

Abstract
Oxytocin (OT)-elicited hypophagia has been linked to neural activity in the nucleus of the solitary tract (NTS). Because plasma OT levels increase after a meal, we hypothesized that circulating OT acts at both peripheral and hindbrain OT receptors (OTRs) to limit food intake. To initially determine whether circulating OT inhibits food intake by acting at hindbrain OTRs, we pretreated rats with an OTR antagonist administered into the fourth ventricle (4V) followed by either central or systemic OT administration. Administration of the OTR antagonist into the 4V blocked anorexia induced by either 4V or ip injection of OT. However, blockade of peripheral OTRs also weakened the anorectic response to ip OT. Our data suggest a predominant role for hindbrain OTRs in the hypophagic response to peripheral OT administration. To elucidate central mechanisms of OT hypophagia, we tested whether OT activates NTS catecholaminergic neurons. OT (ip) increased the number of NTS cells expressing c-Fos, of which 10%–15% were catecholaminergic. Furthermore, electrophysiological studies in mice revealed that OT stimulated 47% (8 of 17) of NTS catecholamine neurons through a presynaptic mechanism. However, OT-elicited hypophagia did not appear to require activation of α1-adrenoceptors, and blockade of glucagon-like peptide-1 receptors similarly did not attenuate anorexia induced by OT. These findings demonstrate that OT elicits satiety through both central and peripheral OTRs and that although catecholamine neurons are a downstream target of OT signaling in the NTS, the hypophagic effect is mediated independently of α1-adrenoceptor signaling.
The neuropeptide oxytocin (OT) suppresses food intake and weight gain, and impaired OT signaling is associated with obesity in both Prader-Willi syndrome (1) and a mutation at the single-minded 1 (SIM1) gene locus (2). Several lines of evidence suggest that the hindbrain nucleus of the solitary tract (NTS) is a key site in OT-elicited hypophagia. The NTS receives direct projections from OT neurons in the hypothalamic paraventricular nucleus (PVN) (3), and intracerebroventricular (icv) administration of OT produces robust c-Fos activation in brain sites that express OT receptors (OTRs), including the NTS (4). Moreover, previous studies implicate a physiological role of endogenous OT to limit meal size (5, 6), likely by a mechanism involving modulation of NTS neural activity because OT enhances the neurotransmission of visceral afferents to the NTS (7) that are implicated in cholecystokinin (CCK)-induced satiety (8–10).
Emerging evidence suggests that both central and peripheral OTRs may contribute to the control of food intake because OTRs are widely distributed in areas of the central nervous system (CNS) linked to the control of food intake (11) as well as in peripheral tissues, including adipocytes (12), the gastrointestinal tract (13), and the nodose ganglion (14). In addition to the activation of hindbrain neurons that influence meal size, meal-related stimuli are also associated with the activation of PVN and supraoptic nucleus OT neurons as well as release of OT into the circulation (15–19). Peripheral administration of blood-brain barrier (BBB)-penetrant OTR antagonists stimulate food intake in mice and rats (20, 21), raising the possibility that both central and peripheral OTRs may participate in the regulation of food intake. The mechanism for the peripheral action of OT on food intake is unknown, however. Reports that systemic OT activates PVN OT neurons (22, 23) and induces OT release within the PVN (21), as well as that CCK elicits c-Fos in parvocellular PVN OT neurons projecting to the dorsal vagal complex (24), suggest that activation of PVN OT neurons may participate in the effects of circulating OT on energy homeostasis.
OT is released from the neurohypophysis after food intake (16, 19, 25), and peripheral administration of OT inhibits food intake (21, 26–30); however, it is not known the extent to which CNS OTRs contribute to the ability of circulating OT to reduce food intake. Central administration of OT is sufficient to reduce food intake (26), but the hypophagic responses after peripheral OT are similar. Furthermore, as with central administration, the systemic administration of OT elicits c-Fos expression in NTS regions in which OTRs are expressed (28). Circulating OT enters the CNS (31) and potentially could access NTS OTRs through a leaky BBB at the area postrema (32), tanycytes (32), or transporters (33, 34).
Thus, a major obstacle to understanding the physiology underlying circulating OT's effects on food intake is determining whether CNS OTRs are involved. Based on the above findings, we hypothesized that although the activation of NTS neurons may contribute to the anorexigenic response to circulating OT, activation of NTS neurons is not necessarily required for the full anorexigenic response to circulating OT. In the present study, we addressed this hypothesis via the pharmacological blockade of central and peripheral OTRs and also identified potential NTS candidate neurons involved in this mechanism. Our data support an important role for hindbrain OTRs in the hypophagic response to peripheral OT administration. We also report that NTS catecholaminergic neurons are among those activated by the systemic and in vitro administration of OT.
Materials and Methods
Animals and surgeries
Adult male Sprague Dawley rats from Charles River Laboratories and adult male C57BL/6J mice from The Jackson Laboratory were individually housed in Plexiglas cages at 22 ± 2°C under a 12-hour light, 12-hour dark cycle with ad libitum access to water and standard rodent chow (Teklad diets; Harlan Laboratories Inc). Rats were acclimated to a daily 6-hour fast preceding the onset of the dark cycle (1:00 pm). All procedures were approved by the Institutional Animal Care and Use Committee at the Veterans Affairs Puget Sound Health Care System. Procedures involving adult transgenic mice expressing enhanced green fluorescent protein (EGFP) driven by the tyrosine hydroxylase (TH) promoter (TH-EGFP) were conducted with the approval of the Animal Care and Use Committee at Washington State University and in accordance with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For third (3V) or fourth ventricle (4V) injection experiments, rats were anesthetized with isoflurane. Guide cannulae (Plastics One Inc) were placed using a stereotaxic apparatus into the 3V and 4V as previously described (35). Animals recovered from surgery for at least 1 week prior to testing. The verification of cannula placement was achieved by administering angiotensin II (20 ng per 1 μL) or bombesin (15 pmol per 1 μL) into the 3V or 4V, respectively. Only rats that drank more than 5 mL water in 30 minutes after the angiotensin II treatment or that demonstrated more than a 20% reduction in feeding within 2 hours after the bombesin treatment were included in subsequent studies.
Drugs
Solutions of OT (Bachem), OTR antagonists, the α1-adrenoceptor agonist, cirazoline (R&D Systems), and the α1-adrenoceptor antagonist, prazosin (R&D Systems) were prepared each experimental day. OT was solubilized in sterile water and diluted with sterile saline. The OTR antagonist [d(CH2)51, Tyr(Me)2, Orn8]-OT (Bachem) was solubilized in sterile water. The OTR antagonist L-371,257 (R&D Systems) was initially solubilized in 100% dimethyl sulfoxide and sonicated for 75 seconds at 47°C before being sequentially diluted in 1:1:8 Tween-80, dimethyl sulfoxide, and sterile saline (1 mg/kg). L-371,257 was serially diluted 1:1 to achieve lower doses (0.25, 0.5 mg/kg). These OT ligands display relatively high affinity for the rat OTR (Supplemental Table 1). Cirazoline was solubilized in sterile saline, and prazosin was gently heated in sterile water and diluted 1:1 with 2 M lactate buffer. Frozen aliquots of bombesin (Bachem) and angiotensin II (Sigma-Aldrich) were thawed and vortexed before the start of experiments. The glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) antagonist, Exendin-3 (9–39) amide (Exendin-9; R&D Systems), the GLP-1R agonists, Exendin-4 (R&D Systems), and GLP-1 (American Peptide Co) were solubilized in sterile saline on the first injection day of an experiment, and aliquots were frozen at −30°C for use on subsequent injection days.
Behavioral testing
Drugs were administered within the final hour of the daily 6-hour fast. Animals were given food at the onset of the dark cycle. Food intake was measured at intervals out to 18 hours. The two-drug treatment was administered sequentially separated by 30 minutes, and the second injection was given 10 minutes prior to food administration. The 3V and 4V injections were administered at 1 μL/min using an injection pump (CMA 100 Syringe Pump; CMA Microdialysis AB) via a 33-gauge injector (Plastics One) connected by polyethylene 20 tubing to a 10-μL Hamilton syringe. Subjects receiving ip injections were acclimated by daily injections of sterile saline for 2–3 days prior to testing (1 mL/kg injection volume). Rats underwent all treatment conditions (unless otherwise noted) in a randomized order separated by at least 48 hours between treatments with the exception of studies in which L-371,257 (2 mL/kg injection volume) was given chronically over 6 successive days. To ensure that drugs elicited effects through neutral antagonism, dose-response studies were conducted to establish doses of OTR, α1-adrenoceptor, and GLP-1R antagonists that were subthreshold to producing independent effects on food intake. Dose-response studies were likewise conducted for corresponding agonists to determine doses that effectively reduce food intake.
Electrophysiology
Hindbrains of both male and female TH-EGFP mice (8–12 wk old) were prepared as previously described (36). Briefly, the hindbrain was removed and placed for 1 minute in cold (0–4°C) artificial (aCSF) cerebrospinal fluid (CSF) composed of 125 mM NaCl, 3 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 10 mM dextrose, and 2 mM CaCl2 and bubbled with 95%O2-5%CO2. The osmolarity was adjusted to 307–310 mOsm using dextrose. The medulla was trimmed to a 2-cm block (rostral-caudal) centered on the obex. A wedge of tissue was removed from the ventral surface to align the solitary tract afferents (ST) with the cutting plane when mounted in a vibrating microtome (Leica VT-1000S). Slices (250 μm thick) cut with a sapphire knife (Delaware Diamond Knives) contained the ST in the same plane as the NTS. Slices were submerged in a perfusion chamber and recordings performed at 32±°C at pH 7.4.
Neurons were observed with an Olympus BX51 upright microscope. Recording electrodes were filled with a solution consisting of 10 mM NaCl, 130 mM K gluconate, 11 mM EGTA, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 2 mM NaATP, 0.2 mM NaGTP at pH 7.3; 297–301 mOsM. Neurons were recorded from NTS within 200 μm rostral or caudal from obex and medial to the ST. Patch electrodes (3.9–4.5 MΩ) were used to make voltage clamp recordings with an Axopatch 700B amplifier (Molecular Devices), Digidata 1440A digitizer (Molecular Devices), and pClamp 10 software (Molecular Devices). Only neurons with holding currents not exceeding 100 pA at VH = −60 mV for the 15-minute control period (input resistance more than 150 MΩ) were studied further. Series resistance was monitored throughout the recordings and neurons were not included in further analysis if it exceeded 20 MΩ or drifted more than 25%. Series resistance did not differ between control (aCSF) and treatment. All drugs were obtained from R&D Systems or Sigma-Aldrich. Recordings were made in aCSF for a 10-minute control period, and then OT was bath applied for 5 minutes followed by a 10-minute wash in aCSF. In the antagonist experiments, we recorded in aCSF for a 10-minute control period, and L-371,257 was then bath applied for 5 minutes prior to coapplication of OT with L-371,257 for 5 minutes followed by a 10-minute wash. Spontaneous excitatory postsynaptic currents (sEPSC) frequency, amplitude, and decay time were analyzed for the 2-minute period after the control period, drug application (or coapplication), and 10-minute wash.
Tissue processing
After a 6-hour fast, rats were given an ip injection of vehicle or OT and deeply anesthetized 90 minutes later with a ketamine cocktail (71.4 mg/kg ketamine hydrochloride, 3.57 mg/kg xylazine, and 1.1 mg/kg acepromazine) or 100 mg/kg sodium pentobarbital for mice. Animals were transcardially perfused with 4% paraformaldehyde in 0.1 M PBS. Brains were removed, stored overnight in fresh fixative at 4°C, and subsequently transferred to 0.1 M PBS containing 25% sucrose for 48 hours and then frozen in isopentane at −80°C and stored at −80°C. Coronal 14-μm cryostat sections were thaw mounted onto slides and stored at −30°C.
Immunocytochemistry
Dual immunocytochemistry with antisera to c-Fos and TH was performed on anatomically matched sections throughout the NTS with tissue from both treatment conditions included in every batch. Briefly, slides were washed 3 × 5 minutes at room temperature with 0.1 M PBS and then incubated in 5% normal goat serum in 1% BSA/0.1 M PBS for 30 minutes. Slides were incubated for 18–20 hours at 4°C with primary antibodies diluted in 1% BSA/0.1 M PBS solution. Primary antibodies were a rabbit polyclonal anti-c-Fos and a mouse monoclonal anti-TH (Table 1). These well-characterized antibodies have been shown to be specific for their target molecules in previous studies (9, 36), and both stained the appropriate cellular and neuronal targets in our studies. Slides were rinsed 6 × 5 minutes in 0.1 M PBS and incubated with secondary antibodies for 30 minutes at room temperature. Secondary antibodies were diluted in 1% BSA/0.1 M PBS. Slides were rinsed 6 × 5 minutes immediately prior to coverslipping using glycerol-based mounting media. Staining specificity with the primary antibodies was assessed by substituting rabbit serum (for anti-c-Fos antibody) and mouse IgG (for anti-TH antibody) at the same concentration used for the primary antibodies and confirming the absence of staining.
Table 1.
Antibodies Used in This Study
| Peptide/Protein Target | Name of Antibody | Manufacturer/ Catalog # | Host | Type | Titer | Incubation h, °C |
|---|---|---|---|---|---|---|
| c-Fos | anti-c-Fos | EMD Millipore; PC38T | Rb | Poly IgG | 1:5000 | 18–20, 4 |
| TH | anti-TH | EMD Millipore; MAB318 | Ms | Mono IgG | 1:1000 for Rat | 18–20, 4 |
| 1:1500 for Ms | 18–20, 4 | |||||
| Rb IgG | Cy3 conjugated IgG (H+L) | Jackson ImmunoResearch | Gt | 1:200 | 0.5, RT | |
| Laboratories | ||||||
| Ms IgG | Alexa Fluor 488 Conjugate IgG (H+L) | Life Technologies | Gt | 1:200 | 0.5, RT | |
| Molecular Probes |
Abbreviations: C, centigrade; Gt, goat; IgG, immunoglobulin G; h, hours; mono, monoclonal; Ms, mouse; Poly, polyclonal; Rb, rabbit; RT, room temperature.
We also verified the specificity of the secondary antibodies for the appropriate primary antibody IgG in the dual-staining protocol. Images of stained preparations were captured as jpeg files and converted to 8-bit jpeg files using a ×20 objective and a COOLSNAPHQ2 Monochrome camera (Photometrics) attached to a Nikon Eclipse 80i fluorescent microscope (Nikon Instruments) and using Nikon NIS-Elements Software. Under conditions used for analysis and image capture, we detected no visible bleed-through of fluorescence between the Cy3 and Alexa 488 emissions. Prior to the analysis, all saved images were adjusted for equal background brightness in Adobe Photoshop CS5 (Adobe Systems). Counting of stained cells was performed manually by a single experimenter blind to treatment conditions. Bilateral counts were taken for cells expressing c-Fos-immunoreactivity (ir), TH-ir, or both, across four coronal sections of the NTS at four different levels, each separated by approximately 280 μm. These levels corresponded to bregma −13.08 mm (level 1), −13.32 mm (level 2), −13.80 mm (level 3), and −14.04 mm (level 4), based on the rat brain atlas by Paxinos and Watson (37), and bregma −7.08 mm (level 1), −7.20 mm (level 2), −7.56 mm (level 3), and −7.76 mm (level 4), based on the mouse brain atlas by Paxinos and Franklin (38).
Statistics
All data are presented as mean ± SEM. For behavioral studies, comparisons were made using repeated-measures ANOVA followed by Fisher's least significant different (LSD) test (SPSS Statistics 21). For immunocytochemical data, repeated-measures ANOVA compared group means across multiple levels of the NTS, followed by post hoc t tests at each level (SPSS). When statistical significance was attained but data failed to display the homogeneity of variance, all levels of the NTS were reanalyzed using nonparametric Mann-Whitney U tests. The outcomes were similar, irrespective of statistical test. For electrophysiological studies, statistical comparisons of drug effects between groups (eg, aCSF vs OT) were made using a one-way ANOVA with a Tukey's honestly significant difference post hoc analysis (SigmaPlot version 11.2). The Kolmogorov-Smirnov test (KS test) was used to determine the significance of the drug effect for individual neurons when analyzing spontaneous EPSCs (Mini Analysis, Synaptosoft Inc.). A P < .05 indicated significant differences.
Results
Fourth ventricular administration of an OTR antagonist blocks 4V OT-elicited hypophagia
Our initial studies sought to determine the extent to which the hindbrain OTRs contribute to the effects of systemically administered OT on food intake. Because hypophagic responses to OT were short lived, being detected primarily within the first 30 minutes upon refeeding after a fast (Figure 1), only 30-minute intakes are reported. To test whether the activation of hindbrain OTRs is sufficient to elicit hypophagia, the rats were pretreated with the 4V OTR antagonist, [d(CH2)51, Tyr(Me)2, Orn8]-OT, or the vehicle prior to 4V OT or vehicle. A significant effect of treatment was observed (F3,18 = 11.557; P < .001). Fourth ventricular administration of OT significantly reduced the mean food intake relative to that after the vehicle (P = .013) or the OTR antagonist alone (P = .001), and this effect was fully blocked with the prior administration of the OTR antagonist (P = .004; Figure 2A).
Figure 1.
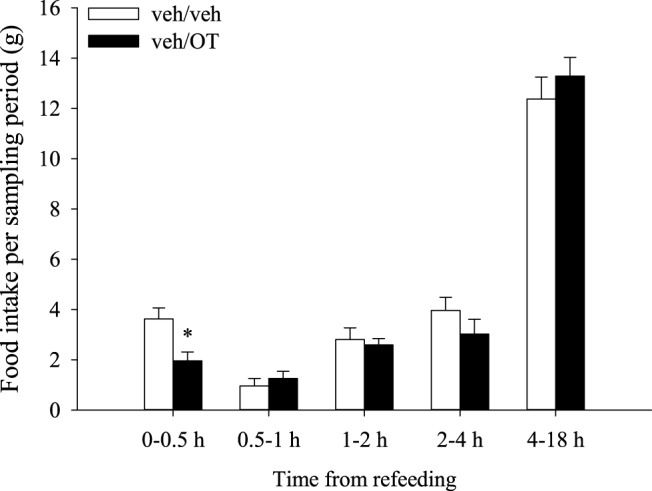
Time course of OT's effects on food intake. Six-hour fasted rats (n = 12/group) were given an ip injection of vehicle, followed 30 minutes later by an ip injection of vehicle or OT (0.5 mg/kg) in a crossover experimental design. Food was returned approximately 10 minutes later at the start of the dark cycle. OT's anorectic effects occurred primarily within the first 30 minutes of refeeding because food intake was significantly reduced at this time point only (t11 = 3.284, P = .007, paired t test). Data are expressed as mean ± SEM.
Figure 2.

Effects of OTR antagonism on OT-elicited hypophagia. Rats were pretreated with 4V vehicle (veh) or OTR antagonist, [d(CH2)51, Tyr(Me)2, Orn8]-OT (ant; 10 μg/2 μL), approximately 30 minutes prior to 4V (n = 7/group) (A) or i.p. veh or OT (OT; 1 μg/1 μL 4V, 0.5 mg/kg ip) (B) in a crossover experimental design (n = 9/group). Intraperitoneal pretreatment with a BBB-penetrable (C; [d(CH2)51, Tyr(Me)2, Orn8]-OT; 10 μg/0.3 mL) (n = 9/group) or nonpenetrable OTR antagonist (D; L-371,257; 0.5 mg/kg) (n = 12/group) preceded an ip injection of veh or OT (0.5 mg/kg) by approximately 30 minutes. For all studies, food was returned at the start of the dark cycle after a 6-hour fast. Data are expressed as mean ± SEM. Different letters above bars denote significant differences between treatments; shared letters are not significantly different from one another (pairwise LSD comparisons, P < .05).
Hindbrain OTRs contribute to systemic OT-elicited hypophagia
The goal of this study was to determine whether hindbrain OTRs also mediate responses to peripherally administered OT. Food intake differed significantly between the four groups (pretreatment with 4V OTR or vehicle followed by the ip injection of either OT or vehicle; F3,24 = 5.467; P = .005). Systemic (ip) administration of OT significantly reduced the mean food intake relative to that after the vehicle (P < .05) or OTR antagonist alone (P < .05). Pretreatment with the 4V OTR antagonist (at the same dose that was sufficient to block 4V OT in the previous study) reversed the anorectic effects of ip OT such that responses did not differ significantly from vehicle (P > .05; Figure 2B).
Peripheral OTRs contribute to systemic OT-elicited hypophagia
To assess the potential contribution of peripherally located OTRs in food intake regulation, both OT and its receptor antagonist were administered systemically. As before, food intake differed significantly with treatment condition (F3,24 = 11.731; P < .001). The systemic (ip) administration of OT significantly reduced the mean food intake relative to that after the vehicle (P < .05) or the OTR antagonist alone (P < .05). Interestingly, pretreatment with a small dose of the OTR antagonist (10 μg per 0.3 mL) was sufficient to attenuate OT's effects on average by 48%, resulting in food intake levels intermediate to those after the vehicle or OT treatment (P < .05; Figure 2C). It is unclear whether this systemic dose of d(CH2)51, Tyr(Me)2, Orn8]-OT crosses the BBB in pharmacologically significant amounts, so we confirmed that this effect was not due to the blockade of central OTRs by repeating this study using an OTR antagonist (L-371,257) that does not penetrate the BBB (39, 40). As before, the ip administration of OT significantly reduced the mean food intake relative to that after the vehicle (P < .05) or the OTR antagonist alone (P < .05; Figure 2D). An intermediate reversal of hypophagia was observed in the presence of the nonpenetrant antagonist, L-371,257 (0.25 mg/kg), as we observed with d(CH2)51, Tyr(Me)2, Orn8]-OT. Similar to what was observed after the systemic administration of d(CH2)51, Tyr(Me)2, Orn8]-OT, pretreatment with L-371,257 was sufficient to attenuate OT's effects on average by 67%, resulting in food intake levels intermediate to those after the vehicle or OT treatment (pairwise comparison: P < .001; omnibus test: F3,33 = 12.867, P < .001).
Systemic administration of nonpenetrant OTR antagonist fails to block 4V OT-elicited hypophagia
To confirm that the blockade of OT-elicited anorexia seen with ip L-371,257 was due to actions at the peripheral OTRs, we examined the effects of the nonpenetrant OTR antagonist, L-371,257 (ip) on the ability of 4V OT (2 μg per 1 μL) to inhibit food intake. We predicted that the ability of 4V OT to suppress food intake would be blocked if L-371,257 entered the hindbrain in sufficient amounts to block hindbrain OTRs. However, 4V OT inhibited food intake similarly, regardless of the presence (1.3 ± 0.4 g) (ip L-371,257+4V OT) or absence of L-371,257 (1.4 ± 0.6 g) (ip vehicle+4V OT) relative to vehicle treatment (4.46 ± 0.6 g; P < .01). Together these data indicate that L-371,257 is unable to enter the hindbrain in sufficient quantities to block hindbrain OTRs and provide further evidence to support a potential role of peripheral OTRs in the regulation of food intake. These data also imply that 4V OT is not likely to leak from the CNS into the periphery in sufficient concentrations to inhibit food intake through the activation of peripheral OTRs.
Systemic administration of nonpenetrant OTR antagonist stimulates food intake
To assess the potential physiological role of peripherally located OTRs in the regulation of food intake, we examined the effects of the nonpenetrant antagonist, L-371,257 (0, 0.5, 1.0 mg/kg, ip) on food intake. With this antagonist, the blockade of peripheral OTRs appeared to increase food intake at 30 minutes (P = .082) and 3 hours (P = .092) at 1 mg/kg and 0.5 mg/kg, respectively (Figure 3A). The L-371,257 (0.5 mg/kg) stimulated food intake relative to vehicle treatment by 4 hours (P < .05). These data are consistent with a potential physiological role of circulating OT at peripheral OTRs in the regulation of food intake.
Figure 3.
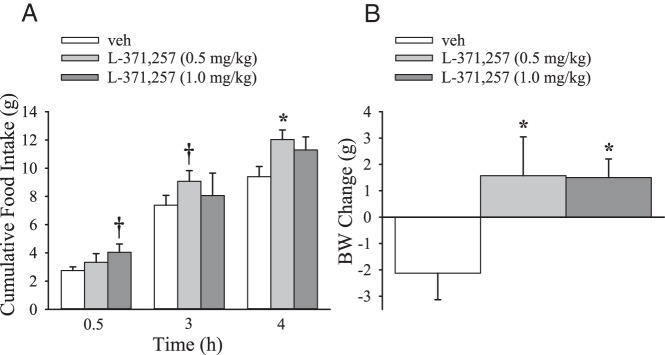
Effects of systemic administration of a nonpenetrant OTR antagonist on food intake and weight gain. Six-hour fasted rats (n = 7–8/group) were given a single ip injection of vehicle or L-371,257 (0.5, 1.0 mg/kg) 30–45 minutes prior to the start of the dark cycle and access to food. A, Food intake was measured at 30 minutes, 3 hours, and 4 hours after access to food. B, BW was measured at 18 hours after access to food. Data are expressed as mean ± SEM (pairwise LSD comparisons). *, P < .05; †, 0.05 < P < .1).
Systemic administration of nonpenetrant OTR antagonist stimulates weight gain
To assess the potential physiological role of peripherally located OTRs in the regulation of body weight (BW), we examined the effects of L-371,257 on weight change when given both acutely and chronically. A single injection of L-371,257 significantly stimulated weight gain at 0.5 and 1.0 mg/kg relative to vehicle treatment (P < .05; Figure 3B). When given repeatedly over 6 days, L-371,257 (0.5 mg/kg, ip) significantly stimulated BW gain (10.5 ± 2.2 g) relative to vehicle treatment (4.7 ± 2.7 g) (P < .05). These data are consistent with a potential physiological role of circulating OT at peripheral OTRs in the regulation of BW.
OT activates NTS catecholamine neurons in rats and mice
We next sought to elucidate the extent to which the caudal NTS catecholamine neurons are activated in response to systemic OT and may therefore represent potential downstream neuronal targets of systemic OT signaling. Initially, we examined the effects of systemic (ip) administration of OT on c-Fos-ir in the NTS in both rats and mice. We selected doses that are anorexigenic in both rats (41) and mice (data not shown). Consistent with previous reports (21, 23, 28, 41), the systemic administration of OT elicited significantly greater c-Fos-ir in the NTS than saline vehicle in rats (F1,16 = 9.249, P = .008) and mice (F1,13 = 25.824, P < .001). In rats, this effect was observed across all four rostrocaudal levels of the NTS examined (Figure 4A), activating over twice as many cells as vehicle injections (153.8 ± 24.8 vs 62.3 ± 17.1). In mice, a similar pattern was observed, although increases in c-Fos induction were statistically significant in only three of the four rostrocaudal levels quantified (P < .05, Mann-Whitney U test; Figure 4B).
Figure 4.

Activation of NTS neurons after OT administration. Rats (A and C) (n = 9/group) or mice (B and D) (n = 7/group) were injected with an ip injection of OT or vehicle (veh) after a 6-hour fast and brains were processed immunocytochemically for c-Fos-ir and TH-ir across four levels of the NTS. A–D, Data are expressed as mean ± SEM. *, P < .05, unpaired t tests. E, left panel, Representative photomicrographs taken at ×20 demonstrate catecholamine neurons (green staining for TH-ir) in the NTS of a vehicle-treated rat; right panel, An OT-treated rat. Activated neurons are labeled by c-Fos-ir (red nuclear staining), and white arrows denote neurons expressing both TH-ir and c-Fos-ir.
To test the hypothesis that NTS noradrenergic neurons are downstream targets of OT signaling, we examined whether neurons expressing TH, a general marker of catecholaminergic neurons, are activated by peripheral OT administration. In rats, we found that TH-ir cells comprised 15% of the total population activated by OT. At the rostral-most levels of NTS quantified (levels 1 and 2), 43% and 36% of TH-ir cells were activated by OT, respectively, resulting in a 3- to 4-fold increase in c-Fos (+) cells over vehicle levels (P < .05, Mann-Whitney U test; Figure 4C). In contrast, c-Fos responses in the TH-positive neurons did not differ significantly between the treatment groups at the caudal levels of the NTS (P > .05; Figure 3C). Similar results were obtained in mice, with 10% of the population of TH-positive neurons being activated by OT and significantly greater colocalization of TH-ir and c-Fos-ir being observed with the OT treatment at all but the caudal-most level (level 4), although a trending increase was observed (P = .07, Mann-Whitney U test; Figure 4D). At levels 1–3, 55%, 42%, and 21% (respectively) of TH-ir neurons expressed c-Fos-ir after the systemic OT injection.
OT increases sEPSCs in NTS TH-EGFP neurons of mice
Having provided evidence to support OT-elicited activation of NTS noradrenergic neurons in rats and mice, we tested whether activation occurs directly on NTS catecholamine neurons. This was accomplished by examining OT's effects on sEPSC frequency using voltage clamp techniques to study labeled TH cells identified from the hindbrain sections from the male and female TH-EGFP mice. Application of OT increased the number of glutamate events in 8 of 17 TH-EGFP neurons in the NTS of mice relative to a control period (P < .05, Kolmogorov-Smirnov test; Figure 5A). This effect was blocked by the prior application of the OTR antagonist L-371,257 (Figure 5B). Neither the average amplitude (Figure 5C) nor the decay time (Figure 5D) of glutamate events was altered during the application of OT relative to aCSF, consistent with a presynaptic mechanism of action. The OT application increased glutamate events (sEPSC frequency) in 6 of 12 non-EGFP-labeled neurons as well [responsive neurons (Hertz): control, 1.1 ± 0.4; OT, 1.8 ± 0.7; wash, 0.8 ± 0.3, n = 6, P < .05]. Nonresponsive neuron (Hertz): Control, 0.9 ± 0.3; OT = 0.9 ± 0.3; Wash = 0.9 ± 0.3, n = 6). As with responsive TH-EGFP neurons, neither the amplitude nor decay time changed in either group (responsive or nonresponsive). These results from the noncatecholamine neurons are similar to those previously reported for OT on rat NTS neurons (7).
Figure 5.

Effects of OT on TH-EGFP neurons in mice. Spontaneous EPSCs were recorded in voltage clamp. A, A comparison of the frequency of sEPSCs between a control period (aCSF) and a 5-minute bath application of OT (500 nM). Responders were neurons in which OT produced a significant increase in sEPSC frequency (P < .05; KS test: 8 of 17 neurons), and nonresponders were neurons in which OT had no significant increase in the sEPSC frequency (P > .05, KS test: 9 of 17 neurons). B, Frequency was compared between a control period (aCSF) and a 5-minute bath application of the antagonist, L-371,257 (500 nM) alone and coapplication of L-371,257 with OT (n = 12). Amplitude (picoamperes [pA]) (C) and decay time (milliseconds) (D) were also measured. Data are expressed as mean ± SEM. Group differences were made with respect to treatments. *, P < .05 (one-way ANOVA).
In addition, we saw a shift in the holding current after the OT application in 6 of the 17 TH-EGFP-positive neurons tested (average change −13.7 pA) and 6 of the 12 TH-EGFP negative neurons tested (average change −16.5 pA), suggesting that OT has additional postsynaptic effects in a subpopulation of both TH-positive and TH negative neurons, similar to the subpopulation of rat NTS neurons that have been shown to display a postsynaptic response (7). For both TH-EGFP-positive and -negative neurons, there was no association with the change in the holding current and the effects of sEPSC frequency; the change in holding current was seen in a subpopulation of both neurons in which OT increased sEPSC frequency and those with no response. These data indicate that both TH-positive and TH-negative neurons in the NTS are targets for the excitatory action of OT.
Blockade of α1-adrenoceptors does not prevent OT-elicited hypophagia
In light of the above findings, we next tested whether neuronal α1-adrenoceptor signaling, which has been implicated in noradrenergic-elicited satiety (42), is required for OT to suppress feeding. In a validation study, we first established an ip dose of the BBB-penetrable (43) α1-adrenoceptor antagonist, prazosin, that can fully rescue food intake after the administration of the selective and BBB-penetrable (43, 44) α1-adrenoceptor agonist, cirazoline, in rats (P = .026; Figure 6A). In pairing ip prazosin pretreatment with ip OT, however, the significant effect of treatment (F3,30 = 16.208; P < .001) was driven primarily by OT administration, and prazosin pretreatment was ineffective in reversing OT hypophagia (P > .05; Figure 6B). Relative to vehicle, therefore, OT reduced food intake in a manner that was not altered by prior α1-adrenoceptor blockade (P < .05; Figure 6B). Thus, OT-induced anorexia is not dependent on signaling via α1-adrenergic receptors.
Figure 6.
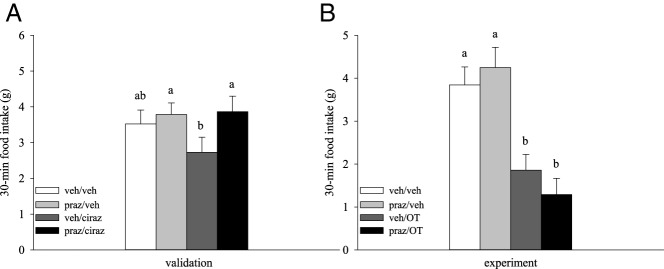
Effects of α1-adrenoceptor blockade on OT-elicited hypophagia. A, A validation study was conducted to establish a dose of α1-adrenoceptor antagonist prazosin (praz) effective in reversing the hypophagic effects of theα1-adrenoceptor agonist cirazoline (ciraz) (n = 12/group). Drugs were administered approximately 30 minutes apart into the peritoneal cavity of 6-hour-fasted rats in a crossover experimental design. B, Pretreatment with this dose of praz was then paired with ip OT in a similar manner (n = 11/group). Data are expressed as mean ± SEM. Different letters above bars denote significant differences between treatments; shared letters are not significantly different from one another (pairwise LSD comparisons, P < .05).
Blockade of GLP-1R does not prevent OT-elicited hypophagia
To identify alternative mechanisms with the potential to mediate OT-induced hypophagia, we tested whether GLP-1R activation plays a role. Neuronal GLP-1R signaling appears to mediate the effect of centrally administered OT because icv administration of a GLP-1R antagonist prevents anorexia elicited by icv OT (45). To determine whether GLP-1Rs mediate OT-elicited hypophagia following peripheral administration, we first established an ip dose of the BBB-penetrable GLP-1R antagonist, Exendin-9 (46), which can rescue food intake after the ip administration of GLP-1R agonist, Exendin-4, which is also capable of crossing the BBB (47) (Figure 7A, left panel). In pairing ip Exendin-9 with ip OT, an overall effect of treatment was observed (F3,15 = 10.814; P < .001) in which ip OT reduced food intake relative to either vehicle or Exendin-9 alone (P < .05; Figure 7A, right panel). However, pretreatment with Exendin-9 prior to OT had no effect on OT's ability to reduce feeding (P > .05). It is not clear from these studies whether Exendin-9 was also able to sufficiently block the GLP-1Rs in the CNS. To exclude the possibility that the above findings reflect the failure of ip Exendin-9 to reach target sites in the brain (48), we conducted an additional study in which Exendin-9 was injected into the 3V at a dose sufficient to block the anorexigenic effects of the 3V GLP-1 (Figure 7B, left panel) prior to ip OT or vehicle. As before, 3V Exendin-9 failed to block the peripheral OT-induced hypophagia (P > .05; overall treatment effect: F3,21 = 8.658; P = .001; Figure 7B, right panel).
Figure 7.
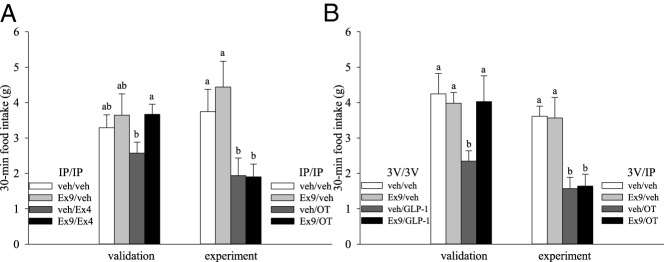
Effects of GLP1-R blockade on OT-elicited hypophagia. A, left panel, The effectiveness of GLP-1R antagonist Exendin-9 (Ex9; 0.1 mg/kg) in reversing hypophagia was verified with ip pretreatment prior to ip administration of GLP-1R agonist Exendin-4 (Ex4; 0.1 mg/kg) or vehicle (veh) in a validation study (n = 9/group). A, right panel, This dose of ip Ex9 was then paired with ip OT (0.5 mg/kg) in experimental studies (n = 6/group). B, The contribution of central GLP-1Rs to ip OT hypophagia was tested in additional studies. B, left panel, A validation study established effectiveness of 3V Ex9 (7.5 μg per 1 μL) in reversing hypophagic effects of 3V GLP-1 (1 μg per 1 μL) (n = 9/group) prior to the experimental study (B, right panel) (n = 8/group) that paired 3V Ex9 (7.5 μg per 1 μL) pretreatment with ip OT (0.5 mg/kg). For all studies, drug injections were separated by approximately 30 minutes and food was returned to 6 hour-fasted rats at the start of the dark cycle. Data are expressed as mean ± SEM. Different letters above bars denote significant differences between treatments; shared letters are not significantly different from one another. Pairwise LSD comparisons were used for all experiments except A, which was conducted between subjects and analyzed with post hoc unpaired t tests. P < .05 was considered significant.
Discussion
These studies revealed that the hypophagic response to systemic OT involves actions at both hindbrain and peripheral OTRs and identified NTS catecholamine neurons as targets of systemically administered OT in both mice and rats. Furthermore, OT activated NTS catecholamine neurons by increasing glutamate release from afferent terminals. Nevertheless, α1-adrenoceptors do not appear to be required for anorexia induced by systemic OT administration and, despite previous reports that OT action in the brain involves neuronal GLP-1R signaling, this did not occur when OT was given systemically. These findings collectively indicate that NTS OTRs contribute to the action of peripheral OT on food intake.
When the OTR antagonist was administered in the 4V, a complete blockade of the satiety response to ip OT ensued, whereas the blockade of peripheral OTRs using a peripheral, nonpenetrant antagonist (39) only attenuated the ability of OT to reduce food intake. These findings suggest that hindbrain OTRs play a predominant, but nonexclusive, role in hypophagia induced by peripheral OT. This evidence of a role for peripheral OTRs in the control of food intake is consistent with our data that reveal an increase in food intake and weight gain after the blockade of peripheral OTRs as well as a previous report of hyperphagia and weight gain in mice after the ip administration of an OTR antagonist (21). Moreover, the secretion of OT into the circulation has been observed to coincide with feeding in rats (25), dogs, and lactating sows (49); consumption of corn oil in women (50); and in response to meal-related humoral signals (25). Although elevations of circulating OT may have functions unrelated to feeding, patterns of observed neurohypophyseal OT secretion coincide with the release of satiety factors such as CCK (17, 25) and may itself participate in inhibiting further consumption. Together these findings indicate the ability of systemic OT to inhibit food intake is largely dependent on hindbrain OTRs but does not exclude a role for peripheral OTRs. However, the physiological mechanisms for the peripheral actions of OT on food intake remain to be elucidated.
We report that the systemic administration of a nonpenetrant OTRs antagonist is associated with a modest increase in food intake but a significant increase in weight gain. These findings are consistent with a potential role of peripheral OTRs in the regulation of BW and extend those from earlier studies that found systemic administration of BBB-permeant OTR antagonists are associated with the stimulation of weight gain (30). Similarly, mice with global loss in OT and OTRs develop adult-onset obesity yet have no impairments in daily food intake (51, 52). These findings are consistent with data showing that weight loss attributed to OT exceeds that from pair-fed control animals (27, 29). The increased weight gain in animals with deficient OT signaling is likely due, in part, to impairments in regulating energy expenditure because OTR knockout- and OT-deficient animals show impairments in sympathetic nervous system activity (51, 52). OT also activates sympathetic preganglionic neurons (53), including the stellate ganglia (54). In addition, systemic administration of OT is associated with increases in lipolysis (27). Impairments in lipolysis (12, 27, 55–57) may therefore contribute to weight gain after deficient OTR signaling as well as that observed with the administration of an OTR antagonist in the present studies. OTRs are expressed on adipocytes (12, 55, 57) and OT elicits direct effects on these cells (56). Furthermore, mice with a global loss in OT or OTRs or that have been treated systemically with a BBB-permeant OTR antagonist have increased fat mass (21, 51, 52). Together these findings indicate that, in addition to increases in food intake, reductions in energy expenditure and/or lipolysis may also contribute to weight gain associated with the blockade of peripheral OTRs.
To determine the precise hindbrain mediators underlying the hypophagic effects of systemic OT, we focused on NTS noradrenergic neurons because OT projections that originate from the PVN are found in anatomical proximity to NTS noradrenergic neurons (58). Moreover, these neurons are downstream targets of CNS OT (45) and are implicated in the ability of OT to enhance the satiety response to CCK-8 (9, 10). In both rats and mice, CCK activates NTS catecholaminergic neurons (59, 60), a subset of which are noradrenergic (localized to the A2 cell group) and project to hypothalamic areas including the PVN (61). Conversely, ablation of NTS noradrenergic neurons attenuates the ability of CCK-8 both to inhibit food intake and to induce c-Fos in PVN OT neurons (62). Lastly, stimulation of PVN α1-adrenoceptors, which are expressed on PVN OT neurons (63), is associated with satiety (44). Together these findings offer a compelling rationale for the importance of determining whether noradrenergic input to the PVN contributes to OT-elicited hypophagia.
In the present studies, rats and mice responded similarly to an ip injection of OT with respect to increasing c-Fos-ir throughout the NTS, albeit with some variation that is to be expected between species. Consistent with our hypothesis that noradrenergic neurons are among those activated by OT, immunocytochemical analyses revealed that TH-expressing neurons were among NTS cells in which the ip administration of OT induced increased c-Fos staining. Confirming this observation, our electrophysiological studies in mice demonstrated that OT activates both TH-expressing and non-TH-expressing neurons.
Despite our finding that OT activates NTS noradrenergic neurons, we found that the pharmacological blockade of α1-adrenoceptors had no effect on OT-elicited satiety in rats. This was somewhat unexpected, given the above findings and the known interaction of noradrenergic signaling with other anorexigenic factors. Although prazosin penetrates the BBB (43), it is possible that when given systemically, it fails to reach PVN α1-adrenoceptors in sufficient concentrations to block OT-elicited release of noradrenaline. Importantly, immunocytochemical and electrophysiological studies detect responsiveness rather than function, and our data suggest that noradrenergic neurons may participate in effects of OT unrelated to the control of food intake, although it remains possible that mediators other than norepinephrine are released after OT stimulation of these neurons and act at a receptor population distinct from α1-adrenoceptors.
Based on this assessment, we shifted our focus to NTS GLP-1 neurons as potential downstream targets of central OT signaling (45). These neurons are among nonadrenergic neurons activated by both OT (45) and CCK (64) that demonstrate similar projection profiles to the PVN. Local infusion of GLP-1 into the PVN suppresses feeding (65), and PVN OT neurons coexpress GLP-1Rs (66). Furthermore, the central blockade of GLP-1Rs abolishes the anorexigenic response to central OT (45). Although these studies support the hypothesis that activation of ascending NTS-PVN GLP-1 neuronal projections contribute to the anorexigenic response to CNS OT, we found that neither 3V nor ip GLP-1R blockade prevented systemic OT-elicited hypophagia. This outcome was surprising because Rinaman and Rothe (45) observed a full blockade of OT-induced hypophagia after pretreatment with a GLP-1R antagonist when both OT and the GLP-1R antagonist were administered into the lateral ventricle, which differs from the routes of administration used presently. Moreover, the GLP-1R antagonist used in the present studies was administered into the 3V at doses intended to target local PVN GLP-1R receptors and presumably would not reach rostral GLP-1 innervated sites such as the nucleus accumbens that can also affect food intake upon direct GLP-1 administration (67, 68). Additional studies examining the blockade of GLP-1R in such reward circuits would help clarify its role in peripheral OT-elicited satiety.
Although we did not find evidence to support a role for α1-adrenoceptors or GLP-1Rs in OT-elicited anorexia, further studies are needed to definitively rule out this possibility as well as to address other downstream targets. We cannot exclude a role for noradrenergic signaling in OT-elicited hypophagia because the present studies specifically examined the contribution of α1-adrenoceptors. It is possible that other receptor types may contribute instead or in conjunction with α1-adrenoceptors, and in fact, most noradrenergic A2 neurons appear to coexpress prolactin-releasing peptide (69) as well as a homolog of the vesicular glutamate transporter-2 (70), suggesting that the preponderance of these neurons are capable of releasing noradrenaline, prolactin-releasing peptide, and/or glutamate.
In conclusion, our present results support an important role for hindbrain OTRs in mediating the effects of circulating OT to reduce food intake in rats. It was not our intent to address the extent to which circulating OT requires hindbrain OTRs to elicit hypophagia in mice or whether α1-adrenoceptor or GLP-1R signaling may contribute to hypophagia in mice because these questions require further investigation. However, because NTS noradrenergic neurons appear to respond to the peripheral administration of OT in both species, the data that we obtained from mice suggest a possible mechanism for activation of rat NTS noradrenergic neurons in response to peripheral OT administration. Our results also support the existence of a peripheral mechanism that appears to serve in a limited capacity to reinforce OT signaling in the hindbrain to regulate food intake in rats. Additional studies are required to address the extent to which OTR signaling in the gastrointestinal tract (13, 14, 71–74), involving the enteric nervous system (14, 71), smooth muscle cells (71, 72), and vagus nerve (14), or in other peripheral tissues, contribute to the anorexigenic response to OT. Even within a defined region such as the NTS, OT may use multiple systems to elicit satiety. As was likely observed in the present studies, eliminating one may not reflect its importance or contribution if its absence is compensated for by other anorectic means. Further investigation is required to elucidate which NTS neuronal populations underlie OT-elicited satiety.
Acknowledgments
This work was supported by the Research and Development Service of the Department of Veterans Affairs and the Cellular and Molecular Imaging Core of the Diabetes Research Center at the University of Washington and supported by National Institutes of Health Grant P30DK017047. The research in our laboratory has been supported by the Department of Veterans Affairs Merit Review Research Program as well as National Institutes of Health Grants P30DK017047-31689, DK083452 (to S.M.A.), and Diabetes, Obesity, and Metabolism Training Grant 2T32DK007247. D.G.B. is the recipient of a Veterans Affairs Senior Research Career Scientist award.
Current address for J.M.H.: Departments of Anesthesiology and Pain Medicine and Medicine, Division of Metabolism, Endocrinology, and Nutrition, University of Washington, Seattle, Washington 98195.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- aCSF
- artificial cerebrospinal fluid
- BBB
- blood-brain barrier
- BW
- body weight
- CCK
- cholecystokinin
- CNS
- central nervous system
- CSF
- cerebrospinal fluid
- EGFP
- enhanced green fluorescent protein
- GLP-1
- glucagon-like peptide-1
- GLP-1R
- GLP-1 receptor
- icv
- intracerebroventricular
- ir
- immunoreactivity
- KS test
- Kolmogorov-Smirnov test
- LSD
- least significant different
- NTS
- nucleus of the solitary tract
- OT
- oxytocin
- OTR
- OT receptor
- PVN
- paraventricular nucleus
- ST
- solitary tract afferent
- sEPSC
- spontaneous excitatory postsynaptic currents
- TH
- tyrosine hydroxylase
- 3V
- third ventricle
- 4V
- fourth ventricle.
References
- 1. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab. 1995;80:573–579 [DOI] [PubMed] [Google Scholar]
- 2. Ramachandrappa S, Raimondo A, Cali AM, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest. 2013;123:3042–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol. 1982;205:260–272 [DOI] [PubMed] [Google Scholar]
- 4. Olson BR, Freilino M, Hoffman GE, Stricker EM, Sved AF, Verbalis JG. c-Fos expression in rat brain and brainstem nuclei in response to treatments that alter food intake and gastric motility. Mol Cell Neurosci. 1993;4:93–106 [DOI] [PubMed] [Google Scholar]
- 5. Blouet C, Jo YH, Li X, Schwartz GJ. Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J Neurosci. 2009;29:8302–8311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamashita M, Takayanagi Y, Yoshida M, Nishimori K, Kusama M, Onaka T. Involvement of prolactin-releasing peptide in the activation of oxytocin neurones in response to food intake. J Neuroendocrinol. 2013;25:455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peters JH, McDougall SJ, Kellett DO, Jordan D, Llewellyn-Smith IJ, Andresen MC. Oxytocin enhances cranial visceral afferent synaptic transmission to the solitary tract nucleus. J Neurosci. 2008;28:11731–11740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baskin DG, Kim F, Gelling RW, et al. A new oxytocin-saporin cytotoxin for lesioning oxytocin-receptive neurons in the rat hindbrain. Endocrinology. 2010;151:4207–4213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blevins JE, Eakin TJ, Murphy JA, Schwartz MW, Baskin DG. Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin. Brain Res. 2003;993:30–41 [DOI] [PubMed] [Google Scholar]
- 10. Olson BR, Drutarosky MD, Stricker EM, Verbalis JG. Brain oxytocin receptor antagonism blunts the effects of anorexigenic treatments in rats: evidence for central oxytocin inhibition of food intake. Endocrinology. 1991;129:785–791 [DOI] [PubMed] [Google Scholar]
- 11. Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81:629–683 [DOI] [PubMed] [Google Scholar]
- 12. Tsuda T, Ueno Y, Yoshikawa T, Kojo H, Osawa T. Microarray profiling of gene expression in human adipocytes in response to anthocyanins. Biochemical Pharmacology. 2006;71:1184–1197 [DOI] [PubMed] [Google Scholar]
- 13. Ohlsson B, Truedsson M, Djerf P, Sundler F. Oxytocin is expressed throughout the human gastrointestinal tract. Regul Pept. 2006;135:7–11 [DOI] [PubMed] [Google Scholar]
- 14. Welch MG, Tamir H, Gross KJ, Chen J, Anwar M, Gershon MD. Expression and developmental regulation of oxytocin (OT) and oxytocin receptors (OTR) in the enteric nervous system (ENS) and intestinal epithelium. J Comp Neurol. 2009;512:256–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnstone LE, Fong TM, Leng G. Neuronal activation in the hypothalamus and brainstem during feeding in rats. Cell Metab. 2006;4:313–321 [DOI] [PubMed] [Google Scholar]
- 16. Lucio-Oliveira F, Franci CR. Effect of the interaction between food state and the action of estrogen on oxytocinergic system activity. J Endocrinol. 2012;212:129–138 [DOI] [PubMed] [Google Scholar]
- 17. Renaud LP, Tang M, McCann MJ, Stricker EM, Verbalis JG. Cholecystokinin and gastric distension activate oxytocinergic cells in rat hypothalamus. Am J Physiol. 1987;253:R661–R665 [DOI] [PubMed] [Google Scholar]
- 18. Ueta Y, Kannan H, Higuchi T, Negoro H, Yamashita H. CCK-8 excites oxytocin-secreting neurons in the paraventricular nucleus in rats—possible involvement of noradrenergic pathway. Brain Res Bull. 1993;32:453–459 [DOI] [PubMed] [Google Scholar]
- 19. Yamashita M, Takayanagi Y, Yoshida M, Nishimori K, Kusama M, Onaka T. Involvement of prolactin releasing peptide in activation of oxytocin neurones in response to food intake. J Neuroendocrinol. 2013;25(5):455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Olszewski PK, Klockars A, Olszewska AM, Fredriksson R, Schioth HB, Levine AS. Molecular, immunohistochemical, and pharmacological evidence of oxytocin's role as inhibitor of carbohydrate but not fat intake. Endocrinology. 2010;151:4736–4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang G, Cai D. Circadian intervention of obesity development via resting-stage feeding manipulation or oxytocin treatment. Am J Physiol Endocrinol Metab. 2011;301:E1004–E1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carson DS, Hunt GE, Guastella AJ, et al. Systemically administered oxytocin decreases methamphetamine activation of the subthalamic nucleus and accumbens core and stimulates oxytocinergic neurons in the hypothalamus. Addict Biol. 2010;15:448–463 [DOI] [PubMed] [Google Scholar]
- 23. Hicks C, Jorgensen W, Brown C, et al. The nonpeptide oxytocin receptor agonist WAY 267,464: receptor-binding profile, prosocial effects and distribution of c-Fos expression in adolescent rats. J Neuroendocrinol. 2012;24:1012–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olson BR, Hoffman GE, Sved AF, Stricker EM, Verbalis JG. Cholecystokinin induces c-fos expression in hypothalamic oxytocinergic neurons projecting to the dorsal vagal complex. Brain Res. 1992;569:238–248 [DOI] [PubMed] [Google Scholar]
- 25. Verbalis JG, McCann MJ, McHale CM, Stricker EM. Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science. 1986;232:1417–1419 [DOI] [PubMed] [Google Scholar]
- 26. Arletti R, Benelli A, Bertolini A. Influence of oxytocin on feeding behavior in the rat. Peptides. 1989;10:89–93 [DOI] [PubMed] [Google Scholar]
- 27. Deblon N, Veyrat-Durebex C, Bourgoin L, et al. Mechanisms of the anti-obesity effects of oxytocin in diet-induced obese rats. PloS One. 2011;6:e25565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maejima Y, Iwasaki Y, Yamahara Y, Kodaira M, Sedbazar U, Yada T. Peripheral oxytocin treatment ameliorates obesity by reducing food intake and visceral fat mass. Aging. 2011;3:1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morton GJ, Thatcher BS, Reidelberger RD, et al. Peripheral oxytocin suppresses food intake and causes weight loss in diet-induced obese rats. Am J Physiol Endocrinol Metab. 2012;302:E134–E144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang G, Bai H, Zhang H, et al. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron. 2011;69:523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Neumann ID, Maloumby R, Beiderbeck DI, Lukas M, Landgraf R. Increased brain and plasma oxytocin after nasal and peripheral administration in rats and mice. Psychoneuroendocrinology. 2013;38:1985–1993 [DOI] [PubMed] [Google Scholar]
- 32. Maolood N, Meister B. Protein components of the blood-brain barrier (BBB) in the brainstem area postrema-nucleus tractus solitarius region. J Chem Neuroanat. 2009;37:182–195 [DOI] [PubMed] [Google Scholar]
- 33. Banks WA. Blood-brain barrier and energy balance. Obesity. 2006;14(suppl 5):234S–237S [DOI] [PubMed] [Google Scholar]
- 34. Banks WA. The blood-brain barrier as a regulatory interface in the gut-brain axes. Physiol Behav. 2006;89:472–476 [DOI] [PubMed] [Google Scholar]
- 35. Blevins JE, Schwartz MW, Baskin DG. Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol. 2004;287:R87–R96 [DOI] [PubMed] [Google Scholar]
- 36. Williams DL, Schwartz MW, Bastian LS, Blevins JE, Baskin DG. Immunocytochemistry and laser capture microdissection for real-time quantitative PCR identify hindbrain neurons activated by interaction between leptin and cholecystokinin. J Histochem Cytochem. 2008;56:285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paxinos G, Watson C. eds. The Rat Brain in Stereotaxic Coordinates. 6th ed Burlington, MA: Academic Press; 2007 [Google Scholar]
- 38. Paxinos G, Franklin KBJ. The Mouse Brain in Sterotaxic Coordinates. 2nd ed San Diego, CA: Academic Press; 2001 [Google Scholar]
- 39. Ring RH, Malberg JE, Potestio L, et al. Anxiolytic-like activity of oxytocin in male mice: behavioral and autonomic evidence, therapeutic implications. Psychopharmacology. 2006;185:218–225 [DOI] [PubMed] [Google Scholar]
- 40. Ring RH, Schechter LE, Leonard SK, et al. Receptor and behavioral pharmacology of WAY-267464, a non-peptide oxytocin receptor agonist. Neuropharmacology. 2010;58:69–77 [DOI] [PubMed] [Google Scholar]
- 41. Morton GJ, Thatcher BS, Reidelberger RD, et al. Peripheral oxytocin suppresses food intake and causes weight loss in diet-induced obese rats. Am J Physiol Endocrinol Metab. 2012;302:E134–E144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wellman PJ, Davies BT, Morien A, McMahon L. Modulation of feeding by hypothalamic paraventricular nucleus α1- and α2-adrenergic receptors. Life Sci. 1993;53:669–679 [DOI] [PubMed] [Google Scholar]
- 43. Guo TZ, Tinklenberg J, Oliker R, Maze M. Central α1-adrenoceptor stimulation functionally antagonizes the hypnotic response to dexmedetomidine, an α2-adrenoceptor agonist. Anesthesiology. 1991;75:252–256 [DOI] [PubMed] [Google Scholar]
- 44. Davies BT, Wellman PJ. Effects on ingestive behavior in rats of the α1-adrenoceptor agonist cirazoline. Eur J Pharmacol. 1992;210:11–16 [DOI] [PubMed] [Google Scholar]
- 45. Rinaman L, Rothe EE. GLP-1 receptor signaling contributes to anorexigenic effect of centrally administered oxytocin in rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R99–R106 [DOI] [PubMed] [Google Scholar]
- 46. Banks WA, During MJ, Niehoff ML. Brain uptake of the glucagon-like peptide-1 antagonist exendin(9–39) after intranasal administration. J Pharmacol Exp Ther. 2004;309:469–475 [DOI] [PubMed] [Google Scholar]
- 47. Kastin AJ, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes Relat Metab Disord. 2003;27:313–318 [DOI] [PubMed] [Google Scholar]
- 48. Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology. 2009;150:1680–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Uvnas-Moberg K, Stock S, Eriksson M, Linden A, Einarsson S, Kunavongkrit A. Plasma levels of oxytocin increase in response to suckling and feeding in dogs and sows. Acta Physiol Scand. 1985;124:391–398 [DOI] [PubMed] [Google Scholar]
- 50. Ohlsson B, Forsling ML, Rehfeld JF, Sjolund K. Cholecystokinin stimulation leads to increased oxytocin secretion in women. Eur J Surg. 2002;168:114–118 [DOI] [PubMed] [Google Scholar]
- 51. Camerino C. Low sympathetic tone and obese phenotype in oxytocin-deficient mice. Obesity. 2009;17:980–984 [DOI] [PubMed] [Google Scholar]
- 52. Takayanagi Y, Kasahara Y, Onaka T, Takahashi N, Kawada T, Nishimori K. Oxytocin receptor-deficient mice developed late-onset obesity. Neuroreport. 2008;19:951–955 [DOI] [PubMed] [Google Scholar]
- 53. Backman SB, Henry JL. Effects of oxytocin and vasopressin on thoracic sympathetic preganglionic neurones in the cat. Brain Res Bull. 1984;13:679–684 [DOI] [PubMed] [Google Scholar]
- 54. Armour JA, Klassen GA. Oxytocin modulation of intrathoracic sympathetic ganglionic neurons regulating the canine heart. Peptides. 1990;11:533–537 [DOI] [PubMed] [Google Scholar]
- 55. Gould BR, Zingg HH. Mapping oxytocin receptor gene expression in the mouse brain and mammary gland using an oxytocin receptor-LacZ reporter mouse. Neuroscience. 2003;122:155–167 [DOI] [PubMed] [Google Scholar]
- 56. Muchmore DB, Little SA, de Haen C. A dual mechanism of action of ocytocin in rat epididymal fat cells. J Biol Chem. 1981;256:365–372 [PubMed] [Google Scholar]
- 57. Schaffler A, Binart N, Scholmerich J, Buchler C. Hypothesis paper: brain talks with fat—evidence for a hypothalamic-pituitary-adipose axis? Neuropeptides. 2005;39:363–367 [DOI] [PubMed] [Google Scholar]
- 58. Llewellyn-Smith IJ, Kellett DO, Jordan D, Browning KN, Travagli RA. Oxytocin-immunoreactive innervation of identified neurons in the rat dorsal vagal complex. Neurogastroenterol Motil. 2012;24:e136–e146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. J Neurosci. 2007;27:13292–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rinaman L, Verbalis JG, Stricker EM, Hoffman GE. Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J Comp Neurol. 1993;338:475–490 [DOI] [PubMed] [Google Scholar]
- 61. Rinaman L, Hoffman GE, Dohanics J, Le WW, Stricker EM, Verbalis JG. Cholecystokinin activates catecholaminergic neurons in the caudal medulla that innervate the paraventricular nucleus of the hypothalamus in rats. J Comp Neurol. 1995;360:246–256 [DOI] [PubMed] [Google Scholar]
- 62. Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci. 2003;23:10084–10092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sands SA, Morilak DA. Expression of α1D adrenergic receptor messenger RNA in oxytocin- and corticotropin-releasing hormone-synthesizing neurons in the rat paraventricular nucleus. Neuroscience. 1999;91:639–649 [DOI] [PubMed] [Google Scholar]
- 64. Rinaman L. Interoceptive stress activates glucagon-like peptide-1 neurons that project to the hypothalamus. Am J Physiol. 1999;277:R582–R590 [DOI] [PubMed] [Google Scholar]
- 65. McMahon LR, Wellman PJ. PVN infusion of GLP-1-(7–36) amide suppresses feeding but does not induce aversion or alter locomotion in rats. Am J Physiol. 1998;274:R23–R29 [DOI] [PubMed] [Google Scholar]
- 66. Zueco JA, Esquifino AI, Chowen JA, Alvarez E, Castrillon PO, Blazquez E. Coexpression of glucagon-like peptide-1 (GLP-1) receptor, vasopressin, and oxytocin mRNAs in neurons of the rat hypothalamic supraoptic and paraventricular nuclei: effect of GLP-1(7–36)amide on vasopressin and oxytocin release. J Neurochem. 1999;72:10–16 [DOI] [PubMed] [Google Scholar]
- 67. Alhadeff AL, Rupprecht LE, Hayes MR. GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. Endocrinology. 2012;153:647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dossat AM, Lilly N, Kay K, Williams DL. Glucagon-like peptide 1 receptors in nucleus accumbens affect food intake. J Neurosci. 2011;31:14453–14457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maruyama M, Matsumoto H, Fujiwara K, et al. Prolactin-releasing peptide as a novel stress mediator in the central nervous system. Endocrinology. 2001;142:2032–2038 [DOI] [PubMed] [Google Scholar]
- 70. Stornetta RL, Sevigny CP, Guyenet PG. Vesicular glutamate transporter DNPI/VGLUT2 mRNA is present in C1 and several other groups of brainstem catecholaminergic neurons. J Comp Neurol. 2002;444:191–206 [DOI] [PubMed] [Google Scholar]
- 71. Monstein HJ, Grahn N, Truedsson M, Ohlsson B. Oxytocin and oxytocin-receptor mRNA expression in the human gastrointestinal tract: a polymerase chain reaction study. Regul Pept. 2004;119:39–44 [DOI] [PubMed] [Google Scholar]
- 72. Qin J, Feng M, Wang C, Ye Y, Wang PS, Liu C. Oxytocin receptor expressed on the smooth muscle mediates the excitatory effect of oxytocin on gastric motility in rats. Neurogastroenterol Motil. 2009;21:430–438 [DOI] [PubMed] [Google Scholar]
- 73. Wu CL, Doong ML, Wang PS. Involvement of cholecystokinin receptor in the inhibition of gastrointestinal motility by oxytocin in ovariectomized rats. Eur J Pharmacol. 2008;580:407–415 [DOI] [PubMed] [Google Scholar]
- 74. Wu CL, Hung CR, Chang FY, Pau KY, Wang PS. Pharmacological effects of oxytocin on gastric emptying and intestinal transit of a non-nutritive liquid meal in female rats. Naunyn Schmiedebergs Arch Pharmacol. 2003,367:406–413 [DOI] [PubMed] [Google Scholar]