that contain programmable DNA-binding domains (Boch et al., 2009; Moscou and Bogdanove, 2009). A typical TALE DNA-binding domain consists of a series of 33- to 35-amino acid repeats that each recognizes a single base pair via two adjacent repeat-variable di-residues (RVDs; Deng et al., 2012; Mak et al., 2012). Toward developing an efficient TALER architecture, we used a library of incrementally truncated TALE domains to select for TALER frameworks that promote highly efficient recombination in bacterial cells (Mercer et al., 2012). We subsequently showed that TALERs also coordinate site-specific recombination of episomal substrates in mammalian cells. While these studies expand the potential targeting capacity of chimeric recombinases, additional experiments are necessary to determine the versatility and flexibility of this system, as well as to evaluate whether TALERs are capable of directing site-specific integration in mammalian cells.
Therapeutic Applications of Evolved Recombinases
Many of the investigations described above relied on selection to help uncover the mechanisms governing DNA recognition and site-specific recombination. The knowledge gained from these studies has enabled researchers to apply these systems to tackle clinically relevant goals, such as using directed evolution to alter Cre specificity for potential therapeutic applications. By taking a combinatorial approach to SLiPE and combining and shuffling individual pre-selected Cre recombinase libraries, Sarker and Buchholz showed that Cre could be evolved to specifically recognize an asymmetric sequence within the HIV-1 long terminal repeat (LTR; Sarkar et al., 2007; Table II). Impressively, this enzyme, called Tre, was shown to excise proviral DNA from the genome of HIV-infected mammalian cells. However, in order to accumulate the mutations necessary to sufficiently shift the specificity of Cre toward the HIV-1 LTR, 126 cycles of selection against a series of six intermediate pseudo-substrates was required. Interestingly, combinatorial addition of these mutations from different libraries led to apparent synergistic effects and, ultimately, the selection of recombinase variants that recognized a target sequence more divergent from loxP than any previously reported. Significantly, much like earlier proof-of-principle efforts, selected variants first lost and then regained their specificity as the evolution progressed. This finding supports earlier observations that artificially selected recombinase specificity may emerge by multistep changes that first relax, and then restrict DNA recognition. In total, Tre contained 19 amino acid mutations when compared to Cre, many of these in regions previously not considered essential for catalysis or DNA binding. However, this evolved recombinase was unable to recombine DNA as efficiently or specifically as wild-type Cre. To address this limitation and increase the practicality of this approach, Buchholz and Pisabarro have recently shown that mutational data obtained from early rounds of evolution can be used to computationally refine Tre specificity (Abi-Ghanem et al., 2013). Alternatively, in a move to eliminate the requirement of evolution for the selection of new Cre variants, numerous groups have shown that genome mining is an effective approach for identifying new, orthogonal site-specific recombination systems that may represent more effective starting points for evolution (Karimova et al., 2013; Nern et al., 2011; Sauer and McDermott, 2004; Suzuki and Nakayama, 2011). While the evolution of Tre suggests that recombinases represent potentially valuable tools for clinical development, several questions remain regarding whether proviral excision of HIV is a viable therapeutic option. In particular, this approach may be rendered ineffective by the evolution of HIV escape variants containing substitutions within the LTR that prevent Tre action. Future recombinase evolutions may thus focus on targeting invariant regions of the HIV genome or the human genes that encode for the HIV co-receptors CCR5 (Gaj et al., 2012; Holt et al., 2010; Perez et al., 2008) and CXCR4 (Yuan et al., 2012).
Another enzyme that has emerged as an attractive tool for site-specific recombination in mammalian cells is the site-specific integrase from bacteriophage φC31 (Groth and Calos, 2004; Groth et al., 2000). φC31 integrase is a member of the large serine recombinase family of enzymes that catalyzes accessory-factor independent recombination between attachment sites from the phage (attP) and host (attB) genomes (Thorpe and Smith, 1998; Fig. 6 and Table I). Notably, because φC31-mediated recombination alters the composition of the central bases within the targeted attachment site, no competing reverse reaction is possible, indicating that φC31 may be a highly effective tool for therapeutic gene transfer. Indeed, Calos et al. have shown that φC31 can integrate into pseudo-attB sites that have partial identity to the native site with efficiencies up to 0.3% in human cells and 5% in mouse cells (Thyagarajan et al., 2001). As such, φC31 has been used to deliver molecular payloads into animals for a variety of uses, including restoration of Factor IX levels in hemophiliac mice (Olivares et al., 2002), site-specific integration of dystrophin for treatment of muscular dystrophy (Bertoni et al., 2006), genetic correction of Red Foot Disease (Junctional epidermolysis bullosa; Ortiz-Urda et al., 2003), and treatment of peripheral vascular disease (Portlock et al., 2006) and rheumatoid arthritis (Keravala et al., 2006) in mice. The φC31 integrase has also been used for genetic correction of dystrophic epidermolysis bullosa in primary patient cells (Ortiz-Urda et al., 2002), as well as for reprogramming somatic cells to pluripotency (Thyagarajan et al., 2008; Ye et al., 2010). More recently, φC31 and the related Bxb1 integrase (Ghosh et al., 2003; Kim et al., 2003; Nkrumah et al., 2006) have emerged as powerful tools for synthetic biology, enabling retrievable data storage within bacterial chromosomes (Bonnet et al., 2012; Siuti et al., 2013) and digital control of gene expression (Bonnet et al., 2013).
However, sequence analysis of nearly 200 independent φC31-mediated integration events in human cells revealed the presence of >100 unique pseudo-recognition sites (Chalberg et al., 2006), indicating that use of φC31 for genome engineering and human gene therapy presents risks of insertional mutagenesis. To address this problem, Calos and co-workers sought to improve the integration specificity of φC31 by using directed evolution to shift its binding preference toward a highly favored pseudo-attP site present on chromosome 8 (Sclimenti et al., 2001; Table II). Relying on a blue/white colony screen to identify active mutants, Calos and colleagues showed that after only two cycles of DNA shuffling, integrase variants demonstrating a >6-fold increase in integration specificity could be identified, suggesting that directed evolution is an effective strategy not only for swapping, but also for improving recombinase specificity. Calos and colleagues achieved further improvement of φC31 through the use of rational design (Keravala et al., 2009). Alanine-scanning mutagenesis led to the identification of several charge-neutralizing mutations (D40A, D44A, and D52A) within the N-terminus that enhanced integration efficiency in murine cells. Similarly, Ehrhardt et al. used alanine-scanning mutagenesis to identify mutations within the DNA-binding domain that enhanced integration into pseudo-attB sites up to 6-fold in human cells (Liesner et al., 2010). Together, these studies suggest that rational mutagenesis can be used to improve site-specific recombination even in the absence of any structural data. Additional experiments are required to assess whether these mutations improve gene transfer efficiency in therapeutic applications, however.
Lastly, although site-specific recombinases are potentially powerful tools for a broad range of therapeutic applications, several questions remain over the safety of these enzymes. In particular, Cre and φC31-mediated recombination between off-target pseudo-recognition sites (Chalberg et al., 2006; Thyagarajan et al., 2000) has been shown to lead to deletions and chromosomal re-arrangements in cultured cells (Ehrhardt et al., 2006; Liu et al., 2006; Loonstra et al., 2001) and mice (Schmidt et al., 2000). Additionally, DNA damage has been observed at genomic targets following φC31 (Malla et al., 2005) and ZFR-mediated recombination (Gaj et al., 2013b). These findings suggest that formation of covalent protein-DNA linkages by these enzymes may activate cellular DNA repair pathways (Wyman and Kanaar, 2006). It remains unknown whether reducing the cellular concentration of these enzymes may lead to lower levels of DNA damage. Finally, the specificity of evolved site-specific recombinases remains an important and unanswered question. Advances in whole-genome sequencing technology (Shendure and Lieberman Aiden, 2012) should allow researchers the opportunity to examine the full scope of off-target modifications (Gabriel et al., 2011). By providing new insight into genome-wide recognition specificity, these approaches may also guide the design of site-specific recombinases with improved targeting capabilities.
Conclusions and Future Directions
The development of new tools capable of achieving targeted genetic manipulations is at the forefront of biotechnology. Although targeted nucleases have emerged as the method of choice for impacting genomic change [reviewed in (Carroll, 2011; Gaj et al., 2013a)], these approaches rely on the activation of cellular repair pathways and mutagenic DNA double-strand breaks to introduce targeted alterations at specific locations. In contrast, site-specific recombinases function autonomously and direct DNA integration, inversion, and excision in a variety of cellular environments. As a result, intense efforts have been directed toward altering the properties of many recombinases by protein engineering. However, numerous questions remain related to the efficacy of many of these approaches. In particular, how does mutation rate impact the speed at which recombinase specificity is recovered following relaxation? The use of selections that enable high-throughput, real-time evaluation of protein parameters may provide insight into this question (Esvelt et al., 2011; Leconte et al., 2013). Additionally, can detailed analysis of the substrate specificity profiles (Hartung and Kisters-Woike, 1998; Whiteson and Rice, 2008) of site-specific recombinases lead to the more seamless evolution of variants with altered specificity, and can mature recombinase variants be generated combinatorially against user-defined targets using broad, pre-selected variant populations? Further, what advances in methods for designing recombinase specificity are needed in order for site-specific recombinases to achieve a level of user-friendly accessibility that allows them to reach their potential for genome engineering? Do post-translational modification sites (e.g., Flp sumoylation; Chen et al., 2005; Xiong et al., 2009) impact the stability and efficiency of these enzymes in human cells? Finally, numerous questions remain regarding the therapeutic potential of these enzymes. In particular, can evolved recombinases demonstrate the requisite specificity to avoid inducing potentially toxic off-target effects, and what are the optimal methods for delivering these enzymes into relevant cell types (Jo et al., 2001; Peitz et al., 2002; Pfeifer et al., 2001; Wang et al., 1996)? Ultimately, these questions will only be answered through continued exploration and analysis of these unique and powerful molecular tools.
Acknowledgments
Contract grant sponsor: National Institutes of Health
Contract grant number: DP1CA174426
Contract grant sponsor: National Institute of General Medicine Sciences fellowship
Contract grant number: T32GM080209
The authors are supported by the National Institutes of Health (Pioneer Award DP1CA174426). T.G. was supported by National Institute of General Medicine Sciences fellowship (T32GM080209). Molecular graphics were generated using PyMol (http://pymol.org). Multiple sequence alignments were generated by STRAP, an editor for the structural alignment of proteins (http://bioinformatics.org/strap). We apologize to those investigators whose important contributions have been omitted due to space constraints.
References
- Abdel-Meguid SS, Grindley ND, Templeton NS, Steitz TA. Cleavage of the site-specific recombination protein gamma delta resolvase: The smaller of two fragments binds DNA specifically. Proc Natl Acad Sci USA. 1984;81(7):2001–2005. doi: 10.1073/pnas.81.7.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Ghanem J, Chusainow J, Karimova M, Spiegel C, Hofmann-Sieber H, Hauber J, Buchholz F, Pisabarro MT. Engineering of a target site-specific recombinase by a combined evolution- and structure-guided approach. Nucl Acids Res. 2013;41(4):2394–2403. doi: 10.1093/nar/gks1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian A, He J, Boocock MR, Stark WM. Chimeric recombinases with designed DNA sequence recognition. Proc Natl Acad Sci USA. 2003;100(15):8688–8691. doi: 10.1073/pnas.1533177100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews BJ, Proteau GA, Beatty LG, Sadowski PD. The FLP recombinase of the 2 micron circle DNA of yeast: Interaction with its target sequences. Cell. 1985;40(4):795–803. doi: 10.1016/0092-8674(85)90339-3. [DOI] [PubMed] [Google Scholar]
- Arnold PH, Blake DG, Grindley ND, Boocock MR, Stark WM. Mutants of Tn3 resolvase which do not require accessory binding sites for recombination activity. EMBO J. 1999;18(5):1407–1414. doi: 10.1093/emboj/18.5.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin EP, Martin SS, Abel J, Gelato KA, Kim H, Schultz PG, Santoro SW. A specificity switch in selected cre recombinase variants is mediated by macromolecular plasticity and water. Chem Biol. 2003;10(11):1085–1094. doi: 10.1016/j.chembiol.2003.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli RR, Barbas CF., III Engineering polydactyl zinc-finger transcription factors. Nat Biotech. 2002;20(2):135–141. doi: 10.1038/nbt0202-135. [DOI] [PubMed] [Google Scholar]
- Beerli RR, Segal DJ, Dreier B, Barbas CF., III Toward controlling gene expression at will: Specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA. 1998;95(25):14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni C, Jarrahian S, Wheeler TM, Li Y, Olivares EC, Calos MP, Rando TA. Enhancement of plasmid-mediated gene therapy for muscular dystrophy by directed plasmid integration. Proc Natl Acad Sci USA. 2006;103(2):419–424. doi: 10.1073/pnas.0504505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005;435(7045):1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Bolusani S, Ma CH, Paek A, Konieczka JH, Jayaram M, Voziyanov Y. Evolution of variants of yeast site-specific recombinase Flp that utilize native genomic sequences as recombination target sites. Nucl Acids Res. 2006;34(18):5259–5269. doi: 10.1093/nar/gkl548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Subsoontorn P, Endy D. Rewritable digital data storage in live cells via engineered control of recombination directionality. Proc Natl Acad Sci USA. 2012;109(23):8884–8889. doi: 10.1073/pnas.1202344109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Yin P, Ortiz ME, Subsoontorn P, Endy D. Amplifying genetic logic gates. Science. 2013;340(6132):599–603. doi: 10.1126/science.1232758. [DOI] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6(1):7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- Broach JR, Hicks JB. Replication and recombination functions associated with the yeast plasmid, 2mu circle. Cell. 1980;21(2):501–508. doi: 10.1016/0092-8674(80)90487-0. [DOI] [PubMed] [Google Scholar]
- Broach JR, Guarascio VR, Jayaram M. Recombination within the yeast plasmid 2 mu circle is site-specific. Cell. 1982;29(1):227–234. doi: 10.1016/0092-8674(82)90107-6. [DOI] [PubMed] [Google Scholar]
- Bruckner RC, Cox MM. Specific contacts between the FLP protein of the yeast 2-micron plasmid and its recombination site. J Biol Chem. 1986;261(25):11798–11807. [PubMed] [Google Scholar]
- Buchholz F, Stewart AF. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat Biotechnol. 2001;19(11):1047–1052. doi: 10.1038/nbt1101-1047. [DOI] [PubMed] [Google Scholar]
- Buchholz F, Ringrose L, Angrand PO, Rossi F, Stewart AF. Different thermostabilities of FLP and Cre recombinases: Implications for applied site-specific recombination. Nucl Acids Res. 1996;24(21):4256–4262. doi: 10.1093/nar/24.21.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz F, Angrand PO, Stewart AF. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol. 1998;16(7):657–662. doi: 10.1038/nbt0798-657. [DOI] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2(1):28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188(4):773–782. doi: 10.1534/genetics.111.131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedrone F, Menez A, Quemeneur E. Tailoring new enzyme functions by rational redesign. Curr Opin Struct Biol. 2000;10(4):405–410. doi: 10.1016/s0959-440x(00)00106-8. [DOI] [PubMed] [Google Scholar]
- Chalberg TW, Portlock JL, Olivares EC, Thyagarajan B, Kirby PJ, Hillman RT, Hoelters J, Calos MP. Integration specificity of phage phiC31 integrase in the human genome. J Mol Biol. 2006;357(1):28–48. doi: 10.1016/j.jmb.2005.11.098. [DOI] [PubMed] [Google Scholar]
- Chen Y, Rice PA. New insight into site-specific recombination from Flp recombinase-DNA structures. Annu Rev Biophys Biomol Struct. 2003;32:135–159. doi: 10.1146/annurev.biophys.32.110601.141732. [DOI] [PubMed] [Google Scholar]
- Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase–Holliday junction complex: Assembly of an active oligomer by helix swapping. Mol Cell. 2000;6(4):885–897. [PubMed] [Google Scholar]
- Chen XL, Reindle A, Johnson ES. Misregulation of 2 microm circle copy number in a SUMO pathway mutant. Mol Cell Biol. 2005;25(10):4311–4320. doi: 10.1128/MCB.25.10.4311-4320.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AA, Lu TK. Synthetic biology: An emerging engineering discipline. Annu Rev Biomed Eng. 2012;14:155–178. doi: 10.1146/annurev-bioeng-071811-150118. [DOI] [PubMed] [Google Scholar]
- Christ N, Corona T, Droge P. Site-specific recombination in eukaryotic cells mediated by mutant lambda integrases: Implications for synaptic complex formation and the reactivity of episomal DNA segments. J Mol Biol. 2002;319(2):305–314. doi: 10.1016/S0022-2836(02)00327-3. [DOI] [PubMed] [Google Scholar]
- Cohen HM, Tawfik DS, Griffiths AD. Altering the sequence specificity of HaeIII methyltransferase by directed evolution using in vitro compartmentalization. Protein Eng Des Sel. 2004;17(1):3–11. doi: 10.1093/protein/gzh001. [DOI] [PubMed] [Google Scholar]
- Conway AB, Chen Y, Rice PA. Structural plasticity of the Flp–Holliday junction complex. J Mol Biol. 2003;326(2):425–434. doi: 10.1016/s0022-2836(02)01370-0. [DOI] [PubMed] [Google Scholar]
- Dale EC, Ow DW. Gene transfer with subsequent removal of the selection gene from the host genome. Proc Natl Acad Sci USA. 1991;88(23):10558–10562. doi: 10.1073/pnas.88.23.10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu JK, Shi Y, Yan N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science. 2012;335(6069):720–723. doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi N, Kumadaki S, Oishi Y, Matsumura N, Yanagawa H. In vitro selection of restriction endonucleases by in vitro compartmentalization. Nucl Acids Res. 2004;32(12):e95. doi: 10.1093/nar/gnh096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorgai L, Yagil E, Weisberg RA. Identifying determinants of recombination specificity: Construction and characterization of mutant bacteriophage integrases. J Mol Biol. 1995;252(2):178–188. doi: 10.1006/jmbi.1995.0486. [DOI] [PubMed] [Google Scholar]
- Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Amacher SL. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26(6):702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF., III Development of zinc finger domains for recognition of the 5′ -ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2001;276(31):29466–29478. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A, Koksch B, Barbas CF., III Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2005;280(42):35588–35597. doi: 10.1074/jbc.M506654200. [DOI] [PubMed] [Google Scholar]
- Ehrhardt A, Engler JA, Xu H, Cherry AM, Kay MA. Molecular analysis of chromosomal rearrangements in mammalian cells after phiC31-mediated integration. Hum Gene Ther. 2006;17(11):1077–1094. doi: 10.1089/hum.2006.17.1077. [DOI] [PubMed] [Google Scholar]
- Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472(7344):499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustaquio AS, Gust B, Galm U, Li SM, Chater KF, Heide L. Heterologous expression of novobiocin and clorobiocin biosynthetic gene clusters. Appl Environ Microbiol. 2005;71(5):2452–2459. doi: 10.1128/AEM.71.5.2452-2459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93(20):10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fen CX, Coomber DW, Lane DP, Ghadessy FJ. Directed evolution of p53 variants with altered DNA-binding specificities by in vitro compartmentalization. J Mol Biol. 2007;371(5):1238–1248. doi: 10.1016/j.jmb.2007.05.099. [DOI] [PubMed] [Google Scholar]
- Fukushige S, Sauer B. Genomic targeting with a positive-selection lox integration vector allows highly reproducible gene expression in mammalian cells. Proc Natl Acad Sci USA. 1992;89(17):7905–7909. doi: 10.1073/pnas.89.17.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29(9):816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Gaj T, Mercer AC, Gersbach CA, Gordley RM, Barbas CF., III Structure-guided reprogramming of serine recombinase DNA sequence specificity. Proc Natl Acad Sci USA. 2011;108(2):498–503. doi: 10.1073/pnas.1014214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF., III Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods. 2012;9(8):805–807. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., III ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013a;31(7):397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Mercer AC, Sirk SJ, Smith HL, Barbas CF., III A comprehensive approach to zinc-finger recombinase customization enables genomic targeting in human cells. Nucl Acids Res. 2013b;41(6):3937–3946. doi: 10.1093/nar/gkt071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersbach CA, Gaj T, Gordley RM, Barbas CF., III Directed evolution of recombinase specificity by split gene reassembly. Nucl Acids Res. 2010;38(12):4198–4206. doi: 10.1093/nar/gkq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersbach CA, Gaj T, Gordley RM, Mercer AC, Barbas CF., III Targeted plasmid integration into the human genome by an engineered zinc-finger recombinase. Nucl Acids Res. 2011;39(17):7868–7878. doi: 10.1093/nar/gkr421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadessy FJ, Ong JL, Holliger P. Directed evolution of polymerase function by compartmentalized self-replication. Proc Natl Acad Sci USA. 2001;98(8):4552–4557. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Kim AI, Hatfull GF. The orientation of mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Mol Cell. 2003;12(5):1101–1111. doi: 10.1016/s1097-2765(03)00444-1. [DOI] [PubMed] [Google Scholar]
- Gordley RM, Smith JD, Graslund T, Barbas CF., III Evolution of programmable zinc finger-recombinases with activity in human cells. J Mol Biol. 2007;367(3):802–813. doi: 10.1016/j.jmb.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Gordley RM, Gersbach CA, Barbas CF., III Synthesis of programmable integrases. Proc Natl Acad Sci USA. 2009;106(13):5053–5058. doi: 10.1073/pnas.0812502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainge I, Jayaram M. The integrase family of recombinase: Organization and function of the active site. Mol Microbiol. 1999;33(3):449–456. doi: 10.1046/j.1365-2958.1999.01493.x. [DOI] [PubMed] [Google Scholar]
- Grindley ND, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- Groth AC, Calos MP. Phage integrases: Biology and applications. J Mol Biol. 2004;335(3):667–678. doi: 10.1016/j.jmb.2003.09.082. [DOI] [PubMed] [Google Scholar]
- Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci USA. 2000;97(11):5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389(6646):40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- Guo J, Gaj T, Barbas CF., III Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J Mol Biol. 2010;400(1):96–107. doi: 10.1016/j.jmb.2010.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JL, Donelson JE. Nucleotide sequence of the yeast plasmid. Nature. 1980;286(5776):860–865. doi: 10.1038/286860a0. [DOI] [PubMed] [Google Scholar]
- Hartung M, Kisters-Woike B. Cre mutants with altered DNA binding properties. J Biol Chem. 1998;273(36):22884–22891. doi: 10.1074/jbc.273.36.22884. [DOI] [PubMed] [Google Scholar]
- Hoess RH, Abremski K. Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol. 1985;181(3):351–362. doi: 10.1016/0022-2836(85)90224-4. [DOI] [PubMed] [Google Scholar]
- Hoess RH, Ziese M, Sternberg N. P1 site-specific recombination: Nucleotide sequence of the recombining sites. Proc Natl Acad Sci USA. 1982;79(11):3398–3402. doi: 10.1073/pnas.79.11.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28(8):839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isalan M, Klug A, Choo Y. A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat Biotechnol. 2001;19(7):656–660. doi: 10.1038/90264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- Jantama K, Haupt MJ, Svoronos SA, Zhang X, Moore JC, Shanmugam KT, Ingram LO. Combining metabolic engineering and metabolic evolution to develop nonrecombinant strains of Escherichia coli C that produce succinate and malate. Biotechnol Bioeng. 2008;99(5):1140–1153. doi: 10.1002/bit.21694. [DOI] [PubMed] [Google Scholar]
- Jo D, Nashabi A, Doxsee C, Lin Q, Unutmaz D, Chen J, Ruley HE. Epigenetic regulation of gene structure and function with a cell-permeable Cre recombinase. Nat Biotechnol. 2001;19(10):929–933. doi: 10.1038/nbt1001-929. [DOI] [PubMed] [Google Scholar]
- Karimova M, Abi-Ghanem J, Berger N, Surendranath V, Pisabarro MT, Buchholz F. Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucl Acids Res. 2013;41(2):e37. doi: 10.1093/nar/gks1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay MA, He CY, Chen ZY. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 2010;28(12):1287–1289. doi: 10.1038/nbt.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keravala A, Portlock JL, Nash JA, Vitrant DG, Robbins PD, Calos MP. PhiC31 integrase mediates integration in cultured synovial cells and enhances gene expression in rabbit joints. J Gene Med. 2006;8(8):1008–1017. doi: 10.1002/jgm.928. [DOI] [PubMed] [Google Scholar]
- Keravala A, Lee S, Thyagarajan B, Olivares EC, Gabrovsky VE, Woodard LE, Calos MP. Mutational derivatives of PhiC31 integrase with increased efficiency and specificity. Mol Ther. 2009;17(1):112–120. doi: 10.1038/mt.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol. 2003;50(2):463–473. doi: 10.1046/j.1365-2958.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- Klippel A, Cloppenborg K, Kahmann R. Isolation and characterization of unusual gin mutants. EMBO J. 1988;7(12):3983–3989. doi: 10.1002/j.1460-2075.1988.tb03286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolot M, Meroz A, Yagil E. Site-specific recombination in human cells catalyzed by the wild-type integrase protein of coliphage HK022. Biotechnol Bioeng. 2003;84(1):56–60. doi: 10.1002/bit.10747. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Leconte AM, Dickinson BC, Yang DD, Chen IA, Allen B, Liu DR. A population-based experimental model for protein evolution: Effects of mutation rate and selection stringency on evolutionary outcomes. Biochemistry. 2013;52(8):1490–1499. doi: 10.1021/bi3016185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesner R, Zhang W, Noske N, Ehrhardt A. Critical amino acid residues within the phiC31 integrase DNA-binding domain affect recombination activities in mammalian cells. Hum Gene Ther. 2010;21(9):1104–1118. doi: 10.1089/hum.2010.034. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13(3):476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Jeppesen I, Nielsen K, Jensen TG. Phi c31 integrase induces chromosomal aberrations in primary human fibroblasts. Gene Ther. 2006;13(15):1188–1190. doi: 10.1038/sj.gt.3302789. [DOI] [PubMed] [Google Scholar]
- Logie C, Stewart AF. Ligand-regulated site-specific recombination. Proc Natl Acad Sci USA. 1995;92(13):5940–5944. doi: 10.1073/pnas.92.13.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci USA. 2001;98(16):9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorbach E, Christ N, Schwikardi M, Droge P. Site-specific recombination in human cells catalyzed by phage lambda integrase mutants. J Mol Biol. 2000;296(5):1175–1181. doi: 10.1006/jmbi.2000.3532. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31(2):294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335(6069):716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malla S, Dafhnis-Calas F, Brookfield JF, Smith MC, Brown WR. Rearranging the centromere of the human Y chromosome with phiC31 integrase. Nucl Acids Res. 2005;33(19):6101–6113. doi: 10.1093/nar/gki922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer AC, Gaj T, Fuller RP, Barbas CF., III Chimeric TALE recombinases with programmable DNA sequence specificity. Nucl Acids Res. 2012;40(21):11163–11172. doi: 10.1093/nar/gks875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller HI, Mozola MA, Friedman DI. int-h: An int mutation of phage lambda that enhances site-specific recombination. Cell. 1980;20(3):721–729. doi: 10.1016/0092-8674(80)90318-9. [DOI] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: The universal reagent for genome tailoring. Genesis. 2000;26(2):99–109. [PubMed] [Google Scholar]
- Nern A, Pfeiffer BD, Svoboda K, Rubin GM. Multiple new site-specific recombinases for use in manipulating animal genomes. Proc Natl Acad Sci USA. 2011;108(34):14198–14203. doi: 10.1073/pnas.1111704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods. 2006;3(8):615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura W, Masuda A, Ohba K, Urabe A, Ito N, Ryo A, Yamamoto N, Tamamura H. Effects of DNA binding of the zinc finger and linkers for domain fusion on the catalytic activity of sequence-specific chimeric recombinases determined by a facile fluorescent system. Biochemistry. 2012;51(7):1510–1517. doi: 10.1021/bi201878x. [DOI] [PubMed] [Google Scholar]
- O'Gorman S, Fox DT, Wahl GM. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science. 1991;251(4999):1351–1355. doi: 10.1126/science.1900642. [DOI] [PubMed] [Google Scholar]
- Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat Biotechnol. 2002;20(11):1124–1128. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- Ortiz-Urda S, Thyagarajan B, Keene DR, Lin Q, Fang M, Calos MP, Khavari PA. Stable nonviral genetic correction of inherited human skin disease. Nat Med. 2002;8(10):1166–1170. doi: 10.1038/nm766. [DOI] [PubMed] [Google Scholar]
- Ortiz-Urda S, Thyagarajan B, Keene DR, Lin Q, Calos MP, Khavari PA. PhiC31 integrase-mediated nonviral genetic correction of junctional epidermolysis bullosa. Hum Gene Ther. 2003;14(9):923–928. doi: 10.1089/104303403765701204. [DOI] [PubMed] [Google Scholar]
- Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: A tool for efficient genetic engineering of mammalian genomes. Proc Natl Acad Sci USA. 2002;99(7):4489–4494. doi: 10.1073/pnas.032068699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: Efficient gene targeting in vivo. Proc Natl Acad Sci USA. 2001;98(20):11450–11455. doi: 10.1073/pnas.201415498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portlock JL, Keravala A, Bertoni C, Lee S, Rando TA, Calos MP. Long-term increase in mVEGF164 in mouse hindlimb muscle mediated by phage phiC31 integrase after nonviral DNA delivery. Hum Gene Ther. 2006;17(8):871–876. doi: 10.1089/hum.2006.17.871. [DOI] [PubMed] [Google Scholar]
- Prorocic MM, Wenlong D, Olorunniji FJ, Akopian A, Schloetel JG, Hannigan A, McPherson AL, Stark WM. Zinc-finger recombinase activities in vitro. Nucl Acids Res. 2011;39(21):9316–9328. doi: 10.1093/nar/gkr652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot C, McPherson AL, Kolb AF, Stark WM. Zinc finger recombinases with adaptable DNA sequence specificity. PLoS ONE. 2011;6(4):e19537. doi: 10.1371/journal.pone.0019537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378(6558):720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25(2):139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Yuan P, Perry K, Sharp R, Van Duyne GD. Attachment site recognition and regulation of directionality by the serine integrases. Nucl Acids Res. 2013 doi: 10.1093/nar/gkt580. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkai E, Dorgai L, Sirot R, Yagil E, Weisberg RA. Analysis of insertion into secondary attachment sites by phage lambda and by int mutants with altered recombination specificity. J Mol Biol. 2003;329(5):983–996. doi: 10.1016/s0022-2836(03)00442-x. [DOI] [PubMed] [Google Scholar]
- Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat Methods. 2011;8(1):67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MR, Freemont PS, Rice PA, Goldman A, Hatfull GF, Grindley ND, Steitz TA. The crystal structure of the catalytic domain of the site-specific recombination enzyme gamma delta resolvase at 2.7 A resolution. Cell. 1990;63(6):1323–1329. doi: 10.1016/0092-8674(90)90427-g. [DOI] [PubMed] [Google Scholar]
- Santoro SW, Schultz PG. Directed evolution of the site specificity of Cre recombinase. Proc Natl Acad Sci USA. 2002;99(7):4185–4190. doi: 10.1073/pnas.022039799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar I, Hauber I, Hauber J, Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007;316(5833):1912–1915. doi: 10.1126/science.1141453. [DOI] [PubMed] [Google Scholar]
- Sauer B, Henderson N. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 1990;2(5):441–449. [PubMed] [Google Scholar]
- Sauer B, McDermott J. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucl Acids Res. 2004;32(20):6086–6095. doi: 10.1093/nar/gkh941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EE, Taylor DS, Prigge JR, Barnett S, Capecchi MR. Illegitimate Cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc Natl Acad Sci USA. 2000;97(25):13702–13707. doi: 10.1073/pnas.240471297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclimenti CR, Thyagarajan B, Calos MP. Directed evolution of a recombinase for improved genomic integration at a native human sequence. Nucl Acids Res. 2001;29(24):5044–5051. doi: 10.1093/nar/29.24.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, Dreier B, Beerli RR, Barbas CF., III Toward controlling gene expression at will: Selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci USA. 1999;96(6):2758–2763. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Lieberman Aiden E. The expanding scope of DNA sequencing. Nat Biotechnol. 2012;30(11):1084–1094. doi: 10.1038/nbt.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz JL, Voziyanova E, Konieczka JH, Voziyanov Y. A genome-wide analysis of FRT-like sequences in the human genome. PLoS ONE. 2011;6(3):e18077. doi: 10.1371/journal.pone.0018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuti P, Yazbek J, Lu TK. Synthetic circuits integrating logic and memory in living cells. Nat Biotechnol. 2013;31(5):448–452. doi: 10.1038/nbt.2510. [DOI] [PubMed] [Google Scholar]
- Smith MC, Thorpe HM. Diversity in the serine recombinases. Mol Microbiol. 2002;44(2):299–307. doi: 10.1046/j.1365-2958.2002.02891.x. [DOI] [PubMed] [Google Scholar]
- Smith AJ, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, Rabbitts P. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat Genet. 1995;9(4):376–385. doi: 10.1038/ng0495-376. [DOI] [PubMed] [Google Scholar]
- Stemmer WP. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc Natl Acad Sci USA. 1994a;91(22):10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994b;370(6488):389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- Surendranath V, Chusainow J, Hauber J, Buchholz F, Habermann BH. SeLOX—A locus of recombination site search tool for the detection and directed evolution of site-specific recombination systems. Nucl Acids Res. 2010;38(Web Server issue):W293–W298. doi: 10.1093/nar/gkq523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E, Nakayama M. VCre/VloxP and SCre/SloxP: New site-specific recombination systems for genome engineering. Nucl Acids Res. 2011;39(8):e49. doi: 10.1093/nar/gkq1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat Biotechnol. 1998;16(7):652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- Tay Y, Ho C, Droge P, Ghadessy FJ. Selection of bacteriophage lambda integrases with altered recombination specificity by in vitro compart-mentalization. Nucl Acids Res. 2010;38(4):e25. doi: 10.1093/nar/gkp1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe HM, Smith MC. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci USA. 1998;95(10):5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B, Guimaraes MJ, Groth AC, Calos MP. Mammalian genomes contain active recombinase recognition sites. Gene. 2000;244(1–2):47–54. doi: 10.1016/s0378-1119(00)00008-1. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP. Site-specific genomic integration in mammalian cells mediated by phage phiC31 integrase. Mol Cell Biol. 2001;21(12):3926–3934. doi: 10.1128/MCB.21.12.3926-3934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B, Liu Y, Shin S, Lakshmipathy U, Scheyhing K, Xue H, Ellerstrom C, Strehl R, Hyllner J, Rao MS, et al. Creation of engineered human embryonic stem cell lines using phiC31 integrase. Stem Cells. 2008;26(1):119–126. doi: 10.1634/stemcells.2007-0283. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87(7):1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Umana P, Gerdes CA, Stone D, Davis JR, Ward D, Castro MG, Lowenstein PR. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat Biotechnol. 2001;19(6):582–585. doi: 10.1038/89349. [DOI] [PubMed] [Google Scholar]
- Van Duyne GD. A structural view of Cre-loxP site-specific recombination. Annu Rev Biophys Biomol Struct. 2001;30:87–104. doi: 10.1146/annurev.biophys.30.1.87. [DOI] [PubMed] [Google Scholar]
- Voziyanov Y, Stewart AF, Jayaram M. A dual reporter screening system identifies the amino acid at position 82 in Flp site-specific recombinase as a determinant for target specificity. Nucl Acids Res. 2002;30(7):1656–1663. doi: 10.1093/nar/30.7.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voziyanov Y, Konieczka JH, Stewart AF, Jayaram M. Stepwise manipulation of DNA specificity in Flp recombinase: Progressively adapting Flp to individual and combinatorial mutations in its target site. J Mol Biol. 2003;326(1):65–76. doi: 10.1016/s0022-2836(02)01364-5. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. Cre-mediated gene deletion in the mammary gland. Nucl Acids Res. 1997;25(21):4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krushel LA, Edelman GM. Targeted DNA recombination in vivo using an adenovirus carrying the cre recombinase gene. Proc Natl Acad Sci USA. 1996;93(9):3932–3936. doi: 10.1073/pnas.93.9.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteson KL, Rice PA. Binding and catalytic contributions to site recognition by flp recombinase. J Biol Chem. 2008;283(17):11414–11423. doi: 10.1074/jbc.M800106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Gumport RI, Gardner JF. Genetic analysis of second-site revertants of bacteriophage lambda integrase mutants. J Bacteriol. 1997;179(12):4030–4038. doi: 10.1128/jb.179.12.4030-4038.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman C, Kanaar R. DNA double-strand break repair: All's well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- Xiong L, Chen XL, Silver HR, Ahmed NT, Johnson ES. Deficient SUMO attachment to Flp recombinase leads to homologous recombination-dependent hyperamplification of the yeast 2 microm circle plasmid. Mol Biol Cell. 2009;20(4):1241–1251. doi: 10.1091/mbc.E08-06-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagil E, Dolev S, Oberto J, Kislev N, Ramaiah N, Weisberg RA. Determinants of site-specific recombination in the lambdoid coliphage HK022. An evolutionary change in specificity. J Mol Biol. 1989;207(4):695–717. doi: 10.1016/0022-2836(89)90238-6. [DOI] [PubMed] [Google Scholar]
- Yagil E, Dorgai L, Weisberg RA. Identifying determinants of recombination specificity: Construction and characterization of chimeric bacteriophage integrases. J Mol Biol. 1995;252(2):163–177. doi: 10.1006/jmbi.1995.0485. [DOI] [PubMed] [Google Scholar]
- Yang W, Steitz TA. Crystal structure of the site-specific recombinase gamma delta resolvase complexed with a 34 bp cleavage site. Cell. 1995;82(2):193–207. doi: 10.1016/0092-8674(95)90307-0. [DOI] [PubMed] [Google Scholar]
- Ye L, Chang JC, Lin C, Qi Z, Yu J, Kan YW. Generation of induced pluripotent stem cells using site-specific integration with phage integrase. Proc Natl Acad Sci USA. 2010;107(45):19467–19472. doi: 10.1073/pnas.1012677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Kurek I, English J, Keenan R. Laboratory-directed protein evolution. Microbiol Mol Biol Rev. 2005;69(3):373–392. doi: 10.1128/MMBR.69.3.373-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan LZ, Rouviere PE, Larossa RA, Suh W. Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng. 2006;8(1):79–90. doi: 10.1016/j.ymben.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL, Gregory PD, Holmes MC, Torbett BE. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol Ther. 2012;20(4):849–859. doi: 10.1038/mt.2011.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen CM, Liu DR. Dissecting protein structure and function using directed evolution. Nat Methods. 2007;4(12):995–997. doi: 10.1038/nmeth1207-995. [DOI] [PMC free article] [PubMed] [Google Scholar]