

Abstract
This chapter presents an overview of the current status of studies on the structural and molecular biology of the retinoid X receptor subtypes α, β, and γ (RXRs, NR2B1–3), their nuclear and cytoplasmic functions, post-transcriptional processing, and recently reported ligands. Points of interest are the different changes in the ligand-binding pocket induced by variously shaped agonists, the communication of the ligand–bound pocket with the coactivator binding surface and the heterodimerization interface, and recently identified ligands that are natural products, those that function as environmental toxins or drugs that had been originally designed to interact with other targets, as well as those that were deliberately designed as RXR-selective transcriptional agonists, synergists, or antagonists. Of these synthetic ligands, the general trend in design appears to be away from fully aromatic rigid structures to those containing partial elements of the flexible tetraene side chain of 9-cis-retinoic acid.
Keywords: Coactivator, corepressor, ligand, ligand-binding domain, nuclear receptor, retinoid X receptor, RXR
1. Introduction
The retinoid X receptor (RXR1) is an intriguing and essential member of the steroid/thyroid hormone superfamily of nuclear receptors (NRs) that predominately function as transcription factors with roles in development, cell differentiation, metabolism, and cell death. This review outlines the accomplishments made in understanding RXR biology from 2004 and also presents an overview of many of the RXR ligands (rexinoids) and their activities reported since 2000.
Briefly, the RXR subtypes or isotypes α–γ (NR2B1–3) (Table 1) are members of the orphan NR family of this NR superfamily because at their discovery natural ligands were unknown. The natural ligand of RXR remains controversial. Although 9-cis-retinoic acid (9-cis-RA in Fig. 1A) was first proposed to have this status, many groups have since been unable to detect endogenous 9-cis-RA in cells either in culture or in vivo unless its isomer, all-transretinoic acid (ATRA), had been present first or added [1,2]. Compounding the uncertainty of its status as the natural ligand of RXR is the instability of the RA tetraene side chain that either in the presence of light or a mercaptan, such as reduced glutathione, can equilibrate to a mixture of double-bond isomers generally containing 80% ATRA, 8–10% 9-cis-RA, and other isomers. Polyunsaturated fatty acids (PUFAs) such as docosahexaenoic acid (DHA) and a saturated metabolite of chlorophyll, phytanic acid (Fig. 1A), were also identified as RXR ligands.
Table 1.
RXR isotypes α, β, and γ (NR2B1–3), RXR isotype isoforms in human (h) or mouse (m), and RXR and ultraspiracle (NR2B4) homologues in other species
| RXR isoform/isotype | Expression site | Sequence | Homology | Role | Ref |
|---|---|---|---|---|---|
| h α | RARα DBD 135–200 61% | [3,18] | |||
| RARα LBD 225–462 27% | |||||
|
| |||||
| m α1 | Predominates in liver | 1–467 | Knock-out is embryolethal | ||
|
| |||||
| m α2 | Predominates in liver and adult testis | 1–439 (28 AA A/B N-terminal deletion) | Major roles in epithelia and liver | ||
| Roles in adipocyte lipogenesis and hypertrophy | |||||
|
| |||||
| m α3 | Predominates in liver | A/B N-terminal deletion (97 AA) | |||
|
| |||||
| m α4 | 1–165 (loss of part of C, and D–E domains | ||||
|
| |||||
| m β | h RXRα DBD 82–147 92% of 61% | ||||
| h RXRα LBD 171–410 88% of 27% | |||||
|
| |||||
| m β1/h β2 | Predominates in CNS | Behavioral and metabolic defects | |||
|
| |||||
| m β2 | 1–520 | ||||
|
| |||||
| m β2E/h | 1–524 with 9 AA insert in D domain (negative function) | ||||
| β3 | |||||
|
| |||||
| β3 | 1–410 (A/B N-deletion) | ||||
|
| |||||
| m γ | h RXRα DBD 139–204 95% | ||||
| h RXRα LBD 229–463 86% | |||||
|
| |||||
| γ1 | Skeletal muscle | 1–463 | Behavioral and metabolic defects | ||
| Brain | |||||
| Pituitary gland | |||||
|
| |||||
| γ2 | Cardiac and skeletal muscle | 1–340 (A/B N-terminal deletion) | Compensates for RXRα in adipocytes | ||
|
| |||||
| Name | Source | Role | Structural differences | Ligands | Ref |
|
| |||||
| dm USP | Drosophila melagaster (Dm) fruit fly | Heterodimeric partner of EcR and DHR38 (NRA4), an ortholog of human NGFI-B | h RXRα DBD 104–169 86% | Phospholipid ligand stabilizes H12 in antagonist conformation | [172,173] |
| H12 in antagonist conformation | h RXRα LBD 233–514 44% | Methoprene (Kd 5.3 nM) | [174,175] | ||
| Methyl farnesoate (Kd 44 nM) | |||||
| Juvenile hormone (JH) (epoxy methyl farnesoate) (Kd 7.6 μM) | |||||
| 9-cis-RA (no binding) | |||||
| Not activated by rexinoids, JH, JH analogs, methyl farnesoate | |||||
|
| |||||
| tc USP | Tribolium castaneum (beetle) | Apo-USP H12 in antagonist conformation | JH, JH analogs, methyl farnesoate | [174] | |
| Not activated by rexinoids | |||||
|
| |||||
| hv USP | Heliothis virescens (moth) | Phopsholipid ligand stabilizes H12 in antagonist conformation | [175] | ||
|
| |||||
| lm RXR | Locusta migratoria | Closer to h RXRγ than USPs | No binding to JH III | [176,177] | |
| Lm RXR-L (DE) with 22 residue insert in H1–H3 loop | IC50s: 9-cis-RA, 108 nM; ATRA, 105 nM; methoprene acid, 8.7 μM; DHA, 4.0 μM | ||||
| Lm RXR-S lacking insert | IC50s: 9-cis-RA, 61 nM; ATRA, 75 nM; methoprene acid, 26 μM; DHA, 2.9 μM | ||||
| h RXRα(DE) | IC50: 9-cis-RA, 74 nM | ||||
|
| |||||
| cp RXR | Celuca pugilator (crab) | h RXRα DBD 85% | [178] | ||
| h RXRα LBD 67% | |||||
|
| |||||
| up RXR | Uca pugilator (fiddler crab) | Expressed in regenerating limb buds and in molt | DBD closer to that of insect USP | [179] | |
| Only isoform having a 33-AA LBD H1–H3 loop insertion | LBD closer to vertebrate RXR | ||||
| Able to heterodimerize with EcR–ecdysone and activate transcription on IRper-1 | |||||
|
| |||||
| ls RXR | Lymnaea stagnalis (mollusk) | Found in developing embryo and adult central neurons | Vertebrate RXRα 80% | 9-cis-RA | [180] |
| RXR agonist PA024 | |||||
Figure 1.
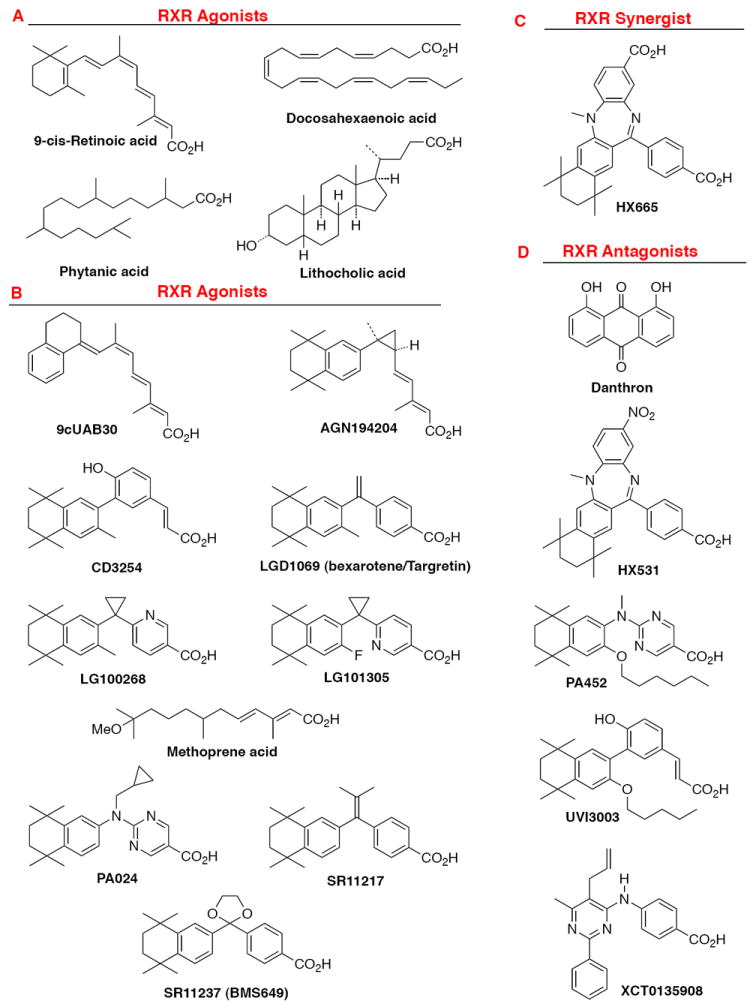
Examples of chemical structures of RXR ligands. A. Natural products that act as RXR ligands: 9-cis-retinoic acid, (E)-5,8,11,14,17,20-docosahexaenoic acid, lithocholic acid, and phytanic acid. B. Synthetic RXR transcriptional agonists; 9cUAB30, AGN194204, CD3254, LG100268, LG101305, methoprene acid, PA024, SR11217, and SR11237 (BMS649). C. Synthetic RXR synergist: HX600. D. Synthetic RXR transcriptional antagonists: HX531, PA452, UVI3003, and XCT0135908.
1.1. Nuclear function of RXR
In the nucleus, RXR functions as a transcription factor by binding to specific six-base-pair sequences of DNA in the promoter regions of genes. In doing so, RXR functions as a dimer with either itself (homodimer) or another NR (heterodimer). Generally, binding by the ligand of the NR partner defines the promoter site (response element or RE) composed of two six base-pair sequences (half-sites) separated by a discrete number of bases to which the RXR–NR heterodimer binds [5′-PuG(G/T)TCA-(X)n-PuG(G/T)TCA-3′] [3]. As indicated by some of the REs listed in Table 2, these sequences can be repeated directly (DR), inverted (IR), everted (ER), palindromic (pal), or disordered depending on the dimer bound. Thus, RXR heterodimers with peroxisome proliferator-activated receptor (PPAR), retinoic acid receptor (RAR), vitamin D receptor (VDR), and thyroid hormone receptor (TR) consist of two directly repeated (DR) half-sites separated by one, two or five, three, and four bases (n), respectively, typically with RXR in the 5′-position. In the case of the RXR heterodimer with RAR bound to a DR-1 response element, RXR can occupy either the 5′ or 3′-position. The RXR homodimer preferentially recognizes two 5′-(A/G)GGTCA-3′ half-sites separated by one base (DR-1).
Table 2.
RXR heterodimeric NR partners with roles in metabolic signaling pathways
| Receptor | RE | Roles | Ligands | Ref. |
|---|---|---|---|---|
| CARa (NR1I3) | DR-3 | High basal (constitutive activity) | TCPOBOP (agonist for m CAR) | [84,182] |
| ER-6 | Response to endobiotic or xenobiotic stress with induction of P450 enzymes | Acetoaminophen (agonist) | ||
| DR-4 | Thyroid hormone and metabolism | Androstanol (antagonist for m CAR) | ||
| Indirect activation through induced nuclear translocation | Chlorpromazine (agonist) | |||
| Phenobarbital (agonist) | ||||
|
| ||||
| FXR (NR1H4) | IR-1 | Rexinoid agonists binding to RXR–FXR act as antagonists to reduce DNA binding and CoA recruitment | Farnesol and its metabolites | [183] |
| Also binds as monomer to AGGTCA half-site | ||||
|
| ||||
| LXRα (NR1H3) | DR-4 | Predominant in adipose tissue, adrenal gland, intestine, kidney, liver (highest), macrophage, and spleen. | Oxysterols such as 22R-OH-cholesterol | [37,58,87,88,136,184] |
| Cholesterol homeostasis by cholesterol transport and catabolism and triacyl glycerol synthesis gene regulation (induces ATP-binding cassette transporters (ABCA1) for efflux of cholesterol and phospholipids from intracellular receptor stores to extracellular acceptors, sterol responsive element binding protein 1c (SREBP-1c), a TF controlling FA synthesis, apolipoproteins ApoD and ApoE, lipoprotein-modifying enzymes such as cholesterol ester and phospholipid transfer proteins (CETP and PLTP) | TO-901317 GW3965 |
|||
| Tangier disease (low HDL-cholesterol and ApoA1 leading to cardiovascular risk) | ||||
| Potential role in Alzheimer’s disease and atherosclerosis | ||||
|
| ||||
| LXRβ (NR1H2) | DR-4 | Ubiquitous | See LXRα | [136,185] |
| For roles see LXRα | ||||
|
| ||||
| NGFI-B/nur77/TR3 (NR4A1) | AAAGGTCA half-site as monomer (NBRE) | Immediate-early gene | Cytosporone B | [186,187] |
| NurRE as a homodimer | T-cell apoptosis | |||
| DR-5 with RXR | Regulation of steroidogenic enzymes in adrenal and gonads/ovary (aromatase) | |||
|
| ||||
| Nurr1 (NR4A2) | AAAGGTCA half-site as monomer (NBRE) | Induces fatty acid-binding protein (FABP) 5 via its promoter NBRE independently of RXR | [188] | |
| NurRE as a homodimer | A box in DBD interacts with A/T 5′ to extended half-site. | |||
| DR-5 with RXR | ||||
|
| ||||
| PPARα (NR1C1) | DR-1 | Expressed mainly in liver, heart, kidney | Fibrates (lowering of blood lipids) | [10,189,190] |
| FA catabolism by peroxisomal β-oxidation and mitochrondrial β- and O-oxidation in liver | Leukotriene B4 | |||
| Fenofibrate activated platelet PPARα and increased cAMP to decrease platelet activation by ADP | Hydroxy-eicosatrienoic acids | |||
|
| ||||
| PPARβ/δ (NR1C2) | DR-1 | Ubiquitous expression | Not as yet used as drug target | [10,189,191] |
| Suggested role similar to PPARα in extrahepatic tissues | FAs | |||
| Placental implantation | Pg J2 | |||
| Wound healing | L-165,041 | |||
| Carcinogenesis | GW501516 (phase II for dyslipidemia) | |||
| Ligands inhibit platelet aggregation | GW0742 | |||
|
| ||||
| PPARγ (1 and 2) (NR1C3) | DR-1 | Expressed mainly in adipose tissue and less in liver, in which it is upregulated by PPARγ agonists | Thiazolidinediones enhance insulin sensitivity in type 2 diabetes; reduce cancer cell proliferation | [11,105,189-191] |
| FA homeostasis | PUFAs, oxidized LDL, 5,8,11,14-eicosatetraynoic acid | |||
| Glucose homeostasis | Pg A1, A2, D2 | |||
| Adipocyte differentiation | 15-deoxy-Δ12,14-Pg J2 | |||
| Placental development | ||||
| Anti-proliferation | ||||
| Anti-inflammation | ||||
| Inducing apoptosis | ||||
| Induction of glutathione S-transferase | ||||
| Agonists inhibit platelet aggegation | ||||
|
| ||||
| PXR / SXR (NR1C2) | DR-3 | Binds DR-3–5 and IR-6, prefers DR-4 | Cisplatin | [115,182,192] |
| ER-6 | Xenobiotic metabolism | Paclitaxel | ||
| DR-4 | Endobiotic homeostasis | Phenobarbitol | ||
| DR-5 | Induces P450 CYP3A4 | Rifampicin | ||
| ER-8 | Induces MDR1 | SR12813 (cholesterol lowering agent) | ||
| IR-6 | Sulforaphane | |||
| Tamoxifen | ||||
| Troglitazone | ||||
| Azole antifungal drugs (bind AF-2 domain as antagonists) | ||||
Abbreviations: CAR, constitutive androstane receptor; DR, direct repeat; ER, everted repeat; FA, fatty acid; FXR, farnesoid X receptor; IR, inverted repeat; LDL, low-density lipoprotein; LXR, liver X receptor; NGFI-B, nerve growth factor I-B; Pg, prostaglandin; PPAR, peroxisome proliferator-activated receptor; PU, polyunsaturated; PXR, pregnane X receptor; RE, response element; RXR, retinoid X receptor; SXR, steroid and xenobiotic receptor.
1.1.1. RXR dimeric status in cells
The status of RXR in cells remains controversial. In addition to forming heterodimers and homodimers in vitro, RXR homotetramers have also been detected. The cellular status of retinoic acid receptor (RAR) ligand-binding domain (LBD)–RXR LBD heterodimers and RXR LBD–RXR LBD homodimers was determined using fluorescence correlation spectroscopy (fluorescence fluctuation brightness analysis) [4]. CV-1 cells were transfected with constructs for yellow fluorescent protein (YFP)-RXR LBD and cyan fluorescent protein (CFP)-RAR LBD. Brightness intensities were then measured. Both YFP and CFP were identically bright after excitation at 905 nm, whereas only YFP fluoresced after excitation at 965 nm. These studies were used to demonstrate that in the transfected cells the labeled RXR existed either as a heterodimer with labeled RAR or as a monomer and not as a homodimer.
1.1.2. DNA-binding status of RXR as a heterodimer
In Table 2 are listed those nuclear receptors that heterodimerize with the RXRs and have roles in regulating genes controlling metabolic signaling pathways, their typical REs and ligands. Among these are the peroxisome proliferator-activated receptor (PPAR) isotypes α, β/δ and γ, which also has roles in cell proliferation and differentiation.
RXR–PPAR
The intracellular behavior of the RXRα–PPAR heterodimer in the presence or absence of a ligand was investigated using the combination of fluoresence recovery after photobleaching (FRAP), fluoresence correlation spectroscopy (FCS), and fluorescence resonance energy transfer (FRET) on transfected enhanced yellow fluorescent protein (eYFP)-PPARα–γ and eYFP-RXRα constructs [5]. Unlike the nuclear patterning exhibited by eYFP-ERα, the fluorescent flakes and foci produced after preliminary transfections of the eYFP-PPARα constructs were considered to be artifacts that were caused by protein over-expression. At lower expression levels, the eYFP-PPARα expression pattern became diffuse in the nuclei of living COS-7 cells. According to FRAP, the eYFP-PPAR proteins in the presence or absence of their ligands were highly mobile in cells that had the diffuse distribution patterns of fluorescence and were unaffected by co-expression of RXRα alone or with added 9-cis-RA. The diffusion pattern for eYFP-RXRα was similar to that of the apo-PPARs. The diffusion constants for the eYFP-PPARs were 4.8–5.5 μm2/sec and decreased to 2.3–3.5 μm2/sec after their ligand bound to suggest that PPAR binding by cofactors had increased to give complexes on the order of 1–2 MKda. The diffusion constant for the eYFP-RXRα was 4.6 μm2/sec. The authors estimated that about 3,600–120,000 fluorescent protein molecules were expressed per cell, whereas the actual number of PPAR target genes was <1,000. Therefore, they speculated that many of the reported interactions of PPARs–RXRα on DNA would have been the consequence of transient or nonspecific interactions at sites resembling authentic PPREs. FRET indicated that PPAR–RXR dimerization occurred prior to ligand binding or DNA binding, however heterodimer binding to DNA was only observed to be stable in vivo after ligand had bound.
RXR–RAR
The RXRs also have major roles in regulating genes controlling cell proliferation and differentiation in the context of their heterodimers with the RARs, thyroid hormone receptors (TRs), and vitamin D receptor (VDR). Both non-denaturing nanoelectrospray ionization (nano-ESI) and high-mass matrix-assisted laser desorption ionization (MALDI) mass spectrometric methods were used to demonstrate that the RXR–RAR heterodimer bound to a DR-5 RARE in the presence of 9-cis-RA and that RXR was upstream (5′), in contrast to the DR-1 RARE in which RAR was upstream [6]. Crosslinking was used to stablilize the complex for MALDI, but did not necessarily stabilize the bound ligand. RAR did not homodimerize in solution but was able to form such a homodimeric complex on the DR-5 in the presence of 9-cis-RA and excess DR-5 to indicate that RXR was not required for RAR to associate with its half-site. Limitations of these methods were also described by the authors. However, the subtypes of the mutant murine retinoid receptors RXRΔA/B and RARΔA/B used in the study were not identified.
1.2. Cytoplasmic function
Recently, RXR has been shown to have cytoplasmic functions that are distinct from its activity as a transcription factor.
1.2.1. Induction of TR3 nuclear export
RXR was reported to shuttle the orphan NR human TR3/mouse Nur77/rat NGFI-B from the nucleus to the cytoplasm, allowing TR3 to interact with mitochondrial Bcl-2 to reverse its anti-apoptotic function to one promoting apoptosis. This activity was first reported by Zhang and colleagues [7,8] and confirmed by Wu and colleagues [9]. Stress induced by treatment of cancer cell lines with a cancer therapeutic agent or an adamantyl-substituted retinoid-related molecule induced TR3 relocalization in several cancer cell lines.
Using MGC80-3 human gastric cancer cells, Wu and colleagues investigated the role of RXRα in inducing TR3 nuclear export [9]. Apo-RXRα was unable to induce export, whereas 1.0 μM 9-cis-RA-treated RXRα effectively did so within 30 min. Deletion analysis indicated that the RXRα DBD contained a nuclear export signal (NES), as did TR3. However, the CRM1-dependent TR3 NES did not appear to be involved in export in this cell line. TR3 lacking its DNA-binding domain (DBD), hinge, and 90 N-terminal residues of the LBD still colocalized in mitochondria, as was shown by staining with the mitochondrial marker Hsp60, and induced apoptosis irrespective of 9-cis-RA treatment, whereas full-length TR3 resided in the nucleus and was unable to migrate out of the nucleus or to induce apoptosis in the absence of RXRα and 9-cis-RA. A mutant lacking the 106 N-terminal residues of the TR3 A/B domain underwent nuclear export to mitochondria in the presence of 9-cis-RA, whereas the C-terminal deletion of 25 residues from the TR3 LBD prevented TR3 interaction with RXRα and nuclear export induced by RXRα and 9-cis-RA.
1.2.2. Platelets
Despite their lack of a nucleus, platelets expressed several NRs, including androgen receptor (AR), estrogen receptors (ERs), glucocorticoid receptor (GR), mineralcorticoid receptor (MR), PPARs, PXR, and RXRs α and β. Platelets are derived from the cytoplasm of megakaryocytes, which have nuclei that express mRNAs for these NRs and have enzymes for their translation [10]. In platelets these NRs were considered to function through nongenomic pathways. For example, platelet aggregation and thromboxane (TX) A2 release were inhibited by the RXR agonists 9-cis-RA and methoprene acid (Fig. 1B). Activated platelets released microparticles that were found to contain RXRα to suggest that RXRα had an extracellular role in modulating the results of platelet activation [11]. Treatment with RXR ligands 9-cis-RA and methoprene acid, but not ATRA, inhibited platelet aggregation induced by TXA2 mimetic U46619 and TXA2 release stimulated by adenosine diphosphate [12]. Inhibition occurred by ligand-bound RXR interacting with G-protein Gq to prevent the activation of the GTPase Rac.
2. RXR general structure
2.1. RXR gene promoter
The human RXRα gene was found to have 10 exons but not a typical TATA transcription initiation site [13,14]. Its promoter sequence has been considered to resemble that of a housekeeping gene because of the high G + C content in its 5′-untranslated region. In keeping with the high G + C content, 17 and 12 putative Sp1 sites were identified upstream and downstream, respectively, of the start site. Putative AP-1, AP-2, AP-4, GATA-1/2, N-Myc, v-myb, SRY, AML-1a, and imperfect DR-0, 3, 4, and 5 sites were also identified. RXR expression in the mouse was induced during the acute-phase response in the heart by cytokines, lipopolysaccharide (LPS), or sepsis and was not cyclical in metabolic tissues except in the liver [15]. RXRα expression was was also induced by ATRA [13]. The human RXRβ gene also has ten exons, is G + C rich, and lacks a TATA motif [16]. In the mouse pituitary, 9-cis-RA downregulated RXRγ1 expression by activating a negative nonconsensus DR-1 site in its promoter [17]. The human RXRα–γ genes are located on chromosomes 9 (band q34.3), 6 (band 21.3), and 1 (band q22–q23), respectively [18].
2.2. RXR isotypes/isoforms
Three isotypes or isoforms (α, β, and γ) of RXR are expressed (Table 1). Their expression levels vary with cell type and differentiation status. Each of these isoforms has several subtypes as the result of alternative splicing. RXRα was found to predominate in the epidermis, intestine, kidney, and liver; RXRβ expression was ubiquitous; and RXRγ was expressed mostly in brain and muscle and was weak in adipose tissue [19]. RXRα or RXRβ deficiency in mice was embryolethal, whereas RXRγ knock-down mice survived and appeared normal.
Nohara and colleagues reviewed the effects of RXR subtype polymorphisms [19]. Those of RXRα were not linked with any metabolic dysfunction, whereas the RXRβ c.51C>T polymorphism was linked to higher body mass, gallstone risk, and bile duct cancer risk. The association of an RXRγ polymorphism with hyperlipidemia was noted. The most common form of hereditary hyperlipidemia is the familial combined type (FCHL), which has been associated with increased very-low density lipoprotein (VLDL) that could be accompanied by increased low-density lipoprotein (LDL). The RXRγ gene is located on chromosome 1q21–q23, which has been termed the “FCHL” locus and linked with higher LDL–cholesterol and triglyceride levels in several families. The RXRγ p.Gly14Ser variant was observed in hyperlipidemic patients, was more common in those with FCHL, and was higher in those with coronary stenosis. Interestingly, the Ser14 variant suppressed lipoprotein lipase (LPL) promoter activity by 60%, whereas the Gly14 variant suppressed LPL by 40%. LPL plays a role in the hydrolysis of VLDL. RXRγ variants have also been linked to free FA and triglyceride levels in familial type 2 diabetes.
2.3. RXR structural and functional domains
Like other NRs, the RXR proteins have six major functional/structural domains (Fig. 2A). Beginning at the NR N-terminus, these domains are: A/B, which contains a ligand-independent activation function (AF)-1 to which coactivator proteins (CoAs) bind; C or DNA-binding domain (DBD), which mediates NR binding to specific sequences of DNA in the promoter regions of genes (half-sites of REs); D or hinge, which connects the DNA and ligand-binding domains; E or ligand-binding domain (LBD) (Fig. 2B–2D) that contains a ligand-dependent AF-2 sequence to which CoAs or corepressors (CoRs) bind to regulate transcriptional activation by the NR; and the F domain, the function of which in RXR remains to be established [18].
Figure 2.
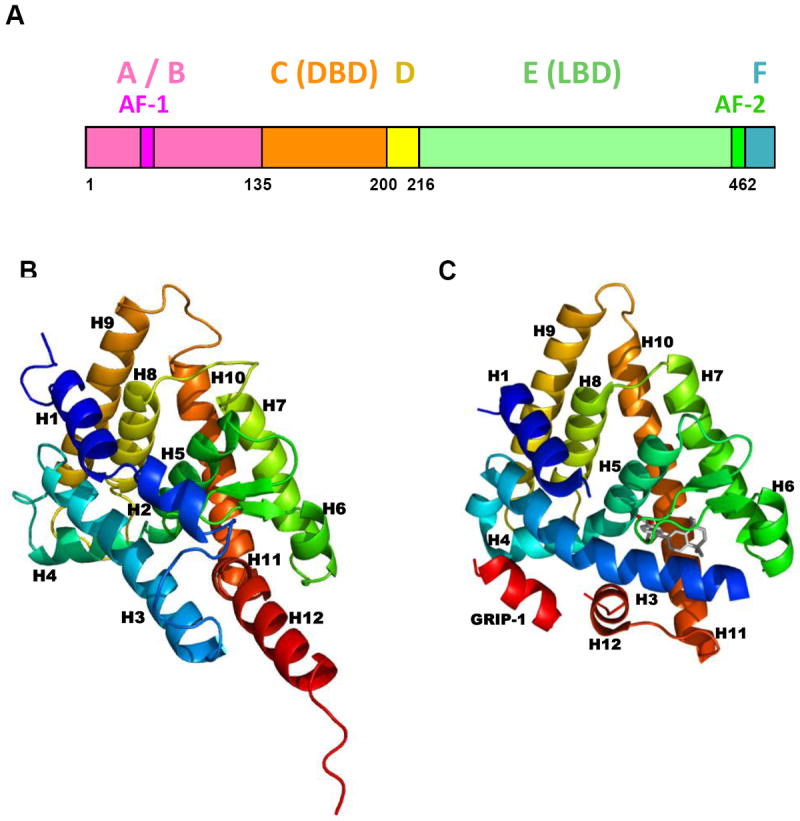
RXRα protein domains and ligand-binding domain structure. A. Map of human RXRα functional domains. Adapted from Ref. [18]. B. Human RXRα ligand-binding domain (LBD) without a bound ligand as found in Protein Data Bank (PDB) crystal structure of the apo-RXRα LBD homodimer 1LBD. C. Human RXRα LBD complexed with transcriptional agonist SR11237 (BMS649, with carbon atoms in gray) as found in PDB crystal structure 1MVC. Protein backbones are shown in ribbon format.
2.3.1. RXR DNA-binding domain and its interaction with DNA
Like other NRs, the RXRα DBD (130–209) has two zinc-finger domains (residues 135–155 and 171–190), each of which complexes a Zn(II) ion through four cysteines [20] (Fig. 3). The Rastinejad group showed that, in the Protein Data Bank (PDB) crystal structure (1BY4 in Table 3) of two RXRα DBD homodimers bound to their DR-1 REs (Fig. 3A) in a 15-base-pair DNA oligomer, the two DBD zinc fingers were separated by a 14-residue sequence that contained the C-terminus of the recognition helix (152–164), which specified the DNA-binding half-site and also included the 152–155 sequence of the first Zn finger (Fig. 3B). In the DNA oligomer, the two DR-1 RE sequences were separated by two base-pairs, which provided an internal DR-2 RE. The two bound DBD homodimers were linerally wrapped around the DNA helix. The DBD recognition helix residues Lys160 and Arg164 interacted with the DNA major groove nucleotide base and phosphate residues. Residues 187–191 of the second zinc finger domain plus the C-terminal residues 192–198 made up helix-II, which was then followed by the T-box in the DBD. The T-box interacted with a zinc finger of the upstream (5′) RXR partner to provide a DBD dimerization interface that permitted cooperative dimerization on DNA. The interface H-bonds that stabilized each DBD homodimer on its DR-1 were made between Arg182 and Arg186 from the Zn finger II loop of the 5′-DBD and Gln208 from the T-box of its of its downstream (3′) partner. In addition, Arg172 and Arg186 from the 3′-RXRα DBD Zn finger II loop of the 5′-homodimer and Glu207 from the T-box of 5′-DBD of the 3′-homodimer formed H-bonds that stabilized the DBD homodimer interface bound to the DR-2. The respective Kd values for the RXRα DBD homodimer binding to the DR-1 and DR-2 REs were approximately 0.35 μM and 0.48 μM. In contrast, binding of an RXRα monomer to its half-site sequence could not be detected at a RXRα concentration as high as 2 μM. The Table in Fig. 3A lists the response elements made up of direct repeats to which RXR binds as a heterodimer and the partners involved. The cartoon in Fig. 3B shows the general structure of the RXRα DBD and its various domains.
Figure 3.
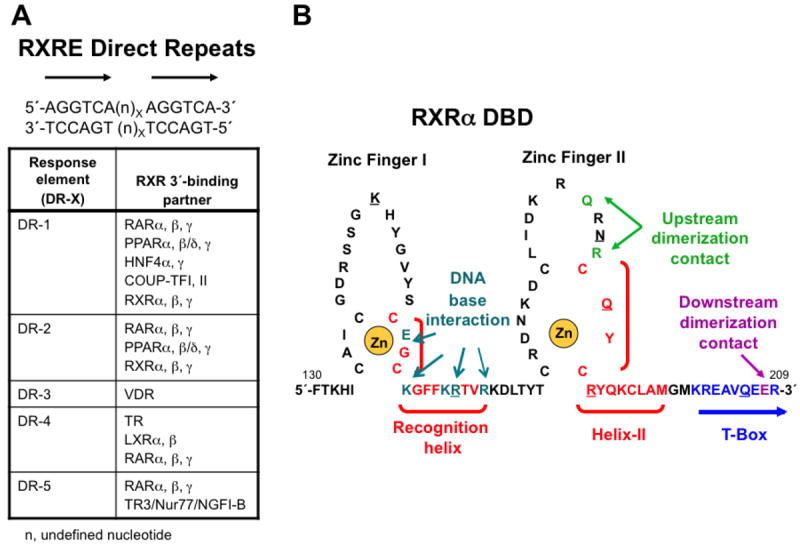
Retinoid X Receptor (RXR) DNA-binding domain (DBD) interaction with DNA. A. Direct repeat (DR) sequence half-sites (5′-AGGTCA-3′) separated by X = 1–5 base-pairs (n) to which RXRα, β, and γ bind as an RXR homodimer or as an RXR heterodimer with retinoic acid receptor (RAR) subtypes 〈, β, and γ, peroxisome proliferator-activated receptors (PPARs) 〈, β/δ, and γ, chick ovalbumin uncoupled protein-transcription factor (COUP-TFs) I and II, vitamin D receptor (VDR), thyroid hormone receptor (TR), liver X receptors (LXRs) 〈 and β, nerve growth factor I beta (NGFI-B/TR3/Nur77) in which RXR is the upstream (5′) binding partner. n, undefined nucleotide base-pair that separates the direct repeats of 5′-AGGTCA-3′. Degenerate sequences also exist. B. Structure of the RXR〈 DBD showing the two zinc fingers (I and II), each of which is stabilized by complexation of a zinc(II) ion (shown in gold) with four of its cysteine sulfhydryl groups (zinc finger I: C135, C138, C152, and C155; and II: C171, C177, C187, and C190). Recognition helix (C152–R164) and helix II (C187–M198) α-helical sequences are shown in red and bracketed. The T-box sequence (K201–R209) is shown in blue. According to NMR studies, when the RXRα DBD monomer was free in solution, its T-box was helical with its E208 residue (magenta) interacting with K160 and R164 (cyan) of the recognition helix. However, according to the crystallographic structure of two RXR〈 DBD homodimers (1BY4), each of which was bound to a direct repeat sequence that was separated by one base-pair—DR-1 (n = A, X = 1) RXRE—that were separated by two residues giving rise to an internal DR-2 ((n)X = GT, X = 2) RXRE, the T-box was a random coil that allowed the R164 side chain to interact with DNA and the K160 side chain to interact with DNA through a water molecule. As a result, the residues K156, E153, and R161 of the most upstream DBD interacted with the most upstream (5′) half-site of the first DR-1 nucleotides 5′-G2, 3′-C3, and 3′-G5, respectively, and residues K22, E19, K26, and R27 of its downstream partner (second DBD) interacted with the downstream DR-1 half-site nucleotides of the first DR-1, namely 5′-G9, 3′-C11, 3′-G12 (through water), and 3′-G12 (directly and through a water), respectively. Interactions of the third RXR〈 DBD with the second DR-1 upstream half-site base-pairs were as follows: K160 with the 3′-C3 and 3′-A4 (both through a water), E153 with 3′-C3, and R161 with 3′-G5 (directly and through a water), and those of its downstream partner (fourth DBD) with the downstream half-site of the second DR-1 were: K156 with 5′- G10 (through a water), K160 with 5′-G10 (directly and through a water), G153 with 3′-C10 (directly) and 3′-A11 (through a water), and R161 with 3′-G12. Thus, contacts of the four RXR〈 DBDs with the nucleotide bases varied and depended on their upstream or downstream position on each DR-1 and to which of the two DR-1 sites they bound. DBD residues R164, N185, R191, R161, and Q188 interacted with phosphate residues of the 5′-G3, 3′-G5, 3′-G5, 3′-T6, and 3′-T6, respectively, whereas Q206 and K145 interacted with phosphates adjacent to 5′-G2 and 5′-A7 from an adjacent subunit, respectively. These residues are underlined. A, adenosine; C, cytosine; G, guanosine; and T, thymidine. Adapted from Ref. [20].
Table 3.
Crystallographic structures containing domains of retinoid X receptor (RXR) or its insect homolog ultraspiracle (USP)
| RSC PDB code |
Components | Ligands | Coactivator peptide source |
Features | Related ref |
|---|---|---|---|---|---|
| 1BY4 | h RXRα DBD–h RXRα DBD–DR-1 15-mer | Zn+2 | Homodimer cooperative assembly induced by DNA DR-1 | [20] | |
| DBD interacts with 3 base pairs of a half-site to imply weaker binding than TR DBD to DR-3 | |||||
|
| |||||
| 1DKF | m RXRα LBD(Phe318Ala)–h RARα LBD | RXR: 9Z-Oleic acid | RXRα mutant is constitutively active | [100] | |
| RAR: 4-[(4,4-Me2-1,2,3,4-tetrahydro-[1,2]binaphthalenyl-7-carbonyl)amino]benzoic acid (BMS614, antagonist) | RARα–LBD H12 in antagonist position | ||||
|
| |||||
| 1DSZ | h RARα DBD–h RXRα DBD–RARE DR-1 (15–mer) | Zn+2 | RXRα is 3′ on DR-1 | [193] | |
|
| |||||
| 1FBY | h RXRα | 9-cis-RA (agonist) | Ligand and H12 not in contact | [27] | |
|
| |||||
| 1FM6 | h RXRα LBD–h PPARγ LBD | RXR: 9-cis-RA | SRC-1 (1 peptide/LBD) | Asymmetric dimer with PPARγ H12 interacts with RXRα H7 and H10 | [99] |
| PPAR: 9-Cl-5-{[4-[2-Me-2-pyridinylamino)ethoxy]phenyl]methyl}-2,4-thiazolidinedione (Rosiglitazone, agonist) | |||||
|
| |||||
| 1FM9 | h RXRα LBD–h PPARγ LBD | RXR: 9-cis-RA | SRC-1 (1 peptide/LBD) | Asymmetric dimer with PPARγ H12 interacts with RXRα H7 and H10 | [99] |
| PPAR: 2-(2-benzoylphenylamino)-3-{4-[2-(5-Me-2-phenyloxazol-4-yl)ethoxy]phenyl}propionic acid (GI262570, agonist) | |||||
|
| |||||
| 1G1U | h RXRα LBD | Inactive tetramer with symmetric dimer H3–H11 interface | [96] | ||
| Each monomer of dimer has H12 binding to AF-2 of partner | |||||
|
| |||||
| 1G2N | hv USP LBD | L-α-Phosphatidyl-β-oleoyl-γ-palmitoyl phosphatidylethanolamine | Heliothis virescens (moth) | [194] | |
| H1–H3 loop prevents canonical agonist conformation | |||||
|
| |||||
| 1G5Y | h RXRα LBD | ATRA | Inactive tetramer with symmetric dimer H3–H11 interface | [96] | |
| Each monomer of dimer has H12 binding to AF-2 of partner | |||||
|
| |||||
| 1H9U | h RXRβ LBD | 6-[1-(3,5,5,8,8-Me5-5,6,7,8-tetrahydronaphthalen-2-yl)cyclopropyl]pyridine-3-carboxylic acid (LDG100268) | H12 does not cap LBP | [56] | |
| Ni+2 | |||||
| Cl–1 | |||||
|
| |||||
| 1HG4 | dm USP LBD | 2-(Hexadecanoyloxy)-1-[phosphonooxy)methyl]ethyl hexadecanoate (1,2-dipalmitoyl-sn-gylcero-3-phosphate) | Drosophila melanogaster | [195] | |
| Inactive conformation | |||||
| LBP partly occupied | |||||
|
| |||||
| 1K74 | h PPARγ LBD–h RXRα LBD | PPAR: 2-(1-Me-3-oxo-3-phenylpropylamino)-3-{4-[2-(5-Me-2-phenyloxazol-4-yl)ethoxy]phenyl}propanoic acid (GW409544) | SRC-1 (1 peptide/LBD) | PPARγ histidine and PPARα tyrosine determine ligand selectivity | [189] |
| RXR: 9-cis-RA | |||||
|
| |||||
| 1LBD | h RXRα LBD | (His)6-tagged domains D + E (hinge + LBD) | [100] | ||
| H1–H3 loop contains H2 LBP | |||||
| H12 extended | |||||
|
| |||||
| 1MVC | h RXRα LBD | 4-[2-(5,5,8,8-Me4-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dioxalan-2-yl]benzoic acid (SR11237, called BMS649) | NCoA-2 | Monomer in agonist conformation | [26] |
|
| |||||
| 1MV9 | h RXRα LBD–h RXRα LBD | (Z)-4,7,10,13,16,19-Docosahexaenoic acid (DHA) | NCoA-2 (SRC-1) | Homodimer in agonist conformation | [26] |
|
| |||||
| 1MZN | h RXRα LBD–h RXRα LBD | 4-[2-(5,5,8,8-Me4-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dioxolan-2-yl]benzoic acid (SR11237, called BMS649, agonist) | NCoA-2 | Homodimer in agonist conformation | [26] |
|
| |||||
| 1RDT | h RXRα LBD–h PPARγ LBD | RXR: (S)-(2E)-3-[4-(5,5,8,8-Me4-5,6,7,8-tetrahydro-2-naphthalenyl)tetrahydro-1-benzofuran-2-yl]-2-propenoic acid (S-46a,b, agonist) | 22-residue peptide with LLxxLL motif (1 peptide/LBD) | [196] | |
| PPAR: 2-(2-Benzoylphenylamino)-3-{4-[2-(5-Me-2- phenyloxaxol-4-yl)ethoxy]phenyl}propionic acid (Farglitazar, agonist) | |||||
|
| |||||
| 1R0N | dm EcR DBD–h RXRα DBD–Ec RE IR-1 (18 mer) | Zn+2 | Compared to EcR DBD–USP DBD–Ec RE (IR-1) | [197] | |
|
| |||||
| 1R0O | dm EcR DBD–dm USP DBD–EcR RE IR-1 (18 mer) | Zn+2 | Compared to EcR DBD–RXRα DBD–Ec RE (IR-1) | [197] | |
|
| |||||
| 1R1K | hv USP LBD–hv EcR LBD | USP: L-α-Phosphatidyl-β-oleoyl-γ-palmitoyl-phosphatidylethanolamine | Heliothis virescens (moth) | [198] | |
| EcR: 2,3,14,20,22-(OH)5-cholest-7-en-6-one (Ponasterone A) | Ligand-dependent EcR LBP size | ||||
|
| |||||
| 1R20 | hv USP LBD–hv EcR LBD | USP: L-α-Phosphatidyl-β-oleoyl-γ-palmitoyl-phosphatidylethanolamine | Heliothis virescens (moth) | [198] | |
| EcR: N-(t-Bu)-3,5-(Me)2-N-[(5-Me-2,3-dihydro-1,4-benzodioxin-6-yl)carbonyl]benzohydrazide (BYI06830, lepidopteran-specific agonist) | Ligand-dependent EcR LBP size | ||||
|
| |||||
| 1RXR | h RXRα DBD(Cys195Ala) | Zn+2 | High-resolution NMR structure similar to that of RXRα DBD–TR DBD on TRE | [199] | |
| C-terminus is helical in solution and stabilized by hydrophobic core | |||||
| 2nd Zn+2 loop disorded in solution | |||||
|
| |||||
| 1UHL | h LXRα LBD–h RXRβ LBD | LXR: N-(2,2,2-F3-Et)-N-{4-[2,2,2-F3-1-OH-1-(CF3)-ethyl]phenyl}benzenesulfonamide (T0901317) | NCoA-2 (GRIP-1) (1 peptide/LBD) | Agonist conformation | [88] |
| Asymmetric structure | |||||
| RXR: (2E,4E)-11-MeO-3,7,11-Me3-dodeca-2,4-dienoic acid (methoprene acid) | RXR H7–LXR L7–8 loop interface | ||||
| LXR LBP 20% smaller | |||||
|
| |||||
| 1XDK | m RARβ2 D–F–m RXRα1 LBD | RAR: 9-cis-RA (agonist) | TRAP220 NR box 2 (1 peptide/LBD) | RARβ2 D (hinge) and 30 C-terminal AA of 40-AA F had low electron density | [64] |
| RXR: 9-cis-RA (agonist) | Both LBDs in agonist conformation | ||||
|
| |||||
| 1XIU | bg RXR LBD | 9-cis-RA bound to each LBD | NCoA-1 (1 peptide/LBD) | Mollusc Biomphalaria glabrata tetramer i | [200] |
| 2 LBD H12s are in agonist and 2 LBD H12s in open conformation to bind partner’s AF-2 | |||||
| Tetramer interfaces between H11 of closed LBD and H6 of open LBD | |||||
|
| |||||
| 1XLS | m CAR LBD–h RXR LBD | CAR: 3,5-(Cl)2-2-{4-[(3,5-(Cl)2-pyridin-2-yl)oxy]phenoxy}pyridine (TCPOBOP) | TIF-2 (NCoA-2 NR box 3 (1 peptide/LBD) | Small CAR LBP | [84] |
| RXR: 9-cis-RA | CAR H10 links to H12 via a 2-turn helix | ||||
| CAR extended H2 interacts with H3 | |||||
| Large heterodimer interface | |||||
|
| |||||
| 1XVP | h CAR LBD–h RXRα LBD | CAR: 6-(4-Cl-phenyl)imidazo[2,1-b][1,3]thiazole-5- carbaldehyde O-(3,4-(Cl)2-benzyl)oxime (CITCO, agonist) | SRC-1 isoform 1 (1 peptide/LBD) | RXR in agonist conformation | [201] |
| RXR: Pentadecanoic acid | RXR ligand over laps with bound 9-cis-RA | ||||
|
| |||||
| 1XV9 | h CAR LBD–h RXRα LBD | CAR: 5β-Pregnane-3,20-dione (agonist) | SRC-1 isoform 1 (1 peptide/LBD) | RXR in agonist conformation | [201] |
| RXR: Pentadecanoic acid | RXR ligand over laps with bound 9-cis-RA | ||||
|
| |||||
| 1YNW | h RXRα DBD–h VDR DBD(Pro61Ala, Phe62Ala, His75Ala)–DR-3 RE | Zn+2 | 18-mer DNA with nonphysiologic LBD polarity on RE | [202] | |
| Mutation of VDR DBD permits heterodimer formation with RXR DBD | |||||
|
| |||||
| 1Z5X | bt USP LBD–bt EcR LBD | EcR: 2,3,14,20,22-(OH)5-cholest-7-en-6-one (Ponasterone A) | Hemipteran Bemisia tabaci (sweet-potato whitefly) | [203] | |
| PO4–3 | |||||
|
| |||||
| 2GL8 | h RXRγ LBD | [204] | |||
|
| |||||
| 2HAN | dm USP DBD–dm EcR DBD–Ec RE (20-mer) | Zn+2 | Drosophila melanogaster Natural pseudopalindromic Ec RE from hsp27 promoter | [205] | |
|
| |||||
| 2NXX | tc USP LBD–tc EcR LBD | EcR: 2,3,14,20,22-(OH)5-cholest-7-en-6-one (Ponasterone A) | Tribolium castaneum (Coleoptera arthropod) USP in apo conformation | [206] | |
| RXR ligands did not bind or activate TcUSP | |||||
|
| |||||
| 2P1T | h RXRα LBD | (2E)-3-[4-OH-3-(3-MeO-5,5,8,8-Me4-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic acid (UVI3007, 2a, agonist) | TIF-2 (NCoA-2 NR box 2) | [207] | |
|
| |||||
| 2Q60 | pm RXR LBD(Ile247Val, Leu257Pro) | Mutant apo-tetramer | Polyandrocarpa misakiensis | [208] | |
|
| |||||
| 2R40 | hv USP LBD–hv EcR LBD | USP: L-α-Phosphatidyl-β-oleoyl-γ-palmitoyl-phosphatidylethanolamine | Heliothis virescens (moth) | [209] | |
| EcR: (2β,3β,5β,22R)-2,3,14,20,22,25-(OH)6-cholest-7- en-6-one (20-OH-Ecdysone) | |||||
| Citrate–2 | |||||
| Glycerol | |||||
| Sulfate–2 | |||||
|
| |||||
| 2ZXZ | h RXRα LBD | 4-[2-(1,1,3,3-Tetramethyl-2,3-dihydro-1H-inden-5-yl)-1,3-dioxolan-2-yl]benzoic acid (agonist, 5a) | GRIP-1 | Comparison to disila-analog explains latter’s increased affinity through interactions with H7 and H11 | [210] |
|
| |||||
| 2ZY0 | h RXRα | 4-[2-(1,1,3,3-Tetramethyl-2,3-dihydro-1H-1,3- benzodisilol-5-yl)-1,3-dioxolan-2-yl]benzoic acid (agonist, 5b) | GRIP-1 | Comparison to dicarbon analog explains disila-analog’s increased affinity through interactions with H7 and H11 | [210] |
|
| |||||
| 3A9E | h RARα LBD–m RXRα LBD | RAR: ATRA (agonist) | NCoA-2 | [211] | |
| RXR: (2E,4E,6Z)-3-methyl-7-(5,5,8,8-tetramethyl-3-propoxy-5,6,7,8-tetrahydronaphthalen-2-yl)octa- 2,4,6-trienoic acid (LG100754, antagonist) | |||||
|
| |||||
| 3DZU | h PPARγ–h RXRα–PPRE DR-1 (20-mer) | PPAR: 2-[(2,4-(Cl)2-benzoyl)amino]-5-(pyrimidin-2-yloxy)benzoic acid (BVT.13) | NCoA-2 (1 peptide/ LBD) | Asymmetric holo-complex on 20-mer containing PPRE DR-1 | [21] |
| RXR: 9-cis-RA | PPAR DBD C-terminal extension (CTE) interacts with DNA and 2 helices (hinge region) connect to LBD. Less structured RXR CTE contacts PPAR DBD + RXR hinge places its DBD and LBD further away | ||||
| Zn+2 | PPAR LBD nestles between RXR LBD and RXR DBD | ||||
| PPAR LBD β-strand (Phe347) interacts with RXR DBD | |||||
| DBD interface is 30 Å2; LBD (H7, H8, H10) interface is 2,160 Å2 | |||||
| DBDs also interact with DR-1 spacer minor groove | |||||
| CoA peptides act independently at the two AF-2 sites | |||||
| A/B domains dynamic | |||||
|
| |||||
| 3DZY | h PPARγ–h RXRα–PPRE DR-1 | PPAR: 9-Cl-5-{[4-[2-Me-2-pyridinylamino)ethoxy]phenyl]methyl}-2,4-thiazolidinedione (Rosiglitazone, agonist) | NCoA-2 (1 peptide/ LBD) | Asymmetric holo-complex on 20-mer containing PPRE DR-1 | [21] |
| RXR: 9-cis-RA | PPAR DBD C-terminal extension (CTE) interacts with DNA and 2 helices (hinge region) connect to LBD. Less structured RXR CTE contacts PPAR DBD + RXR hinge places its DBD and LBD further away | ||||
| Zn+2 | PPAR LBD nestles between RXR LBD and RXR DBD | ||||
| PPAR LBD β-strand (Phe347) interacts with RXR DBD | |||||
| DBD interface is 30 Å2 ; LBD (H7, H8, H10) interface is 2,160 Å2 | |||||
| DBDs also interact with DR-1 spacer minor groove | |||||
| CoA peptides act independently at the two AF-2 sites | |||||
| A/B domains dynamic | |||||
|
| |||||
| 3E00 | h PPARγ–h RXRα–PPRE DR-1 (20-mer) | PPAR: 2-Cl-5-NO2-N-phenylbenzamide (GW9662) | NCoA-2 (1 peptide/ LBD) | Asymmetric holo-complex on 20-mer containing PPRE DR-1 | [21] |
| RXR: 9-cis-RA | PPAR DBD C-terminal extension (CTE) interacts with DNA and 2 helices (hinge region) connect to LBD. Less structured RXR CTE contacts PPAR DBD + RXR hinge places its DBD and LBD further away | ||||
| Zn+2 | PPAR LBD nestles between RXR LBD and RXR DBD | ||||
| PPAR LBD β-strand (Phe347) interacts with RXR DBD | |||||
| DBD interface is 30 Å2; LBD (H7, H8, H10) interface is 2,160 Å2 | |||||
| DBDs also interact with DR-1 spacer minor groove | |||||
| CoA peptides act independently at the two AF-2 sites | |||||
| A/B domains dynamic | |||||
|
| |||||
| 3E94 | h RXRα LBD | (n-Bu)3Sn (agonist, endocrine disruptor) OAc−1 | TIF-2 (NCoA-2) | Activates RXRα–PPARα–γ through RXR | [41] |
| Canonical agonist conformation | |||||
| H11 Cys432 in two different conformations (7:3) with weak covalent bond to Sn | |||||
| Contacts, including that of Leu436, compare with that of 9-cis-RA | |||||
|
| |||||
| 3EYB | amphi RXR LBD | Apo-tetramer from invertebrate cephalochordate amphioxus Branchiostoma floridae | [212] | ||
| Apo-antagonist conformation with H11 in LBP | |||||
| Activated by retinoids | |||||
|
| |||||
| 3FAL | h RXRα LBD–m LXRα LBD | RXR: 9-cis-RA | Structure used with that of LXRβ–19 complex to design LXRβ-selective ligands | [213] | |
| LXR: 2-{4-[Butyl-(3-Cl-4,5-(MeO)2-benzyl)amino]phenyl}-1,1,1,3,3,3-F6-propan-2-ol (19, agonist binding IC50 α/β ratio: 165/25) | Separation of anti-inflammatory and lipogenic activities in h LXRβ ligand GSK9772 | ||||
|
| |||||
| 3FC6 | h RXRα LBD–m LXRα LBD | RXR: 9-cis-RA | [184] | ||
| LXR: [4-(3-{[2-Cl-3-(CF3)benzyl](2,2-diphenylethyl)amino}propoxy)-1H-indol-1-yl]acetic acid (SB786875, agonist) | |||||
|
| |||||
| 3FUG | h RXRα LBD | (2E)-3-[4-Hydroxy-3-(3,5,5,8,8-Me5-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]prop-2-enoic acid (CD3254; agonist) | NCoA-2 NR box 2 | Impact of 3′-alkoxy side chain on activity | [214] |
|
| |||||
| 3H0A | h PPARγ LBD–h RXRα LBD | PPAR: [(4-{[2-(Pent-2-yn-1-yloxy)-4-{[4-(CF3)-phenoxy]methyl}phenyl]sulfanyl}-5,6,7,8-tetrahydronaphthalen-1-yl)oxy]acetic acid (partial agonist) | NCoA-1 | Comparison with the agonist-bound PPARγ LBD facilitated design of full PPARδ agonist with partial PPARγ agonist activity | [215] |
| RXR: 4-[1-(3,5,5,8,8-Me5-5,6,7,8-tetrahydronaphthalen-2-yl)ethenyl]benzoic acid (agonist) | |||||
|
| |||||
| 3HOO | h RXRα LBD | Apo-tetramer structure withdrawn | [216] | ||
| See 3NSP | |||||
|
| |||||
| 3HOQ | h RXRα LBD | Danthron (chrysazine, 1,8-(OH)2-anthra-9,10-quinone (irritant, mutagen, RXR antagonist) | Antagonist-bound tetramer structure withdrawn | [216] | |
| See 3NSQ | |||||
|
| |||||
| 3KWY | h RXRα LBD | Ph3Sn (agonist, endocrine disruptor) OAc−1 | NCoA-2 NR box 2 | Sn interacts with H11 Cys432 | [217] |
|
| |||||
| 3NSP | h RXRα LBD | Dimer | [216] | ||
|
| |||||
| 3NSQ | h RXRα LBD | Danthron, (1,8-[OH]2-anthracene-9,10-dione) antagonist | Dimer | [216] | |
|
| |||||
| 3OAP | h RXRα LBD | 9-cis-RA | GRIP-1 (NCoA-2) | Interactions of GRIP-1 peptide with H12 Glu453 and Glu456 and H4 Arg302 | [66] |
| HDX conducted | |||||
|
| |||||
| 3P1U | h RXRα LBD | (2E)-3-[4-OH-3-(3-EtO-5,5,8,8-Me4-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic acid (UVI3004, 2b, partial agonist) | TIF-2 (NCoA-2 NR box 2) | [207] | |
|
| |||||
| 3P1V | h RXRα LBD | (2E)-3-[4-OH-3-(3-PrO-5,5,8,8-Me4-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic acid (UVI3002, 2c, antagonist) | TIF-2 (NCoA-2NR box 2) | [207] | |
|
| |||||
| 3R29 | h RXRα LBD | SMRT2 CoR peptide | Tetramer composed of four CoR-bound monomers | [97] | |
|
| |||||
| 3R2A | h RXRα LBD | Rhein (3-carboxy-1,8-(OH)2-9,10-anthraquinone) | SMRT2 peptide | Tetramer composed of two rhein-bound monomers and two CoR-bound monomers, each of which differs in H3 and H12 positions | [97] |
Abbreviations: ATRA, all-trans-retinoic acid; CAR, constitutively active receptor; CoA, coactivator; CoR, coreprressor; DBD, DNA-binding domain; DHA, docosahexaenoic acid; DR, direct repeat; EcR, ecdysone receptor; GRIP, glucocorticoid receptor interacting protein; H, helix; h, human; IR, inverted repeat; LBD, ligand-binding domain; LBP, ligand-binding pocket; LXR, liver X receptor; m, mouse; NCoA, nuclear receptor coactivator; NR, nuclear receptor; Ph, phenyl; PPAR, peroxisome proliferator-activated receptor; PXR, pregnane receptor/sterol and xenobiotic receptor (SXR); RA, retinoic acid; RAR, retinoic acid receptor; RARE, retinoic acid response element; RE, response element; RXR, retinoid X receptor; SRC, steroid receptor coactivator; TIF, transcriptional intermediary factor; TRAP, thyroid hormone receptor-associated protein; USP, ultraspiracle nuclear receptor.
Summarized from the RCSB Protein Data Bank (http://www.rcsb,org/pdb)
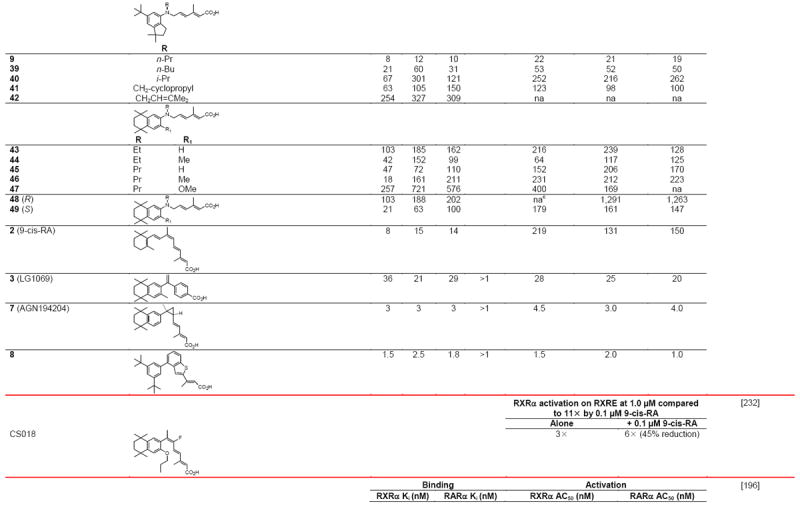
The crystal and solution structured of the RXRa DBD have been compared [20]. NMR studies of the RXRα DBD in solution indicated that its T-box was helical with its Glu208 having interactions that masked the DNA recognition helix residues Lys160 and Arg164. In contrast in the crystal structure, the downstream DBD T-box in the homodimer DBD–DR-1 complex was extended and disordered. The authors postulated that in the latter conformation, the 3′-DBD Lys160 and Arg164 were unmasked and interacted with DNA and the T-box Glu208. As a result, Glu208 interacted with the 5′-DBD’s Gln182 and Arg186 to stabilize the position of the zinc finger II loop. Unlike other NR DBDs, which recognized all six base-pairs of an RE half-site, each recognition helix in the RXRα DBD homodimer only recognized three base-pairs of the half-site. The authors suggested that the reduced contact in the homodimer led to preferential RXR heterodimerization on DNA that allowed more contacts and, thus, stronger interactions with DNA. In the RXRα DBD–RARα DBD heterodimer, the RXRα DBD T-box formed an interface with the second Zn finger of the RARα DBD. In the RXRα–RARα heterodimers bound to the DR-2 and DR-5 REs, RXR was upstream, whereas binding polarity was reversed on the DR-1 RE with RAR being upstream. The DR-1 RE was also bound by RXR homodimers and by the complex of PPARγ(102–505)–RXRα(11-462) bound to rosiglitazone and 9-cis-RA, respectively, and two NCoA2 (SRC-2) peptides, as found in the PDB crystal structure 3DZY (Fig. 4) [21].
Figure 4.
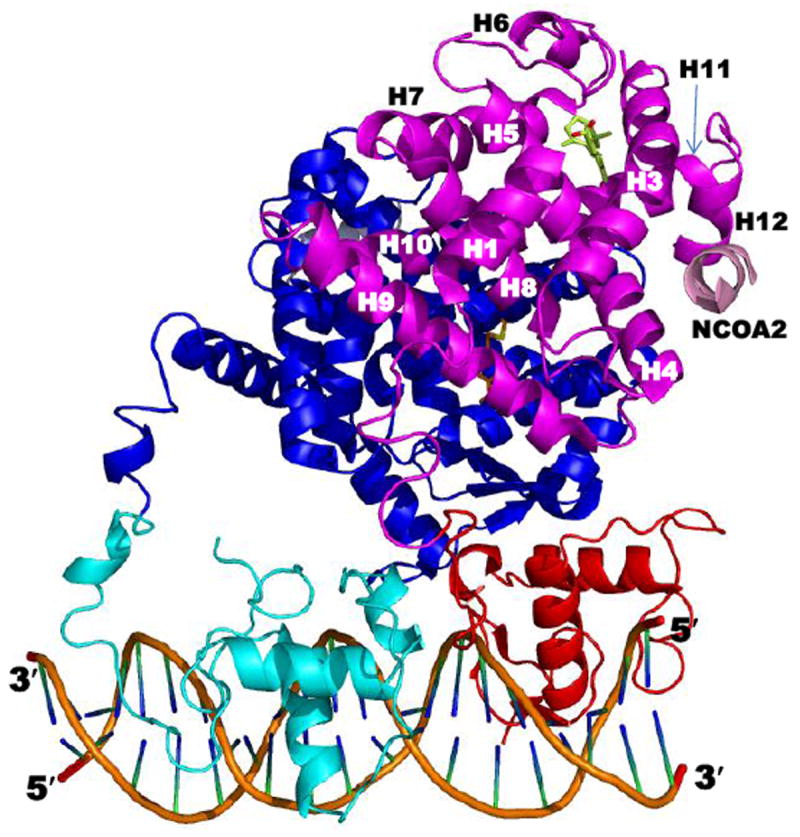
Structure of the rosiglitazone–PPARγ–RXRα–9-cis-RA complex bound to two coactivator (CoA) NCoA2 peptides and DNA (PDB 3DZY). Components are colored as follows: PPARγ LBD (blue), DBD (cyan), ligand (carbons in orange), and CoA peptide (gray); RXRα LBD (magenta), DBD (red), ligand (Cs in yellow-green), and CoA peptide (pink); and DNA backbone (orange), nucleosides (blue and green). The RXRα LBD helices are numbered. Note that the RXRα hinge was not defined in 3DZY due to absence of electron density. Adapted from Ref. [21].
RXRα export from the nucleus is mediated by the binding of its DBD recognition helix, which is adjacent to the first zinc finger, to the calcium-binding protein calreticulin, whereas mutation of two conserved phenylalanines (158 and 159) in this helix abrogates its export. The RXRα nuclear localization sequence (NLS) is also located in this region between residues 160–165 (Lys-Arg-Tyr-Val-Arg-Lys) [22]. Unlike several other NRs, RXRα lacks NLS sequences in its hinge or LBD. The results using RXRα and vitamin D receptor (VDR) chimeras with fluorescent proteins led the authors to conclude that RXRα: (i) dynamically shuttled between nucleus and cytoplasm; (ii) heterodimerized with VDR in the cytoplasm regardless of calcitriol (vitamin D3) binding status; and (iii) facilitated the apo-VDR nuclear residence time.
2.3.2. Hinge
Deletion of 7, 14, or 28-amino acids from the C-terminus of the human RXRα hinge domain (residues 200–229) demonstrated that the RXRα mutants with 7- and 14-residue deletions retained 90% and 70% transcriptional activity, respectively, of the native RXRα on a DR-1-driven-luciferase (Luc) reporter in response to 0.1 μM 9-cis-RA, whereas the 28-residue deletion mutant was inactive [23]. Mutants having such deletions from the N-terminus of the hinge were inactive. Heterodimers of the RXRαΔA/B(223–229), RXRαΔA/B(216–229), and RXRαΔA/B(209–229) mutants with the VDRΔA/B retained the ability to bind to the VDRE DR-3 (binding constants of 88, 79, and 92 nM) although their affinities were lower than that of the RXRαΔA/B–VDRΔA/B (71 nM). The authors concluded that the RXRα hinge was very flexible, and this flexibility permitted RXRα to bind to various REs in its heterodimers with a variety of NRs [24]. The flexibility of the RXRα hinge was fully demonstrated in the crystal structure (3DZY) of the rosiglitazone–PPARγ–RXRα–9-cis-RA complex bound to a DNA 20-base oligomer containing a PPRE [21]. The flexible hinge permitted the RXRα LBD to shift to the opposite side of the DNA helix from its DBD to provide sufficient space for the PPARγ LBD to reside between the RXRα LBD and RXRα DBD (Fig. 4).
2.3.3. RXR ligand-binding domain
Of the RXR domains, the LBD has been the most extensively studied. Crystallography has been used to investigate this domain in depth either alone, agonist bound, and in complexes with the LBDs of its dimeric NR partners and/or CoA peptides. These structures and those of the RXR DBD bound to DNA, and the complex of holo-RXRα–holo-PPARγ bound to CoA peptides and DNA that have been deposited in the PDB as of 2011 are listed in Table 3.
The LBD structure, which is conserved among members of the steroid/thyroid hormone NR superfamily, consists of 12 α-helices and a small β-sheet between helices H5 and H6 that are arranged in what is termed a barrel or an anti-parallel helical sandwich in which H4, H5, H8, H9, and H11 are between layers formed by H1–H3 and H6, H7, and H10 (Fig. 2B–2D). In the absence of a ligand, all 12 helices of the RXR LBD (apo form) are present (Fig. 2B). To accompany the structural changes in the LBD (holo form) induced by the binding of a rexinoid that induces gene transcription (transcriptional agonist), H2 unwound to provide a longer loop between helices H1 and H3 that permitted H3 to undergo a 13 Å tilt to form a surface with H4 and H12 that accommodated the binding of a CoA or the related peptide containing the NR box motif (Fig. 2C). The full CoA, when bound, could then recruit other regulatory proteins that include the transcriptional protein complex that connects the NR–RE complex with the transcription start site. The functional domains of the RXRα LBD are shown in Fig. 5. These include the ligand-binding pocket (LBP) (Fig. 5A), the coactivator surface consisting of residues from H3, H4, and H12 to which CoA proteins and and certain repressors bind (Fig. 5B), and the dimerization interface between the RXRα and PPARγ LBDs consisting of predominantly residues from H10 and some from H7–H9 (Fig. 5C).
Figure 5.
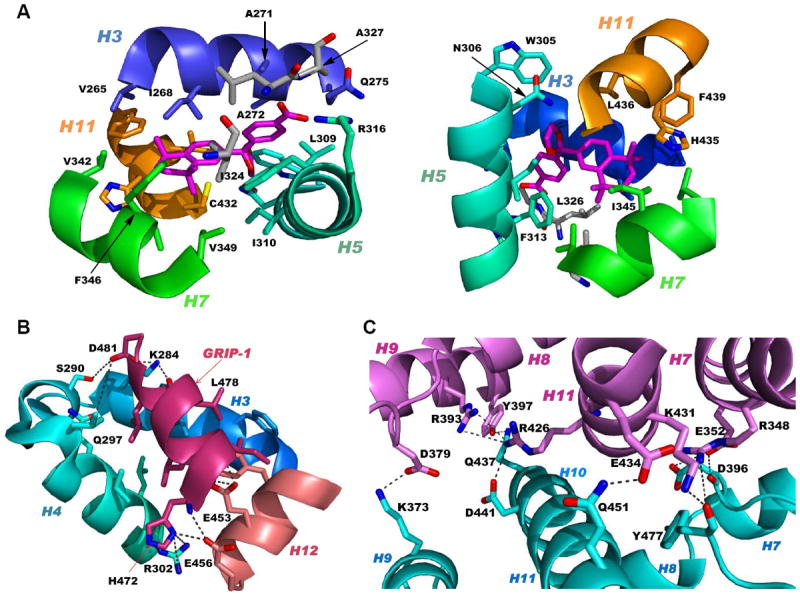
RXRα ligand-binding domain (LBD) functional domains. A. Structure of RXRα LBD ligand-binding pocket showing contacts between pocket residues and RXR agonist SR11237 (BMS64) as found in the PDB structure 1MVC. Two views of the residues in helices H3 (backbone and residue side-chain Cs in blue), H5 (Cs in cyan), H7 (Cs in green), and H11 (Cs in orange) and β-sheet (Cs in gray) contacting SR11237 (Cs in magenta). B. RXRα LBD CoA surface residues (cyan backbone and residue side-chain Cs) forming salt-bridge contacts that stabilize binding of the CoA GRIP-1 peptide (rose) as shown in 1MVC. C. PPARγ LBD–RXRα LBD heterodimer interface as found in PDB 3DZY with RXRα residue contacts labeled. PPARγ and RXRα backbones and side-chain Cs in pink and cyan, respectively. Ns (blue), Os (red), and S (yellow). A, alanine; C, cysteine; D, aspartate; E, glutamate; F, phenylalanine; H, histidine; I, isoleucine; L, leucine; N, asparagine; Q, glutamine; R, arginine; V, valine; W, tryptophan; and Y, tyrosine.
Crystal structures reveal that the conformations assumed by the RXR LBD H12 were often undefined and atypical of NRs because, even when the RXR LBD was bound by a transcriptional agonist, the position of its H12 varied. Only the combined binding by the agonist and CoA peptide committed H12 to an agonist conformation with H3 and H4 to create a stabilized groove or surface to which the CoA-derived peptide bound. The entire CoA surface has not been rigorously defined because structural work has focused on bound small peptides that contain the CoA binding motif (NR box) of Leu-XX-Leu-Leu (X = unspecified residue) plus adjacent residues that define CoA binding specificity rather than the full CoA sequence. In addition, CoAs can have multiple NR boxes that can interact with the AF-1 and AF-2 sites on the same NR or its partner.
Normally, RXR is diffusely localized in the nucleoplasm in normal human mammary epithelial cells and in ATRA-sensitive MCF-7 breast cancer cells, whereas RARα is both dispersed diffusely in the nucleoplasm and localized in microspeckles with PML bodies [25]. In contrast, in the ATRA-resistant MDA-MB-231 breast cancer cell line, RXRα exhibited a punctate pattern in the splicing factor compartment (SFC), as was indicated by immunostaining with antibodies for RXRα and SFC components SC-35 and p105. In MDA-MB-231 cells, RXRα was not associated with DNA or RNA and did not participate in gene transcription. The C-terminal deletion mutant RXRαΔ(417–462) showed reduced localization in the speckles, whereas a peptide corresponding to this deletion localized in the SFC. These results suggested that the RXRα E domain participated in SFC localization. In tumor samples from five of 12 invasive breast cancer patients, RXRα was also localized in the SFC regardless of treatment with 9-cis-RA or RXR agonist AGN194204 (Fig. 1B).
2.3.4. F domain
Whether any of the RXR isotypes has a functional F domain has yet to be established. This domain is usually included with the RXR LBD as E/F. For example, human RXRα is typically considered to be a 462 residue protein with the E domain ending at 462. However, other NRs such as estrogen receptor (ER) α and hepatic nuclear factor (HNF) 4α have large F domains with specific sequences that modulate gene transcription.
3. RXR ligands
3.1. Ligand binding alters the ligand-binding pocket conformation
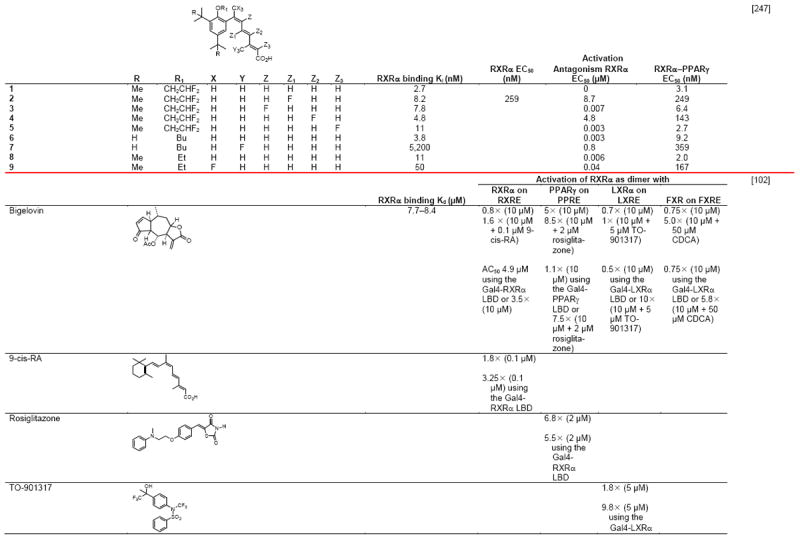
Notable differences in complexes of the RXRα LBDs with the agonists 9-cis-RA, DHA (docasahexaenoic acid in Fig. 1A), and BMS649 (SR11237 in Fig. 1B) included their respective ligand-binding pocket (LBP) volumes (494, 528, and 472–480 Å3), LBP volume occupied by ligand (74, 81, and 86–88%), and van der Waals/polar contacts (71/6, 89/6, and 89–92/6–7), which variously impacted their respective ligand-binding affinities to the RXRα LBD (2, 50–100, and 5–10 nM) [26,27]. Overlap of these ligands in their bound conformations showed that the agonist DHA, which had the lowest affinity, more closely overlapped 9Z-oleic acid, which was considered to confer an antagonist conformation to the RXRα LBD in its heterodimer with the RARα LBD–antagonist BMS614 (1DKF), whereas the structures of transcriptional agonists 9-cis-RA and SR11237 differed from those of DHA and 9Z-oleic acid and overlapped more closely with each other. Interestingly, the SR11237 1,3-dioxalane ring and the 9-cis-RA 19-methyl group occupied the same region of the LBP. The authors accounted for the 10% decrease in pocket volume when SR11237 was bound by RXRα to the repositioning of its H5 Gln306 into the LBP from the LBD surface, where it was located in the 9-cis-RA and DHA complexes. They termed the corner of the L pocket the “hinge” region. They noted that binding by SR11237 revealed unoccupied subpockets around H5 Tyr305 and Gln306 and around H3 Ile268 and H5 Phe313 that could be exploited in ligand design and postulated that this “hinge” provided maximal adaptability and flexibility to the LBP.
3.2. Ligand-induced communication with the AF-2 core to form the CoA binding groove or surface
Second-order Möller–Plesset perturbation analysis of molecular interactions by the 9-cis-RA–RXRα complex (PDB 1FBY), in which missing regions had been replaced with those from the human 9-cis-RA–RXRα LBD–SRC-1 peptide complex (1FM9), suggested the residues through which 9-cis-RA interacted to stabilize the seven-residue activation function-2 core (AF-2C, H12 450–456) in its AF-2 core-binding pocket (AF-2CBP, containing 10-residues from H3–H5, H10, and H11) to produce the H12 canonical agonist conformation [28]. The polar side chains of H12 Glu453 and Glu456, which resided outside the AF-2CBP, were considered to have a role in CoA recruitment. The other five residues are hydrophobic and were locked within the AF-2CBP by van der Waals interactions. On binding, 9-cis-RA assumed an L-shaped conformation. Of the residues surrounding 9-cis-RA, 19 were within 4.2 Å and so considered to make contacts with the ligand. H5 Tyr305 and H11 Leu436 were closest to the 9-cis-RA 19-methyl group, which was located at the corner of the 9-cis-RA L-shaped conformation, and also participated in forming the AF-2CBP. Thus, 9-cis-RA did not interact directly with H12, but was ≥ 5.9 Å from the AF-2 core residues. The authors concluded that Tyr305 and Leu436 provided the means for communication between the ligand and the AF-2 core.
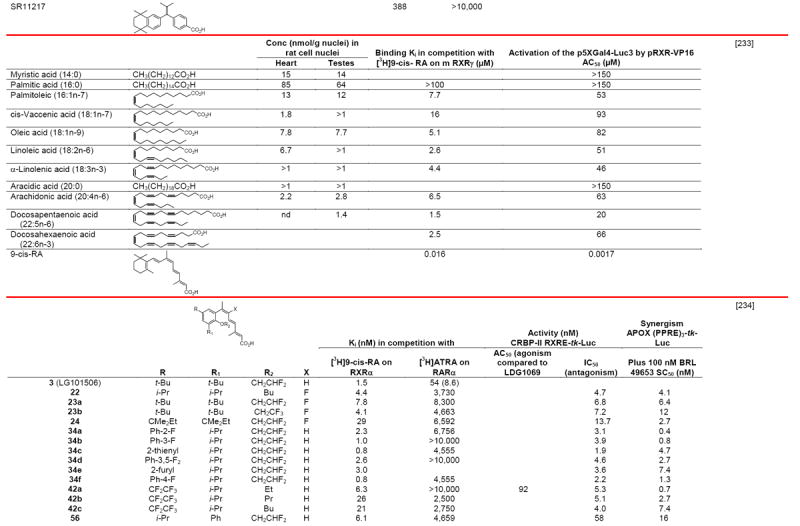
3.3. Natural ligands
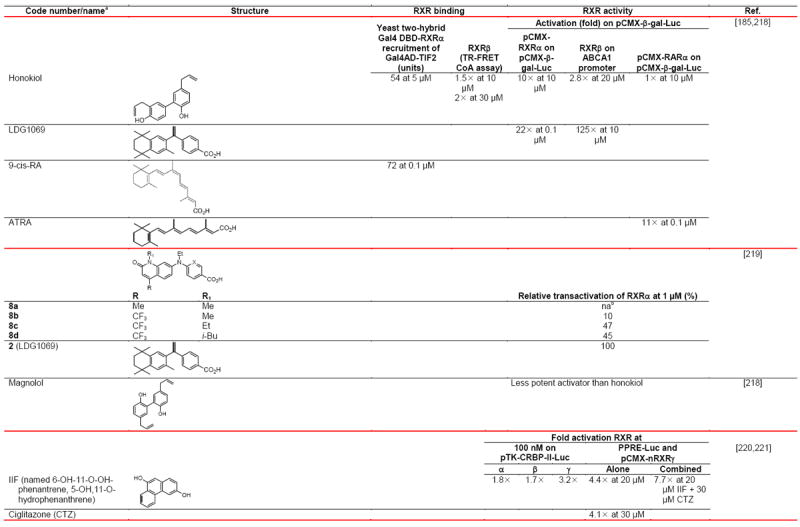
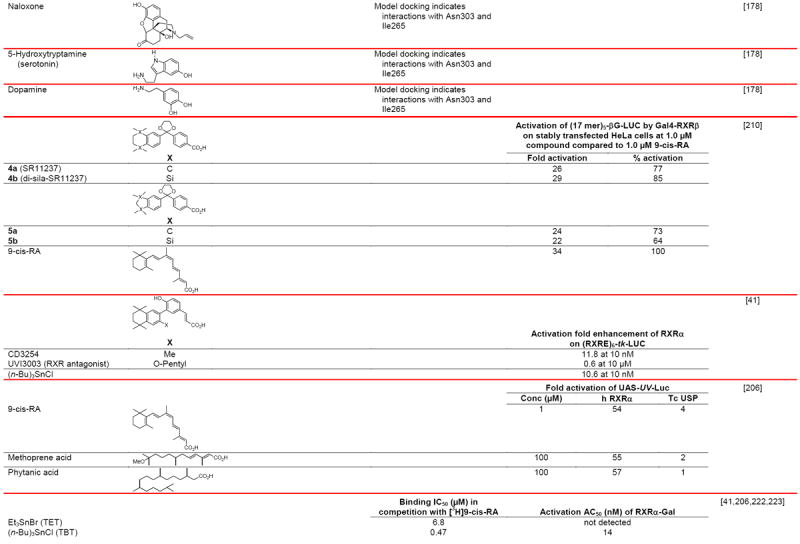
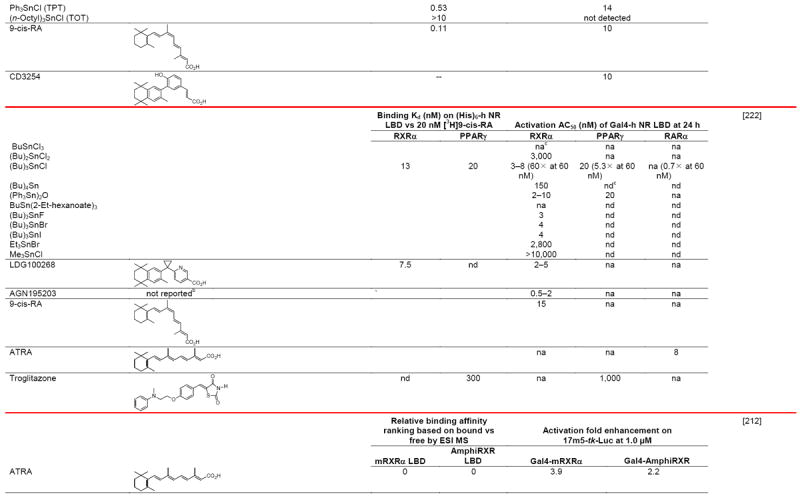
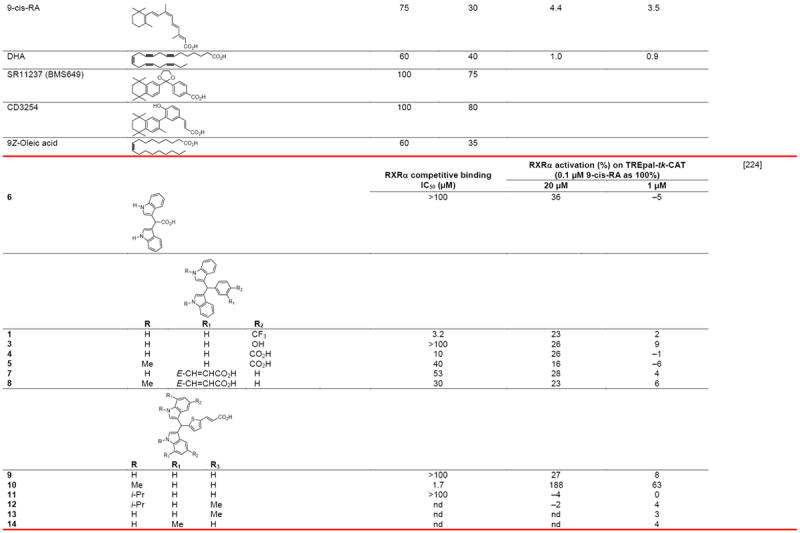
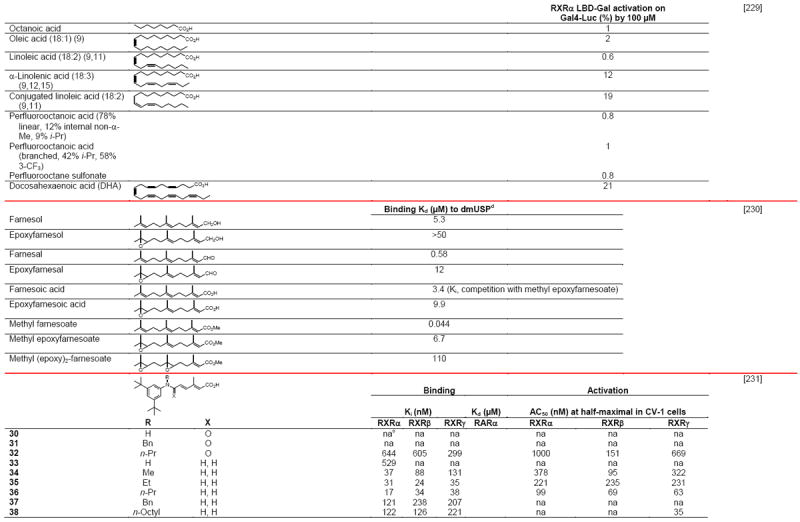
Negative ion electrospray mass spectrometry of the lipids, which were isolated from recombinant RXRα LBD protein that had been incubated with brain-conditioned medium, was used to identify the unsaturated fatty acids (FAs) docosahexaenoic acid (22:6) (DHA), arachidonic acid (20:4) (AA), and oleic acid (18:1) (OA) as most highly bound [29]. AC50 values for their induction of RXRα activation on the ApoA1 RXRE-tk-Luc in transfected cells were 5–10 μM. Docosapentaenoic acid (22:5) was slightly less potent than DHA or AA, and all were more potent than linolenic acid (18:3) and linoleic acid (18:2), whereas OA had lower potency (see Table 4 for structures). Arachidic acid (20:0) and stearic acid (18:0) were inactive. DHA reduced mouse YAMC colonocyte proliferation and activated a DR-1 RE-reporter in YAMC and normal human NMC460 colon cells to suggest a potential use in cancer prevention [30].
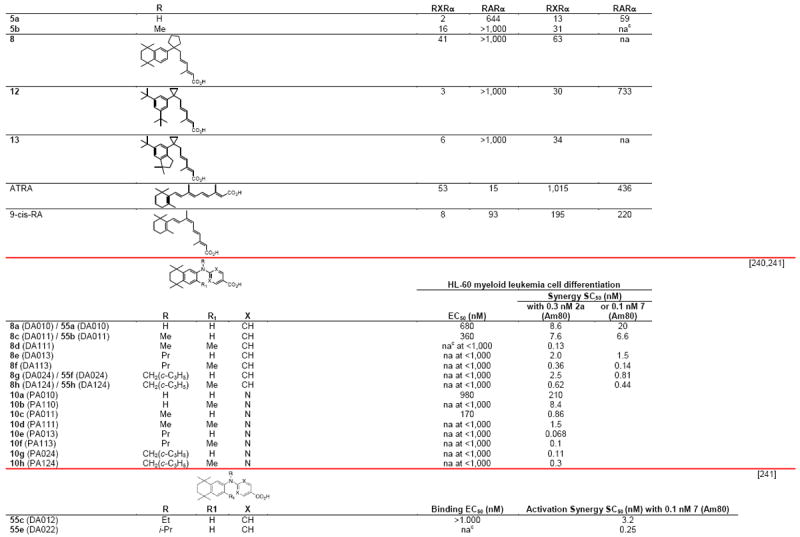
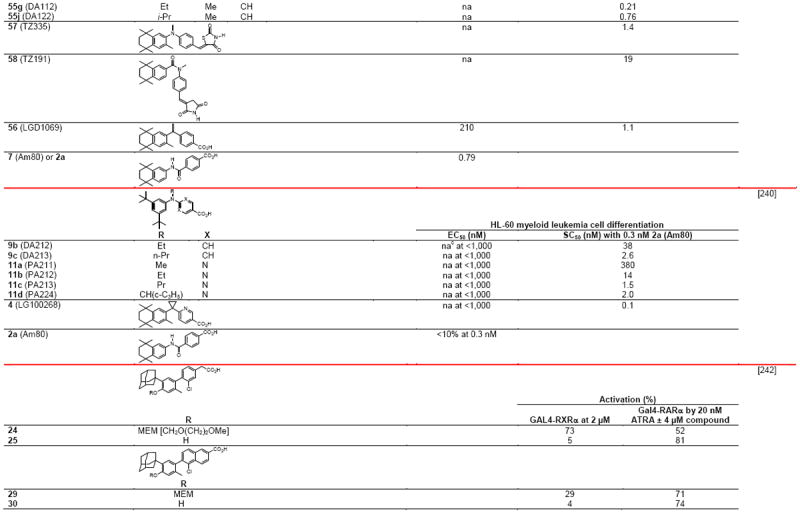
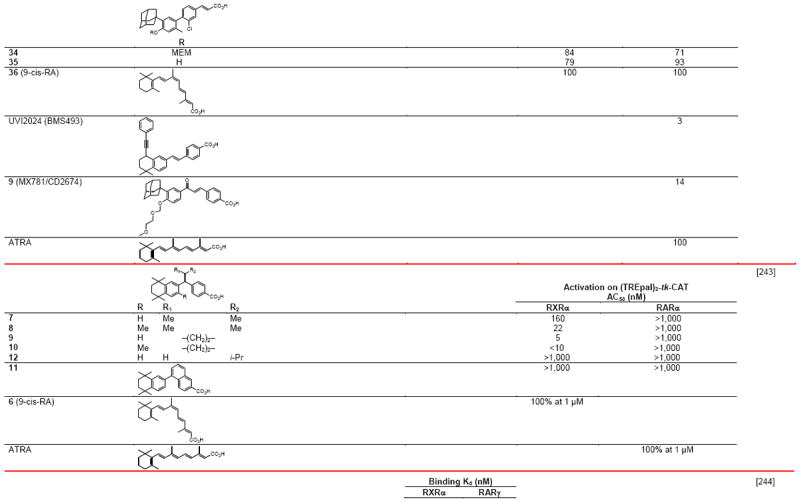
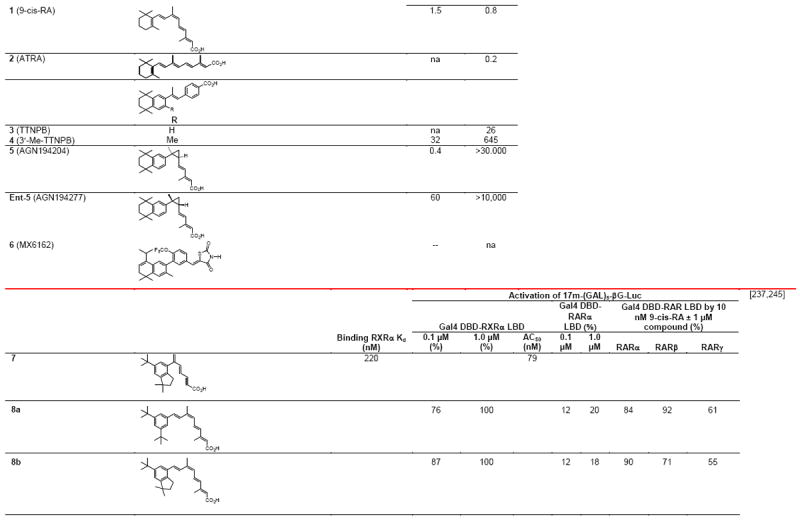
Table 4.
RXR transcriptional agonists and synergists
|
|
|
|
|
|
|
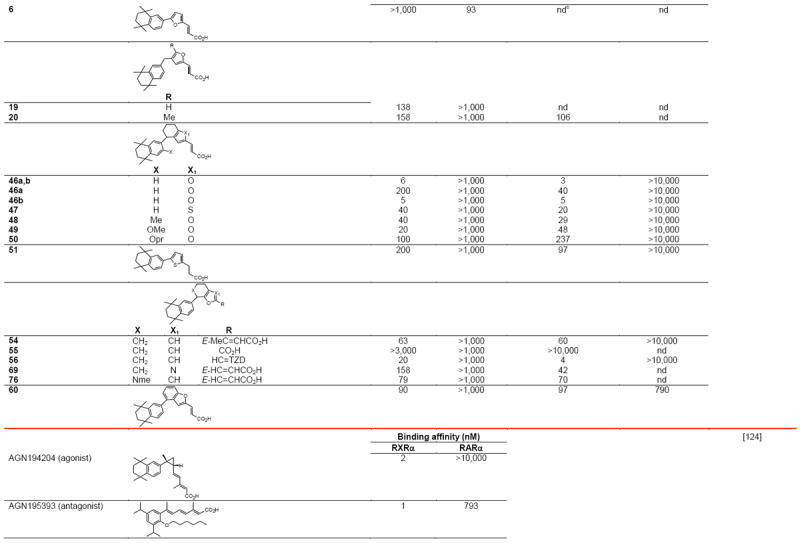
|
|
|
|
|
|
|
|
|
|

|
Compound names and numbers refer to those used in the cited references.
Searches of PubMed, SciFinder and Google databases did not provide a structure for AGN195203.
Abbreviations: na, not active; nd, not determined.
Excitation of Trp at 290 nM and fluorescence at 340 nM.
Concentrations undefined in Ref. [246].
RXRα was found to be expressed and activated in the rostral spinal cord of Xenopus laevis tadpoles beginning at developmental stage 24/25, which is the time of primary neuron formation, and then declined at the swimming stage [31]. These results suggested to the authors that an endogenous RXR ligand had a role in frog development.
3.4. Synthetic RXR ligands and their developmental status
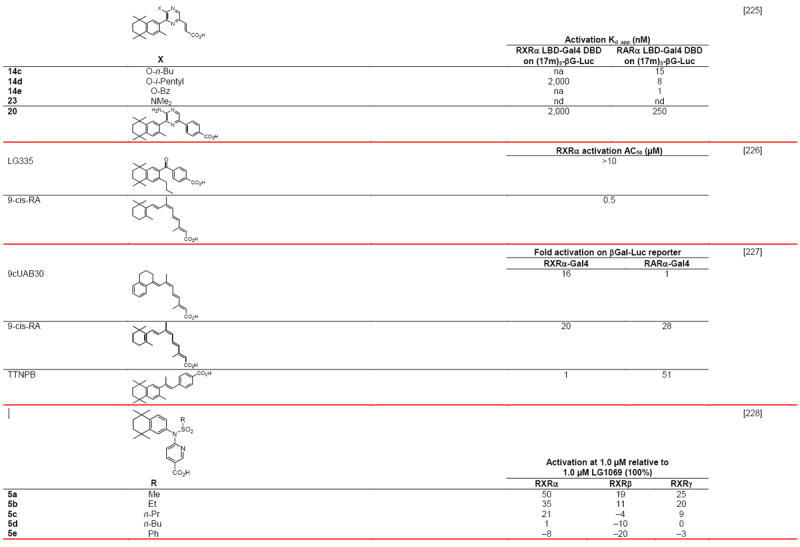
The lack of significant therapeutic efficacy in cancer patients partipating in clinical trials using bexarotene (Targretin™, LGD1069) accompanied by their experiencing severe adverse events has dampened enthusiasm for the development of more potent or more RXR-selective rexinoids for treatment of cancer or diabetes by such pharmaceutical companies as Bristol-Myers-Squibb, Hoffman-La Roche, and Lilly. Limited accessibility to rexinoids for research purposes or use of evaluative assays due to laborious material transfer agreements or concerns of litigation has also restricted retinoid research by the U.S. academic community.
The U.S. National Cancer Institute has initiated clinical trials on 9cUAB (Fig. 1B) as a rexinoid agonist for use in cancer prevention. 9cUAB was first reported by Muccio and colleagues at the University of Alabama at Birmingham [32]. One preliminary report described a single-dose pilot study in 14 volunteers, who were given 5, 10, or 20 mg of 9cUAB30 orally [33]. No grade 3 or 4 toxicities occurred although one volunteer had grade 2 symptoms and seven had grade 1 (predominately headache, which the authors suggest might have been related to their lack of caffeine consumption). Tmax was at 2–3 h after dosing, and t1/2 was 2.8–7.2 h. Maximum plasma concentration at the 20-mg dose was 70 ng/mL. In a 6-month study in mice dosed daily by gavage at 30, 100, or 300 mg/kg, dose-related hepatomegaly was observed in both sexes, which the authors associated with induction of liver enzymes [34].
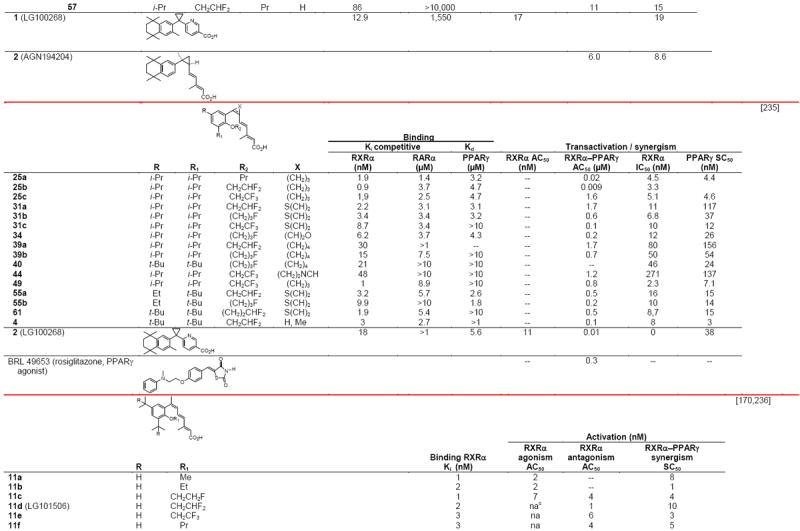
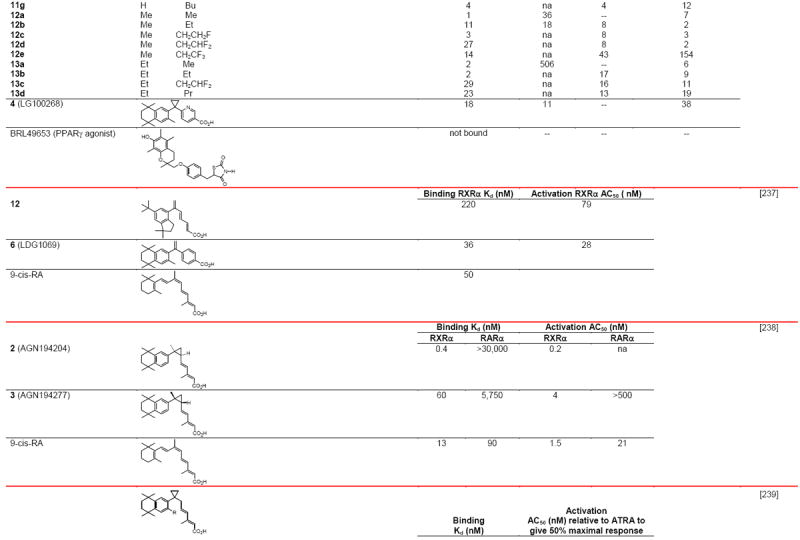
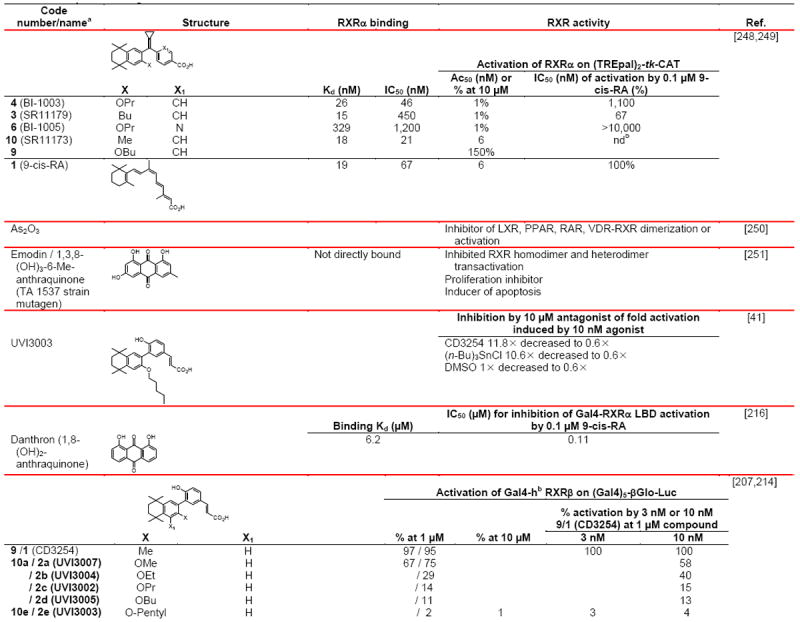
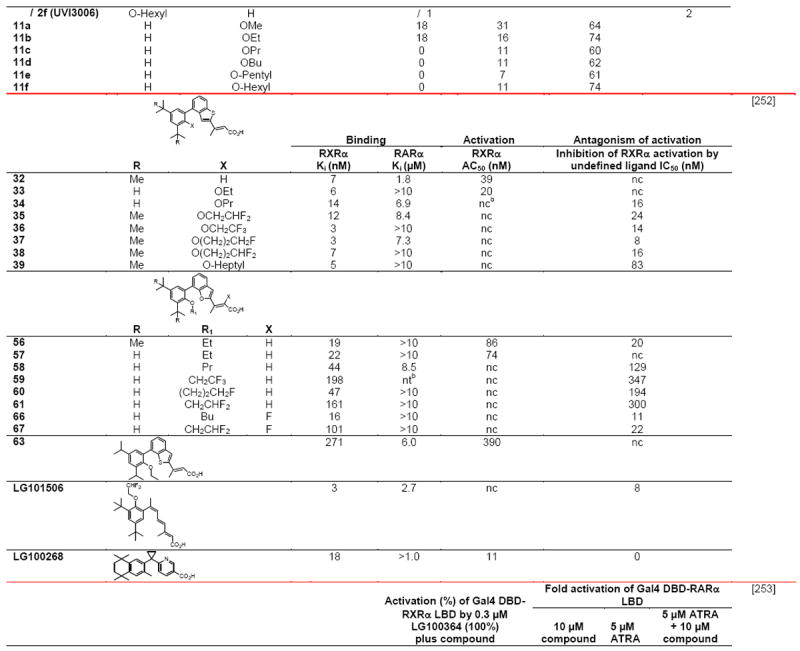
A recent analysis of the relationship between an RXR polymorphism and successful outcome in a recurrence of head-and-neck cancer prevention trial using 13-cis-RA [35] suggests a potential use in targeting RXR in one particular patient subpopulation. Despite the major hiatus of rexinoid research currently ongoing in the U.S., groups in Europe and Japan have been very productive. In Tables 4 and 5 are listed many of the retinoid agonists and antagonists, respectively, reported in the open literature for the period 2000–2010. Many were the result of the ongoing productive collaboration between the Gronemeyer and de Lera groups in Europe.
Table 5.
RXR transcriptional antagonists
|
|
|
|
Compound numbers refer to those cited in the references.
Abbreviations: h, human; nc, not conducted; nd, not determined; nt, not tested.
3.5. Impact of RXR ligand binding on its apo or holo-NR partner and other proteins
3.5.1. Ligand-induced RXR–androgen receptor crosstalk
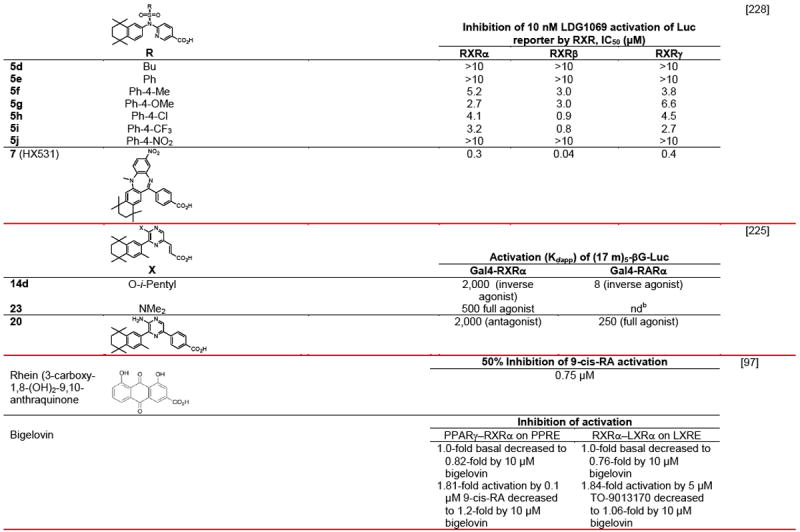
RXRα was shown to crosstalk with androgen receptor (AR) in prostate cancer cells [36]. At 1.0 μM, both 9-cis-RA and the rexinoid agonist LGD101305, which is the 3′-fluoro analog of LG100268, repressed the activation of the AR on the MMTV-ARE-Luc reporter construct by 10 nM dihydrotesterone (DHT) in transfected PC-3 prostate cancer cells. Repression of DHT-induced activation on the p(ARE)4-Luc and prostate specific antigen (PSA)-Luc reporter constructs by 9-cis-RA was observed in androgen-independent PC-3 and androgen-dependent LNCaP prostate cancer cells. Similarly, 10 nM DHT plus transfected AR repressed the activation of the pCRBP-II DR-1-Luc reporter by RXRα and 1.0 μM 9-cis-RA in co-transfected PC-3 cells, whereas 9-cis-RA inhibited the interaction between AR and its ARE. Co-immunoprecipitation and GST-pull-down indicated interaction between RXRα and AR. Mutational deletions suggested that the interaction interface was between the RXRα A/B plus LBD H4–H6 domains and the AR LBD H7 (772–800). The authors suggested that one of the mechanisms by which rexinoids prevented androgen-dependent cancer cell growth was by the binding of the RXR–rexinoid complex to the apo- or holo-AR homodimer, which then blocked the AR homodimer from binding to AREs to prevent DHT-induced gene transcription.
3.5.2. Importance of RXR ligand on coactivator and dimeric partner binding
The binding of the CoA SRC-1 peptide containing the NR box Leu-His-Arg-Leu-Leu motif to RXRβ complexed with RXR agonist 9-cis-RA or PA024 (Fig. 1B and Table 4) was detected using surface plasmon resonance (SPR) [37]. The CoA peptide-biotin conjugate was immobilized on streptavidin chips, which were then treated with recombinant RXRβ that had been pre-incubated with 9-cis-RA or PA024 alone or in the presence of RXR antagonist HX531 (Fig. 1D [38] and Table 5). The Kd value for the SRC-1 peptide binding to the RXRβ–9-cis-RA complex was 59 nM and the Ka was 1.7 × 107 M−1. Kd values for the binding of the apo-LXRα and apo-LXRβ LBDs to the chip-immobilized RXRα LBD were 0.29 and 1.1 μM, respectively, and decreased to 2.2 nM and 2.3 nM, respectively, in the presence of the LXR ligand 22R-hydroxycholesterol, providing evidence that the bound LXR ligand enhanced the strength of the LBD–CoA bond [37]. Respective Kd values of apo-RXRβ binding to immobilized LXRα and LXRβ were 0.62 μM and 0.78 μM, and in the presence of 9-cis-RA decreased to 0.53 pM and 0.41 nM. Thus, ligand binding was considered to strengthen the heterodimer LBD interface between the RXRβ LBD and that of its NR partner.
3.6. Rexinoid synergists
3.6.1. Synergists reveal differential roles for RXR isotypes in heterodimers with NGFI-B
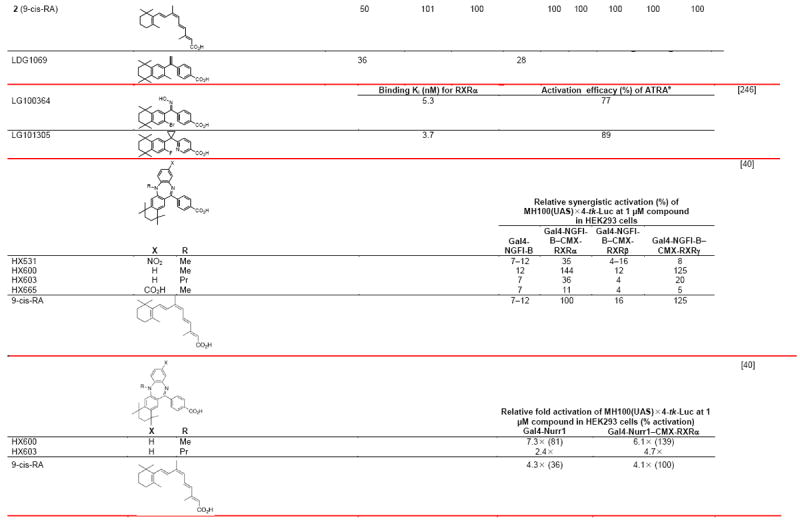
Several retinoids reported by Shudo, Kagechika, and coworkers were termed RXR synergists because of their inability to induce robust HL-60 myeloid leukemia cell differentiation alone but to enhance that induced by RAR agonists [39]. HX600 functioned as a weak RXR agonist, whereas HX531 (Fig. 1D), HX603, and HX665 (Table 4) were unable to activate RXR in a reporter assay. Only HX603 inhibited reporter activation induced by 9-cis-RA and so behaved as a rexinoid antagonist in this context. Unlike 9-cis-RA, the RXR synergists HX531, HX600, HX603, and HX665 (Fig. 1C) at 1.0 μM were unable to activate Gal4-RXRα–γ LBD chimeras on the MH100(UAS)×4-tk-Luc reporter in transfected HEK293 cells [40]. Except for the decrease in reporter response induced by HX603, the other compounds at 1 μM did not affect the level of activation of Gal4-RXRα by 0.1 μM 9-cis-RA. Neither the synergists nor 9-cis-RA alone induced Gal4-NGFI-B to activate its reporter construct. However, 9-cis-RA or HX600 at 0.1 or 1.0 μM induced robust reporter activation when Gal4-NGFI-B was cotransfected with RXRα or RXRγ, while reporter activation by HX603 at 1.0 μM was weaker. In contrast, these rexinoids were unable to activate the reporter when Gal4-NGFI-B was transfected with RXRβ. Co-transfection of both wild-type RXRα and NGFI-B with the DR-5 NX3′X3-tk-Luc reporter produced a greater than additive response on treatment with 1.0 μM 9-cis-RA, HX600, or HX603 compared with transfection with each receptor alone. Morita and colleagues attributed the activation mediated by the binding of the synergists to the RXR–NGFI-B heterodimer to be allosteric. The structures and activities of these compounds are shown in Table 4.
3.6.2. Effects of synergists on RXRα–orphan NR Nurr1 heterodimer
RXR synergists HX600 and HX603 activated the Gal4-Nurr1–RXRα heterodimer on the MH100(UAS)×4-tk-Luc reporter [40]. At 1.0 μM, HX600 alone activated Gal4–Nurr1 on this reporter, and further enhanced reporter activation in the presence of cotransfected RXRα. However, HX600 was unable to effect the activation of Gal4 constructs with NRs NOR1, DHR38, FXR, PPARγ, TRβ, and RARα on this reporter construct or that of full-length LXRα, LXRβ, or FXR on the inverted repeat (IR)-1×3-tk-Luc. These results suggested an unusual allosteric interaction between RXR and Nurr1 that was not observed with other NR partners.
3.7. Other agents functioning as RXR ligands
3.7.1. Organotins
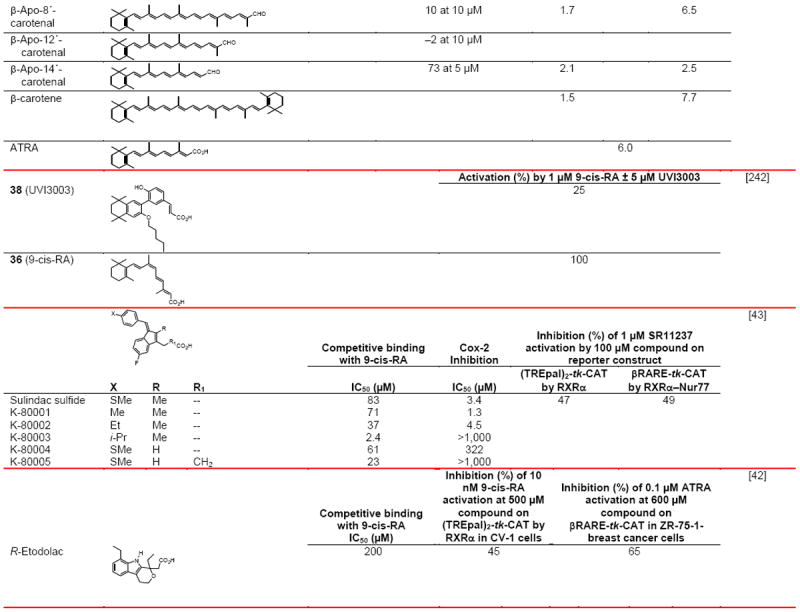
As Table 4 indicates, the organotins that are used as anti-fouling agents on ship hulls and other marine structures are potent RXR ligands [41]. They are also disruptors of marine life. Notably, they cause sex reversal and so lead to species decline.
3.7.2. Nonsteroidal anti-inflammatory agents
The nonsteroidal anti-inflammatory drugs R-etodolac and sulindac sulfide were reported to interact with RXR as transcriptional antagonists [42,43]. Administration of R-etodolac induced the ubiquitination of RXRα, decreased RXRα levels in prostate tissues of TRAMP mice, and reduced their incidences of gross urogenital mass and metastasis to 12% and 29% compared to 25% and 58% in the nontreated control mice [42]. R-etodolac also inhibited β-catenin-mediated signaling on a TCF–LEF-dependent reporter through interaction with the PPARγ–RXRα heterodimer [44]. Sulindac sulfide at 75 μM induced >80% apoptosis of F9 teratocarcinoma cells at 24 h [43]. Apoptosis was independent of the ability of these drugs to inhibit the activity of cyclooxygenase-2 but dependent on their interaction with RXRα or its truncated form (tRXRα).
3.8. Effects of combinations of rexinoid and the ligand of the NR partner
3.8.1. PPARγ
RXR agonist LGD1069 (Fig. 1B) and the PPARγ agonist rosiglitazone (Table 4), which were combined at 0.5 μM each, up-regulated the expression of 20 genes ≥3.8-fold at 24 h in A375(DRO) melanoma cells [45]. The four most highly up-regulated genes were TIE1 (122×), S100A2 (69×), IL1B (40×), and ANGPTL4 (32×). At 1.0 μM, LGD1069 and PPARγ agonist pioglitazone individually up-regulated S100A2 3.4× and 4.9×, respectively, to indicate that the combination was synergistic. Note that the authors either used two different agents or misnamed one. The finding that S100A2 expression was higher in premalignant nevi than in primary melanoma tumors or their metastases suggested to the authors that the loss of the calcium-binding protein S100A2 had a role in transformation. Knock-down of S100A2 expression by shRNA was accompanied by reduced anti-proliferative responses to LGD1069 and rosiglitazone either alone or combined, while in nontransfected wild-type cells inhibition of proliferation was enhanced by treatment by either ligand or their combination.
Combination treatment with 100 nM PPARγ agonist rosiglitazone (BRL 49653) and 50 nM 9-cis-RA decreased MCF-7, tamoxifen-resistant MCF-7 TR1, SKBR-3, and T47D breast cancer cell viability to a greater extent than would be expected from the additive effects of the agents [46]. MCF-7 cells were shown to undergo apoptosis. In contrast, the combination or either agent alone had no effect on the viability of the immortalized normal breast epithelial cell line MCF-10A. The combination induced the expression of p53 in MCF-7 cells, whereas each agent alone did not. Deletion analysis of the p53 promoter indicated that its NFκB site was responsible for this effect. The electromobility shift assay (EMSA) was used to demonstrate that PPARγ–RXRα bound to this promoter site. High expression of RXRα in PPARγ-co-transfected CV-1 monkey kidney cancer cells attenuated the response of the PPARγ–RXRα on the PPRE DR-1-tk-Luc promoter to troglitazone [47]. The nontransfected control cells did not express PPARγ or RXRα in the presence or absence of troglitazone. The authors suggested that the excess uncomplexed RXRα had bound essential cofactors that were necessary for activation of reporter expression by the PPARγ–RXRα hetrodimer.
The combination of the PPARγ ligand 15-deoxy-Δ12,14-PgJ2 or rosiglitazone with the RXR agonist 9-cis-RA induced the expression of glutathione S-transferase (GST), a phase II liver enzyme with a role in carcinogen detoxification via its formation of glutathione conjugates of carcinogens or carcinogen metabolites, which could then be excreted in the bile [48]. The concentration of glutathione in liver is 10 mM, and primary hepatocytes express PPARγ1. The authors explained that the GST promoter had binding sites for the NF-E2-related factor (Nrf) and CCAAT/enhancer binding protein (C/EBP) β, both of which were required for GST expression, and that both transcription factors had a PPRE in their promoters.
3.8.2. RAR
Treatment of human colon adenocarcinoma CaCo-2 cells with 0.1 μM RARα-selective Am580 produced a 2.5-fold induction of mRNA for the breast cancer resistance protein (BCRP), which acts as an ABC transporter of the benzo[a]pyrene sulfate metabolite [49]. This induction was enhanced to 10.7-fold by 0.01 μM RXR agonist CD2608/LGD1069, which alone induced BCRP expression 5.0-fold. Note that the rexinoid was named incorrectly but its chemical structure was correct in Fig. 1 of the cited paper. Differentiation of human NB4 myeloid leukemia cells to granulocytes was induced by RARα-selective agonist BMS753; however, both the RARα agonist and an RXR-selective agonist BMS649 (SR11237) were required to induce the expression of the cytochrome P450 26A1 enzyme, which was also induced by ATRA [50], which can isomerize to produce 9-cis-RA. Cyp26A1, which functions as an RA 4-hydroxylase, has a role in the development of retinoid resistance. Thus, the authors concluded that combination therapy with RXR and RAR-selective retinoids could have either positive or negative effects on cancer cell response.
3.8.3. Synergism with anti-cancer drugs
Treatment with LGD1069 (bexarotene) alone at 10 μM produced only minimal growth inhibition (≤ 25%) of Calu3, EKVX, H358M, H441, HOP62, HOP92, and SKMES1 non-small cell lung cancer (NSCLC) cell lines, and 30–50% inhibition of A427, A549, and H322M NSCLC cell lines although all lines expressed RXRβ and only SK-MES-1 failed to express RXRα [51]. However, co-treatment of the cells with LGD1069 (bexarotene) at 1 μM was able to reduce the IC50 values of pacitaxel by 32–54% and vinorelbine by 18–48%. The IC50 value for 50% cell growth inhibition by paclitaxel was reduced 0.4 log and that by vinorelbine by 0.45 log in the presence of 1 μM LGD1069. The combination of 20 mg/kg of paclitaxel or 2.5 mg/kg of vinolrebine with 100 mg/kg of LGD1069 reduced Calu-6 tumor volume in treated mice to 32% and 59%, respectively, of that of the nontreated tumor-bearing mice, and these values were greater than the reductions produced by either agent alone. LGD1069 (4 μM) was also reported to synergize with paclitaxel (40 nM) in NMU-417 rat mammary cancer cells, which had been derived from an N-methylnitrosourea (NMU)-induced rat mammary cancer, leading to their increased apoptosis (8-fold combined vs 1.03-fold and 3-fold alone, respectively) [52]. The complete regression of existing NMU-induced mammary tumors (≥75 mm2) in the rat was increased to 80% after 6 weeks by combined dosing of oral LGD1069 (100 mg/kg) daily and intraperitoneal paclitaxel (20 mg/kg) weekly compared to either agent alone (6% and 54%, respectively).
In a phase III trial comparing the addition of bexarotene to a cisplatin and vinorelbine combination with the two anti-cancer drug combination alone in patients with advanced or metastatic NSCLC (n = 623), no significant difference in survival time was observed in the treatment arms except in a subgroup of patients, who were male smokers that had experienced a weight loss ≥ 5% in the past 6 months, stage IV disease, and grade 3/4 triglyceridemia [53]. The group on the bexarotene plus drug combination had a 12.3-month median survival time compared to the 9.2-month median survival of the anti-cancer drug combination alone-treated group. In another phase III trial comparing the addition of bexarotene to carboplatin plus paclitaxel combination with the two anti-cancer drug combination in advanced or metastatic NSCLC patients (n = 612), no significant difference was observed in the treatment arms overall [54]. However, a similarly categorized group that had grade 3/4 triglyceridemia survived 3.2 months longer than the anti-cancer drug combination alone-treated group, which had a 9.2-month median survival [54]. These results suggest a more personalized approach to using RXR ligands for NSCLC cancer treatment.
4. RXR Interaction Partners
Protein partners of the RXRs include (i) transcription factors, including the NRs—CAR, EAR2, FXR, LXRs α and β, NGFI-B/Nur77/TR3, Nurr1, PPARs α, β/δ, and γ, PNR, PXR/SXR, RARs α–γ, RXRs α–γ, SHP, TRs α and β, and VDR; the circadian rhythm transcription factors Arntl/Bmal1, Clock, and nPAS2/MOP4; and others such as Bcl3, integrin β3-binding protein, MyoD, NFκB-1, NFκB-1B, Oct1/POU2F1, Oct2/POU2F2, RelA, SMAD-2, SP1, and TATA-binding protein; (ii) transcriptional cofactors including CoAs such as BRD8, CNOT1, EDF1, Med24/Trap100, Med25, NCoAs 1–3, 6, and 62/SNW1, NRBF2, PGC-1α, PNRC2, and TIF-1α/TRIM24; and CoRs such as the histone deacetylases (HDACs) 3 and 4, NCoR2, and RIP-140/NRIP1; (iii) DNA-modifying agents such as DNTTIP2, FUS, GADD45s α and γ, MPG, and T:G mismatch-specific thymine DNA glycosylase (TDG); and (iv) other proteins, including CTLS1, Cyp27B1, DAND5, insulin growth factor-binding protein (IGFBP) 3, importin β, PRKD2, RNF8, TAGT-12/TMPRSS3, and ubiquitin ligase N4. These proteins are listed in Fig. 2 in a review article by Lefebvre et al. [13]. Insulin growth factor-binding protein (IGFBP)-3 was also found to interact with RXRα in a GST pull-down assay [55].
4.1. Cofactors
Generally, during cofactor binding the NR LBD H12 shifts to associate with H3 and H4 to generate domains for binding by CoAs or CoRs. The NR interaction domains of these coregulatory proteins differ. The CoA two-turn α-helix Leu-XX-Leu-Leu or NR box motif and the CoR three-turn α-helix Leu-XXX-Ile-XXX-Leu sequences (X is unspecified) allow them to dock into hydrophobic pockets consisting of the H12 AF-2 and residues from H3 and H4, which then are stabilized by two charge clamps between the cofactor and its binding pocket. The longer length of the CoR motif is considered to sterically hinder H12 from adopting an agonist conformation. Whether the CoR binding surface is larger than that of the CoA or whether other structural changes occur has not been investigated for the RXRs in the context of their complexes with the native cofactors.
Two-hybrid recruitment assays indicated that the apo-RXRβ, RARα, and TR LBDs bound the CoR silencing mediator of retinoid and thyroid hormone receptors (SMRT) and that SMRT dissociated from RARα and TR after treatment with their respective agonists (Am580 and T3), whereas SMRT did not dissociate on treatment of RXRβ with its agonist LG100268 (Fig. 1B) [56]. Dissociation of SMRT from the LG100268–RXRβ LBD complex required the presence of the CoA ACRT. The crystal structure 1H9U of the LG100268–RXRβ LBD homodimer complex revealed that H12 occupied two positions, one of which had H12 in the canonical agonist conformation (AF-2 surface), whereas in the other H12 was located below the LBD. Schwabe and colleagues concluded that agonist binding alone to the RXR LBD was unable to induce transcriptional agonism but required the bound CoA. The authors observed that, unlike the conformation of 9-cis-RA in the LBP, in which 9-cis-RA did not contact H12, the 3′-methyl group of LG100268 would contact the side chain of RXRβ H12 Leu522 (RXRα H12 Leu451) and through this contact would stabilize the contact between H12 and the CoA. As a result, LG100268 would produce higher transactivation activity than that observed on binding of 9-cis-RA. Other reseasons given for the higher transactivation activity of LG100268 were its greater volume (355 Å3) compared to that of 9-cis-RA (315 Å3) that would lead to additional contacts with the LBP surface and its greater rigidity that would entropically favor binding.
Cofactors of RXR have been reviewed by Wei [57].
4.1.1. Coactivators
NR box structure
Structural analyses of NR LBD–CoA peptide complexes revealed that the CoA Leu-X-X-Leu-Leu motif had a two-turn α-helical structure. On CoA peptide binding to the hydrophobic NR LBD AF-2 surface such as that of LXR, which was comprised of residues from H3, H4, and H12, conserved H3 lysine and H12 glutamate residues formed a charge clamp to stabilize the binding by the CoA motif, for example, Leu-Ser-Gln-Leu-Leu in the CoA activating signal co-integrator-2 (ASC-2) [58]. The three leucines in this motif interacted with the AF-2 hydrophobic surface, and its three upstream (N-terminus) residues (Pro-Thr-Ser in ASC-2) played an essential role in determining binding selectivity.
Coactivator selectivity
Mutational analysis of the NR box regions of the CoAs steroid receptor coactivator (SRC) 1 and CREB-binding protein (CBP/p300) demonstrated that RXR preferentially bound the former and PPARγ the latter [59]. Of the three NR boxes in SRC-1, LG100268-bound RXR had the highest affinity for NR box III followed by I and then II, and none for the CBP NR box. Of the NR box III residues conferring binding selectivity, the +2 and 3+ positions of the sequence 749-Leu-Arg-Tyr-Leu-Leu-753 played important roles, as did the immediate upstream (−1) and downstream (+6) residues (Leu and Glu, respectively).
Phantom effect
As a dimeric partner of a NR, RXR played a role in its transcriptional activity. The peptide Co4aN (1429–1511) derived from ASC-2 (also called NRC/RAP250/TRBP/PRIP/gene product of AIB3), which contained the ASC-2 NR box 2, interacted with LXRα LBD residues from H3–H5 and H12. Binding by the peptide to the 22R-hydroxycholesterol–LXRα LBD complex was enhanced five-fold by the dimerization of the holo-LXRα LBD with the apo-RXR LBD and further enhanced two-fold in the presence of RXR agonist 9-cis-RA (0.1 μM) [58]. This RXR heterodimerization-mediated allosteric enhancement that required the presence of the RXR H12 (AF-2) was termed the “phantom effect”. The authors posited that the asymmetric interaction between the LXRα LBD and RXR LBD stabilized the LXRα LBD H12 in a more favorable position that allowed the CoA peptide to bind even in the presence of the apo-LXRα.
RFP8
The ring finger protein (RFP) 8 was found to interact with the apo-RXRα by using a two-hybrid assay on a human liver cDNA library [60]. RFP8 stimulated RXRα transactivation activity on CRBP-II and RARβ RARE-linked reporter constructs, which was then augmented by 9-cis-RA (1 μM). The RFP8 and RXRα proteins colocalized in the nucleus of transfected COS-7 cells. The interaction site with RFP8 was predominantly in the N-terminal 28 residues of the RXRα A/B domain. Although RFP8 had previously been found to interact with the ubiquitin-conjugating enzyme UBE2E2, which mediates RXRα ubiquitination in hepatoma cells, RFP8 did not enhance RXRα degradation through the ubiquitin pathway.
PCAF
The CoA p300/CBP-associating factor (PCAF) interacted with the apo-RARα LBD–apo-RXRβ LBD heterodimer and that interaction was enhanced by the RXR agonist AGN194204 (Fig. 1B) in GST pull-down assays [61]. The apo-RARα LBD also interacted with PCAF but that interaction was not enhanced by AGN194204, whereas interaction between PCAF and the apo-RXRβ LBD was low and enhanced in the presence of the RXR agonist. In contrast, the interaction of the NR DBDs with PCAF was constitutive. Agonist binding induced the formation of 1:1 stoichometric complexes of PCAF–RARα LBD–RXRβ LBD. The efficacy of PCAF binding to the full heterodimer was enhanced five-fold in the presence of RXR agonist AGN194204 (Kd 47.2 nM vs 9.4 nM, respectively) with the differences in the PCAF off-rate having the higher impact. A similar enhancement occurred with the heterodimer bound to the βRARE.
4.1.2. Coactivator–NR interaction relationships
Similarity cluster analyses of NR holo-LBP and CoA binding sites were conducted using the crystallographic structures of 177 human NRs from the PDB [62]. On the basis of the hydrophobic and polar contact preferences of 35 CoA-binding sites, RXRα and RXRβ of the RXRs (α, β, and γ, which were represented by respective PDB structures 1MV9, 1UHL, and 2GL8) were most closely related. The RXRs were next most closely related to CAR, followed by RARs β and γ, PPARγ, and LXRβ. When both ligand-binding and CoA site fields were combined, RXRs α and β were again more closely related to each other than to RXRγ. The RXRs were next most closely related to HNF4α and HNF4γ.
Dynamics methods were used to model the binding of 9-cis-RA and/or a TRAP220 CoA peptide to the apo- and holo-RXRα LBDs as represented in crystal structures 1LBD and 1XDK, respectively, as a means investigating agonist–NR–CoA peptide interactions [63]. A dynamics comparison of the two structures revealed that of 217 residues, the positions of 71 were conserved (rmsd = 0.3 Å), whereas 19 LBD and 16 AF-2 site residues had rmsds of 3.0 Å and 14.8 Å, respectively, to indicate less positional conservation in the LBP and even less in the AF-2. Protein dynamics and thermodynamics on the Cα and Cβ backbone atoms and pseudoatoms that represented the centers of the residue side-chains were used to study whether ligand and CoA binding was sequential or simultaneous. The outcome being in agreement with the crystal structures led the authors to conclude that ligand and CoA binding to the RXRα LBD occurred sequentially; however, their analysis was unable to predict the binding order.
4.1.3. CoA–AF-2 Interaction
An ab initio fragment molecular orbital study of the RXRα LBD–9-cis-RA complex with the CoA steroid receptor coactivator (SRC) 1 peptide NR box motif was performed to investigate the role of the transcriptional AF-2 core [24]. AF-2 Glu453 and Glu456 were separately mutated to lysine to investigate the role of each glutamate. These studies suggested that the strength of the transcriptional response paralleled that of the strength of the AF-2–CoA interaction.
Aromatic clamp locks H12 in the agonist conformation
Comparison of holo-RXR structures alone and their complexes with CoA NR box peptides demonstrated the roles of three phenylalanine residues (RXRα LBD H3 Phe282, H11 Phe442, and H12 Phe455) in facilitating CoA peptide binding [64]. In the mouse apo-RXRα1 LBD(227–467) these phenylalanines were exposed to solvent and did not interact with other residues. After CoA TRAP220 NR box 2 peptide bound to the mouse 9-cis-RA–RARβ2 LBD–RXRα1 LBD complex, the side chains of the phenylalanines reoriented to come into close contact with H12 and the TRAP220 peptide. Comparison of this complex with that of 9-cis-RA–RXRα LBD showed that TRAP220 peptide binding produced a 110° rotation about the H3 Phe282 Cα–Cβ bond that led to a 104° pivot of H12 Phe455 and a 2-Å shift of H12 towards both H3 and the CoA peptide that resulted in the stabilization of Phe455 in a hydrophobic pocket comprised of H3 Leu281, Phe282, and Leu285, H12 Met459, and two leucines and a methionine of the peptide. H11 Phe442 shifted by 112° to contact H12 Leu460 and H3 Phe282 to form the aromatic clamp that correctly positioned H12. While not inhibiting the binding efficacy of 9-cis-RA, mutation of Phe282 or Phe442 to alanine blocked the transactivational activity of the 9-cis-RA–RXRα homodimer on the DR-1-tk-CAT reporter, thereby demonstrating the importance of these residues in stabilizing CoA binding.
Role of RXRα LBD H12 in CoA recruitment by a non-permissive heterodimer (apo-RXRα–apo-RARα)
Reconstitution experiments in yeast, which did not express the CoA SRC-3 (pCIP/AIB-1/RAC-3/TRAM-1/ACTR) or the CoR SMRT, reporter assays, and EMSAs were used to demonstrate that binding of the RXR agonist LG100268 to the wild-type RXRα–RARα heterodimer induced the recruitment of the SRC-3 receptor interacting domain (RID) peptide and activation of a DR-5 reporter construct [65]. However, this complex was unable to induce the release of the SMRT RID peptide, which only occurred in the presence of the RAR agonist ATRA, which also had recruited the CoA peptide [65]. The RXRαΔ(403) mutant lacked the RXR H11–H12 loop and H12. The RXRαΔ(403)–RARα heterodimer remained responsive to ATRA-mediated CoA recruitment, whereas RXR agonist LG100268 weakly impaired CoA recruitment but did not impact CoR binding. On the DR-1 reporter construct, the wild-type RXRα–RARα heterodimer was activated by ATRA to recruit SRC-3 and lose SMRT, whereas LG100268 only weakly inhibited SRC-3 binding and had no effect on SMRT dissociation. Similar results were observed with the wild-type and mutant RXR heterodimers with TRβ. Three-hybrid assays in yeast showed that the mutant RXR heterodimer had higher affinity (two-fold) for SRC-3 than the wild-type heterodimer. The authors concluded that the RXR LBD H12 inhibited CoA association in the apo-heterodimer but was essential for heterodimer activation by a rexinoid agonist. Supporting the role of the RXRα H12, was the finding that Xenopus laevis embryos expressing a mutant with a deletion of the RXR LBD H12, Gal4-RXRΔ(470–488), were unable to activate the 14×UAS-E1b-Luc reporter in the presence of LG100268 [31].
4.1.4. Impact of CoA binding on bound ligand
Muccio and colleagues conducted a thorough investigation of the binding of the CoA glucocorticoid receptor-interacting protein (GRIP) 1 NR box 2 peptide by the 9-cis-RA–RXRα LBD homodimer complex [66]. Their analysis was based on comparisons of the crystal structure of this complex with that of 9-cis-RA–RXRα LBD complex (PDB 1FBY) and their thermodynamic properties, which had been determined by isothermal calorimetry (ITC), hydrogen/deuterium exchange mass spectrometry (HDX MS), and UV spectroscopy of their complexes in solution [66]. The 9-cis-RA–RXRα LBD–GRIP-1 structure revealed changes typical of an holo-RXR–CoA peptide complex, namely the H1—H3 loop in which H2 was unwound and the GRIP-1 peptide bound as a two-turn helix in the CoA pocket formed by residues from H3, H4, and H12. The RXRα LBD stabilized GRIP-1 peptide binding through the formation of charge clamps. Its H12 Glu453 carboxylate group formed one such charge clamp through three H-bond interactions with GRIP-1 backbone NHs and its H3 Lys284 amino group formed the second charge clamp by a salt bridge with the GRIP-1 histidine ring. Binding was further stabilized by the GRIP-1 NR box 2 motif leucines and the–1 upstream isoleucine interacting with hydrophobic residues from H3, H4, and H12. These interactions required changes in the conformations of RXRα LBD H3, H4 and H12 residues. For example, H4 Arg302 was induced to shift towards H12 to permit a salt bridge between its guanidinium group and the H12 Glu453 and Glu456 carboxylate Os, which stabilized the H12 position. H-bonds also formed between the Arg302 guanidinium group Ns and the peptide backbone.
Most interesting, just as bound rexinoid agonists affected LBD geometry to facilitate CoA binding, the binding of the CoA impacted the positions of the LBP residues and 9-cis-RA geometry [66]. The authors showed that the bound GRIP-1 peptide (Kd = 0.9 μM by circular dichroism and 0.6 μM by ITC) reduced the dynamics of residues responsible for CoA binding and those near the bound agonist. The authors observed that binding by the GRIP-1 peptide had to be exothermic to overcome the entropy opposing binding. Peptide binding did not produce large changes in the positions of the H6, H7, H8, H9, H10, and β-sheet backbones (rmsd = 0.16 Å for 229 residues), although changes occurred near the CoA pocket and extended to H11 residues interacting with the 9-cis-RA hydrophobic terminus. Peptide binding induced a 2-Å shift in the H12 C-terminus towards the CoA peptide and H11 and the rotation of H11 Phe437 towards the H12 Leu455. As a result, the conformation about 9-cis-RA was altered so that its 16-methyl group had reduced contact with H11 His435 and Leu436, lost contact with H3 Ile268 and Cys269, but now contacted H11 Cys432 and His435. Contacts of the 9-cis-RA 17-methyl and 18-methyl groups and 2, 3, and 4-methylene groups with pocket residues from H3, H7, and H11 were also altered. The 9-cis-RA 16,17,18-trimethylcyclohexenyl ring was flipped to the alternative half-chair conformation and the surface area contact between 9-cis-RA and H11 increased from 118 Å2 to 140 Å2, whereas the 9-cis-RA 6–7 torsion angle decreased from −70° to −20°. The contact between the 9-cis-RA side chain 19-methyl and H11 Leu436 increased, whereas that between its 20-methyl and H3 decreased. The important stabilizing ionic and H-bond interactions between the 9-cis-RA 15-carboxylate and the H5 Arg316 were maintained because the 9-cis-RA carboxylate shifted only 0.6 Å away. Thus, 9-cis-RA binding continued to be stabilized, although its LBP surface contacts had been reduced by 30%. The authors concluded that GRIP-1 peptide binding stabilized both the GRIP-1–RXRα interface and interior structure one layer from the bound ligand. The importance of the H11 Phe437 in transmitting these conformational changes between ligand and peptide was noted.
4.1.5. Heterodimeric partner affects CoA Binding to RXR
The dissociation constants (Kds) for binding of a CoA-derived NR box to the RXRα LBD were compared to those of the RARα LBD. Using fluorescently tagged peptides, the binding affinities of the SCR-1 NR box 1–3 peptide and the TRAP220 NR box 2 peptide were found to be lower for RXRα alone on the basis of their Kd values (53 and 24, respectively) than those for RARα alone (8.2 and 10.9, respectively) [64]. Similarly, in the presence of their respective agonists (CD3254 in Fig. 1B and Am80), peptide affinities were also lower for holo-RXRα (1.5 and 1.9, respectively) than holo-RARα (0.58 and 0.47, respectively). In the context of the RXRα–RARα heterodimer, RARα-selective Am80 enhanced affinity for the NR boxes 1 and 3 and NR box 2 peptides (0.40, 0.40, and 0.44, respectively) compared to those of the apo-heterodimer (4.8 and 3.0, respectively), which were not appreciably altered by presence of the RXR antagonist UVI3003 (Fig. 1D) (0.48 and 0.43, respectively). Interestingly, the RXR agonist CD3254 exerted effects on recruitment of the peptides to the heterodimer (0.4 and 1.0, respectively) and its complexes with Am80 (0.13 and 0.39, respectively) and RARα antagonist BMS614 (1.2 and 1.9. respectively). Notably, the impact of CD3254 on NR box 2 peptide recruitment to the heterodimer was diminished in the presence of Am80 (0.39). These values were determined using fluorescence anisotropy measurements at ligand concentrations above saturation levels and assumed one peptide bound by a monomer or a dimer. These results suggested to the authors that the agonist-bound LBDs acted independently in recruiting the CoA peptides. However, peptide recruitment to the RARα–RXRα–CD3254 complex in the presence of RARα agonist Am80 or antagonist BMS614 was lower than that induced by the apo-RARα alone to suggest that both heterodimerization and the structure of the RARα ligand played roles. In the context of recruitment of the native CoAs with multiple NR boxes cooperativity was observed between the two LBDs.
4.1.6. Corepressors
The general dogma of CoAs binding NRs complexed with transcriptional agonists and CoRs binding to antagonist complexes has been expanded to include the findings that agonist binding can also lead to the recruitment of certain CoRs, which interact with NRs through their CoRNR motifs (Leu/Ile-X-X-Ile/Val-Ile) [67].
Dec1 and Dec2
The basic helix-loop-helix DEC1 (BHLH E40 or B2/Stra13/Sharp2) and DEC 2 (BHLH E41 or B3/Sharp 2) transcription factors functioned as ligand-dependent RXR CoRs in the context of the RXR homodimer or its heterodimers with LXR, FXR, RAR, and VDR [68]. DEC2 was more potent than DEC1 in repressing 9-cis-RA-activated RXRα. Unlike DEC1, DEC2 was resistant to inhibition by the HDAc inhibitor trichostatin A and prevented CoA DRIP205 binding by RXRα. Interaction of the DECs with RXR was direct, mediated through the DEC N-terminal Leu-Lys-Asp-Leu-Leu CoA motif, and enhanced by 9-cis-RA binding. DEC2 suppressed the activation of Gal4-RXRs by 9-cis-RA, whereas the effects of DEC1 on the 9-cis-RA-bound Gal4-RXRα, β, and γ constructs were respectively modest, ineffective, and weak. The role of DEC in circadian rhythm was suggested by its attenuation of RXR–LXR signaling in the liver.
COPR1 and COPR2
The comodulators of PPAR and RXR (COPRs) 1 and 2 were detected in normal human keratinocytes and MCF-7 breast cancer cells and found to function as partial CoRs because of their proline-rich autonomous activation domains [69]. Both COPRs bound to the RXRα AF-2 site through their NR box motif Leu-Leu-Tyr-Leu-Leu. COPR binding and corepression were enhanced in the 9-cis-RA–RXRα complex. Interactions of COPR1 and COPR2 with RXRα were stronger than those with the apo-PPAR and 9-cis-RA–RARα and RARγ complexes. Interaction of RXRα with COPR1 was higher than that with COPR2, which has a 50-residue insert N-terminal to the NR box.
RIP140
The rexinoid agonist AGN194204 (Fig. 1B) only had a minor impact on binding of the RARα–RXRβ heterodimer by the CoR RIP140 (NRIP1) C-terminus (RIP-C) peptide because the Kd value was only decreased to 2.0 nM compared to the apo-Kd of 3.6 nM [61]. The C-terminus of RIP140 has an agonist-dependent motif, Leu-Tyr-Tyr-Met-Leu, whereas full-length RIP140 has nine traditional NR box motifs [67]. RIP140 binding led to the recruitment of HDAc to silence transcription in the presence of bound agonist. Moreover, binding by RIP-C or RIP140 to the heterodimer was stronger in either the presence or absence of the rexinoid compared to CoA PCAF binding (Kd of 2 nM for RIP-C versus 9.4 nM for PCAF in the presence of AGN194294 as shown by competition assays, SPR, co-immunoprecipitation, or altered histone acetylation levels [61]. Kinetics revealed that the Kon rate for RIP-C was 19-fold higher (142 × 103/Msec) than that for PCAF. Interestingly, the authors observed that the rexinoid could even induce the recruitment of PCAF to the apo-RARα–RXRα heterodimer.
4.2. Insulin-like growth factor binding protein-3
IGFBP-3 was found to inhibit activation of an RARE by ATRA but to enhance that of an RXRE by a rexinoid [70]. Treatment of 22RV1 human prostate cancer cells with IGFBP-3 or 0.5 μM RXR agonist VTP194204 (AGN194204 in Fig. 1B) for 72 h inhibited proliferation by 22% and 37%, respectively, whereas their combination inhibited proliferation by 55%, a value slightly below the additive effect of 59%. The combination of LGD1069 (Fig. 1B) and IGFBP-3 had higher than additive effects on the induction of human LAPC-4 and LNCaP prostate cancer cell apoptosis in vitro at both 24 h and 48 h (2.36-fold vs 1.32-fold and 1.57-fold, and 1.90-fold vs 1.27-fold and 1.26-fold, respectively). Induction of caspase-3/7 activity at 24 h was also above additive. Neither agent alone had significant effects on tumor weight or serum prostate specific antigen (PSA) level when administered by intraperitoneal injection in a murine LAPC-4 prostate cancer xenograft model, but the combination of IGFBP-3 (4 mg/kg/day) and rexinoid (4 mg/kg/day) reduced tumor weight by 54% and PSA levels by 40% compared to the saline-treated control mice.
Transfection of IGFBP-3 into HL-60 and NB4 acute myeloid leukemia cells, which normally do not express this protein, was observed to enhance differentiation-induced by RXR agonist AGN194204 and down-regulate signaling by RAR and VDR that was induced by ATRA and vitamin D3 analog EB 1089, respectively [71].
4.3. β-Catenin
RXRα interacted with β-catenin, which mediates Wnt signaling in embryogenesis and tumorigenesis [72]. β-Catenin has an extra-nuclear role in cell adherent complexes, whereas in the nucleus it functions as a CoA of the transcription factor T-cell factor/lymphoid enhancer factor (TCF/LEF) family through one or more of its five NR box motifs. RXRα agonist AGN194204 inhibited β-catenin-induced TCF/LEF-tk-Luc reporter activity. RXR agonists AGN194204, AGN195362, AGN195741, AGN196060, and 9-cis-RA induced β-catenin degradation through the proteasome pathway, which was blocked by the RXR antagonist AGN195393. Both the RXRα A/B domain and H12 were required for interaction with β-catenin. Wnt/β-catenin signaling was found to be constitutively activated in colorectal cancers and cancer stem cells [73]. Nuclear β-catenin was found to be expressed in 74% of 214 colorectal cancer samples. Its level of expression and distribution did not correlate with levels of LEF-1 (26%) or TCF4 (46%), which were considered as predictors of longer or shorter survival, respectively.
5. Dimerization
Increasing evidence indicates that RXR does not play a passive role as a heterodimeric partner but impacts the responses of its NR partner, regardless of its permissive, non-permissive, or conditionally permissive status.
5.1. Dimeric partner classification
5.1.1. Non-permissive versus permissive
Heterodimeric partners of RXRs have been classified as either non-permissive NRs (TRs and VDR), permissive NRs—farnesoid X receptor (FXR), liver X receptors (LXRs), and PPARs, or conditionally permissive NRs (RARs). The cartoons in Fig. 6 illustrate how RXR behaves in each of these heterodimers. In non-permissive heterodimers, RXR activity was classified as being subordinated to that of its partner, although RXR was still capable of binding its transcriptional agonists and interacting with CoAs [74]. Thus, a non-permissive NR in the context of an RXR heterodimer would only be activated in response to its own agonist, while the binding of RXR to a rexinoid agonist would not enhance the transactivational response induced by the NR–agonist complex on the NR response element (Fig. 6A). In such a heterodimer, a CoR would only be released by the binding of an agonist of the partnering NR, but not by the rexinoid agonist binding to RXR [75]. In permissive NR–RXR heterodimers, transactivation would occur through the binding by a transcriptional agonist to either partner or by agonists binding to both partners (Fig. 6B). Binding by both agonists could have additive or synergistic affects. A CoR protein would also be released when a rexinoid agonist bound to RXR [75]. The RAR heterodimers have a mixed status in that, while this activation of the apo-heterodimer would not be not affected by an RXR agonist, binding by an RAR agonist would induce both transactivation and permissivity so that an RXR agonist could then bind and enhance the activation by the RAR agonist (Fig. 6C).
Figure 6.
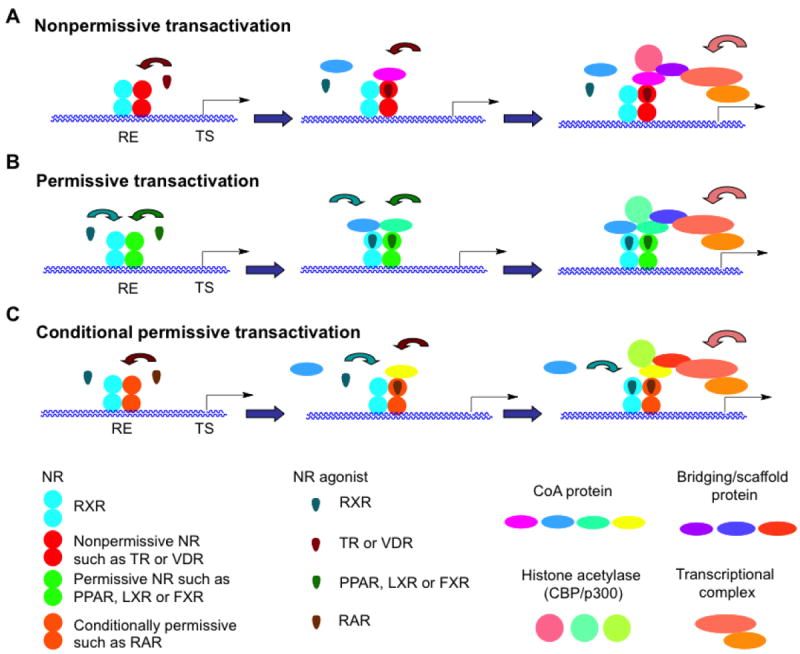
Transcriptional activation by nonpermissive, permissive, and conditionally permissive heterodimers of RXR and another nuclear receptor (NR). A. Transcriptional activation by a nonpermissive heterodimeric partner such as thyroid hormone receptor (TR) or vitamin D receptor (VDR). The nonpermissive NR is dominant so that binding by its agonist (T3 or VD3) controls the recruitment of a coactivator protein (CoA) to the TR or VDR AF-2 surface of the RXR–TR or VDR complex bound to its response element (TRE or VDRE) in the promoter region of a T3 or VD3-activated gene. The bound CoA could then recruit a histone acetylase, a bridging or scaffolding protein, and the transcriptional complex to initiate gene transcription from the nonpermissive-ligand responsive gene transcriptional start site (TS). Binding of an RXR agonist would not enhance the response induced by the bound TR or VDR agonist. B. Transcriptional activation by a permissive RXR heterodimeric partner such as farnesoid (bile acid) X receptor (FXR), liver (oxysterol) X receptor (LXR), and peroxisome proliferator-activated receptor (PPAR). An agonist of either partner in the heterodimeric pair such as RXR–PPAR could bind its own NR to initiate the recruitment of a CoA to the RXR–NR (RXR–PPAR) complex bound to its NRE (PPRE) in the promoter region of an NR (PPAR) agonist-responsive gene. Thus, either agonist-bound RXR or NR (PPAR) could recruit a coactivator (CoA), a histone acetylase, a bridging or scaffolding protein, and the transcriptional complex to initiate gene transcription. Binding of an agonist to the second NR in the dimer would enhance the transcriptional response induced by first NR–agonist complex either additively or synergistically. C. Transcriptional activation by the conditionally permissive heterodimeric partner RAR. Binding of the RAR agonist would control the transcriptional response and also permit the binding of an RXR agonist. Thus, the RAR–agonist complex would be permissive. The RXR–agonist complex would then enhance the transcriptional response induced by the RAR agonist. Adapted from Refs. [13] and [75].
Heterodimer structure was considered to define permissivity [75]. The conditionally permissive RXRα LBD–RARα LBD heterodimer was symmetric with no interaction occurring between the RARα H12 (AF-2 helix) and the RXRα LBD, whereas the permissive RXRα LBD–PPARγ LBD heterodimer was asymmetric with the PPARγ LBD H12 interacting with the RXRα LBD helices H7 and H10 even in the absence of a PPARγ ligand. Although the longer length of H12 in the NR partner has been suggested as facilitating this permissive interaction, H12 lengths in the permissive Nurr1 (NR4A2) and NGFI-B (NR4A1) orphan NRs were found to be shorter than those in the permissive PPAR and LXR LBDs and the same length as the non-permissive TRβ LBD H12.
Mutational analysis was used to show that the heterodimer interface between RXR and its NR partner did not mediate permissivity and the interaction with a CoR, but that the N-terminus of the LBD of the NR partner controlled the heterodimer classification status [75]. The dimerization-deficient RXRα(Ala416Lys) mutant, which could not heterodimerize with Nurr1, a CoR-binding-deficient RXRα(Leu294Arg) mutant, which could not interact with the CoR SMRT, and RXR chimeric constructs with the RARγ LBD were used to demonstrate that RXR recruited SMRT to the permissive RXR–Nurr1 heterodimer without altering the ability of Nurr1 to interact with SMRT or NCoR. Nurr1, itself, was unable to interact with a CoA and interacted only weakly with a CoR. Thus, binding by 9-cis-RA or the rexinoid agonist SR11237 [76] (Fig. 1B) to RXR inhibited RXR interaction with SMRT. In contrast, binding by RAR agonist TTNPB (Ro13-7410) [77], but not SR11237, inhibited the interaction between SMRT and RARγ in the non-permissive RXR–RARγ heterodimer.
5.1.2. Exceptions
Interestingly, exceptions to the standard permissivity definition [75] are increasing and appear to depend on the cell type, the RE of the NR partner, and cofactors present. Exceptons include the interactions of RXR with the non-permissive TRs and the permissive LXRs and PXR. Thus, categorization of the transcriptional response is becoming increasingly complex.
Nagy and coworkers conducted microarray profiling of genes regulated by 9-cis-RA and LG100268 when bound to RXR that was partnered with permissive LXRs and PPARs and non-permissive RARα and VDR and their agonists in human monocyte-derived dendritic cells [74]. These primary dendritic cells expressed mRNAs for RXRα, LXRs α and β, PPARs β/δ and γ, RARα, and VDR but did not, or only at very low levels, express mRNAs for CAR, FXR, PPARα, PXR, RARs β and γ, and TRs α and β. Treatment with an RXR agonist enhanced the original low expression of LXRα and PPARβ/δ. The authors found that the RXR agonists were unable to completely replicate the expression pattern induced by the permissive NR agonists—LXRα/β agonist GW3965, PPARβ/δ agonist GW1516, and PPARγ agonist rosiglitazone—on LXR and PPAR target genes as indicated by the incomplete overlap of altered gene sets at 12 h after treatment and the dose-response curves for LXR-dependent genes for ABCA1, ABCG1, LXRα, and SCD and for PPARβ/δ or γ-dependent genes for ADRP, CD36, FABP4, and PDK4. They concluded that in myeloid cells the RXR agonists and LXR or PPAR agonists employed different pathways to down-regulate gene expression and that RXR permissiveness for gene up-regulation was impaired. The RXR agonists were able to modulate only 14% and 8% of the respective genes regulated by RARα agonist Am580 and VDR agonist 1α,25-(OH)2-vitamin D3 (VD3). However, they did detect ATRA (2.2 nM), but not 9-cis-RA, in the cells, which could have modulated the expression induced by the RXR agonist. The differential modulation of LXR and PPAR-signaling pathways by RXR ligands suggested to these authors that RXR homodimer signaling was involved and that permissivity was defined by cell type and cofactors present. They concluded that RXR signaling was mediated both by multiple permissive heterodimers and independently of such heterodimers.
RXRα–TR
The non-permissive RXRα–chick TRβ heterodimer had the ability to function permissively [78]. The distal enhancer region upstream of the prolactin proximal promoter has a non-conserved DR-4. Transactivation on this site was activated by the RXRα–TRβ heterodimer in the presence of TR agonist T3 or RXRα agonist 9-cis-RA (Fig. 1A) or LG100268 (Fig. 1B), or the combination of T3 and either RXR agonist. Each ligand also enhanced CoA recruitment. Permissive activation occurred in pituitary cells and more weakly in GH4C1 cells but not in CV-1 cells in which T3 alone was responsible for CoA recruitment. The AF-2 domains of both receptors were required to recruit the CoA. The authors suggested that the availability of CoAs also affected the transactivational response.
Samuels and colleagues provided results that supported this finding [14]. Apo-RXRα inhibited T3-induced activation of the Gal4-TRα on a pCM100 reporter construct, whereas the 9-cis-RA–RXRα complex enhanced activation of the TRE-DR-4-CAT reporter. RXRβ alone or with 9-cis-RA had no effect. Deletion of the RXRα or RXRβ A/B domain indicated that the RXRα A/B was responsible for inhibition of Gal4-TR activity. The authors suggested that the RXRα A/B directly impacted the interaction of TR with T3. In addition, 9-cis-RA–RXR binding to the Gal4-TR inhibited T3 induced transactivation on the G5-tk-CAT but enhanced that of T3–TR on the TRE DR-4. In earlier work, this group showed that while 9-cis-RA was not able to activate the RXR–TR, it did induce the dissociation of a CoR from TR in the heterodimer [79]. The prolactin promoter, which has a DR-4 RE in its distal enhancer, is responsive to 9-cis-RA, which induces CoA recruitment to the heterodimer and activation, and also enhances activation induced by T3 in transfected pituitary cells [78]. These authors also reported that in transfected CV-1 cells, 9-cis-RA normally is unable to activate the RXR–TR unless unless high levels of an exogenous CoA are co-expressed.
RXR–pregnane X receptor heterotetramer
RXRα played a subordinate roll in the RXRα LBD–PXR LBD heterodimer. Molecular dynamics, quasiharmonic, and normal moded analyses were conducted on the pregnane X receptor (PXR) LBD in the context of its heterodimer and heterotetramer with RXRα LBD [80]. Models were derived from the PXR LBD–RXRα LBD heterodimer (1ILG) and two PPARγ LBD–RXRα LBD structures (1RDT and 1FM6). In the heterotetramer, residues in each PXR LBD β-turn-β motif (β-sheet) contributed to formation of an aromatic zipper that functioned as an essential homodimerization interface, which was necessary for interaction with the CoA SCR-1, without interfering with heterotetramer localization, RXR heterodimerization, or ligand and DBD interactions. This PXR LBD–PXR LBD interface (β1, β1′, β3, β4), which was 30 Å from the PXR LBD AF-2 surface, formed a strongly correlated unit with H1, H3, H3′, H4, and H9 that led to their highly correlated motion in the same direction. The motion of adjacent helices including H12 was also correlated. The authors suggested that that the correlations indicated that H3 acted as the conduit through which the stabilizing effects of the PXR homodimer were communicated to the AF-2 surface (H3, H3′, H4, and AF-2), which facilitated interactions with a CoA and induction of transcription. In contrast, in the RXR–PXR heterodimer and heterodimer subunit interactions of the PXR β-sheet with the AF-2 surface exhibited only disjointed and smaller motions that were anti-correlated. Correlated AF-2 domain motions were also seen in the PPARγ LBD of the PPARγ LBD–RXRα LBD complex.
RXR–LXR
LGD1069 was able to activate activate hepatic lipogenic genes leading to increased triglycerides via the RXR–LXRα/β heterodimer [81]. This activity was confirmed in wild-type but not LXRα/β-knockout mice. However, bexarotene was unable to similarly increase cholesterol homeostasis as demonstrated by lack of increased plasma total cholesterol. In contrast the LXR ligand TO-901317 could activate lipogenic genes—sterol regulatory element-binding protein (SREBP) 1c, stearoyl-CoA desaturase 1 (SCD1), and fatty acid synthase (FAS)—and cholesterol homeostasis genes—the ATP-binding cassette transporters ABCGFs 1, 5, and 8 and ABCA1.
5.2. Allosteric interactions between RXR and its dimeric partner
Ranganathan, Mangelsdorf and colleagues employed statistical coupling analyses of NR LBDs to identify interior communication pathways of residues (allosterism) that linked the four NR functional surfaces: (i) ligand-binding pocket; (ii) AF-2 motif; (iii) dimerization interface; and (iv) cofactor binding-surface [82]. The authors indicated that the transcriptional response of the RXR–NR heterodimers to the NR ligands defined their functional status as non-permissive, in which the NR’s ligand was solely responsible for transactivation (i.e., NR = TRs and VDR); conditionally permissive, in which the RXR ligand alone had only a minimal effect but enhanced transcription induced by the partner’s ligand (NR = RAR); and permissive, in which, either the RXR ligand or the NR’s ligand induced transcription and their combination behaved additively or synergistically (NR = FXR, LXR, Nurr1, and PPARs). Mapping of the allosteric network identified by the cluster analysis of the NR LBDs onto the RXR LBD identified 27 coupled residues in H1, H1–H3 loop, H3, H4, H5, β-turn, H8, H9, H10, H11, and H12 that interacted either internally (H1 with H3, H1–H3 loop internally and with H3 and H4, H3 with H4, H4 with H5 and the H8–H9 loop, and H5 with H10) or externally with the ligand (H5 Trp305 with 9-cis-RA), the CoA peptide (H3 Lys284, H4 Gln297, and H12 Phe450), and the dimeric partner’s LBD interface (H8–H9 loop Asp379, H9 Arg393, and H10 Leu419 and Arg426). Thus, about half of the residues had roles in LBD interactions and the others formed the allosteric network with the NR partner.
Alanine mutational analysis of LBP residues in permissive heterodimers of RXR with FXR, LXR, and PPARα showed that their LBP residues were energetically linked to their heterodimerization interface [82]. Mutation of the allosteric network in the permissive partner had a greater impact on the response of RXR to its agonist than the comparable mutation in RXR and decreased responses to RXR agonists, whereas similar mutations in the context of non-permissive RARα and TRβ heterodimers with RXR did not. This finding suggested to the authors that the permissive partner of RXR had the greater functional dominance. The authors noted that the non-permissive heterodimers were involved in endocrine signalling, whereas the permissive ones regulated metabolic pathways in response to lipid or lipid products, and that the responsive flexibility of the permissive heterodimers permitted them to function as sensors of dietary factors to activate metabolism. For example, Nurr1(Glu445Ala) having a mutation in the LBD N-terminus had enhanced constitutive activity but exhibited a reduced response in the presence of an RXR ligand [75].
5.2.1. RXR–CAR
The orphan NR constitutive androgen receptor (CAR) is considered to be constitutively activated although functional ligands have been identified. The behavior of RXR–CAR heterodimers varied with cell type and ligand [83]. While 9-cis-RA enhanced interaction of RXR–CAR with the CoA SRC-3, it inhibited the CAR agonist 1,4-[3,3′,5,5′-(Cl)4-bispyridyloxy]benzene (TCPOBOP) from enhancing CoA recruitment and the CAR antagonist androstanol from decreasing it. In a DR-5 reporter assay, 9-cis-RA or LGD1069 alone was inactive but either blocked activation by the CAR ligand. However, on other RXR–CAR REs both rexinoids enhanced heterodimer transactivation. The authors concluded that RXR agonist LGD1069 blocked CAR agonist activity but not the inhibitory activity of the CAR antagonist in HepG2 hepatoma cells.
Heterodimerization of the RXRα LBD with the agonist TCPOBOP–mouse CAR LBD complex increased the potency of CAR agonist-induced recruitment of the CoA TIF-II NR box 3 peptide from 243 nM to 83 nM (four-fold) and increased that for CAR antagonist androstanol-induced TIF-II dissociation from an IC50 value of 183 nM to 60 nM as determined by luminescence proximity assays [84]. Recruitment of the TIF-II peptide by monomeric apo-CAR was increased approximately three-fold in the context of the CAR heterodimer with apo-RXRα (IC50 reduced from 10 μM to 3 μM). IC50 binding affinity values of the TCPOBOP–CAR complex for the peptide decreased from an IC50 value of 0.5 μM in the monomer to 0.1 μM in the RXRα heterodimer.
5.2.2. RXR–TR allosterism
Although allosterism between RXR and its partner was first considered the purview of the permissive receptors, increasing evidence indicates communication also occurs with non-permissive partners.
Allosteric interactions between RXR and TR were explored using isothermal scanning calorimetry (ITC) [85]. Conclusions drawn from this study were that (i) the TR response element (TRE) induced conformational changes in the TR DBD that decreased interactions with the TR LBD; (ii) T3–TRα–TRE bound the CoA SRC-1 peptide with three-fold higher affinity than T3–TRα; (iii) T3–TRα–RXR also bound the SRC-1 peptide with three-fold higher affinity than T3–TRα; whereas (iv) T3–TRα–RXR–TRE bound with only two-fold higher affinity to indicate that binding by the complex components was not additive and that the TRE in the complex attenuated the enhancement of SRC-1 binding by RXR. Interestingly, binding of 9-cis-RA to RXR further reduced SRC-1 affinity. The authors called this type of interaction “a negative permissive effect”.
The affinities of the RXR–TRα to the DR-4 and inverted palindrome (IP)-6 TREs were not affected by the binding of 9-cis-RA or T3 [85]. Levels of transcriptional activation induced on either TRE by 9-cis-RA or by T3 were the same; however, activation by the ligand combination produced a higher response on the DR-4 RE. Binding by both ligands eliminated the negative effect of the TRE on SCR-1 binding and produced equivalent transactivation from either RE when both ligands were added sequentially. The authors concluded that RXR functioned allosterically to enhance the binding of T3 by TR.
5.2.3. RXR–VDR allosterism
Even in the absence of a RXR agonist, in the context of the RXR–VDR binding by a VDR agonist was found to allosterically induce a change in the RXR conformation from that of the nonliganded (apo) receptor to the liganded (holo) agonist conformation [86]. In this reverse “phantom effect”, RXR became capable of recruiting the CoAs SRC-1, TIF-II, and ABC-1.
5.3. Tail wrapping by RXR LBD H12
5.3.1. RXR–LXR heterodimer
The LXRα and LXRβ LBDs form homodimers in solution and on crystallization; however, a physiologic role for the LXR homodimer has not been confirmed in vivo. Overlap of the agonist GW3965–LXRα LBD homodimer with the RXRα LBD–LXRα LBD heterodimer indicated a <0.8 Å rmsd in LXRα backbone carbons [87]. Comparison of the structures of the NCoA2 peptide-bound agonist N-(2,2,2-trifluoroethyl)-N-[4-(2,2,2-trifluoro-1-hydroxy-CF3-ethyl)phenyl]benzenesulfonamide–LXRα LBD–RXRβ LBD–agonist methoprene acid heterodimer complex (1UHL) [88] and the NCoA1 (SRC-1) peptide-bound agonist 3-Cl-4-{[3-(7-propyl-3-CF3-1,2-benzisoxazol-6-yl)oxy]propylsulfanyl} phenylacetic acid (TO-901317)–LXRα LBD homodimer complex (3IPS) [87] showed that several LXRα LBD residues in the former had rotated from their homodimeric positions to form strong heterodimer salt bridges with RXRβ (LXRα His390–RXRβ Glu472 and LXRα Glu465–RXRβ Arg406) to favor heterodimer formation. In the crystal structures of the agonist—1-benzyl-3-(4-MeO-phenylamino)-4-phenylpyrrrole-2,5-dione (SB313987), (4-{3-[(2-Cl-3-CF3-benzyl)(2,2-Ph2-ethyl)amino]propoxy}-1H-indol-1-yl)acetic acid (SB786875), and 2-{4-[(butyl)(3-Cl-4,5-(MeO)2-benzyl)amino]phenyl}-1,1,1,3,3,3-F6-propan-2-ol (GSK2186)—LXRα LBD–RXRα LBD–9-cis-RA heterodimer complexes without bound cofactor peptides (2ACL, 3FC6, and 3FAL, respectively), the RXRα C-terminus including its H12 protruded around LXRα to bind the LXRα AF-2 or CoA surface [87]. In order to do so, the RXRα H11 became disrupted at Phe437 and Phe438. Binding of the RXRα H12 C-terminus was stabilized by a charge clamp with LXRα Lys273, while LXRα Glu441 bound the RXRα H12 Phe450 backbone. Hydrophobic interactions (RXRα H11–H12 loop and H12 Pro446, Phe450, Leu451, and Leu455) also occurred and masked another stabilizing salt bridge between LXRα H3 Lys291–RXRα H12 Asp448–LXRα H12 Val445 carbonyl (tail wrap conformation). LXRα H3 Lys291 position was found to be conserved in those NRs forming heterodimers with RXR—FXR, LXRs, and Nurr1 (in which case lysine was replaced by glutamate), PPARs, PXR, RARs α and β, RORα, and TRα) whereas in the RXR isotypes H12 Arg448 was conserved. In its tail-wrap conformation, the conserved RXR H12 Glu453 was located on the LXRα AF-2 surface and, thus, able to reverse the LXRα AF-2 surface charge, thereby repelling binding by a similarly charged CoA and preventing LXRα signaling even when an LXR agonist was bound. The authors proposed that in the presence of sufficient levels of an RXR agonist and a CoA, the RXR tail would shift to form an RXR CoA surface as was seen in the N-(2,2,2-F3-Et),N-[4-(1,1,1,3,3,3-F6-2-OH-2-propyl)phenyl]benzenesulfonamide (TO-901317)–LXRα LBD–RXRβ LBD–methoprenic acid–complex bound by two CoA GRIP-1 peptides (1UHL). Thus, RXR activation was required before LXR could be activated. They also cited supportive studies that included the tail-wraps found in the RXRα LBD homotetramer that were stabilized by the H12 Asp448–H4 Arg302 salt bridge.
5.4. Dimer interface between LBDs of RXR and its NR partner
5.4.1. RXR homodimers
Moras and colleagues compared the structures and sequences of NRs forming homodimers (class I) with those forming heterodimers with RXRs (class II) [89]. They found that differentially expressed conserved residues formed salt bridges to stabilize the homodimer interface. Thus, in the RXR LBD homodimer, H1 Glu239 formed a salt bridge with H8 Arg371 and H8 Glu366 formed another salt-bridge with the H10 N-terminus Arg414 to stabilize the H9–H10 loop adjacent to the homodimer interface. The authors suggested that the proximity of H1 to the CoA surface provided a means for possible communication with the interface. In addition, a conserved H4 Tyr-305 was found to participate in the LBP. RXR homodimers were reported to bind the malic enzyme promoter PPRE site in vivo and induce its transactivation in the presence of 9-cis-RA and the CoA SRC-1 [90].
RXR tetramer-to-dimeric (oligomeric) DNA switch
In addition to its role as a conventional dimeric NR transcription factor, Noy and collaborators presented evidence that RXR can function as a DNA architectural switch through its ability to interconvert from nonliganded homotetramers to homodimers [91,92]. Because the RXR homotetramers have their DBDs exposed, they retained the ability to complex with RXREs. Binding by the two nonliganded homodimeric components of the homotetramer to two RXREs that were reasonably close caused DNA to form a loop. Looping was cleverly established by demonstrating that the binding of the apo-RXR homotetramer to two RXREs, which were separated by 250 base-pairs in a 382 base-pair sequence, facilitated DNA ligation to form a circular sequence. Circularization was inhibited by the binding of either the RXR agonist 9-cis-RA or 4-[5,6,7,8-tetrahydro-3,5,5,8,8-Me5-2-naphthalenyl)-2-methylpropenyl]benzoic acid (SR11345) or the RXR antagonist 2-[(3-n-hexyloxy-5,6,7,8-tetrahydro-5,5,8,8-Me4-2-naphthalenyl)(methyl)amino]pyrimidyl-5-carboxylic acid (PA452 in Fig. 1D) [93]. The RXR ligands caused the homotetramer to dissociate into ligand-bound homodimers [91]. Using a transcriptionally inactive RXRα mutant lacking the LBD H12 (RXRΔH12), which formed homotetramers, bound DNA, and dissociated into homodimers on agonist or antagonist binding, but was transcriptionally inactive by being unable to bind CoAs, they established that the mutant homotetramer induced DNA looping. This dominant-negative mutant was also capable of inducing the expression of genes involved in cell-cycle progression and apoptosis in transfected MCF-7 breast cancer cells treated with SR11345. Up-regulated genes included Btg2 (2.8-fold), procaspase-9 (2.8-fold), GADDs 34, 45α, 45β, and 45γ (2.4–3.7-fold), TNF 2 (3.1-fold), and TR3/Nur77/NGFI-B (4.5-fold). The RAR antagonist LE540 did not have any significant affect on Btg2 and procaspase-9 expression induced by either rexinoid but blocked that induced by RAR agonist TTNPB (Ro13-7410). Moreover, in MCF-7 cells transfected with the constitutively active homotetrameric RXR mutant RXR(Phe321Ala), which was unable to dissociate to homodimers in the presence of a ligand, only very low levels of Btg2 and procaspase-9 were induced by agonist SR11345 or antagonist PA452, whereas in similarly treated cells transfected with the constitutively active dimeric RXR mutant RXR(Phe318Ala) high levels of these genes were expressed. In the presence of 1 μM SR11345 or PA452, MCF-7 cells that had been transfected with wild-type RXRα, RXRαΔH12, or RXR(Phe318Ala), which could form homodimers, underwent apoptosis, whereas similarly treated cells that had been transfected with constitutive homotetrameric RXR(Phe321Ala) mutant did not. The authors concluded that the RXR homotetramer had a physiologic role in this cell line.
Impact of ligand and CoA
Overlap of the structures of the 9-cis-RA–RXRα LBD and LG100268–RXRβ LBD as found in the crystal structures of their homodimers (Table 3) showed that their backbones comprising the homodimeric interfaces were identical [56]. This finding suggested to the authors that the agonists were unable to transmit their structural differences, as was evidenced by their inducing different LBP shapes, and thus did not exert major allosteric differences on the RXR homodimeric interfaces.
However, HDX MS was used to demonstrate that 9-cis-RA binding to the RXRα LBD homodimer decreased exchange at the homodimer interface, which was comprised of interacting residues from H7, H10, H11, and the H7–H8 and H10–H11 loops that were not in direct contact with the ligand, in addition to the decreased exchange observed in those residues directly contacting the ligand [94]. These results supported the dynamics studies of Moras and colleagues, which indicated that ligand binding produced a more compact RXR homodimer [95].
Both ligand and CoA binding by RXR produced changes in the dimer interface. HDX MS was also used to show that after 9-cis-RA binding to the RXRα LBD, exchange by residues was most suppressed in the area surrounding the LBP and was followed by that of residues from mid-H10 (Leu419) to mid-H11 (Cys432) [66]. The H7 Val354–MeT362 sequence, which did not contact 9-cis-RA, was also protected from exchange. Binding by the CoA GRIP-1 peptide further suppressed the level of exchange by these same residues. Exchange was also suppressed in the H11 Leu433–Phe438 and H12 Phe450–Thr462 sequences, which would have been masked by the bound GRIP-1.
5.4.2. Structure of the RXR LBD homotetramer
The structures of the RXRα LBD homotetramer alone (1G1U), the homotetramer complexed with four CoR SMRT2 peptides (3R29), that with two inactive isomers of retinoic acid, most likely ATRA in which the the 9E,11E-double bonds are cisoid (1G5Y), and that with two SMRT2 peptides and two molecules of the antagonist rhein (3R2A) have been determined (see Table 3). The RXRα LBD tetramer structures are shown in cartoon format in comparison to that of the agonist bigelovin–RXRα LBD–GRIP-1 dimer (3OZJ) in Fig. 7. What is most notable about these tetramers compared to that of the RXRα LBD dimer complexed with its agonist 9-cis-RA (3OAP) or bigelovin is the position of H12. In the agonist-bound dimers, H12 is tucked under the LBD to form the AF-2 surface with H3 and H4 to facilitate GRIP-1 or SRC-1 CoA peptide binding (Fig. 7 and Fig. 8).
Figure 7.
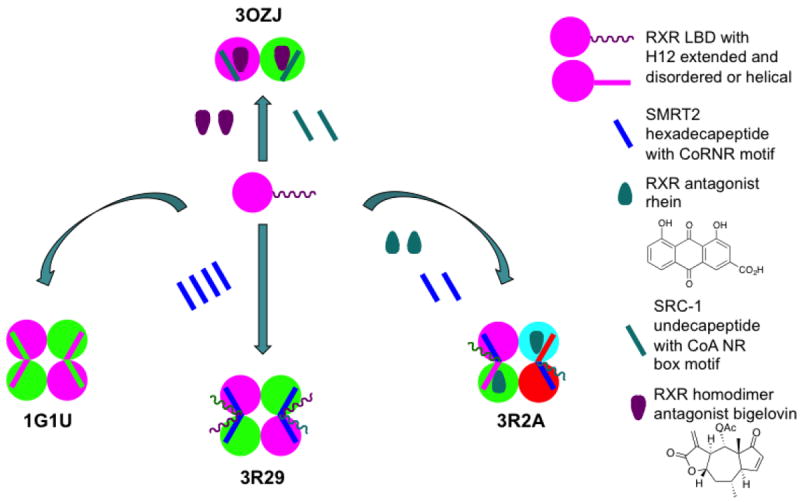
Cartoon demonstrating behavior of RXRα ligand-binding domain (LBD) helix H12 in the context of its homodimeric complex with bigelovin and coactivator (CoA) SRC-1 peptide (PDB structure 3OZJ), the apo-tetramer (1G1U), the tetramer bound with four corepressor (CoR) SMRT2 peptides (3R29), and the tetramer bound with two molecules of antagonist rhein and two CoR SMRT2 peptides (3R2A). In 3OZJ, H12 of each monomer formed an AF-2 surface with its H3 and H4 to bind the CoA peptide despite bigelovin demonstrating antagonist behavior by repressing wild-type RXRα basal transactivation activity in a reporter assay (Ref. [102]). In 1G1U, H12 of each monomer of the tetramer formed a surface with helices H3 and H4 of the monomer from the opposing homodimer to repress transactivation. In 3R29, H12 of each monomer formed a CoR surface with H3 and H4 of a monomer from the opposing dimer to bind one CoR peptide. In 3R2A, the homotetramer was composed of two homodimers, each containing one monomer bound with rhein and the other bound with a CoR peptide. The CoR surface of the monomer that bound the CoR peptide was composed of its own H3 and H4 with H12 from the monomer to which rhein was bound in the adjacent dimer. The backbones of each monomer differed as did the structures of the CoR peptides and the positions of the rhein ligands. The more ordered CoR peptide-bound monomer dimerized with the more disordered ligand-bound monomer and vice-versa. Cartoon based on Refs. [97] and [102].
Figure 8.
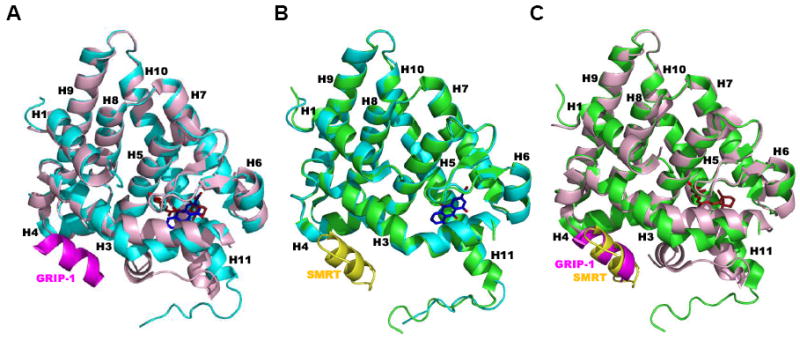
Major differences occur in the human RXRα ligand-binding domain (LBD) structures when complexed as apo-tetramers and corepressor (CoR) peptide-bound tetramers compared to the agonist and antagonist-bound structures. A. Structure of one RXRα LBD monomer (light-pink backbone) complexed with transcriptional agonist 9-cis-RA (C atoms in brown) and coactivaor (CoA) GRIP-1 peptide (magenta backbone) (PDB 3OAP homodimeric LBD–9-cis-RA complex) of the superposed with that of one RXRα LBD monomer (cyan backbone) complexed with the antagonist rhein (Cs, blue) (PDB 3R2A tetrameric LBD complex containing two rhein molecules and two peptides). B. RXRα LBD monomer (green backbone) complexed with CoR SMRT2 peptide (yellow backbone) (PDB 3R29 tetrameric LBD–SMRT2 complex) superposed with one RXRα LBD monomer (cyan backbone) complexed with antagonist rhein (Cs, blue) (PDB 3R2A tetramer complex. C. RXRα LBD monomer (light-pink backbone) complexed with 9-cis-RA (Cs, brown) and GRIP-1 peptide (magenta backbone) (PDB 3OAP) superposed with one RXRα LBD monomer (green backbone) complexed with SMRT2 peptide (yellow backbone) (PDB 3R29 tetramer complex). RXRα LBD helices are numbered. Ligand Os in red.
In the structures of the apo-RXRα LBD homotetramer 1G1U and the RXRα LBD–ATRA tetramer (1G5Y), Gampe Jr. and colleagues observed two symmetric dimers (A1–B1 and B2–A2) that formed major stabilizing interactions between like helices, namely A1 H3–B2 H3, A1 H11–B2 H11, A2 H3–B1 H3, and A2 H11–B1 H11, with H12 of each monomer extending onto the monomeric CoA surface of the adjacent symmetric dimer to function as a CoA NR box motif would to block CoA binding (Fig. 7) [96]. The authors termed the function of H12 in this position as autorepression. In these interfaces, only the interactions between H11 residues Leu436 and Phe437 of adjacent monomers from opposing dimers were symmetric, whereas those for H3 in adjacent monomers involved a symmetric interaction between their two Cys269s and one asymmetric interaction between, for example, the A2 Asp273 carboxylate with the B1 Val265 NH and Tyr266 OH. Also observed was a short helix (labeled H3′) between H3 and H4.
Shen and coworkers reported two different RXRα LBD tetramers to which the CoR SMRT2(2237–2352) peptide was bound [97]. Binding of one CoR peptide to each monomer in the RXRα LBD tetramer structure 3R29 caused a rearrangement compared to their positions in the apo-tetramer with one of the dimers was rotated 30°. This rotation led to different interlocking interactions with the H11 (termed H10 by the Shen group) Ile442 carbonyl of the first monomer hydrogen-bonding with the H7 Arg348 of a second adjacent monomer and its H11 Thr444 OH hydrogen-bonding with the H4 Arg302 of the other adjacent monomer. H11 residues from each monomer formed a hydrophobic core to stabilize the tetramer. Thus, H11 Phe438 of the first monomer interacted with the H11 Phe438 of the second monomer and H11 Phe437 of the first monomer interacted with H11 Phe437 of the third adjacent monomer and so forth around this core. The four H11 Leu441s protruded into the center of the core to further stabilize binding. As a result the tetramer interface was found to increase from 1,476 Å2 in the apo-tetramer to 2,180 Å2 in the SMRT2 peptide-bound tetramer. H12 of one monomer contacted the AF-2 surface of the adjacent monomer, which was comprised of residues from H3, H3′, and H4, to allow binding of the SMRT2 peptide. The N-terminus of H3 was also bent but more sharply than that observed in LBD–agonist structure (Fig. 8).
Rhein (1,8-dihydroxy-3-carboxy-9,10-anthroquinone) from rhubarb roots is an ingredient in Chinese medicines used to treat gastrointestinal disorders and is reported to have anti-tumor and anti-angiogenic activities [98]. Shen and coworkers found that rhein functioned as an antagonist in the contexts of the RXR homodimer on the RXRE and the RXR heterodimers with PPARγ, LXR, and LXRα on their REs by inhibiting transactivation induced by their respective agonists 9-cis-RA, rosiglitazone, TO-901317, and chenodeoxycholic acid (concentrations not given) in cotransfection assays [97]. The IC50 value for inhibition of 9-cis-RA-induced RXRα transactivation by rhein was 0.75 μM.
Crystallization of the RXRα LBD with excess rhein and SMRT2 peptide by Shen and colleagues produced an asymmetric tetramer (3R2A) [97]. Inspection of this structure (3R2A) revealed two different dimers, each containing one monomer bound with rhein and one bound with SMRT2 peptide (Figure 9). Rhein formed H-bonds with the H11 Cys432 carbonyl O of the LBDs. Both monomers binding rhein and both monomers binding the SMRT2 peptide differed in structure. Interestingly, the more ordered rhein-bound monomer dimerized with the more disordered SMRT2-bound monomer and vice-versa. The N-terminus of H3 in the rhein bound monomer underwent an outward shift and could not stabilize the binding of a SMRT2 peptide. Instead, H12 of the SMRT2 peptide-bound monomer from the adjacent dimer covered the rhein bound in the LBP and the SMRT2-binding site of the first monomer. In the other rhein-bound monomer the H3 N-terminus was not helical. One SMRT peptide was disorded and the other helical. Other major structural differences in the LBDs were in H3 and H12 backbones and the length of the β-sheet. The authors concluded that neither the antagonist nor the CoR peptide alone was fully capable of stabilizing a fully symmetric LBD tetramer structure. The authors also concluded that the H12 AF-2 motif assumed different conformations on interacting with an agonist or antagonist and different positions to mediate the recruitment of a CoA or CoR [97]. These various RXR LBD structures are shown in the overlaps in Fig. 8.
Figure 9.
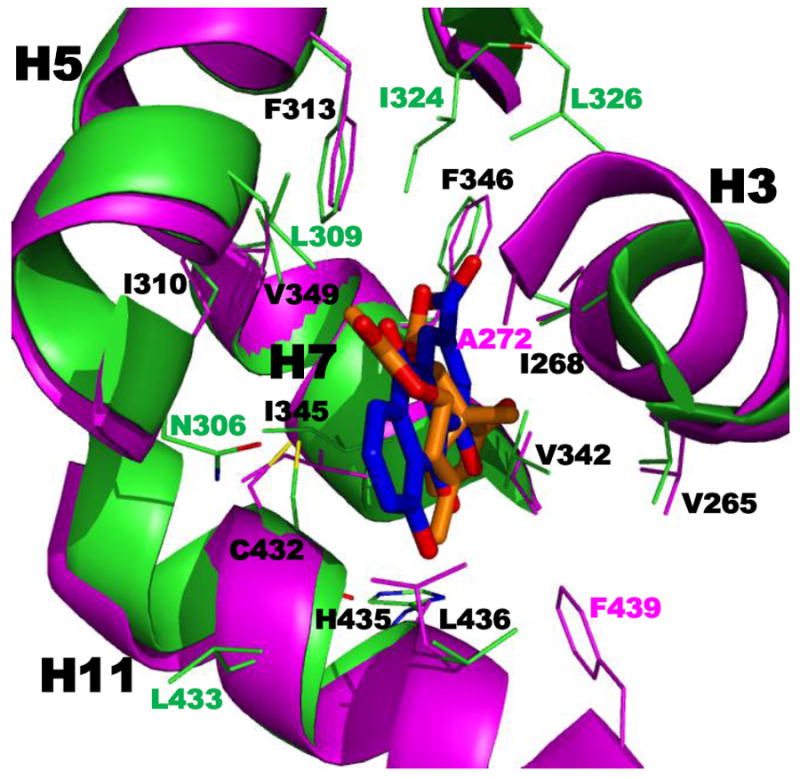
Overlap of structures of human RXRα ligand-binding domains (LBDs) complexed with transcriptional antagonists that occupy the hydrophobic portion of the L-shaped RXRα ligand-binding pocket (LBP). View of the LBP (green backbone and residue side-chain Cs) in the more-ordered monomer bound with antagonist rhein (blue Cs) as found in the tetrameric LBD complex containing two molecules of rhein and two corepressor (CoR) SMRT2 peptides (PDB 3R2A). This view is overlaid with that of the LBP (magenta backbone and residue side-chain Cs) bound with bigelovin (orange Cs) as found in the homodimeric LBD complex with two coactivator (CoA) SRC-1 peptides (PDB 3OZJ). LBP residues within 4.0 Å of the ligand are shown. Helices H3, H5, H7, and H11 and their side chains having similar positions are labeled in black. Those side chains occupying different positions in the two structures are labeled according to their backbone colors. See H11 F439, for example. Although F349 is reported to have a major role in tetramerization, its position in 3OZJ appears not to support participation. The 1-OH and 8-OH O atoms of rhein were within 3–4 Å of the C432 backbone O (red bond above the H435 label), whereas similar H-bond stabilization for bigelovin was not observed. O atoms in red; Ns, blue; Ss, yellow.
In the more ordered of the LBDs, rhein occupied the hydrophobic region of the LBD to overlap a portion of the cyclohexenyl ring and C-7 and C-8 of the tetraene side chain of 9-cis-RA as found in the LBD–9-cis-RA complex 3OAP (Fig. 8A). The most notable differences were in the positions of H12 and the sharpness in the bending of H3 toward the bound ligand with that conferred by 9-cis-RA being more gradual. The overlaps of the more ordered rhein-bound and SMRT2-bound LBDs were highly similar with H12 extended and disordered and H3 bent to the same degree (Fig. 7B). Inspection of the LBP showed van der Waals interactions between rhein and the side chains of the residues in its hydrophobic arm. It will be interesting to determine whether an antagonist more closely resembling the more conventional RXR antagonists behaves similarly.
5.4.3. Heterodimer interface
Crystallography demonstrated that RXR LBD–NR LBD heterodimers were asymmetric with the NR’s C2 symmetry axis rotated (10°) from that of RXR [99,100]. Residues in H7, H9, H10, and the H9–H10 loop from both the RXR LBD and that of its partner participated in forming the heterodimer interface. The H10 N-terminus in both LBDs has a conserved dimerization motif ΘΨKΨΨΨKΨΨΣRΨΨ (Θ = Lys, Phe, and Trp; Ψ = Ala, Ile, Leu, Met, Pro, and Val; Σ = Asp and Glu) [87] that formed a hydrophobic coiled coil core that was generally surrounded by H-bonds between the H7s of each partner and the H9–H10 loop residues of the other partner [99,100]. This structure is found in the agonist farglitazar (GI-262570)–PPARγ LBD–RXRα–LBD–9-cis-RA heterodimer complexed with the SRC-1 peptide (1FM9) [99]. Both H12s were in the active conformation. In contrast to the linear conformation of H7 (Val342–MeT360) in the apo-RXRα LBD structure [100], H7 in the heterodimer unwound at Glu352 (E insert) to allow a helical bend that permitted the strengthening of the heterodimer interface. As a result, the H7 Glu352 formed a salt bridge with the H7 Arg 348 that brought it close to the H10 Lys431, which then formed a salt bridge with the PPARγ H12 C-terminus (Tyr477) to stabilize the PPARγ H12 on the PPARγ AF-2 surface for CoA recruitment. These interactions increased the heterodimer interface surface area by 10%. Formation of a corresponding salt bridge between the RXRα H12 terminus and PPARγ H7 was not possible. Another heterodimer stabilizing interaction occurred between the RXRα H9 Arg393 and PPARγ H10 Asp441.
Comparison of RXRα LBD interfaces formed in heterodimers with PPARγ and CAR LBDs revealed major differences that accounted for the 10% larger LBD interface surface that RXRα made with CAR (995 Å) [84,99]. The overlap between these heterodimeric structures was high for the RXR backbones, but substantial differences occured between the PPARγ and CAR backbones. These included a 16° tilt in the CAR H9 axis that allowed such interface changes as a one helical-turn rotation in the CAR H10 so that its H9–H10 loop became included in the interface core and H-bond formation between the CAR H7 and the RXR H9–H10 loop, which were parallel. The authors did not address the impact of the RXRα H7 Glu352 (E insert).
5.4.4. Impact of ligand on heterodimeric interface
RXRα–RAR
Ligand binding to the RXR LBD did not introduce major changes in its backbone in the heterodimer interface of the RXRα LBD–RARα LBD [64]. Overlapping of the structures of the RARα antagonist BMS614–RARα LBD–RXRα LBD–9Z-oleic acid complex (1DKF), in which RXRα was considered to have an antagonist conformation, with that of the 9-cis-RA–RARβ(D–F)–RXRα LBD–TRAP220 peptide complex (1XDK) and measurement of differences in the positions of their 90 dimer interface H7–H10 Cα atoms produced a rmsd of 0.4 Å to indicate that the two RXR backbones were almost identically positioned. The interface backbones for RARα and RARβ were similarly close (rmsd 0.59 Å for 89 Cα atoms). Overlap of the RXR backbones as found in the apo-RXRα LBD homodimer and the holo-RXR heterodimer again indicated low deviations between interface backbone Cαs (rmsd 0.48Å) to indicate highly similar backbone structures at the heterodimer interface. However, side chain positions did change. The authors concluded that allosteric effects would only be reflected as very subtle changes in receptor conformation at the heterodimer interface.
RXRα–LXRα
Molecular dynamics simulations on the apo-RXRα LBD–apo-LXRα LBD, the 22R-hydroxycholesterol–LXRα LBD, and the 9-cis-RA–RXRα LBD–apo-LXRα LBD complexes suggested that the increase in heterodimer binding strength induced by the bound ligand had a greater impact on the H-bond interactions at the heterodimer interface than it did on the hydrophobic interactions [37]. Binding by 9-cis-RA to RXRα or 22R-hydroxycholesterol to LXRα increased the number of interface H-bonds from three to six or 10, respectively; whereas the respective hydrophobic interface interactions decreased by five or increased by one.
RXR–VDR
Time-dependent HDX MS was conducted on the apo- and holo-VDR LBDs and the holo-VDR–RXR complex [101]. These studies revealed that the apo-VDR–RXR was sufficiently stable for this type of analysis and that the exchange profiles for the ligand bound-VDR LBD were similar to those for the holo-heterodimer complex, except that the dimer interface was more highly protected from exchange after ligand binding.
RXR heterodimers with PPARγ, LXRα and FXR
Shen and coworkers reported the structure (3OZJ) of the dimeric bigelovin–RXRα LBD–SRC-1 CoA peptide complex [102] (Fig. 7). The sesquiterpene bigelovin is another ingredient found in Chinese herbal medicine and is reported to induce leukemia cell growth arrest and apoptosis [103]. Bigelovin had an AC50 value of 4.9 μM for activating the GAL4-RXRα LBD in a luciferase reporter assay in HEK293T cells and a Kd value for binding the RXRα LBD of 8.7 μM [102]. It slightly enhanced activation of the GAL4-PPARγ LBD by rosiglitazone and GAL4-FXR LBD by chenodeoxycholic acid in this reporter assay but not that of the GAL4-LXRα LBD by TO-901317. In cotransfection assays using the wild-type receptors, bigelovin at 10 μM appeared to function as a weak antagonist of RXRα on the RXRE (approx. 80% activation of the control) but was able to activate the RXRα–PPARγ on the PPRE compared to the control and slightly enhanced activation induced by rosiglitazone. Bigelovin at 10 μM functioned as a weak antagonist of the RXRα–LXRα on the LXRE and of the RXRα–FXR on the FXRE. However, bigelovin only reduced the transactivation induced by the LXR agonist TO-01317 but not that by FXR agonist chenodeoxycholic acid. Bigelovin did not bind to the PPARγ, LXRα, and FXR LBDs. Interestingly, bigelovin only occupied the hydrophobic arm of the RXRα LBP to overlap with most of the antagonist rhein except for the C-5 to C-7 portion of the rhein scaffold (Fig. 9). The acetyl group of bigelovin overlapped the carboxylate of rhein. Again, the major ligand interactions observed in the LBP were hydrophobic [102]. On the basis of its transactivation behavior, the question arises as to why the SRC-1 CoA peptide would bind the bigelovin–RXRα LBD complex. Overlapping of the structures of the LBD complexes with bigelovin and rhein (Fig. 7) indicated that the major differences in LBP side chain positions were for H5 Trp305 and H11 Phe439, which resided much closer to bigelovin than to rhein. Although neither compound bound to the RXRα LBD as potently as 9-cis-RA or many synthetic analogs, their analogues may offer another means of manipulating the functions of RXRs.
6. Posttranscriptional processing
6.1. Phosphorylation
Phosphorylation is the most extensively studied post-translational modification of RXRα. Rochette-Egly and colleagues reported that ATRA-induced activation of jun-terminal kinase (JNK) led to the phosphorylation of AF-1 domain residues (Ser61, Ser75, and Thr87) in mouse RXRα, which then allowed cooperation between the AF-1 and AF-2 domains in the RXRα–RARγ heterodimer leading to enhanced transactivation activity and was necessary for the antiproliferative effect of ATRA [104]. They also showed that stress (ribotoxic anisomycin or UV irradiation)-induced signaling pathways led to the activation of JNK1/2, which resulted in the phosphorylation of Ser265 in the LBD H1–H3 loop, to interfere with retinoid-responsive gene activation. RXRα Ser61, Ser65, and Thr87 were also phosphorylated. RARα and RARγ were not affected. The responses of wild-type RXRα and its Ser265Ala and Ser61/75/Thr87Ala mutants to anisomycin-induced phosphorylation in the context of their heterodimers with RARγ and RARα on both DR-5 and mRARβ2 response elements in the presence of combinations of their ligands (RARα-selective BMS753 or RARγ-selective BMS961 plus RXR-selective BMS649) showed that the transcriptional effects of Ser265 phosphorylation were modulated by both the NR and RE. In contrast, phosphorylation of Ser265 in the LBD H1–H3 loop (omega-loop) of mouse RXRα (Ser260 of human RXRα) by the ras–raf–MAPK(ERK) pathway mediated VD3 and ATRA-resistance in hepatocellular cancers and attenuated CoA recruitment [105]. Higher RXRα Ser260/265 phosphorylation levels were found to be associated with colon, hepatic, and pancreatic cancers, the ATRA-resistant HL-60R myeloid leukemia line, and leiomyoma cells. Phosphorylation of Ser260 lowered RXRα transactivation activity and retarded its ubiquitin-proteasomal degradation, but was blocked by RXRα heterodimerization with RARβ. Phosphorylation of the human Ser260, which was adjacent to H12, by a mitogen-activated kinase impaired RXR–RAR transactivation in HuH7 hepatoma cells and its degradation by proteolysis so that RXRα was able to continue functioning as a dominant-negative protein [106].
Using RXRα(Thr82Asp/Ser260Asp) and (Thr82Ala/Ser260Ala) double-mutants, the Moriwaki group established the role of phosphorylation at these positions [107]. In the presence of 9-cis-RA, the alanine double-mutant exhibited enhanced transcriptional activity on the CRBP-II-RARE-tk-Luc and CRBP-II-RXRE-tk-Luc reporters and more effectively inhibited growth and induced apoptosis of transfected Huh7 human hepatocarcinoma or HEK-293T human embryonic kidney epithelial cells. In contrast, the phosphomimetic aspartate double-mutant did not exhibit enhanced transcriptional activity and was unable to inhibit cell growth or induce apoptosis. These findings suggested to the authors that phosphorylation inhibited the ability of RXRα to homodimerize or heterodimerize with RARβ.
The RXRα LBD Ser260 is located close to both the CoA and CoR interaction surfaces [108]. An anti-RXRα LBD antibody that did not recognize the phosphorylated version of Ser260, the constitutively active RXRα(Ser260Ala) mutant, and MAPK kinase inhibitors were used in the human keratinocyte cell line HPK1A and its ras-transformed derivation to demonstrate that in RXRα heterodimers with PPARγ, RARα, TRβ, and VDR and in RXRα homodimers, phosphorylation of the RXRα Ser260 attenuated CoA recruitment and RE binding by the heterodimeric partner. On this basis the authors proposed that the conformation of the RXRα AF-2 domain was modulated by the adjacent phosphorylated H1–H3 loop (Asn-Met-Gly-Leu-Asn-Pro-Ser-Ser260-Pro-Asn-Asp-Pro-Val-Thr-Asn) through the interaction of the Pro264 and Asn262 loop residues with the H12 AF-2 residues Phe450 and Glu453.
Agonist AGN194204-bound RXRα was found to interact with and be phosphorylated by casein kinase 1α (CK1α) on a serine residue [109]. Phosphorylation did not affect RXRα homodimer or RXRα–RARα heterodimer activity on their respective RXRE and RARE sites in luciferase reporter assays, but did cause RXRα relocalization to the clusters of interchromatin granules. RXRα relocalization was accompanied by resistance to both rexinoid-induced cell growth inhibition and apoptosis.
Rochette-Egly and colleagues also reported that knock-out of RXRα blocked the induction of endodermal differentiation of mouse F9 teratocarcinoma cells by ATRA and the ATRA-mediated induction of several genes [110]. In F9 cells, RXRα A domain Ser 22 was constitutively phosphorylated. Wild-type, RXRα−/−/RXRα-transfected, and RXRα−/−/RXRα(Ser22Ala)-transfected F9 cells were used to demonstrate that phosphorylation of Ser22 had no effect on F9 endodermal differentiation but was required for induction of several RA-responsive genes—HNF3α, CRABP-II, Hoxβ-1, and HNF1β—but not that of Hoxa-1 or Stra6. Ser22 did have a role in ATRA-induced inhibition of cell proliferation and G1 arrest caused by RA-induced down-regulation of the cyclin-dependent kinase inhibitor p27KIP and the removal of p21CIP protein by enhanced proteosomal degradation. Other phosphorylation sites in the mouse RXRα LBD included Tyr249 in the H1–H3 loop and H9 Ser397 [111].
Rochette-Egly and colleagues also investigated the interaction of the AF-1 and AF-2 domains in the RXRα–RARγ2 heterodimer by using deletion and alanine mutants and wild-type mouse RXRα [112]. They concluded that the AF-1 phosphorylation sites and the AF-2 domain of RXRα cooperated with RARγ2 to recruit cofactors or activate NR degradation. The RARγ2Δ(AF-1) mutant was activated by RXRα plus RXR agonist BMS649 (SR11237 in Fig. 1B) to 42% of the wild-type response as measured by the induction of HNF3α mRNA expression. Similarly, deletion of either the RXRα AF-1 or AF-2 did not significantly affect the DR-5-tk-CAT reporter response induced by retinoic acid (stereochemistry undefined) or the combination of the RARγ agonist BMS961 and RXR agonist in the presence of the wild-type RARγ2 or the RARγ2Δ(AF-1). However, transactivation was reduced and autonomous RXR degradation was lost when the RXRα AF-1 domain serines were mutated to alanines to suggest a loss of synergy between the receptors. Phosphorylation of the RXRα AF-1 domain at Ser61, Thr75, and Ser87 by MAPKs in F9 and COS-1 cells was induced by BMS649 within 2 h, but did not mediate RXRα degradation or that of its NR partner; however, deletion of the AF-1 or mutation of its phosphorylation sites reduced RXRα degradation induced by its agonist [112].
6.2. Truncation
RXRα was cleaved by calcium-dependent calpain in the liver to an N-terminal truncated 44-kDa protein (tRXRα) that could localize in mitochondria [113-115]. There, tRXRα heterodimerized with TR to enhance mRNA levels in response to 9-cis-RA or T3. In the cytoplasm, tRXRα interacted with Akt to activate the Akt/PI3K pathway. Zhang and colleagues recently reported that tRXRα interaction with the Akt p85α subunit and subsequent Akt activation were inhibited by sulindac sulfide and several analogs, when combined with TNFα [43]. The RXRα–p85α interaction domain was located between RXRα residues 80–100. The authors observed that tRXRα was highly expressed in breast, liver, prostate, and thyroid cancer tissues but not in adjacent normal tissues.
6.3. RXR-mediated degradation of its NR partner
LGD1069 and LG100268 (Fig. 1B), which typically function as RXR agonists, at 10 μM were found to be weak transcriptional activators of human PXR/SXR on the (CYP3A1 DR-3)2-tk-CAT (four-fold compared to eight-fold for rifamicin and 21-fold for SR12813) [115] and antagonized the binding of the RXR–PXR heterodimer to the MDR1 and CYP3A4 PXREs, its CoA recruitment, and the activation of PXR by its agonists rifampicin and SR12813 in MCF-7 breast cancer and HepG2 hepatocarcinoma cells [115]. Competitive binding to human PXR using [3H]SR12813 by 10 μM SR12813 (Kd = 41 nM) and rexinoids LGD1069 and LG100268 gave displacements of 100%, 85, and 87%, respectively [116]. The rexinoids enhanced the degradation of both PXR and RXR by calpain within 1 h of treatment, but did not alter their mRNA levels [115]. Rifampicin did not enhance NR degradation. Whereas the proteasome inhibitor lactacystin blocked the degradation of RXR, it had no effect on that of PXR, and calpeptin, a selective inhibitor of calpains, blocked the degradation of PXR but not RXR. LGD1069 enhanced the protease activity of calpain in LS180 cells by an unestablished mechanism. These studies suggested to the authors that the administration of rexinoids with PXR ligands could modulate drug metabolism. The authors noted that LGD1069 also induced the cleavage of c-Jun by calpain. Other calpain targets that were suggested for modification in the presence of rexinoids were p53, c-Myc, and androgen receptor (AR). Antagonism by RXR agonists was also observed in the context of the RXR–FXR heterodimer [117].
6.4. Sequestration
Gene translocations producing fusions of PLZF, NuMA, NPM, and STAT5b with RARα account for 5% of acute promyelocytic leukemia cases. Patients having these translocations do not respond to ATRA therapy as effectively as those with the PML-RARα translocation [118]. Proteins translated from these ATRA-nonresponsive translocations also homodimerized and formed RXR heterodimers that bound to RAREs to prevent RARα-induced gene activation. Their heterodimerization with RXR led to RXR relocalization and sequestering that resulted in reduced RXR intracellular activity. These higher order oligomeric complexes with the RARα fusion proteins were found to recruit corepressors, bind RAREs, and play a role in transformation [119]. Treatment with RXR panagonist SR11237 suppressed transformation or induced apoptosis of transformed cells.
7. Therapeutic potential
7.1. Atherosclerosis
While cholesterol metabolism and its peripheral distribution are mediated by the liver, dysfunctional processing of cholesterol by macrophages leads to the development of hypercholesterolemia and cholesterol-containing atherosclerotic plaques and lesions (reviewed in [120]). PPARγ activated by 9- and 13-OH-octadienoic acids plays a role in macrophage differentiation, uptake of oxidized low-density lipoprotein (LDL), and the expression of LXRα, which, in turn, induces the expression of such lipid transporters as ATP-binding cassette (ABC) A1 and ABCG1. Rexinoids were able to activate permissive heterodimers of RXR with PPARγ and LXRα, which have respective roles in lipid and sterol processing. Macrophage-expressing and lesion-containing cytochrome P450 CYP17A1 was induced by agonists of PPARγ [121], RXR, and RAR, but not by LXR agonists [120]. This P450 enzyme hydroxylates cholesterol to produce 17-OH-cholesterol, which is an LXR agonist, and, thus, has a role in the sterol elimination. Activation of RXR–FXR by rexinoids led to the repression of the CYPA1 and CYPB1 hydroxylases, which have roles in the synthesis of bile acids that are responsible for uptake of fats by the intestine. The activation of RXR–LXR by bile acids in intestinal enterocytes induced the expression of ABC-1 that transports free cholesterol from these cells into the intestinal lumen.
7.2. Cancer treatment/prevention challenges
The potential of retinoids as cancer preventive and therapeutic agents and the role of RXR have been reviewed [122]. Loss of RXR leads to hyperplasia, whereas its overexpression plus treatment with an RXR agonist leads to inhibition of proliferation and/or induction of differentiation. Causative factors involved in the loss of RXR function included its relocalization and the overexpression of RAR leading to RAR–RXR heterodimer formation at the expense of RXR homodimers so that p21 expression via two RXREs in its promoter could no longer be upregulated.
In malignant MDA-MB-231 breast cancer cells, RXRα protein expression levels remained the same as those of normal mammary epithelial and retinoid-responsive MCF-7 breast cancer cells [25]. However, unlike MCF-7 and normal cells, where RXRα protein was diffusely distributed in the nucleoplasm, in retinoid-resistant MDA-MB-231 breast cancer cells RXRα localized in the nucleus in a punctate pattern that colocalized as nuclear speckles with splicing factor compartment (SFC) proteins SC-35 and p105, a SFC substructural component [25]. As a result, RXRα signaling in response to 9-cis-RA or AGN194204 was silenced in MDA-MB-231 cells. The distribution of RXRβ and RXRγ remained normal. In the SFC, RXRα was resistant to digestion by DNase and RNase. The RXRα C-terminus (441–454) peptide inhibited RXRα localization to the SFC. The authors considered that the relocalization of RXRα was due to a protein–protein interaction mediated by the RXRα C-terminus. In contrast, RXRα was normally localized in MDA-MB-435, BT-20, and BT-549 breast cancer, SK-OV-3 ovarian cancer, HeLa cervical cancer, and ras-transformed NIH 3T3 cell lines. Immunochemistry showed RXRα localized in the SFC of mesenchymal cells from five of 12 human invasive breast carcinoma samples, but not in normal tissue or benign hyperplasia. The authors suggested that the compartmentalization of RXR in the SFC could have produced the limited results observed in clinical trials using the rexinoid LG1069 (bexarotene in Fig. 1B) for prevention or treatment of cancer [25].
In a screen comparing RXR protein expression in thyroid tumor samples to that in tissues from their normal margins, nuclear levels of RXRα and RXRβ were decreased 0–50% and 0–30%, respectively, in the thyroid carcinomas (anaplastic, follicular, medullary, and papillary), whereas nuclear levels in the adenomas were decreased about 30% [123]. RXRγ levels were decreased to 0–35% in the carcinomas and to 40% in the adenomas. Levels of cytoplasmic RXRα were reduced only to 50–80% of normal in the carcinoma cells and to 30% in the adenomas. Cytoplasmic RXRβ and RXRγ were decreased in carcinomas and adenomas to 0–15% of normal. The authors suggested that these decreases may have impacted the negative outcome in two trials using retinoids to treat thyroid cancers by inducing the redifferentiation of transformed cells.
The RXR agonist AGN194204 (Fig. 1B) alone was able to inhibit the proliferation of pancreatic cancer cells [124]. After 7 days, cell growth inhibition by 1.0 μM AGN194204 was about 50% in Mia PaCA-2, 40% in BxPC-3, and 20% in AsPC-1 cell lines. Levels of cyclin E and cdk6 had decreased, and p27 had increased in retinoid-treated MIA PaCa-2 cells to suggest that inhibition of the cell cycle was involved. MIA PaCa-2 cells did not undergo apoptosis as was indicated by lack of procaspase-3, 8, and 9 cleavage.
The only selective rexinoid available for cancer treatment is bexarotene (Targretin™, LGD1069), which is FDA-approved for oral treatment of cutaneous T-cell lymphoma. Despite its evaluation alone and in combination with various potent cancer therapeutic agents, clinical trials against solid tumors such as non-small cell lung cancers have not demonstrated sufficient efficacy against the tumor and/or have caused dose-related severe adverse effects. Its far more RXR-selective analog, LG100268 (Fig. 1B), has not advanced into cancer treatment trials despite promising results in animal models such as a triply transgenic mouse model of pancreatic cancer [PDX-1-Cre, Kras(Gly12Asp), and p53(Arg172His] reported by Sporn and colleagues [125]. Of mice treated with LG100268 (45 mg/kg diet) 44% survived at 24 weeks (average life-span 23.7 ± 1.7 weeks) compared to 32% of the control group (average life-span 20.5 ± 0.9 weeks); however, at 40 weeks all mice had died. LG100268 combined with the terpenoid CDDO-Me (60 mg/kg diet) extended lifetime by 4 additional weeks.
On the basis of its efficacy against breast and lung cancer xenograft growth in mice, a Phase I clinical trial on AGN194204 (NRX194204 or NRX4204) was launched against breast and NSCL cancers by NuRx Pharmaceuticals (Irvine, CA) in 2008 [126].
Recently, the acyclic retinoid (polyprenoic acid, 4,5-didehydrogeranylgeranoic acid, (E)-3,7,11,15-tetramethyl-2,4,6,10,14-hexadecapentaenoic acid, peretinoin, NIK-333, or E5166) was evaluated against recurrence of liver cancer in subjects at risk [127]. Liver carcinogenosis is associated with deactivation of RXRα function due to Ras-MAPK-mediated hyperphosphorylation [128]. According to subgroup analyses of data from an up to 96-week phase II/III trial evaluating NIK-333 against recurrence of hepatitis C virus-positive hepatocellular carcinoma, treated patients with mild liver impairment (cirrhosis status Child-Pugh A), who had undergone curative therapy, (N = 310) or those with Child-Pugh impairment and having the major tumor of < 20-mm diameter before treatment (N = 110) had promising results [129]. NIK-333 at 600 mg daily (N = 100) reduced risk of recurrence or death to about 40% of the placebo control (N = 106) in the first group, whereas risk in the second treated group (N = 49) was reduced to 62% compared to the placebo control (N = 49). Adverse events included tolerable albuminuria, headache, and hypertension [129]. Whether NIK-333 isomerizes like ATRA to produce an isomer with properties similar to those of 9-cis-RA appears not to have been investigated. Polyprenoic acid (E5166) is cited as having RXR activity [130] and was first reported by Moriwaki, Muto, and coworkers [131]. E5166 was found to activate the CRBP-II RXRE-CAT reporter in transfected HuH-7 hepatoma cells, which constitutively express RXRα [132].
E5166 was evaluated for prevention of tumor recurrence or a second primary tumor in patients previously treated for hepatoma by surgical resection or percutaneous injection of ethanol into the tumor [133]. Beginning no later than 8 months after curative therapy, patients were treated with 300 mg of E5166 (N = 44) or peanut oil (N = 45) twice daily for 48 weeks. At the 38-month follow-up, five (11%) in the treatment group had disease recurrence and seven (16%) developed a second primary tumor (well-differentiated hepatocellular carcinoma grade I–II), whereas in the placebo group the respective values were two (4%) and 20 (44%). At the median 62-month follow-up, the estimated 6-year survival was 74% in the treatment group and 46% in the placebo group according to Kaplan-Meier estimates (P = 0.04) [134]. A subsequent study indicated that up to 38 weeks after treatment, the probability of treatment failure did not differ between groups, but began to differ between 51–199 weeks with that for the treatment group being lower, after which time failure in the treatment group increased [135]. Increased serum levels of an α-fetoprotein isoform (AFP) L3 and prothrombin-induced vitamin K absence II protein (PIVKA-II) were determined to be predictive of recurrence and biomarkers of the presence of latent hepatocellular carcinoma cells in the treatment and placebo groups (N = 21 each). For example, comparison of biomarker levels at the beginning and end of the 48-week treatment revealed that in the placebo group AFP-L3 positivity rose from 19% to 57% and its serum levels increased; whereas in the treatment group AFP-L3 positivity decreased from 24% to 5% and its serum levels decreased. At 48 weeks, AFP-L3 levels between the groups differed by 680-fold. Respective differences in incidence of PIVKA-II were 29% and 10%, and PIVKA-II levels differed by 5.2-fold. At the 7-year point after beginning treatment, second primary tumors had been detected in 23 of the treatment group and 28 of the placebo group.
7.3. Dementia
In a small case study comparing patients with (N = 70) and without (N = 18) dementia, levels of RXRα gene and protein expression in the inferior temporal gyrus (area 20) were higher in the group with dementia (P < 0.0005 and P = 0.007, respectively) and significantly associated with higher clinical dementia rating (CDR) scores (P = 0.005) [136]. Higher levels of RXRα gene expression in the hippocampus were also correlated with higher CDR scores in individuals with dementia and Alzheimer’s disease-associated neuropathology (P = 0.008) but not with Braak neuropathological staging or neuronal plaque density. RXRα protein expression levels were highly correlated with its message levels in the hippocampus (P < 0.0005). RXRα expression also correlated with expression levels of genes for ApoE, which is the principle CNS cholesterol transporter, and ABCA-1, which modulates cholesterol and phospholipid efflux from cells into plasma (P < 0.0001). Significant correlations with levels of LXRβ expression were not found. These results suggest that an RXR ligand could play a role in modulating the effects of dementia.
In Parkinson’s disease, midbrain dopaminergic neurons degenerate. A method for treating this disease would involve using a rexinoid to induce the differentiation of induced pluripotent stem (IPS) cells to functional neurons if IPS-related teratoma formation could be avoided [137]. RXR ligand DHA activated Nurr1 expression and signaling in IPS cells leading to their differentiation to tyrosine hydroxylase-producing neurons and increased dopamine release and behavior in an animal model of the disease. Primary dopamine neurons isolated from ventral midbrain obtained from embryonic day (E) 14.5 rat embryos, E14.1-stage induced mouse embryonic stem cells (ESCs), or sonic hedgehog and FGF-induced nestin-responsive Lmx1α-overexpressing mouse ESCs were protected from the neurodegeneration-inducing toxin 6-OH-dopamine by pretreatment with 1 μM RXR agonist LG100268 or RXR–Nurr1-selective ligand XCT0139508 (Fig. 1D) [138]. This toxin at 10 and 15 μM for primary neurons or at 150 and 500 μM for mESC-derived neurons induced cell death and neurotransmitter depletion by first inducing oxidative stress and hypoxia (0–1% O2), which were accompanied by an increase in tyrosine hydroxylase-positive cells. However, neither rexinoid protected the neurons from stress induced by the glutamate analogue kainic acid (100 or 500 μM) or hydrogen peroxide (80 or 110 μM). Protective effects by LG100268 or DHA pretreatment only occurred in cells that expressed Nurr1. In the absence of cell stress, the rexinoid did not affect cell numbers. These results suggest that RXR ligands functioning through the RXR–Nurr1 heterodimer have a neuroprotective effect. Nurr1 (NRA42) has been considered to function as a true orphan nuclear receptor on the basis of its LBP being filled with aromatic residues in the crystal structure of its LBD [139]. However, weakly binding Nurr1 ligands (Kd = 20 μM) have been detected by 19F NMR-based library screening [140] and missing NMR signals in two regions of the putative LBP also support a flexible pocket capable of interaction with a ligand [141].
7.4. Diabetes
The roles of RXR in modulating the effects of insulin resistance observed in diabetes patients were reviewed by Szanto et al. [120] and are summarized as follows. In addition to enhancing the activity of the PPARγ-selective thiazolidinedione drugs as insulin sensitizers, rexinoids functioned through other signaling pathways. The accumulation of saturated fatty acids (FAs) in skeletal muscle cells in diabetic patients was found to interfere with their insulin signaling and glucose uptake, whereas rexinoids increased both uptake and oxidation of saturated FAs by these cells. Rexinoid agonists such as LG100268 sensitized human skeletal muscle to insulin-dependent glucose disposal by enhancing the expression of genes for the FA transporter CD36, UCP3, which increases mitochondrial FA oxidation, and PDK4, which enhances fat disposal. Rexinoids also induced the expression of stearoyl-CoA desaturase (SCD) 1, which converts saturated FAs to monounsaturated FAs for storage as triglycerides, thereby reducing the levels of the saturated FAs that would be converted to diacylglycerol and ceramide. Both of these lipids reduced insulin signaling and increased the conversion of glucose to glycogen for storage.
In type 2 diabetes, the decrease in slow-twitch muscle fibers, which derive energy from FA oxidation and glycolysis, contributed to loss of insulin sensitivity, whereas rexinoids increased the expression of such slow-twitch muscle markers as myoglobin and troponin I in skeletal muscle. Rexinoids also attenuated insulin resistance, which is caused by normally sensitive skeletal muscle cells becoming increasingly unable to respond to stimulation of glucose transport by insulin and adipocytes failing to suppress lipolysis (lipid ester cleavage) and increasing their release of FAs. The increased availability of FAs resulted in suppressesd gluconeogenesis and increased glucose production by the liver. The PPARγ-selective thiazolidinediones reduced the release of FAs by adipocytes, increased their levels of the insulin-sensitizing adipokine, adiponectin, and reduced that of insulin resistance-inducing TNFα and resistin. Rexinoids contributed by enhancing PPARγ activity through the PPARγ–RXR heterodimer-mediated activation of the c-Cbl-associated protein (CAP)/c-Cbl pathway to increase CAP expression and c-Cbl phosphorylation. Rexinoids alone stimulated the insulin receptor substrate/Akt pathway to enhance glucose use in skeletal muscle by decreasing the phosphorylation of insulin receptor substrate (IRS) 1. Unfortunately, rexinoids had the adverse effect of causing triglyceridemia by suppressing lipoprotein lipase expression in both skeletal muscle and heart and so their translation into useful drugs for the treatment of diabetes has not been realized.
Micrroarray analysis of a doxycycline-inducible dominant-negative RXRβ(ΔLBD C-terminus 20 AA) that was expressed in MIN6 mouse insuloma cells, which also expressed SV40-T antigen, showed that the dysregulated genes included down-regulated Hes5 (Notch signaling effector), Hk1 (hexokinase), Mki67, Nr2f6, and Nkx6-2 (role in pancreatic endocrine development) [142]. All down-regulated genes were confirmed by RT-PCR. cMaf, which induces insulin expression, was also down-regulated. Genes with roles in mitochondrial function, protein synthesis or trafficking, signal transduction, and transcription were up-regulated. The authors concluded that these findings supported the use of 9-cis-RA plus high-dose rosiglitazone or the over-expression of both RXR and PPARγ to inhibit insulin secretion by INS-1 cells stimulated by high glucose.
7.5. Innate inflammatory response
Innate immunity is a nonspecific method by which the host rapidly defends itself against pathogens by recruiting immune cells to the infection site, by producing cytokines, and by activating the complement cascade, processes that remove dead cells, bacteria, and other foreign matter, and the adaptive immune system [143]. Leukocyte activation occurs when the Gram-negative bacterial cell-wall component lipopolysaccharide (LPS) complexes with serum LPS binding protein to bind the cell-surface marker CD14, which is associated with toll-like receptor 4 (TLR4), to activate a signaling process leading to increases in intracellular Ca(II) levels, tyrosine kinase phosphorylation, and NFκB activation that then leads to increased synthesis of such cytokines and chemokines as TNFα and interleukins (ILs) 1, 6, and 8 [144]. Gram-positive bacterial peptidoglycans and lipoproteins bind TLR2 to activate a similar process.
Inflammation due to recruitment of immune cells can become dysregulated during sepsis and cause the release of excessively high levels of proinflammatory mediators into the blood stream that produce organ failure. Sepsis caused by an inbalance of the host inflammatory response is characterized by the systemic inflammatory response syndrome in which the patient has two of the following symptoms: temperature of <35 °C or >38.5 °C, heart rate >90 beats/min, respiratory rate of >20 breaths/min, white blood cell count >12,000/mm3 or <4,000/mm3, or >10% immature cells of total leukocytes, and has confirmed presence of infection [145]. The studies described below suggest two pathways by which an RXR-selective retinoid could function in mediating the immune response to infection. Additional work will be needed in this area to discern the actual signaling processes involved.
7.5.1. Down-regulation of cytokine expression
Conditional knock-out (KO) of RXRα in mouse peritoneal macrophages led to down-regulation of the levels of chemokines CCL6 and CCL9 and the attenuation of leukocyte recruitment to sites of acute inflammation in a chemically induced mouse peritonitis model [143]. Interestingly, these KO mice, if suffering from sepsis induced by cecal ligation and puncture or by LPS-induced septic shock, experienced prolonged survival. CCL6 and CCL9 function as chemoattractants that induce monocyte/macrophage and granulocyte migration and the secretion of IL-6 and MCP-1. Treatment with 9-cis-RA or rexinoid LG100268 up-regulated CCL expression in wild-type macrophages and induced the activation of a RXR homodimer-responsive DR-1 RXRE in the CCL promoter in transfected mouse macrophage RAW 264.7 cells, whereas the RXR homodimer antagonist and PPARγ–RXR and RAR–RXR agonist LG100754 inhibited the induction of CCL6 and CCL9 mRNA expression by either 9-cis-RA or LG100268. The authors suggested that their results support RXR as a target for treatment of sepsis and inflammatory diseases by preventing the release of chemokines and leukocyte migration. On the basis of these results, patients should be treated with an RXR antagonist in the context of RXR functioning as a homodimer. Other studies suggest the involvement of RXR heterodimeric partners LXR and PPARs in the hepatic inflammatory acute phase response, which has been reviewed by Treuter and coworkers [146].
7.5.2. Immune response to bacterial challenge
Pathogenic bacteria causing anthrax, plague, and Salmonella and Shigella infections induced macrophage apoptosis through the TLR4 pathway in order to elude the innate immune response [147]. Activation of the RXR–LXR heterodimer by the LXR ligands 24S,25-epoxycholesterol, TO-901317, and GW3965 alone or combined with 9-cis-RA induced the expression of anti-apoptotic genes such as AIM/CT2/Api6, Bcl-XL, and Birc1a/NeuroAIP and attenuated the expression of pro-apoptotic genes such as procaspases-1, 4/11, 7, and 12, FasL, and DNases 1I3/DNase γ and Cidewa. These results suggested a role for a rexinoid that behaved as a selective RXR–LXR agonist in treatment of bacterial infection.
7.6. Memory Deficit
Rats were treated orally with RARα-selective agonist Am80 (5 mg/kg) daily and RXR-selective synergist HX630 (20 mg/kg) daily either alone or in combination at 3 h before being injected with scopolamine, which induced amnesia [148]. The rats were tested 30 min later for how long their passive avoidance lasted each day for a 7-day period. This duration test measured the time delay before trained rats moved from their lighted enclosure into a dark one, where they recieved an electric shock. At the 7-day test point, the scolopamine-treated control delay time (latency period) was 3.1 sec compared to 158 sec for the Am50-treated group, 192 sec for the HX630 group, and 269 sec for the combination, whereas the delay time for the nontreated trained group (blank) was 272 sec. Time-responsive restoration of memory was noted for both the blank and retinoid-treated groups. The results suggested than an RXR-selective retinoid would potentiate the effects of the RARα-selective retinoid at memory restoration.
7.7. Obesity
LG100268 was found to decrease body weight and food consumption in obese, insulin-resistant Zucker rats [149]. Rats that had been orally dosed with LG100268 for 6 weeks had a six-fold increase in apoptosis of subcutaneous fat and a decrease in that fat mass. Dosed rats also had the adverse effects of triglyceride levels being elevated two-fold and 75% total thyroid hormone T4 suppression. Direct injection of the rexinoid into the brain at 30 μg daily reduced food intake, weight gain, and insulin levels in the rats without inducing triglyceridemia to suggest separate pathways for energy balance and lipid homeostasis by rexinoids.
However, the potential use of a rexinoid for weight reduction may be limited because of the ability of an RXR ligand to activate the PPARγ–RXR heterodimer. Activation of this heterodimer by the environmental obesogen tri(n-butyl)tin chloride was found to induce human and mouse white adipose-derived multipotent stromal stem cells to differentiate into adipocytes. As a result, cellular lipid content and expression of adipogenic genes increased [150,151]. The authors suggested that the increase in adipocytes in utero would lead to increased adipose mass in the offspring. In addition to activating RXR, tri(n-butyl)tin chloride functions as a PPARγ agonist [152].
7.8. Severe chronic hand eczema
Eczema is considered to be caused by a dysregulation of the cutaneous innate immune system that leads to prolonged inflammation in response to environmental stimuli and frequently afflicts those having occupations that expose them to frequent contact with water such as bakers, hairdressers, and masons [153]. Its incidence has been estimated at 5–10% of the population [154,155]. This condition affects both the hands and feet and is thought to have a genetic component. Hyperactive cytokine secretin by type-2 T-helper lymphocytes of the adaptive immune system is considered as one component of this condition. Keratinocytes are also involved. They express the surface receptors CD14 and TLR4, both of which increase on exposure to LPS [144]. Epidermal and dermal dendritic cells also express TLR2 and TLR4 [153]. Lipoteichoic acid and peptidoglycan from Gram-positive bacteria such as Staphylococcus areus activate TLR2 [153], whereas TLR4 is activated by Gram-negative bacterial LPS. Their activation results rapid intracellular Ca(II) response, NFκB nuclear-translocation, secretion of proinflammatory chemokines and cytokines [153,156]. In addition, mitogen-activated protein kinase signaling is induced leading to synthesis of IL-1β, TNFα, inducible nitric oxide synthetase, and reactive oxygen species. The cutaneous innate immune first-line defense also includes acid mammalian chitanase that degrades microbial chitans, and antimicrobial peptides such as leukocyte and epithelial cathelicidin-cleavage product hCAP18/LL-37, beta-defensins, and psoriasin. While the latter peptides rise in eczema lesions, this study indicated that levels of LL-37 were lower in eczema and so could be used as a biomarker for childhood eczema severity [153]. Interestingly, 67% and 39%, respectively, of 144 children with eczema had coexisting allergic rhinitis and asthma.
Oral aliretinoin (9-cis-RA) was recently approved in Europe for treatment of severe cases of chronic hand eczema. Results from a muticenter randomized trial in standard treatment-refractory patients (N =1,032) demonstrated efficacy and safety [157]. Patients were treated for up to 24 weeks with 30 mg or 10 mg of 9-cis-RA (N = 310 each) or placebo daily and followed up for 24 weeks. Respective response rates based on clearing or almost clearing were 195 of 409 (48%), 115 of 418 (28%), and 34 of 205 (17%). Respective median reduction in disease extent was 75%, 50%, and 33%. Median time to relapse was 5.5, 6.2, and 5.4 months. In the high-dose group, headache was the most frequent adverse event (20%) followed by erythema (7%), and abnormal laboratory results were elevated cholesterol (14%) and triglyceride levels (8%) and decreased thyroid-stimulating hormone (7%). Results from a smaller trial in dermatitis patients using topical 1% bexarotene gel twice or three times daily were 39% for ≥90% clearance and 79% for ≥50% clearance [158]. Most responders achieved ≥50% clearance after 5 weeks and ≥90% clearance after 10–12 weeks.
The role of RXR in innate immune response suggests why 9-cis-RA would be effective in treating this disease. The dimeric partner(s) involved in the cutaneous innate immune response remain to be conclusively identified. In keratinocytes, RXR dimeric partner VDR complexed with VD3 and the CoA SRC-3 have a role in the synthesis and processing of the lipids involved in skin permeability barrier function and triggering of the innate immune response [159]. On activation of toll-like receptors by their ligands, VD3 synthesis increased leading to increased expression of cathelicidin and TLR2 [153,160]. The RXR heterodimeric partner PPARγ is reported to have a role innate immunity by regulating AP-1 and p38 MAP kinase activity [161].
8. Background reading (reviews)
Since 2000, many excellent reviews describing RXR transcriptional function have appeared. In 2010, Lefebvre and colleagues provided a thought-provoking overview of research on the signaling mechanisms of the RXR isotypes (α–γ) with emphasis on the research issues that should be addressed and the unique role that the RXRs have in transcriptional signaling [13]. They stressed the need for RXR isotype-selective rexinoids for mechanistic studies and pharmacological use on the basis of differences observed in isotype responses, particularly that of RXRβ [13]. Several such isotype selective rexinoids are listed in Table 4. They also highlighted several intriguing behaviors of the RXRs, including (i) the ability of the RXR tetramer to contribute to promoter looping, which was first recognized by Noy and colleagues [92]; (ii) the identity of heterodimer regulatory sites (REs) to which RXR was prebound that were located significantly upstream from transcription start sites; (iii) the possible relationship between RXR permissivity and metabolic regulation; and (iv) interaction of RXRs with non-NR transcription factors such as nPAS2/MOP4 and Clock, which act as regulators of circadian feedback. As described above, the versatility of RXR in effecting diverse signaling pathways is evidenced by their listing of its interacting proteins.
Several authors provided a clear step-by-step overview of the transcription process induced by the retinoid NRs that includes basic structure, binding to REs, ligand binding, CoA recruitment, transcriptional machinery recruitment, heterodimer degradation by the ubiquitin–proteasome pathway, and modulation of transcription by phosphorylation of RXR, RAR, and cofactors [57,111,162]. Reviews on the therapeutic and preventive potential of rexinoids in cancer [163,164] and multiple sclerosis [165] have appeared, as have reviews describing RAR and RXR-selective retinoids [166,167] and the roles of RXR in metabolic regulation [168]. The adverse effects of the only FDA-approved rexinoid (bexarotene) have been reviewed [169].
9. Conclusions and Future Directions
Despite entensive clinical trials, only two RXR agonists have been approved for drug use and then only for limited indications, namely topical and systemic treatment of CTCL using bexarotene (LGD1069) and topical treatment of Kaposi’s sarcoma and systemic treatment of refractory chronic hand eczema using aliretinoin (9-cis-RA). Both drugs have significant adverse effects that may be due to their abilities to activate the RARs. Their effects on other nuclear receptors such as those that bind fatty acids (PPARs and HNF4α) should also be examined. In their review, Lefebvre et al. describe several rexinoids that were reported to show selective activities in the context of a particular heterodimer [13]. These included LG101506, which activated RXR–PPARα, but did not activate RXR heterodimers with PPARβ/δ, PPARγ, LXRα/β, RARα, and FXRα in reporter assays [170]. However, LG101506 did synergize with ligands for PPARβ/δ and PPARγ, but not that for RARα. Results for synergy with other receptors were not reported. Unlike LG100268 at 30 mg/kg daily for 14 days, LG101506 did not raise triglyceride levels in normal rats but did so in both lean and obese Zucker rats treated for 7 days at 1 and 3 mg/kg daily. Rexinoids HX630 and TZ335 enhanced HL60 leukemia cell differentiation induced by RARα-selective Am80, whereas rexinoid PA024 enhanced both differentiation and apoptosis of HL60 cells [171]. Their effects on gene expression in HL60 cells also differed. In combination with Am80, HX630 or TZ335 induced the expression of 50 predictor genes that resembled those induced by ATRA in a DNA microarray assay, whereas Am80 plus PA024 induced a pattern similar to that of 9-cis-RA. Whether these RXR synergists affected other RXR heterodimers in HL60 cells was not explored.
Acknowledgments
Research from the groups of Drs. Xiao-Kun Zhang and Marcia I. Dawson that is described in this review was partially supported by U.S. National Cancer Institute Grants P01 CA51993, R01 CA107039, and R01 CA109345, which is gratefully acknowledged. We also thank Ms. Laura Nelson for assistance with the manuscript.
Footnotes
Abbreviations: 9-cis-RA, 9-cis-retinoic acid; AA, arachidonic acid; ABCA1, ATP-binding cassette 1 transporter A1; AC50, concentration of ligand required for 50% of maximum gene activation by ligand’s NR bound to its NR RE; ADRP, adipose differentiation-related protein; AF, activation function; ASC, activating signal cointegrator 2; ATRA, all-trans-retinoic acid; bg, Biomphalaria glabrata; βG, β-galactosidase; bt, Bemisia tabaci; CAR, constitutive androgen receptor; CAT, chloramphenicol acetyl transferase gene; CBP, core-binding pocket; CCL6, chemokine (C–C motif) ligand 6; CD36, class B scavenger receptor family member on macrophage to which oxidized low-density lipoprotein binds; cdk6, cyclin-dependent kinase 6; cDNA, circular DNA; CDR, clinical dementia rating; CoA, coactivator; CoR, corepressor; cp, Celuca pugilator; CRABP-II, cytosolic retinoic acid-binding protein II; CRBP, cytosolic retinol-binding protein; Cyp or CYP, cytochrome P450; ΔA/B. deletion of the A/B domain; DBD, DNAbinding domain; DEC, deleted in esophageal cancer; DHA, docosahexaenoic acid; DHT, dihydrotestosterone; dm, Drosophila melanogaster; DR, direct repeat; EAR2, ν-erbA-related receptor 2; Ec, ecdysone; eCFP, enhanced cyan fluorescent protein; EMSA, electromobility shift assay; ER, estrogen receptor or everted repeat; ERK, extracellular signal-regulated kinase; esi, electrospray ionization; eYFP, yellow fluorescent protein; FA, fatty acid, FABP, fatty acid-binding protein; FCHL, familial combined type hereditary hyperlipidemia; FCS, fluorescence correlation spectroscopy; FRAP, fluorescence recovery after photobleaching; FRET, fluorescence resonance energy transfer; FXR, farnesoid X receptor; Gal, galactosidase gene; GFP, green fluorescent protein; GR, glucocorticoid receptor; GRIP-1, glucocorticoid receptor-interacting protein 1; GST, glutathione S-transferase; h, human; H, helix; HDAC, histone deacetylase; HDX MS, hydrogen/deuterium exchange mass spectrometry; HNF, hepatic nuclear factor; hv, Heliothis virescens; IGFBP, insulin-like growth factor-binding protein; IL-6, interleukin-6; IR, inverted repeat; ITC, isothermal scanning calorimetry; JNK, jun terminal kinase; LBD, ligand-binding domain; LBP, ligand-binding pocket; LDL, low-density lipoprotein; LEF, lymphoid enhancer-binding factor; lm, Locusta migratoria; LPS, lipopolysaccharide; ls, Lymnaea stagnalis; Luc, luciferase gene; LXR, liver X receptor; m, mouse; MALDI, matrix-assisted laser desorption ionization; MAPK, mitogen-activated phosphokinase; MDR1, multi-drug resistance 1; mESC, murine embryonic stem cell; MMTV, murine mammary tumor virus; MR, mineralcorticoid receptor; na, not active; NBRE, Nurr1 and NGFI-B monomer response element; nc, not conducted; NCoR, nuclear receptor corepressor; nd, not determined; NES, nuclear export signal; NGFI-B, nerve growth factor IB, rat NR4A1; NMR, nuclear magnetic resonance spectrometry; NMU, N-nitrosomethylurea; nPAS2, neuronal PAS domaincontaining protein 2; NR, nuclear receptor; NSCLC, non-small cell lung cancer; nt, not tested; Nurr77, mouse NR4A1; OA, oleic acid; pal, palindromic response element; PCAF, p300/CBP-associating factor; PDB, Protein Data Bank; PDK4, pyruvate dehydrogenase kinase 4; Pg, prostaglandin; pm, Polyandrocarpa misakiensis; PML, promyeloleukemia protein; PNR, photoreceptor-specific nuclear receptor; PPAR, peroxisomal proliferator-activated receptor; PPRE, PPAR response element; PSA, prostate specific antigen; PUFA, polyunsaturated fatty acid; PXR, pregnane X receptor; RAR, retinoic acid receptor; RARE, RAR response element; RE, response element; RFP, ring finger protein; RID, receptor-interacting domain; RIP140, receptor-interacting protein 140; ROR, retinoid-related orphan receptor; RXR, retinoid X receptor; SC50, concentration of RXR synergist required to enhance response of its dimeric partner’s ligand to 50% of the maximal response induced; SCD, stearoyl-coenzyme A desaturase; SFC, splicing factor compartment; SHP, small heterodimer partner NR; shRNA, short hairpin RNA; SMRT, silencing mediator of retinoid and thyroid hormone receptors; SPR, surface plasmon resonance; SRC, steroid receptor coactivator; SXR, steroid and xenophobic receptor; T3, triiodothyronine; T4, thyroxine; tc, Tribolium castaeneum; TCF, T-cell factor; TCPOBOB, 1,4-(3,3′,5,5′-tetrachlorobispyridyloxy)benzene; TIF-II, transcription intermediary factor II; tk, thymidine kinase promoter; TNFα, tumor necrosis factor α; TR, thyroid hormone receptor; TRE, TR response element; TRAP220, thyroid hormone receptor-associated protein complex component; tRXR, truncated RXR; TR3, testicular receptor 3, human NR4A1; TTNPB, (E)-4-[2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid; UAS, upstream activation sequence; UCP3, uncoupling protein 3; up, Uca pugilator; USP, ultraspiracle; VDR, vitamin D receptor.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mic FA, Molotkov A, Benbrook DM, Duester G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc Natl Acad Sci USA. 2003;100:7135–7140. doi: 10.1073/pnas.1231422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolf G. Is 9-cis-retinoic acid the endogenous ligand for the retinoic acid-X receptor? Nutr Rev. 2006;64:532–538. doi: 10.1111/j.1753-4887.2006.tb00186.x. [DOI] [PubMed] [Google Scholar]
- 3.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 4.Chen Y, Wei L-N, Müller JD. Unraveling protein-protein interactions in living cells with fluorescence fluctuation brightness analysis. Biophys J. 2005;88:4366–4377. doi: 10.1529/biophysj.105.059170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feige JN, Gelman L, Tudor C, Engelborghs Y, Wahli W, Desvergne B. Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J Biol Chem. 2005;280:17880–17890. doi: 10.1074/jbc.M500786200. [DOI] [PubMed] [Google Scholar]
- 6.Bich C, Bovet C, Rochel N, Peluso-Iltis C, Panagiotidis A, Nazabal A, Moras D, Zenobi R. Detection of nucleic acid-nuclear hormone receptor complexes with mass spectrometry. J Am Soc Mass Spectrom. 2010;21:635–645. doi: 10.1016/j.jasms.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Cao X, Liu W, Lin F, Li H, Kolluri SK, Lin B, Han Y-H, Dawson MI, Zhang X-K. Retinoid X receptor regulates Nur77/TR3-dependent apoptosis [corrected] by modulating its nuclear export and mitochondrial targeting. Mol Cell Biol. 2004;24:9705–9725. doi: 10.1128/MCB.24.22.9705-9725.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X-K. Targeting Nur77 translocation. Expert Opin Ther Targets. 2007;11:69–79. doi: 10.1517/14728222.11.1.69. [DOI] [PubMed] [Google Scholar]
- 9.Lin X-F, Zhao B-X, Chen H-Z, Ye X-F, Yang C-Y, Zhou H-Y, Zhang M-Q, Lin S-C, Wu Q. RXRα acts as a carrier for TR3 nuclear export in a 9-cis retinoic acid-dependent manner in gastric cancer cells. J Cell Sci. 2004;117:5609–5621. doi: 10.1242/jcs.01474. [DOI] [PubMed] [Google Scholar]
- 10.Bishop-Bailey D. The platelet as a model system for the acute actions of nuclear receptors. Steroids. 2010;75:570–575. doi: 10.1016/j.steroids.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Ray DM, Spinelli SL, Pollock SJ, Murant TI, O’Brien JJ, Blumberg N, Francis CW, Taubman MB, Phipps RP. Peroxisome proliferator-activated receptor γ and retinoid X receptor transcription factors are released from activated human platelets and shed in microparticles. Thromb Haemost. 2008;99:86–95. doi: 10.1160/TH07-05-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moraes LA, Swales KE, Wray JA, Damazo A, Gibbins JM, Warner TD, Bishop-Bailey D. Nongenomic signaling of the retinoid X receptor through binding and inhibiting Gq in human platelets. Blood. 2007;109:3741–3744. doi: 10.1182/blood-2006-05-022566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab. 2010;21:676–683. doi: 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Li D, Yamada T, Wang F, Vulin AI, Samuels HH. Novel roles of retinoid X receptor (RXR) and RXR ligand in dynamically modulating the activity of the thyroid hormone receptor/RXR heterodimer. J Biol Chem. 2004;279:7427–7437. doi: 10.1074/jbc.M311596200. [DOI] [PubMed] [Google Scholar]
- 15.Feingold K, Kim MS, Shigenaga J, Moser A, Grunfeld C. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;286:E201–E207. doi: 10.1152/ajpendo.00205.2003. [DOI] [PubMed] [Google Scholar]
- 16.Numasawa T, Koga H, Ueyama K, Maeda S, Sakou T, Harata S, Leppert M, Inoue I. Human retinoic X receptor β: Complete genomic sequence and mutation search for ossification of posterior longitudinal ligament of the spine. J Bone Miner Res. 1999;14:500–508. doi: 10.1359/jbmr.1999.14.4.500. [DOI] [PubMed] [Google Scholar]
- 17.McDermott NB, Gordon DF, Kramer CA, Liu Q, Linney E, Wood WM, Haugen BR. Isolation and functional analysis of the mouse RXRγ1 gene promoter in anterior pituitary cells. J Biol Chem. 2002;277:36839–36844. doi: 10.1074/jbc.M203689200. [DOI] [PubMed] [Google Scholar]
- 18.Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- 19.Nohara A, Kobayashi J, Mabuchi H. Retinoid X receptor heterodimer variants and cardiovascular risk factors. J Atheroscler Thromb. 2009;16:303–318. doi: 10.5551/jat.no786. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Q, Chasse SA, Devarakonda S, Sierk ML, Ahvazi B, Rastinejad F. Structural basis of RXR-DNA interactions. J Mol Biol. 2000;296:509–520. doi: 10.1006/jmbi.1999.3457. [DOI] [PubMed] [Google Scholar]
- 21.Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-γ–RXR-α nuclear receptor complex on DNA. Nature. 2008;456:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prüfer K, Barsony J. Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol Endocrinol. 2002;16:1738–1751. doi: 10.1210/me.2001-0345. [DOI] [PubMed] [Google Scholar]
- 23.Shaffer PL, McDonnell DP, Gewirth DT. Characterization of transcriptional activation and DNA-binding functions in the hinge region of the vitamin D receptor. Biochemistry. 2005;44:2678–2685. doi: 10.1021/bi0477182. [DOI] [PubMed] [Google Scholar]
- 24.Ito M, Fukuzawa K, Mochizuki Y, Nakano T, Tanaka S. Ab initio fragment molecular orbital study of molecular interactions between liganded retinoid X receptor and its coactivator; Part II: Influence of mutations in transcriptional activation function 2 activating domain core on the molecular interactions. J Phys Chem A. 2008;112:1986–1998. doi: 10.1021/jp075430r. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka T, Dancheck BL, Trifiletti LC, Birnkrant RE, Taylor BJ, Garfield SH, Thorgeirsson U, De Luca LM. Altered localization of retinoid X receptor α coincides with loss of retinoid responsiveness in human breast cancer MDA-MB-231 cells. Mol Cell Biol. 2004;24:3972–3982. doi: 10.1128/MCB.24.9.3972-3982.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egea PF, Mitschler A, Moras D. Molecular recognition of agonist ligands by RXRs. Mol Endocrinol. 2002;16:987–997. doi: 10.1210/mend.16.5.0823. [DOI] [PubMed] [Google Scholar]
- 27.Egea PF, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRα ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19:2592–2601. doi: 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito M, Fukuzawa K, Ishikawa T, Mochizuki Y, Nakano T, Tanaka S. Ab initio fragment molecular orbital study of molecular interactions in liganded retinoid X receptor: Specification of residues associated with ligand inducible information transmission. J Phys Chem B. 2008;112:12081–12094. doi: 10.1021/jp803369x. [DOI] [PubMed] [Google Scholar]
- 29.Lengqvist J, Mata de Urquiza A, Bergman A-C, Willson TM, Sjövall J, Perlmann T, Griffiths WJ. Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor α ligand-binding domain. Mol Cell Proteomics. 2004;3:692–703. doi: 10.1074/mcp.M400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Fan Y-Y, Spencer TE, Wang N, Moyer MP, Chapkin RS. Chemopreventive n-3 fatty acids activate RXRα in colonocytes. Carcinogenesis. 2003;24:1541–1548. doi: 10.1093/carcin/bgg110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luria A, Furlow JD. Spatiotemporal retinoid-X receptor activation detected in live vertebrate embryos. Proc Natl Acad Sci USA. 2004;101:8987–8992. doi: 10.1073/pnas.0307053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muccio DD, Brouillette WJ, Breitman TR, Taimi M, Emanuel PD, Zhang X, Chen G, Sani BP, Venepally P, Reddy L, Alam M, Simpson-Herren L, Hill DL. Conformationally defined retinoic acid analogues. 4. Potential new agents for acute promyelocytic and juvenile myelomonocytic leukemias. J Med Chem. 1998;41:1679–1687. doi: 10.1021/jm970635h. [DOI] [PubMed] [Google Scholar]
- 33.Kolesar JM, Hoel R, Pomplun M, Havighurst T, Stublaski J, Wollmer B, Krontiras H, Brouillette W, Muccio D, Kim K, Grubbs CJ, Bailey HE. A pilot, first-inhuman, pharmacokinetic study of 9cUAB30 in healthy volunteers. Cancer Prev Res. 2010;3:1565–1570. doi: 10.1158/1940-6207.CAPR-10-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapetanovic IM, Horn TL, Johnson WD, Cwik MJ, Detrisac CJ, McCormick DL. Murine oncogenicity and pharmacokinetics studies of 9-cis-UAB30, an RXR agonist, for breast cancer chemoprevention. Int J Toxicol. 2010;29:157–164. doi: 10.1177/1091581809360070. [DOI] [PubMed] [Google Scholar]
- 35.Lee JJ, Wu X, Hildebrandt MAT, Yang H, Khuri FR, Kim E, Gu J, Ye Y, Lotan R, Spitz MR, Hong WK. Global assessment of genetic variation influencing response to retinoid chemoprevention in head and neck cancer patients. Cancer Prev Res. 2011;4:185–193. doi: 10.1158/1940-6207.CAPR-10-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuang K-H, Lee Y-F, Lin W-J, Chu C-Y, Altuwaijri S, Wan Y-JY, Chang C. 9-cis-Retinoic acid inhibits androgen receptor activity through activation of retinoid X receptor. Mol Endocrinol. 2005;19:1200–1212. doi: 10.1210/me.2004-0181. [DOI] [PubMed] [Google Scholar]
- 37.Yue L, Ye F, Gui C, Luo H, Cai J, Shen J, Chen K, Shen X, Jiang H. Ligand-binding regulation of LXR/RXR and LXR/PPAR heterodimerizations: SPR technology-based kinetic analysis correlated with molecular dynamics simulation. Protein Sci. 2005;14:812–822. doi: 10.1110/ps.04951405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanayasu-Toyoda T, Fujino T, Oshizawa T, Suzuki T, Nishimaki-Mogami T, Sato Y, Sawada J, Inoue K, Shudo K, Ohno Y, Yamaguchi T. HX531, a retinoid X receptor antagonist, inhibited the 9-cis retinoic acid-induced binding with steroid receptor coactivator-1 as detected by surface plasmon resonance. J Steroid Biochem Mol Biol. 2005;94:303–309. doi: 10.1016/j.jsbmb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Ebisawa M, Inoue N, Fukasawa H, Sotome T, Kagechika H. Thiazolidinediones with thyroid hormone receptor agonistic activity. Chem Pharm Bull (Tokyo) 1999;47:1348–1350. doi: 10.1248/cpb.47.1348. [DOI] [PubMed] [Google Scholar]
- 40.Morita K, Kawana K, Sodeyama M, Shimomura I, Kagechika H, Makishima M. Selective allosteric ligand activation of the retinoid X receptor heterodimers of NGFI-B and Nurr1. Biochem Pharmacol. 2005;71:98–107. doi: 10.1016/j.bcp.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 41.le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, Balaguer P, Bourguet W. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 2009;10:367–373. doi: 10.1038/embor.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolluri SK, Corr M, James SY, Bernasconi M, Lu D, Liu W, Cottam HB, Leoni LM, Carson DA, Zhang X-K. The R-enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid X receptor and induces tumor-selective apoptosis. Proc Natl Acad Sci USA. 2005;102:2525–2530. doi: 10.1073/pnas.0409721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou H, Liu W, Su Y, Wei Z, Liu J, Kolluri SK, Wu H, Cao Y, Chen J, Wu Y, Yan T, Cao X, Gao W, Molotkov A, Jiang F, Li W-G, Lin B, Zhang H-P, Yu J, Luo S-P, Zeng J-Z, Duester G, Huang P-Q, Zhang X-K. NSAID sulindac and its analog bind RXRα and inhibit RXRα-dependent AKT signaling. Cancer Cell. 2010;17:560–573. doi: 10.1016/j.ccr.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu D, Cottam HB, Corr M, Carson DA. Repression of β-catenin function in malignant cells by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci USA. 2005;102:18567–18571. doi: 10.1073/pnas.0509316102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klopper JP, Sharma V, Bissonnette R, Haugen BR. Combination PPARγ and RXR agonist treatment in melanoma cells: Functional importance of S100A2. PPAR Res. 2010;2010 doi: 10.1155/2010/729876. Article ID 729876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonofiglio D, Cione E, Qi H, Pingitore A, Perri M, Catalano S, Vizza D, Panno ML, Genchi G, Fuqua SAW, Andò S. Combined low doses of PPARγ and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. Am J Pathol. 2009;175:1270–1280. doi: 10.2353/ajpath.2009.081078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monden T, Yamada M, Nihei Y, Kishi M, Tomaru T, Ishii S, Hashida T, Shibusawa N, Hashimoto K, Satoh T, Kasai K, Mori M. Unliganded RXR acts as an inhibitory factor on troglitazone-induced activation. Life Sci. 2004;76:731–741. doi: 10.1016/j.lfs.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 48.Park EY, Cho IJ, Kim SG. Transactivation of the PPAR-responsive enhancer module in chemopreventive glutathione S-transferase gene by the peroxisome proliferator-activated receptor-γ and retinoid X receptor heterodimer. Cancer Res. 2004;64:3701–3713. doi: 10.1158/0008-5472.CAN-03-3924. [DOI] [PubMed] [Google Scholar]
- 49.Hessel S, Lampen A. All-trans retinoic acid enhances the transport of phase II metabolites of benzo[a]pyrene by inducing the breast cancer resistance protein expression in Caco-2 cells. Toxicol Lett. 2010;197:151–155. doi: 10.1016/j.toxlet.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 50.Idres N, Marill J, Chabot GG. Regulation of CYP26A1 expression by selective RAR and RXR agonists in human NB4 promyelocytic leukemia cells. Biochem Pharmacol. 2005;69:1595–1601. doi: 10.1016/j.bcp.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 51.Hermann TW, Yen W-C, Tooker P, Fan B, Roegner K, Negro-Vilar A, Lamph WW, Bissonnette RP. The retinoid X receptor agonist bexarotene (Targretin) synergistically enhances the growth inhibitory activity of cytotoxic drugs in non-small cell lung cancer cells. Lung Cancer. 2005;50:9–18. doi: 10.1016/j.lungcan.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 52.Yen W-C, Prudente RY, Lamph WW. Synergistic effect of a retinoid X receptor-selective ligand bexarotene (LGD1069, Targretin) and paclitaxel (Taxol) in mammary carcinoma. Breast Cancer Res Treat. 2004;88:141–148. doi: 10.1007/s10549-004-1426-5. [DOI] [PubMed] [Google Scholar]
- 53.Ramlau R, Zatloukal P, Jassem J, Schwarzenberger P, Orlov SV, Gottfried M, Rodriguez Pereira J, Temperley G, Negro-Vilar R, Rahal S, Zhang JK, Negro-Vilar A, Dziewanowska ZE. Randomized phase III trial comparing bexarotene (L1069-49)/cisplatin/vinorelbine with cisplatin/vinorelbine in chemotherapy-naïve patients with advanced or metastatic non-small-cell lung cancer: SPIRIT I. J Clin Oncol. 2008;26:1886–1892. doi: 10.1200/JCO.2007.12.2614. [DOI] [PubMed] [Google Scholar]
- 54.Blumenschein GR, Jr, Khuri FR, von Pawel J, Gatzemeier U, Miller WH, Jr, Jotte RM, Le Treut J, Sun S-L, Zhang JK, Dziewanowska ZE, Negro-Vilar A. Phase III trial comparing carboplatin, paclitaxel, and bexarotene with carboplatin and paclitaxel in chemotherapy-naïve patients with advanced or metastatic non-small-cell lung cancer: SPIRIT II. J Clin Oncol. 2008;26:1879–1885. doi: 10.1200/JCO.2007.12.2689. [DOI] [PubMed] [Google Scholar]
- 55.Schedlich LJ, O’Han MK, Leong GM, Baxter RC. Insulin-like growth factor binding protein-3 prevents retinoid receptor heterodimerization: implications for retinoic acid-sensitivity in human breast cancer cells. Biochem Biophys Res Commun. 2004;314:83–88. doi: 10.1016/j.bbrc.2003.12.049. [DOI] [PubMed] [Google Scholar]
- 56.Love JD, Gooch JT, Benko S, Li C, Nagy L, Chatterjee VKK, Evans RM, Schwabe JWR. The structural basis for the specificity of retinoid-X receptor-selective agonists: New insights into the role of helix H12. J Biol Chem. 2002;277:11385–11391. doi: 10.1074/jbc.M110869200. [DOI] [PubMed] [Google Scholar]
- 57.Wei L-N. Retinoid receptors and their coregulators. Annu Rev Pharmacol Toxicol. 2003;43:47–72. doi: 10.1146/annurev.pharmtox.43.100901.140301. [DOI] [PubMed] [Google Scholar]
- 58.Son YL, Park OG, Kim GS, Lee JW, Lee YC. RXR heterodimerization allosterically activates LXR binding to the second NR box of activating signal co-integrator-2. Biochem J. 2008;410:319–330. doi: 10.1042/BJ20070837. [DOI] [PubMed] [Google Scholar]
- 59.Shao G, Heyman RA, Schulman IG. Three amino acids specify coactivator choice by retinoid X receptors. Mol Endocrinol. 2000;14:1198–1209. doi: 10.1210/mend.14.8.0495. [DOI] [PubMed] [Google Scholar]
- 60.Takano Y, Adachi S, Okuno M, Muto Y, Yoshioka T, Matsushima-Nishiwaki R, Tsurumi H, Ito K, Friedman SL, Moriwaki H, Kojima S, Okano Y. The RING finger protein, RNF8, interacts with retinoid X receptor α and enhances its transcription-stimulating activity. J Biol Chem. 2004;279:18926–18934. doi: 10.1074/jbc.M309148200. [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Hu X, Wei LN. Molecular interaction of retinoic acid receptors with coregulators PCAF and RIP140. Mol Cell Endocrinol. 2004;226:43–50. doi: 10.1016/j.mce.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 62.Wohlfahrt G, Sipilä J, Pietilä L-O. Field-based comparison of ligand and coactivator binding sites of nuclear receptors. Biopolymers. 2009;91:884–894. doi: 10.1002/bip.21273. [DOI] [PubMed] [Google Scholar]
- 63.Kurcinski M, Kolinski A. Theoretical study of molecular mechanism of binding TRAP220 coactivator to retinoid X receptor alpha, activated by 9-cis retinoic acid. J Steroid Biochem Mol Biol. 2010;121:124–129. doi: 10.1016/j.jsbmb.2010.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pogenberg V, Guichou J-F, Vivat-Hannah V, Kammerer S, Pérez E, Germain P, de Lera ÁR, Gronemeyer H, Royer CA, Bourguet W. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. J Biol Chem. 2005;280:1625–1633. doi: 10.1074/jbc.M409302200. [DOI] [PubMed] [Google Scholar]
- 65.Liu H, Shaw C-K, Reineke EL, Liu Y, Kao H-Y. Retinoid X receptor α (RXRα) helix 12 plays an inhibitory role in the recruitment of the p160 co-activators by unliganded RXRα/retinoic acid receptor α heterodimers. J Biol Chem. 2004;279:45208–45218. doi: 10.1074/jbc.M408033200. [DOI] [PubMed] [Google Scholar]
- 66.Xia G, Boerma LJ, Cox BD, Qiu C, Kang S, Smith CD, Renfrow MB, Muccio DD. Structure, energetics and dynamics of binding coactivator peptide to human retinoid X receptor α ligand binding domain complex with 9-cis-retinoic acid. Biochemistry. 2010;50:93–105. doi: 10.1021/bi101288y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Y, Kerimo A, Khan S, Wei L-N. Real-time analysis of molecular interaction of retinoid receptors and receptor-interacting protein 140 (RIP140) Mol Endocrinol. 2002;16:2528–2537. doi: 10.1210/me.2002-0124. [DOI] [PubMed] [Google Scholar]
- 68.Cho Y, Noshiro M, Choi M, Morita K, Kawamoto T, Fujimoto K, Kato Y, Makishima M. The basic helix-loop-helix proteins differentiated embryo chondrocyte (DEC) 1 and DEC2 function as corepressors of retinoid X receptors. Mol Pharmacol. 2009;76:1360–1369. doi: 10.1124/mol.109.057000. [DOI] [PubMed] [Google Scholar]
- 69.Flores AM, Li L, Aneskievich BJ. Isolation and functional analysis of a keratinocyte-derived, ligand-regulated nuclear receptor comodulator. J Invest Dermatol. 2004;123:1092–1101. doi: 10.1111/j.0022-202X.2004.23424.x. [DOI] [PubMed] [Google Scholar]
- 70.Liu B, Lee K-W, Li H, Ma L, Lin GL, Chandraratna RAS, Cohen P. Combination therapy of insulin-like growth factor binding protein-3 and retinoid X receptor ligands synergize on prostate cancer cell apoptosis in vitro and in vivo. Clin Cancer Res. 2005;11:4851–4856. doi: 10.1158/1078-0432.CCR-04-2160. [DOI] [PubMed] [Google Scholar]
- 71.Ikezoe T, Tanosaki S, Krug U, Liu B, Cohen P, Taguchi H, Koeffler HP. Insulin-like growth factor binding protein-3 antagonizes the effects of retinoids in myeloid leukemia cells. Blood. 2004;104:237–242. doi: 10.1182/blood-2003-07-2203. [DOI] [PubMed] [Google Scholar]
- 72.Xiao J-H, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, Cordrey A, Zhao Y, Chandraratna RAS. Adenomatous polyposis coli (APC)-independent regulation of β-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278:29954–29962. doi: 10.1074/jbc.M304761200. [DOI] [PubMed] [Google Scholar]
- 73.Kriegl L, Horst D, Reiche JA, Engel J, Kirchner T, Jung A. LEF-1 and TCF4 expression correlate inversely with survival in colorectal cancer. J Transl Med. 2010;8:123. doi: 10.1186/1479-5876-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Széles L, Póliska S, Nagy G, Szatmari I, Szanto A, Pap A, Lindstedt M, Santegoets SJAM, Rühl R, Dezsö B, Nagy L. Research resource: Transcriptome profiling of genes regulated by RXR and its permissive and nonpermissive partners in differentiating monocyte-derived dendritic cells. Mol Endocrinol. 2010;24:2218–2231. doi: 10.1210/me.2010-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lammi J, Perlmann T, Aarnisalo P. Corepressor interaction differentiates the permissive and non-permissive retinoid X receptor heterodimers. Arch Biochem Biophys. 2008;472:105–114. doi: 10.1016/j.abb.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 76.Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP, Haefner P, Dawson MI, Pfahl M. Retinoids selective for retinoid X receptor response pathways. Science. 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- 77.Loeliger P, Bollag W, Mayer H. Arotinoids, a new class of highly active retinoids. Eur J Med Chem -Chim Ther. 1980;15:9–15. [Google Scholar]
- 78.Castillo AI, Sánchez-Martínez R, Moreno JL, Martínez-Iglesias OA, Palacios D, Aranda A. A permissive retinoid X receptor/thyroid hormone receptor heterodimer allows stimulation of prolactin gene transcription by thyroid hormone and 9-cis-retinoic acid. Mol Cell Biol. 2004;24:502–513. doi: 10.1128/MCB.24.2.502-513.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li D, Li T, Wang F, Tian H, Samuels HH. Functional evidence for retinoid X receptor (RXR) as a nonsilent partner in the thyroid hormone receptor/RXR heterodimer. Mol Cell Biol. 2002;22:5782–5792. doi: 10.1128/MCB.22.16.5782-5792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teotico DG, Frazier ML, Ding F, Dokholyan NV, Temple BRS, Redinbo MR. Active nuclear receptors exhibit highly correlated AF-2 domain motions. PLoS Comput Biol. 2008;4:e1000111. doi: 10.1371/journal.pcbi.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lalloyer F, Pedersen TÅ, Gross B, Lestavel S, Yous S, Vallez E, Gustafsson J-Å, Mandrup S, Fiévet C, Staels B, Tailleux A. Rexinoid bexarotene modulates triglyceride but not cholesterol metabolism via gene-specific permissivity of the RXR/LXR heterodimer in the liver. Arterioscler Thromb Vasc Biol. 2009;29:1488–1495. doi: 10.1161/ATVBAHA.109.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell. 2004;116:417–429. doi: 10.1016/s0092-8674(04)00119-9. [DOI] [PubMed] [Google Scholar]
- 83.Tzameli I, Chua SS, Cheskis B, Moore DD. Complex effects of rexinoids on ligand dependent activation or inhibition of the xenobiotic receptor. CAR Nucl Recept. 2003;1:2. doi: 10.1186/1478-1336-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suino K, Peng L, Reynolds R, Li Y, Cha J-Y, Repa JJ, Kliewer SA, Xu HE. The nuclear xenobiotic receptor CAR: Structural determinants of constitutive activation and heterodimerization. Mol Cell. 2004;16:893–905. doi: 10.1016/j.molcel.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 85.Putcha B-DK, Fernandez EJ. Direct interdomain interactions can mediate allosterism in the thyroid receptor. J Biol Chem. 2009;284:22517–22524. doi: 10.1074/jbc.M109.026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bettoun DJ, Burris TP, Houck KA, Buck DW, II, Stayrook KR, Khalifa B, Lu J, Chin WW, Nagpal S. Retinoid X receptor is a nonsilent major contributor to vitamin D receptor-mediated transcriptional activation. Mol Endocrinol. 2003;17:2320–2328. doi: 10.1210/me.2003-0148. [DOI] [PubMed] [Google Scholar]
- 87.Fradera X, Vu D, Nimz O, Skene R, Hosfield D, Wynands R, Cooke AJ, Haunso A, King A, Bennett DJ, McGuire R, Uitdehaag JCM. X-ray structures of the LXRα LBD in its homodimeric form and implications for heterodimer signaling. J Mol Biol. 2010;399:120–132. doi: 10.1016/j.jmb.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 88.Svensson S, Östberg T, Jacobsson M, Norström C, Stefansson K, Hallén D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L. Crystal structure of the heterodimeric complex of LXRα and RXRβ ligand-binding domains in a fully agonistic conformation. EMBO J. 2003;22:4625–4633. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brelivet Y, Kammerer S, Rochel N, Poch O, Moras D. Signature of the oligomeric behaviour of nuclear receptors at the sequence and structural level. EMBO Rep. 2004;5:423–429. doi: 10.1038/sj.embor.7400119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.IJpenberg A, Tan NS, Gelman L, Kersten S, Seydoux J, Xu J, Metzger D, Canaple L, Chambon P, Wahli W, Desvergne B. In vivo activation of PPAR target genes by RXR homodimers. EMBO J. 2004;23:2083–2091. doi: 10.1038/sj.emboj.7600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yasmin R, Kannan-Thulasiraman P, Kagechika H, Dawson MI, Noy N. Inhibition of mammary carcinoma cell growth by RXR is mediated by the receptor’s oligomeric switch. J Mol Biol. 2010;397:1121–1131. doi: 10.1016/j.jmb.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yasmin R, Yeung KT, Chung RH, Gaczynska ME, Osmulski PA, Noy N. DNA-looping by RXR tetramers permits transcriptional regulation “at a distance”. J Mol Biol. 2004;343:327–338. doi: 10.1016/j.jmb.2004.08.070. [DOI] [PubMed] [Google Scholar]
- 93.Hashimoto Y, Miyachi H. Nuclear receptor antagonists designed based on the helix-folding inhibition hypothesis. Bioorg Med Chem. 2005;13:5080–5093. doi: 10.1016/j.bmc.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 94.Yan X, Broderick D, Leid ME, Schimerlik MI, Deinzer ML. Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry. 2004;43:909–917. doi: 10.1021/bi030183c. [DOI] [PubMed] [Google Scholar]
- 95.Egea PF, Rochel N, Birck C, Vachette P, Timmins PA, Moras D. Effects of ligand binding on the association properties and conformation in solution of retinoic acid receptors RXR and RAR. J Mol Biol. 2001;307:557–576. doi: 10.1006/jmbi.2000.4409. [DOI] [PubMed] [Google Scholar]
- 96.Gampe RT, Jr, Montana VG, Lambert MH, Wisely GB, Milburn MV, Xu HE. Structural basis for autorepression of retinoid X receptor by tetramer formation and the AF-2 helix. Genes Dev. 2000;14:2229–2241. doi: 10.1101/gad.802300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang H, Chen L, Chen J, Jiang H, Shen X. Structural basis for retinoic X receptor repression on the tetramer. J Biol Chem. 2011;286:24593–24598. doi: 10.1074/jbc.M111.245498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang Q, Lu G, Shen HM, Chung MCM, Ong CN. Anti-cancer properties of anthraquinones from rhubarb. Med Res Rev. 2007;27:609–630. doi: 10.1002/med.20094. [DOI] [PubMed] [Google Scholar]
- 99.Gampe RT, Jr, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE. Asymmetry in the PPARγ/RXRα crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cel. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 100.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Natur. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 101.Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Garcia-Ordonez RD, Pascal BD, Burris TP, Dodge JA, Griffin PR. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure. 2010;18:1332–1341. doi: 10.1016/j.str.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang H, Li L, Chen L, Hu L, Jiang H, Shen X. Structure basis of bigelovin as a selective RXR agonist with a distinct binding mode. J Mol Biol. 2011;407:13–20. doi: 10.1016/j.jmb.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 103.Zeng G-Z, Tan N-H, Ji C-J, Fan J-T, Huang H-Q, Han H-J, Zhou G-B. Apoptosis inducement of bigelovin from Inula helianthus-aquatica on human leukemia U937 cells. Phytother Res. 2009;23:885–891. doi: 10.1002/ptr.2671. [DOI] [PubMed] [Google Scholar]
- 104.Bruck N, Bastien J, Bour G, Tarrade A, Plassat J-L, Bauer A, Adam-Stitah S, Rochette-Egly C. Phosphorylation of the retinoid X receptor at the omega loop, modulates the expression of retinoic-acid-target genes with a promoter context specificity. Cell Signal. 2005;17:1229–1239. doi: 10.1016/j.cellsig.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 105.Shimizu M, Moriwaki H. Synergistic effects of PPARγ ligands and retinoids in cancer treatment. PPAR Res. 2008 doi: 10.1155/2008/181047. Article ID 181047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matsushima-Nishiwaki R, Okuno M, Adachi S, Sano T, Akita K, Moriwaki H, Friedman SL, Kojima S. Phosphorylation of retinoid X receptor α at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res. 2001;61:7675–7682. [PubMed] [Google Scholar]
- 107.Yoshimura K, Muto Y, Shimizu M, Matsushima-Nishiwaki R, Okuno M, Takano Y, Tsurumi H, Kojima S, Okano Y, Moriwaki H. Phosphorylated retinoid X receptor α loses its heterodimeric activity with retinoic acid receptor β. Cancer Sci. 2007;98:1868–1874. doi: 10.1111/j.1349-7006.2007.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Macoritto M, Nguyen-Yamamoto L, Huang DC, Samuel S, Yang XF, Wang TT, White JH, Kremer R. Phosphorylation of the human retinoid X receptor α at serine 260 impairs coactivator(s) recruitment and induces hormone resistance to multiple ligands. J Biol Chem. 2008;283:4943–4956. doi: 10.1074/jbc.M707517200. [DOI] [PubMed] [Google Scholar]
- 109.Zhao Y, Qin S, Atangan LI, Molina Y, Okawa Y, Arpawong HT, Ghosn C, Xiao J-H, Vuligonda V, Brown G, Chandraratna RAS. Casein kinase 1α interacts with retinoid X receptor and interferes with agonist-induced apoptosis. J Biol Chem. 2004;279:30844–30849. doi: 10.1074/jbc.M404651200. [DOI] [PubMed] [Google Scholar]
- 110.Bastien J, Adam-Stitah S, Plassat JL, Chambon P, Rochette-Egly C. The phosphorylation site located in the A region of retinoic X receptor α is required for the antiproliferative effect of retinoic acid (RA) and the activation of RA target genes in F9 cells. J Biol Chem. 2002;277:28683–28689. doi: 10.1074/jbc.M203623200. [DOI] [PubMed] [Google Scholar]
- 111.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 112.Gianni M, Tarrade A, Nigro EA, Garattini E, Rochette-Egly C. The AF-1 and AF-2 domains of RARγ2 and RXRα cooperate for triggering the transactivation and the degradation of RARγ2/RXRα heterodimers. J Biol Chem. 2003;278:34458–34466. doi: 10.1074/jbc.M304952200. [DOI] [PubMed] [Google Scholar]
- 113.Casas F, Daury L, Grandemange S, Busson M, Seyer P, Hatier R, Carazo A, Cabello G, Wrutniak-Cabello C. Endocrine regulation of mitochondrial activity: involvement of truncated RXRα and c-Erb Aα1 proteins. FASEB J. 2003;17:426–436. doi: 10.1096/fj.02-0732com. [DOI] [PubMed] [Google Scholar]
- 114.Matsushima-Nishiwaki R, Shidoji Y, Nishiwaki S, Moriwaki H, Muto Y. Limited degradation of retinoid X receptor by calpain. Biochem Biophys Res Commun. 1996;225:946–951. doi: 10.1006/bbrc.1996.1276. [DOI] [PubMed] [Google Scholar]
- 115.Pettersson F, Hanna N, Lagodich M, Dupéré-Richer D, Couture MC, Choi C, Miller WH., Jr Rexinoids modulate steroid and xenobiotic receptor activity by increasing its protein turnover in a calpain-dependent manner. J Biol Chem. 2008;283:21945–21952. doi: 10.1074/jbc.M710358200. [DOI] [PubMed] [Google Scholar]
- 116.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NCO, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: A promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 117.Kassam A, Miao B, Young PR, Mukherjee R. Retinoid X receptor (RXR) agonist-induced antagonism of farnesoid X receptor (FXR) activity due to absence of coactivator recruitment and decreased DNA binding. J Biol Chem. 2003;278:10028–10032. doi: 10.1074/jbc.M208312200. [DOI] [PubMed] [Google Scholar]
- 118.Dong S, Stenoien DL, Qiu J, Mancini MA, Tweardy DJ. Reduced intranuclear mobility of APL fusion proteins accompanies their mislocalization and results in sequestration and decreased mobility of retinoid X receptor α. Mol Cell Biol. 2004;24:4465–4475. doi: 10.1128/MCB.24.10.4465-4475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zeisig BB, Kwok C, Zelent A, Shankaranarayanan P, Gronemeyer H, Dong S, So CWE. Recruitment of RXR by homotetrameric RARα fusion proteins is essential for transformation. Cancer Cel. 2007;12:36–51. doi: 10.1016/j.ccr.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 120.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJA, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11(Suppl. 2):S126–S143. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 121.Quinn CM, Jessup W, Wong J, Kritharides L, Brown AJ. Expression and regulation of sterol 27-hydroxylase (CYP27A1) in human macrophages: a role for RXR and PPARγ ligands. Biochem J. 2005;385:823–830. doi: 10.1042/BJ20041776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tanaka T, De Luca LM. Therapeutic potential of “rexinoids” in cancer prevention and treatment. Cancer Res. 2009;69:4945–4947. doi: 10.1158/0008-5472.CAN-08-4407. [DOI] [PubMed] [Google Scholar]
- 123.Takiyama Y, Miyokawa N, Sugawara A, Kato S, Ito K, Sato K, Oikawa K, Kobayashi H, Kimura S, Tateno M. Decreased expression of retinoid X receptor isoforms in human thyroid carcinomas. J Clin Endocrinol Metab. 2004;89:5851–5861. doi: 10.1210/jc.2003-032036. [DOI] [PubMed] [Google Scholar]
- 124.Balasubramanian S, Chandraratna RAS, Eckert RL. Suppression of human pancreatic cancer cell proliferation by AGN194204, an RXR-selective retinoid. Carcinogenesis. 2004;25:1377–1385. doi: 10.1093/carcin/bgh122. [DOI] [PubMed] [Google Scholar]
- 125.Liby KT, Royce DB, Risingsong R, Williams CR, Maitra A, Hruban RH, Sporn MB. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev Res. 2010;3:1427–1434. doi: 10.1158/1940-6207.CAPR-10-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bass T. More mileage out of myelin. SciBX. 2011;4:6–7. [Google Scholar]
- 127.Morgan TR. Chemoprevention of hepatocellular carcinoma in chronic hepatitis C. Recent Results Cancer Res. 2011;188:85–99. doi: 10.1007/978-3-642-10858-7_7. [DOI] [PubMed] [Google Scholar]
- 128.Shimizu M, Sakai H, Moriwaki H. Chemoprevention of hepatocellular carcinoma by acyclic retinoid. Front Biosci. 2011;16:759–769. doi: 10.2741/3718. [DOI] [PubMed] [Google Scholar]
- 129.Okita K. Peretinoin may reduce de novo carcinogenesis. Gastrointestinal Cancers Symposium 2011, American Society of Clinical Oncology; San Francisco, CA. January 11, 2011; Kowa Company, Ltd.; 2011. Abstract #165. http//www.businesswire.com/news/home/20110123005006/e. [Google Scholar]
- 130.Tsujino T, Nagata T, Katoh F, Yamasaki H. Inhibition of Balb/c 3T3 cell transformation by synthetic acyclic retinoid NIK-333; possible involvement of enhanced gap junctional intercellular communication. Cancer Detect Prev. 2007;31:332–338. doi: 10.1016/j.cdp.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 131.Muto Y, Moriwaki H, Omori M. In vitro binding affinity of novel synthetic polyprenoids (polyprenoic acids) to cellular retinoid-binding proteins. Gann. 1981;72:974–977. [PubMed] [Google Scholar]
- 132.Araki H, Shidoji Y, Yamada Y, Moriwaki H, Muto Y. Retinoid agonist activities of synthetic geranyl geranoic acid derivatives. Biochem Biophys Res Commun. 1995;209:66–72. doi: 10.1006/bbrc.1995.1471. [DOI] [PubMed] [Google Scholar]
- 133.Muto Y, Moriwaki H, Ninomiya M, Adachi S, Saito A, Takasaki KT, Tanaka T, Tsurumi K, Okuno M, Tomita E, Nakamura T, Kojima T. Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma for the Hepatoma Prevention Study Group. N Engl J Med. 1996;334:1561–1567. doi: 10.1056/NEJM199606133342402. [DOI] [PubMed] [Google Scholar]
- 134.Muto Y, Moriwaki H, Saito A. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. N Engl J Med. 1999;340:1046–1047. doi: 10.1056/NEJM199904013401315. [DOI] [PubMed] [Google Scholar]
- 135.Takai K, Okuno M, Yasuda I, Matsushima-Nishiwaki R, Uematsu T, Tsurumi H, Shiratori Y, Muto Y, Moriwaki H. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. Updated analysis of the long-term follow-up data. Intervirology. 2005;48:39–45. doi: 10.1159/000082093. [DOI] [PubMed] [Google Scholar]
- 136.Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V. Increased expression of RXRα in dementia: An early harbinger for the cholesterol dyshomeostasis? Mol Neurodegener. 2010;5:36. doi: 10.1186/1750-1326-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang Y-L, Chen S-J, Kao C-L, Hung S-C, Ding D-C, Yu C-C, Chen Y-J, Ku H-H, Lin C-P, Lee K-H, Chen Y-C, Wang J-J, Hsu C-C, Chen L-K, Li H-Y, Chiou S-H. Docosahexaenoic acid promotes dopaminergic differentiation in induced pluripotent stem cells and inhibits teratoma formation in rats with Parkinson-like pathology. Cell Transplant. 2011;20 doi: 10.3727/096368911X580572. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 138.Friling S, Bergsland M, Kjellander S. Activation of retinoid X receptor increases dopamine cell survival in models for Parkinson’s disease. BMC Neurosci. 2009;10:146. doi: 10.1186/1471-2202-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, Xu H, Walker NPC, Perlmann T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 140.Poppe L, Harvey TS, Mohr C, Zondlo J, Tegley CM, Nuanmanee O, Cheetham J. Discovery of ligands for Nurr1 by combined use of NMR screening with different isotopic and spin-labeling strategies. J Biomol Screen. 2007;12:301–311. doi: 10.1177/1087057106299161. [DOI] [PubMed] [Google Scholar]
- 141.Michiels P, Atkins K, Ludwig C, Whittaker S, van Dongen M, Günther U. Assignment of the orphan nuclear receptor Nurr1 by NMR. Biomol NMR Assign. 2010;4:101–105. doi: 10.1007/s12104-010-9210-4. [DOI] [PubMed] [Google Scholar]
- 142.Miyazaki S, Taniguchi H, Moritoh Y, Tashiro F, Yamamoto T, Yamato E, Ikegami H, Ozato K, Miyazaki J. Nuclear hormone retinoid X receptor (RXR) negatively regulates the glucose-stimulated insulin secretion of pancreatic β-cells. Diabete. 2010;59:2854–2861. doi: 10.2337/db09-1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Núñez V, Alameda D, Rico D, Mota R, Gonzalo P, Cedenilla M, Fischer T, Boscá L, Glass CK, Arroyo AG, Ricote M. Retinoid X receptor α controls innate inflammatory responses through the up-regulation of chemokine expression. Proc Natl Acad Sci US. 2010;107:10626–10631. doi: 10.1073/pnas.0913545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Song PI, Park Y-M, Abraham T, Harten B, Zivony A, Neparidze N, Armstrong CA, Ansel JC. Human keratinocytes express functional CD14 and toll-like receptor 4. J Invest Dermatol. 2002;119:424–432. doi: 10.1046/j.1523-1747.2002.01847.x. [DOI] [PubMed] [Google Scholar]
- 145.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–874. [PubMed] [Google Scholar]
- 146.Venteclef N, Jakobsson T, Steffensen KR, Treuter E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol Metab. 2011;22:333–343. doi: 10.1016/j.tem.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 147.Valledor AF, Hsu L-C, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci USA. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shudo K, Kagechika H, Yamazaki N, Igarashi M, Tateda C. A synthetic retinoid Am80 (tamibarotene) rescues the memory deficit caused by scopolamine in a passive avoidance paradigm. Biol Pharm Bull. 2004;27:1887–1889. doi: 10.1248/bpb.27.1887. [DOI] [PubMed] [Google Scholar]
- 149.Ogilvie KM, Saladin R, Nagy TR, Urcan MS, Heyman RA, Leibowitz MD. Activation of the retinoid X receptor suppresses appetite in the rat. Endocrinology. 2004;145:565–573. doi: 10.1210/en.2003-0907. [DOI] [PubMed] [Google Scholar]
- 150.Janesick A, Blumberg B. Minireview: PPARγ as the target of obesogens. J Steroid Biochem Mol Biol. 2011 doi: 10.1016/j.jsbmb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kirchner S, Kieu T, Chow C, Casey S, Blumberg B. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24:526–539. doi: 10.1210/me.2009-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li X, Ycaza J, Blumberg B. The environmental obesogen tributyltin chloride acts via peroxisome proliferator activated receptor gamma to induce adipogenesis in murine 3T3-L1 preadipocytes. J Steroid Biochem Mol Biol. 2011 doi: 10.1016/j.jsbmb.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schauber J, Oda Y, Büchau AS, Yun Q-C, Steinmeyer A, Zügel U, Bikle DD, Gallo RL. Histone acetylation in keratinocytes enables control of the expression of cathelicidin and CD14 by 1,25-dihydroxyvitamin D3. J Invest Dermatol. 2008;128:816–824. doi: 10.1038/sj.jid.5701102. [DOI] [PubMed] [Google Scholar]
- 154.Fowler J. Chronic hand eczema: A prevalent and challenging skin condition. Cutis. 2008;82(4 Suppl):4–8. [PubMed] [Google Scholar]
- 155.Meding B. Epidemiology of hand eczema in an industrial city. Acta Derm Venereol Suppl (Stockh) 1990;153:1–43. [PubMed] [Google Scholar]
- 156.Pivarcsi A, Kemény L, Dobozy A. Innate immune functions of the keratinocytes. A review. Acta Microbiol Immunol Hung. 2004;51:303–310. doi: 10.1556/AMicr.51.2004.3.8. [DOI] [PubMed] [Google Scholar]
- 157.Ruzicka T, Lynde CW, Jemec GBE, Diepgen T, Berth-Jones J, Coenraads PJ, Kaszuba A, Bissonnette R, Varjonen E, Holló P, Cambazard F, Lahfa M, Elsner P, Nyberg F, Svensson A, Brown TC, Harsch M, Maares J. Efficacy and safety of oral alitretinoin (9-cis retinoic acid) in patients with severe chronic hand eczema refractory to topical corticosteroids: results of a randomized, double-blind, placebo-controlled, multicentre trial. Br J Dermatol. 2008;158:808–817. doi: 10.1111/j.1365-2133.2008.08487.x. [DOI] [PubMed] [Google Scholar]
- 158.Hanifin JM, Stevens V, Sheth P, Breneman D. Novel treatment of chronic severe hand dermatitis with bexarotene gel. Br J Dermatol. 2004;150:545–553. doi: 10.1046/j.1365-2133.2003.05729.x. [DOI] [PubMed] [Google Scholar]
- 159.Bikle DD, Teichert A, Arnold LA, Uchida Y, Elias PM, Oda Y. Differential regulation of epidermal function by VDR coactivators. J Steroid Biochem Mol Biol. 2010;121:308–313. doi: 10.1016/j.jsbmb.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Büchau AS, Schauber J, Hultsch T, Stuetz A, Gallo RL. Pimecrolimus enhances TLR2/6-induced expression of antimicrobial peptides in keratinocytes. J Invest Dermatol. 2008;128:2646–2654. doi: 10.1038/jid.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dai X, Sayama K, Tohyama M, Shirakata Y, Hanakawa Y, Tokumaru S, Yang L, Hirakawa S, Hashimoto K. PPARγ mediates innate immunity by regulating the 1α,25-dihydroxyvitamin D3 induced hBD-3 and cathelicidin in human keratinocytes. J Dermatol Sci. 2010;60:179–186. doi: 10.1016/j.jdermsci.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 162.Wei L-N. Retinoids and receptor interacting protein 140 (RIP140) in gene regulation. Curr Med Chem. 2004;11:1527–1532. doi: 10.2174/0929867043365017. [DOI] [PubMed] [Google Scholar]
- 163.Bushue N, Wan Y-JY. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285–1298. doi: 10.1016/j.addr.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shimizu M, Takai K, Moriwaki H. Strategy and mechanism for the prevention of hepatocellular carcinoma: Phosphorylated retinoid X receptor α is a critical target for hepatocellular carcinoma chemoprevention. Cancer Sci. 2009;100:369–374. doi: 10.1111/j.1349-7006.2008.01045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Baron-Van Evercooren A, Chambon P, ffrench-Constant C, Franklin RJM. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14:45–53. doi: 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dawson MI, Zhang X-K. Discovery and design of retinoic acid receptor and retinoid X receptor class- and subtype-selective synthetic analogs of all-trans-retinoic acid and 9-cis-retinoic acid. Curr Med Chem. 2002;9:623–637. doi: 10.2174/0929867023370789. [DOI] [PubMed] [Google Scholar]
- 167.de Lera AR, Bourguet W, Altucci L, Gronemeyer H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat Rev Drug Discov. 2007;6:811–820. doi: 10.1038/nrd2398. [DOI] [PubMed] [Google Scholar]
- 168.Desvergne B. RXR: From partnership to leadership in metabolic regulations. Vitam Horm. 2007;75:1–32. doi: 10.1016/S0083-6729(06)75001-4. [DOI] [PubMed] [Google Scholar]
- 169.Dawson MI. Retinoids. In: Rotella D, editor. Burger’s Medicinal Chemistry, Discovery and Develpment. Wiley Online Library, John Wiley & Sons; New York: 2010. pp. 1–119. [Google Scholar]
- 170.Leibowitz MD, Ardecky RJ, Boehm MF, Broderick CL, Carfagna MA, Crombie DL, D’Arrigo J, Etgen GJ, Faul MM, Grese TA, Havel H, Hein NI, Heyman RA, Jolley D, Klausing K, Liu S, Mais DE, Mapes CM, Marschke KB, Michellys P-Y, Montrose-Rafizadeh C, Ogilvie KM, Pascual B, Rungta D, Tyhonas JS, Urcan MS, Wardlow M, Yumibe N, Reifel-Miller A. Biological characterization of a heterodimer-selective retinoid X receptor modulator: Potential benefits for the treatment of type 2 diabetes. Endocrinology. 2006;147:1044–1053. doi: 10.1210/en.2005-0690. [DOI] [PubMed] [Google Scholar]
- 171.Ishida S, Shigemoto-Mogami Y, Kagechika H, Shudo K, Ozawa S, Sawada J, Ohno Y, Inoue K. Clinically potential subclasses of retinoid synergists revealed by gene expression profiling. Mol Cancer Ther. 2003;2:49–58. [PubMed] [Google Scholar]
- 172.Henrich VC, Sliter TJ, Lubahn DB, MacIntyre A, Gilbert LI. A steroid/thyroid hormone receptor superfamily member in Drosophila melanogaster that shares extensive sequence similarity with a mammalian homologue. Nucl Acids Res. 1990;18:4143–4148. doi: 10.1093/nar/18.14.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Oro AE, McKeown M, Evans RM. Relationship between the product of the Drosophila ultraspiracle locus and the vertebrate retinoid X receptor. Nature. 1990;347:298–301. doi: 10.1038/347298a0. [DOI] [PubMed] [Google Scholar]
- 174.Beck Y, Delaporte C, Moras D, Richards G, Billas IML. The ligand-binding domains of the three RXR-USP nuclear receptor types support distinct tissue and ligand specific hormonal responses in transgenic Drosophila. Dev Biol. 2009;330:1–11. doi: 10.1016/j.ydbio.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 175.Sasorith S, Billas IML, Iwema T, Moras D, Wurtz J-M. Structure-based analysis of the ultraspiracle protein and docking studies of putative ligands. J Insect Sci. 2002;2:25. doi: 10.1093/jis/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hayward DC, Dhadialla TS, Zhou S, Kuiper MJ, Ball EE, Wyatt GR, Walker VK. Ligand specificity and developmental expression of RXR and ecdysone receptor in the migratory locust. J Insect Physiol. 2003;49:1135–1144. doi: 10.1016/j.jinsphys.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 177.Nowickyj SM, Chithalen JV, Cameron D, Tyshenko MG, Petkovich M, Wyatt GR, Jones G, Walker VK. Locust retinoid X receptors: 9-Cis-retinoic acid in embryos from a primitive insect. Proc Natl Acad Sci USA. 2008;105:9540–9545. doi: 10.1073/pnas.0712132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nagaraju GPC, Prasad GLV, Taliaferro-Smith L, Aruna BV, Naik BR, Sekhar YN. Computational analysis of the structural basis of ligand binding to the crustacean retinoid X receptor, Comp Biochem Physiol Part D: Genom. Proteo. 2010;5:314–324. doi: 10.1016/j.cbd.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 179.Wu X, Hopkins PM, Palli SR, Durica DS. Crustacean retinoid-X receptor isoforms: distinctive DNA binding and receptor receptor interaction with a cognate ecdysteroid receptor. Mol Cell Endocrinol. 2004;218:21–38. doi: 10.1016/j.mce.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 180.Carter CJ, Farrar N, Carlone RL, Spencer GE. Developmental expression of a molluscan RXR and evidence for its novel, nongenomic role in growth cone guidance. Dev Biol. 2010;343:124–137. doi: 10.1016/j.ydbio.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 181.Mangelsdorf DJ, Umesono K, Evans RM. The retinoid receptors: Biology, chemistry, and medicine. In: Sporn MB, Roberts AB, Goodman DS, editors. The Retinoid Receptors. Raven Press; New York: 1994. pp. 319–349. [Google Scholar]
- 182.Gao J, Xie W. Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab Dispos. 2010;38:2091–2095. doi: 10.1124/dmd.110.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kemper JK. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim Biophys Acta. 2011;1812:842–850. doi: 10.1016/j.bbadis.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Washburn DG, Hoang TH, Campobasso N, Smallwood A, Parks DJ, Webb CL, Frank KA, Nord M, Duraiswami C, Evans C, Jaye M, Thompson SK. Synthesis and SAR of potent LXR agonists containing an indole pharmacophore. Bioorg Med Chem Lett. 2009;19:1097–1100. doi: 10.1016/j.bmcl.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 185.Jung C-G, Horike H, Cha B-Y, Uhm K-O, Yamauchi R, Yamaguchi T, Hosono T, Iida K, Woo J-T, Michikawa M. Honokiol increases ABCA1 expression level by activating retinoid X receptor beta. Biol Pharm Bull. 2010;33:1105–1111. doi: 10.1248/bpb.33.1105. [DOI] [PubMed] [Google Scholar]
- 186.Pearen MA, Muscat GEO. Minireview: Nuclear hormone receptor 4A signaling: Implications for metabolic disease. Mol Endocrinol. 2010;24:1891–1903. doi: 10.1210/me.2010-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhao Y, Bruemmer D. NR4A orphan nuclear receptors: Transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol. 2010;30:1535–1541. doi: 10.1161/ATVBAHA.109.191163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Volakakis N, Joodmardi E, Perlmann T. NR4A orphan nuclear receptors influence retinoic acid and docosahexaenoic acid signaling via up-regulation of fatty acid binding protein 5. Biochem Biophys Res Commun. 2009;390:1186–1191. doi: 10.1016/j.bbrc.2009.10.116. [DOI] [PubMed] [Google Scholar]
- 189.Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, Oplinger JA, Kliewer SA, Gampe RT, Jr, McKee DD, Moore JT, Willson TM. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 2001;98:13919–13924. doi: 10.1073/pnas.241410198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bugge A, Mandrup S. Molecular mechanisms and genome-wide aspects of PPAR subtype specific transactivation. PPAR Res. 2010 doi: 10.1155/2010/169506. Article ID 169506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gustafsson MCU, Knight D, Palmer CNA. Ligand modulated antagonism of PPARγ by genomic and non-genomic actions of PPARδ. PLoS ONE. 2009;4:e7046. doi: 10.1371/journal.pone.0007046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ekins S, Kholodovych V, Ai N, Sinz M, Gal J, Gera L, Welsh WJ, Bachmann K, Mani S. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Mol Pharmacol. 2008;74:662–672. doi: 10.1124/mol.108.049437. [DOI] [PubMed] [Google Scholar]
- 193.Rastinejad F, Wagner T, Zhao Q, Khorasanizadeh S. Structure of the RXR–RAR DNA-binding complex on the retinoic acid response element DR1. EMBO J. 2000;19:1045–1054. doi: 10.1093/emboj/19.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Billas IML, Moulinier L, Rochel N, Moras D. Crystal structure of the ligand-binding domain of the ultraspiracle protein USP, the ortholog of retinoid X receptors in insects. J Biol Chem. 2001;276:7465–7474. doi: 10.1074/jbc.M008926200. [DOI] [PubMed] [Google Scholar]
- 195.Clayton GM, Peak-Chew SY, Evans RM, Schwabe JWR. The structure of the ultraspiracle ligand-binding domain reveals a nuclear receptor locked in an inactive conformation. Proc Natl Acad Sci USA. 2001;98:1549–1554. doi: 10.1073/pnas.041611298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Haffner CD, Lenhard JM, Miller AB, McDougald DL, Dwornik K, Ittoop OR, Gampe RT, Jr, Xu HE, Blanchard S, Montana VG, Consler TG, Bledsoe RK, Ayscue A, Croom D. Structure-based design of potent retinoid X receptor α agonists. J Med Chem. 2004;47:2010–2029. doi: 10.1021/jm030565g. [DOI] [PubMed] [Google Scholar]
- 197.Devarakonda S, Harp JM, Kim Y, Ozyhar A, Rastinejad F. Structure of the heterodimeric ecdysone receptor DNA-binding complex. EMBO J. 2003;22:5827–5840. doi: 10.1093/emboj/cdg569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Billas IML, Iwema T, Garnier J-M, Mitschler A, Rochel N, Moras D. Structural adaptability in the ligand-binding pocket of the ecdysone hormone receptor. Nature. 2003;426:91–96. doi: 10.1038/nature02112. [DOI] [PubMed] [Google Scholar]
- 199.Holmbeck SMA, Foster MP, Casimiro DR, Sem DS, Dyson HJ, Wright PE. High-resolution solution structure of the retinoid X receptor DNA-binding domain. J Mol Biol. 1998;281:271–284. doi: 10.1006/jmbi.1998.1908. [DOI] [PubMed] [Google Scholar]
- 200.de Groot A, de Rosny E, Juillan-Binard C, Ferrer J-L, Laudet V, Pierce RJ, Pebay-Peyroula E, Fontecilla-Camps JC, Borel F. Crystal structure of a novel tetrameric complex of agonist-bound ligand-binding domain of Biomphalaria glabrata retinoid X receptor. J Mol Biol. 2005;354:841–853. doi: 10.1016/j.jmb.2005.09.090. [DOI] [PubMed] [Google Scholar]
- 201.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. A structural basis for constitutive activity in the human CAR/RXRα heterodimer. Mol Cel. 2004;16:919–928. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 202.Shaffer PL, Gewirth DT. Structural analysis of RXR–VDR interactions on DR3 DNA. J Steroid Biochem Mol Biol. 2004;89-90:215–219. doi: 10.1016/j.jsbmb.2004.03.084. [DOI] [PubMed] [Google Scholar]
- 203.Carmichael JA, Lawrence MC, Graham LD, Pilling PA, Epa VC, Noyce L, Lovrecz G, Winkler DA, Pawlak-Skrzecz A, Eaton RE, Hannan GN, Hill RJ. The X-ray structure of a hemipteran ecdysone receptor ligand-binding domain: Comparison with a lepidopteran ecdysone receptor ligand-binding domain and implications for insecticide design. J Biol Chem. 2005;280:22258–22269. doi: 10.1074/jbc.M500661200. [DOI] [PubMed] [Google Scholar]
- 204.Schuetz A, Min JR, Loppnau P, Weigelt J, Sundstrom M, Edwards AM, Arrowsmith CH, Bochkarev A, Plotnikov AN. Human retinoic acid receptor RXR-γ ligand-binding domain. RCSB protein data bank PDB 2GL8 (deposited 2006, modified 2009) [Google Scholar]
- 205.Jakób M, Kołodziejczyk R, Orłowski M, Krzywda S, Kowalska A, Dutko-Gwóźdź J, Gwóźdź T, Kochman M, Jaskólski M, Ożyhar A. Novel DNA-binding element within the C-terminal extension of the nuclear receptor DNA-binding domain. Nucl Acids Res. 2007;35:2705–2718. doi: 10.1093/nar/gkm162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Iwema T, Billas IML, Beck Y, Bonneton F, Nierengarten H, Chaumot A, Richards G, Laudet V, Moras D. Structural and functional characterization of a novel type of ligand-independent RXR-USP receptor. EMBO J. 2007;26:3770–3782. doi: 10.1038/sj.emboj.7601810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nahoum V, Pérez E, Germain P, Rodríguez-Barrios F, Manzo F, Kammerer S, Lemaire G, Hirsch O, Royer CA, Gronemeyer H, de Lera AR, Bourguet W. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc Natl Acad Sci USA. 2007;104:17323–17328. doi: 10.1073/pnas.0705356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Borel F, de Groot A, Juillan-Binard C, de Rosny E, Laudet V, Pebay-Peyroula E, Fontecilla-Camps JC, Ferrer J-L. Crystal structure of the ligand-binding domain of the retinoid X receptor from the ascidian Polyandrocarpa misakiensis. Proteins. 2009;74:538–542. doi: 10.1002/prot.22294. [DOI] [PubMed] [Google Scholar]
- 209.Browning C, Martin E, Loch C, Wurtz J-M, Moras D, Stote RH, Dejaegere AP, Billas IML. Critical role of desolvation in the binding of 20-hydroxyecdysone to the ecdysone receptor. J Biol Chem. 2007;282:32924–32934. doi: 10.1074/jbc.M705559200. [DOI] [PubMed] [Google Scholar]
- 210.Lippert WP, Burschka C, Götz K, Kaupp M, Ivanova D, Gaudon C, Sato Y, Antony P, Rochel N, Moras D, Gronemeyer H, Tacke R. Silicon analogues of the RXR-selective retinoid agonist SR11237 (BMS649): Chemistry and biology. ChemMedChem. 2009;4:1143–1152. doi: 10.1002/cmdc.200900090. [DOI] [PubMed] [Google Scholar]
- 211.Sato Y, Ramalanjaona N, Huet T, Potier N, Osz J, Antony P, Peluso-Iltis C, Poussin-Courmontagne P, Ennifar E, Mely Y, Dejaegere A, Moras D, Rochel N. The “phantom effect” of the rexinoid LG100754: Structural and functional insights. PLoS ONE. 2010;5:e15119. doi: 10.1371/journal.pose.0015119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tocchini-Valentini GD, Rochel N, Escriva H, Germain P, Peluso-Iltis C, Paris M, Sanglier-Cianferani S, Van Dorsselaer A, Moras D, Laudet V. Structural and functional insights into the ligand-binding domain of a nonduplicated retinoid X nuclear receptor from the invertebrate chordate amphioxus. J Biol Chem. 2009;284:1938–1948. doi: 10.1074/jbc.M805692200. [DOI] [PubMed] [Google Scholar]
- 213.Chao EY, Caravella JA, Watson MA, Campobasso N, Ghisletti S, Billin AN, Galardi C, Wang P, Laffitte BA, Iannone MA, Goodwin BJ, Nichols JA, Parks DJ, Stewart E, Wiethe RW, Williams SP, Smallwood A, Pearce KH, Glass CK, Willson TM, Zuercher WJ, Collins JL. Structure-guided design of N-phenyl tertiary amines as transrepression-selective liver X receptor modulators with anti-inflammatory activity. J Med Chem. 2008;51:5758–5765. doi: 10.1021/jm800612u. [DOI] [PubMed] [Google Scholar]
- 214.Pérez Santín E, Germain P, Quillard F, Khanwalkar H, Rodríguez-Barrios F, Gronemeyer H, de Lera ÁR, Bourguet W. Modulating retinoid X receptor with a series of (E)-3-[4-hydroxy-3-(3-alkoxy-5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic acids and their 4-alkoxy isomers. J Med Chem. 2009;52:3150–3158. doi: 10.1021/jm900096q. [DOI] [PubMed] [Google Scholar]
- 215.Connors RV, Wang Z, Harrison M, Zhang A, Wanska M, Hiscock S, Fox B, Dore M, Labelle M, Sudom A, Johnstone S, Liu J, Walker NPC, Chai A, Siegler K, Li Y, Coward P. Identification of a PPARδ agonist with partial agonistic activity on PPARγ. Bioorg Med Chem Lett. 2009;19:3550–3554. doi: 10.1016/j.bmcl.2009.04.151. [DOI] [PubMed] [Google Scholar]
- 216.Zhang H, Zhou R, Li L, Chen J, Chen L, Li C, Ding H, Yu L, Hu L, Jiang H, Shen X. Danthron functions as a retinoic X receptor antagonist by stabilizing tetramers of the receptor. J Biol Chem. 2011;286:1868–1875. doi: 10.1074/jbc.M110.166215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.le Maire A, Bourguet W, Balaguer P. A structural view of nuclear hormone receptor: endocrine disruptor interactions. Cell Mol Life Sci. 2010;67:1219–1237. doi: 10.1007/s00018-009-0249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kotani H, Tanabe H, Mizukami H, Makishima M, Inoue M. Identification of a naturally occurring rexinoid, honokiol, that activates the retinoid X receptor. J Nat Prod. 2010;73:1332–1336. doi: 10.1021/np100120c. [DOI] [PubMed] [Google Scholar]
- 219.Yamada S, Ohsawa F, Fujii S, Shinozaki R, Makishima M, Naitou H, Enomoto S, Tai A, Kakuta H. Fluorescent retinoid X receptor ligands for fluorescence polarization assay. Bioorg Med Chem Lett. 2010;20:5143–5146. doi: 10.1016/j.bmcl.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 220.Orlandi M, Mantovani B, Ammar K, Avitabile E, Dal Monte P, Bartolini G. Retinoids and cancer: Antitumoral effects of ATRA, 9-cis RA and the new retinoid IIF on the HL-60 leukemic cell line. Med Princ Pract. 2003;12:164–169. doi: 10.1159/000070753. [DOI] [PubMed] [Google Scholar]
- 221.Papi A, Rocchi P, Ferreri AM, Guerra F, Orlandi M. Enhanced effects of PPARγ ligands and RXR selective retinoids in combination to inhibit migration and invasiveness in cancer cells. Oncol Rep. 2009;21:1083–1089. doi: 10.3892/or_00000327. [DOI] [PubMed] [Google Scholar]
- 222.Grün F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T, Blumberg B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 223.Nakanishi T, Nishikawa J, Hiromori Y, Yokoyama H, Koyanagi M, Takasuga S, Ishizaki J, Watanabe M, Isa S, Utoguchi N, Itoh N, Kohno Y, Nishihara T, Tanaka K. Trialkyltin compounds bind retinoid X receptor to alter human placental endocrine functions. Mol Endocrinol. 2005;19:2502–2516. doi: 10.1210/me.2004-0397. [DOI] [PubMed] [Google Scholar]
- 224.Dawson MI, Ye M, Cao X, Farhana L, Hu QY, Zhao Y, Xu LP, Kiselyuk A, Correa RG, Yang L, Hou T, Reed JC, Itkin-Ansari P, Levine F, Sanner MF, Fontana JA, Zhang X-K. Derivation of a retinoid X receptor scaffold from peroxisome proliferator-activated receptor γ ligand 1-di(1H-indol-3-yl)methyl-4-trifluoromethylbenzene. ChemMedChem. 2009;4:1106–1119. doi: 10.1002/cmdc.200800447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Garcia J, Khanwalkar H, Pereira R, Erb C, Voegel JJ, Collette P, Mauvais P, Bourguet W, Gronemeyer H, de Lera ÁR. Pyrazine arotinoids with inverse agonist activities on the retinoid and rexinoid receptors. Chembiochem. 2009;10:1252–1259. doi: 10.1002/cbic.200900030. [DOI] [PubMed] [Google Scholar]
- 226.Schwimmer LJ, Rohatgi P, Azizi B, Seley KL, Doyle DF. Creation and discovery of ligand–receptor pairs for transcriptional control with small molecules. Proc Natl Acad Sci USA. 2004;101:14707–14712. doi: 10.1073/pnas.0400884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Jiang W, Deng W, Bailey SK, Nail CD, Frost AR, Brouillette WJ, Muccio DD, Grubbs CJ, Ruppert JM, Lobo-Ruppert SM. Prevention of KLF4-mediated tumor initiation and malignant transformation by UAB30 rexinoid. Cancer Biol Ther. 2009;8:289–298. doi: 10.4161/cbt.8.3.7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Morishita K, Yakushiji N, Ohsawa F, Takamatsu K, Matsuura N, Makishima M, Kawahata M, Yamaguchi K, Tai A, Sasaki K, Kakuta H. Replacing alkyl sulfonamide with aromatic sulfonamide in sulfonamide-type RXR agonists favors switch towards antagonist activity. Bioorg Med Chem Lett. 2009;19:1001–1003. doi: 10.1016/j.bmcl.2008.11.086. [DOI] [PubMed] [Google Scholar]
- 229.Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-α, -β, and -γ, liver X receptor-β, and retinoid X receptor-α. Toxicol Sci. 2006;92:476–489. doi: 10.1093/toxsci/kfl014. [DOI] [PubMed] [Google Scholar]
- 230.Jones G, Jones D, Teal P, Sapa A, Wozniak M. The retinoid-X receptor ortholog, ultraspiracle, binds with nanomolar affinity to an endogenous morphogenetic ligand. FEBS J. 2006;273:4983–4996. doi: 10.1111/j.1742-4658.2006.05498.x. [DOI] [PubMed] [Google Scholar]
- 231.Farmer LJ, Marron KS, Canan Koch SS, Hwang CK, Kallel EA, Zhi L, Nadzan AM, Robertson DW, Bennani YL. Aza-retinoids as novel retinoid X receptor-specific agonists. Bioorg Med Chem Lett. 2006;16:2352–2356. doi: 10.1016/j.bmcl.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 232.Deng T, Shan S, Li Z-B, Wu Z-W, Liao C-Z, Ko B, Lu X-P, Cheng J, Ning Z-Q. A new retinoid-like compound that activates peroxisome proliferator-activated receptors and lowers blood glucose in diabetic mice. Biol Pharm Bull. 2005;28:1192–1196. doi: 10.1248/bpb.28.1192. [DOI] [PubMed] [Google Scholar]
- 233.Goldstein JT, Dobrzyn A, Clagett-Dame M, Pike JW, DeLuca HF. Isolation and characterization of unsaturated fatty acids as natural ligands for the retinoid-X receptor. Arch Biochem Biophys. 2003;420:185–193. doi: 10.1016/j.abb.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 234.Michellys P-Y, Boehm MF, Chen J-H, Grese TA, Karanewsky DS, Leibowitz MD, Liu S, Mais DA, Mapes CM, Reifel-Miller A, Ogilvie KM, Rungta D, Thompson AW, Tyhonas JS, Yumibe N, Ardecky RJ. Design and synthesis of novel RXR-selective modulators with improved pharmacological profile. Bioorg Med Chem Lett. 2003;13:4071–4075. doi: 10.1016/j.bmcl.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 235.Michellys P-Y, Ardecky RJ, Chen J-H, D’Arrigo J, Grese TA, Karanewsky DS, Leibowitz MD, Liu S, Mais DA, Mapes CM, Montrose-Rafizadeh C, Ogilvie KM, Reifel-Miller A, Rungta D, Thompson AW, Tyhonas JS, Boehm MF. Design, synthesis, and structure activity relationship studies of novel 6,7-locked-[7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta]-2,4,6-trienoic acids. J Med Chem. 2003;46:4087–4103. doi: 10.1021/jm020401k. [DOI] [PubMed] [Google Scholar]
- 236.Michellys P-Y, Ardecky RJ, Chen J-H, Crombie DL, Etgen GJ, Faul MM, Faulkner AL, Grese TA, Heyman RA, Karanewsky DS, Klausing K, Leibowitz MD, Liu S, Mais DA, Mapes CM, Marschke KB, Reifel-Miller A, Ogilvie KM, Rungta D, Thompson AW, Tyhonas JS, Boehm MF. Novel (2E,4E,6Z)-7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta-2,4,6-trienoic acid retinoid X receptor modulators are active in models of type 2 diabetes. J Med Chem. 2003;46:2683–2696. doi: 10.1021/jm020340q. [DOI] [PubMed] [Google Scholar]
- 237.Dominguez B, Vega MJ, Sussman F, de Lera AR. Synthesis and characterization of a new RXR agonist based on the 6-tert-butyl-1,1-dimethylindanyl structure. Bioorg Med Chem Lett. 2002;12:2607–2609. doi: 10.1016/s0960-894x(02)00477-8. [DOI] [PubMed] [Google Scholar]
- 238.Vuligonda V, Thacher SM, Chandraratna RAS. Enantioselective syntheses of potent retinoid X receptor ligands: Differential biological activities of individual antipodes. J Med Chem. 2001;44:2298–2303. doi: 10.1021/jm0100584. [DOI] [PubMed] [Google Scholar]
- 239.Farmer LJ, Zhi L, Jeong S, Lamph WW, Osburn DL, Croston G, Flatten KS, Heyman RA, Nadzan AM. Retinoic acid receptor ligands based on the 6-cyclopropyl-2,4-hexadienoic acid. Bioorg Med Chem Lett. 2003;13:261–264. doi: 10.1016/s0960-894x(02)00924-1. [DOI] [PubMed] [Google Scholar]
- 240.Ohta K, Kawachi E, Inoue N, Fukasawa H, Hashimoto Y, Itai A, Kagechika H. Retinoidal pyrimidinecarboxylic acids. Unexpected diaza-substituent effects in retinobenzoic acids. Chem Pharm Bull (Tokyo) 2000;48:1504–1513. doi: 10.1248/cpb.48.1504. [DOI] [PubMed] [Google Scholar]
- 241.Kagechika H. Novel synthetic retinoids and separation of the pleiotropic retinoidal activities. Curr Med Chem. 2002;9:591–608. doi: 10.2174/0929867024606975. [DOI] [PubMed] [Google Scholar]
- 242.Pérez-Rodríguez S, Ortiz MA, Pereira R, Rodríguez-Barrios F, de Lera AR, Piedrafita FJ. Highly twisted adamantyl arotinoids: Synthesis, antiproliferative effects and RXR transactivation profiles. Eur J Med Chem. 2009;44:2434–2446. doi: 10.1016/j.ejmech.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Dawson MI, Hobbs PD, Jong L, Xiao D, Chao W-R, Pan C, Zhang X-K. sp2-bridged diaryl retinoids: Effects of bridge-region substitution on retinoid X receptor (RXR) selectivity. Bioorg Med Chem Lett. 2000;10:1307–1310. doi: 10.1016/s0960-894x(00)00243-2. [DOI] [PubMed] [Google Scholar]
- 244.Sussman F, de Lera AR. Ligand recognition by RAR and RXR receptors: Binding and selectivity. J Med Chem. 2005;48:6212–6219. doi: 10.1021/jm050285w. [DOI] [PubMed] [Google Scholar]
- 245.Alvarez R, Vega MJ, Kammerer S, Rossin A, Germain P, Gronemeyer H, de Lera AR. 9-cis-Retinoic acid analogues with bulky hydrophobic rings: new RXR-selective agonists. Bioorg Med Chem Lett. 2004;14:6117–6122. doi: 10.1016/j.bmcl.2004.08.072. [DOI] [PubMed] [Google Scholar]
- 246.Claudel T, Leibowitz MD, Fievet C, Tailleux A, Wagner B, Repa JJ, Torpier G, Lobaccaro JM, Paterniti JR, Mangelsdorf DJ, Heyman RA, Auwerx J. Reduction of atherosclerosis in apolipoprotein E knockout mice by activation of the retinoid X receptor. Proc Natl Acad Sci USA. 2001;98:2610–2615. doi: 10.1073/pnas.041609298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gernert DL, Ajamie R, Ardecky RA, Bell MG, Leibowitz MD, Mais DA, Mapes CM, Michellys P-Y, Rungta D, Reifel-Miller A, Tyhonas JS, Yumibe N, Grese TA. Design and synthesis of fluorinated RXR modulators. Bioorg Med Chem Lett. 2003;13:3191–3195. doi: 10.1016/s0960-894x(03)00703-0. [DOI] [PubMed] [Google Scholar]
- 248.Cavasotto CN, Liu G, James SY, Hobbs PD, Peterson VJ, Bhattacharya AA, Kolluri SK, Zhang X-K, Leid M, Abagyan R, Liddington RC, Dawson MI. Determinants of retinoid X receptor transcriptional antagonism. J Med Chem. 2004;47:4360–4372. doi: 10.1021/jm030651g. [DOI] [PubMed] [Google Scholar]
- 249.Lu J, Dawson MI, Hu QY, Xia Z, Dambacher JD, Ye M, Zhang X-K, Li E. The effect of antagonists on the conformational exchange of the retinoid X receptor alpha ligand-binding domain. Magn Reson Chem. 2009;47:1071–1080. doi: 10.1002/mrc.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Padovani AMS, Molina MF, Mann KK. Inhibition of liver X receptor/retinoid X receptor-mediated transcription contributes to the proatherogenic effects of arsenic in macrophages in vitro. Arterioscler Thromb Vasc Biol. 2010;30:1228–1236. doi: 10.1161/ATVBAHA.110.205500. [DOI] [PubMed] [Google Scholar]
- 251.He FL, Wang L, Zhang X-K, Zeng JZ. Emodin induces apoptosis of cancer cells and inhibits retinoid X receptor transcriptional activity. Yao Xue Xue Bao. 2008;43:350–355. [PubMed] [Google Scholar]
- 252.Michellys P-Y, D’Arrigo J, Grese TA, Karanewsky DS, Leibowitz MD, Mais DA, Mapes CM, Reifel-Miller A, Rungta D, Boehm MF. Design, synthesis and structure–activity relationship of novel RXR-selective modulators. Bioorg Med Chem Lett. 2004;14:1593–1598. doi: 10.1016/j.bmcl.2003.12.089. [DOI] [PubMed] [Google Scholar]
- 253.Ziouzenkova O, Orasanu G, Sukhova G, Lau E, Berger JP, Tang G, Krinsky NI, Dolnikowski GG, Plutzky J. Asymmetric cleavage of β-carotene yields a transcriptional repressor of retinoid X receptor and peroxisome proliferator-activated receptor responses. Mol Endocrinol. 2007:77–88. doi: 10.1210/me.2006-0225. [DOI] [PubMed] [Google Scholar]