

Abstract
With age, growth plate cartilage undergoes programmed senescence, eventually causing cessation of bone elongation and epiphyseal fusion. Estrogen accelerates this developmental process. We hypothesized that senescence occurs because progenitor cells in the resting zone are depleted in number and that estrogen acts by accelerating this depletion. To test this hypothesis, juvenile ovariectomized rabbits received injections of estradiol cypionate or vehicle for 5 weeks, and then were left untreated for an additional 5 weeks. Exposure to estrogen accelerated the normal decline in growth plate height and in the number of proliferative and hypertrophic chondrocytes. Five weeks after discontinuation of estrogen treatment, these structural parameters remained advanced, indicating an irreversible advancement in structural senescence. Similarly, transient estrogen exposure hastened epiphyseal fusion. Estrogen also caused a more rapid decline in functional parameters of growth plate senescence, including growth rate, proliferation rate, and hypertrophic cell size. However, in contrast to the structural parameters, once the estrogen treatment was discontinued, the growth rate, chondrocyte proliferation rate, and hypertrophic cell size all normalized, suggesting that estrogen has a reversible, suppressive effect on growth plate function. In addition, estrogen accelerated the normal loss of resting zone chondrocytes with age. This decrease in resting zone cell number did not appear to be due to apoptosis. However, it was maintained after the estrogen treatment stopped, suggesting that it represents irreversible depletion. The findings are consistent with the hypothesis that estrogen causes irreversible depletion of progenitor cells in the resting zone, thus irreversibly accelerating structural senescence and hastening epiphyseal fusion. In addition, estrogen reversibly suppresses growth plate function.
Longitudinal bone growth occurs at the growth plate by endochondral bone formation. The growth plate consists of 3 principal zones: resting, proliferative, and hypertrophic. The resting zone, adjacent to the epiphyseal bone, contains progenitor cells that provide new chondrocytes to the proliferative zone (1). The proliferative zone contains replicating chondrocytes arranged in columns parallel to the long axis of the bone. The proliferative chondrocytes located farthest from the resting zone stop replicating and enlarge to become hypertrophic chondrocytes (2–4). The hypertrophic chondrocytes produce a matrix rich in collagen X as well as factors that attract blood vessels and bone cells, which remodel the newly formed cartilage into bone (5, 6). The net result is bone elongation.
The rate of this process, and therefore the rate of longitudinal bone growth, declines with age. This decline is due, in large part, to a decline in chondrocyte proliferation and a decrease in hypertrophic cell size (7–9). These functional senescent changes are accompanied by structural senescent changes; there is a gradual decline in the overall growth plate height, proliferative zone height, and hypertrophic zone height, as well as the number of cells in each zone (7, 9, 10). As a final step in this program, the growth plate undergoes epiphyseal fusion. Growth plate senescence appears to be caused by a mechanism intrinsic to the growth plate (8). Growth-inhibiting conditions slow the rate of senescence, suggesting that growth plate senescence is not simply a function of age, but depends on growth (7, 10, 11). Thus, the prior growth history of the growth plate affects subsequent growth plate function and structure. This long-term information about prior growth is likely retained in the resting zone because nonprogenitor chondrocytes do not remain in the growth plate in the long term; proliferative zone chondrocytes differentiate into hypertrophic zone chondrocytes and then undergo programmed cell death (12). Consistent with this hypothesis, we previously showed evidence that growth plate senescence is associated with a decline in the number of resting zone chondrocytes and in the rate of proliferation of these cells, suggesting that senescence is caused by loss of progenitor cell number and proliferative capacity (13).
Estrogen hastens epiphyseal fusion and cessation of longitudinal bone growth. In children with precocious puberty, linear growth stops and epiphyseal fusion occurs at an earlier age than normal (14), whereas in individuals with hypogonadism (15) or selective deficiency of estrogen (16) or estrogen receptor α (17, 18), cessation of linear growth and onset of epiphyseal fusion occur at a later age than normal. Similarly in mice, estrogen receptor α deletion delays growth deceleration (19).
We previously showed evidence that estrogen hastens the cessation of longitudinal bone growth and the onset of epiphyseal fusion by accelerating the program of growth plate senescence, including both functional and structural changes (20). Because senescence appears to involve the depletion of resting zone progenitor cells, we hypothesized that estrogen accelerates senescence by accelerating this depletion. To test this hypothesis, in the current study, ovariectomized rabbits were exposed to physiological doses of estradiol or vehicle for 5 weeks to determine the effects on the number of resting zone cells. We also sought to determine whether any resulting diminution in the number of these chondrocytes would reverse after the estrogen treatment stopped, and thus represent a transient suppressive effect of estrogen or whether the depletion would persist after cessation of estrogen, and thus represent an irreversible depletion of the progenitor cells, consistent with our hypothesis. We therefore treated additional sets of animals with estradiol or vehicle for 5 weeks, after which we stopped the treatment and determined the number of resting zone chondrocytes after an additional 5-week period. In this latter experiment, we also assessed other structural and functional components of growth plate senescence to determine whether the accelerating effects of estrogen on other components of growth plate senescence were reversible or irreversible.
Materials and Methods
Animals
Forty-seven New Zealand female rabbits were ovariectomized at 9 weeks of age (Covance Research Products). All animals received National Institutes of Health open formula rabbit ration (NIH 32) and water ad libitum. Animal care was in accordance with the Guide for the Care and Use of Laboratory Animals (21). The protocol was approved by the Animal Care and Use Committee (National Institute of Child Health and Human Development, National Institutes of Health).
Study design
We selected rabbits as the animal model because, like humans, but unlike mice and rats, rabbits undergo epiphyseal fusion at the time of sexual maturation, in response to rising estrogen levels (20). Beginning at 11 weeks of age, animals received weekly im injections of either 70 μg/kg estradiol cypionate or vehicle (cottonseed oil) for 5 consecutive weeks. We previously demonstrated that this estradiol treatment regimen produced serum estradiol levels similar to that of intact mature female rabbits (20). Before (age 11 weeks), at the end (16 weeks), and 5 weeks after treatment (21 weeks), groups of animals (n = 8–10 per treatment group per time point) were killed by pentobarbital overdose. Bromodeoxyuridine (BrdU; 50 mg/kg ip; Sigma-Aldrich Co. LLC) was administered 6 hours and again 1 hour before the animals were killed. Distal femurs, distal radii, and proximal and distal tibias were preserved for histologic analysis and BrdU immunohistochemistry.
Growth rate determination
In order to assess the growth rate of the proximal tibial (PT) growth plates, metal pins were inserted below the proximal growth plate of the right tibia of 10 vehicle- and 10 estradiol-treated animals as previously described (22). Radiographs of right tibias were obtained on day 0 (before the first injection) and repeated every 2.5 weeks. For radiographs, animals were sedated and positioned on a horizontal cassette containing x-ray film, with the right hind leg on the cassette. Two radiographs of the right tibia were obtained from each rabbit at each time point. The rabbits were repositioned in between the radiographs. Using these radiographic images, the growth rate of the PT growth plate was assessed by measuring the maximal distance between the most proximal surface of the tibia to the inner tip of the proximal pin. All measurements were performed in triplicate by a single observer blinded to age and treatment, using a digital vernier caliper (Mitutoyo Inc). We averaged measurements for individual animals at each time point and calculated the growth velocity at 2.5-week intervals.
Tissue processing
Tibias, femurs, and radii were fixed in 10% phosphate-buffered formalin for 48 hours, placed in 70% ethanol for storage, decalcified in formic acid, and embedded in paraffin. Sagittal 5-μm sections of the distal femoral, PT, and distal tibial growth plates were obtained from the center of the bone.
Chondrocyte proliferation by BrdU immunohistochemistry
BrdU-labeled cells were visualized by immunohistochemical staining for BrdU using a commercial kit containing biotinylated monoclonal mouse anti-BrdU (HCS24, Oncogene Research Products) per the manufacturer's protocol except that the anti-BrdU antibody was diluted 1:1 with PBS. Labeled and unlabeled cells were counted in approximately 20 chondrocyte columns near the center of each growth plate section. Proliferation rate was defined as the number of BrdU-labeled cells per proliferative column.
Assessment of apoptosis in the resting zone
Apoptotic cells were identified using the TUNEL (terminal deoxynucleotide transferase mediated dUTP nick end labeling) assay (FragELTM DNA Fragmentation Detection Kit, EMD Millipore), according to the manufacturer's protocol. Briefly, after treatment with 20 μg/mL proteinase K in Tris, pH 8, for 20 minutes at room temperature, sections were incubated with the Fluorescein-FragELTM TdT Labeling Reaction Mix for 1 hour at 37°C, rinsed, and mounted with Fluorescein FragELTM Mounting Media, containing 4′,6-diamidino-2-phenylindole counterstain. The total number of positive and negative resting-zone chondrocyte cells was counted by an observer blinded to the treatment groups on 2 sections for each animal (n = 6 per group), and the percent positive cells was calculated.
Quantitative histology
Histologic evaluations were performed on Masson Trichrome-stained tissue sections using a light microscope with a VIA-100 video measurement system (Boeckeler). At the end of the study, all PT, distal femoral, and distal radial growth plates were still unfused. In the distal tibia, the fraction of unfused growth plate was assessed along the entire width of the growth plate in 3 different sections and averaged. All other histologic measurements were performed in the central two-thirds of the growth plate sections. Heights were measured parallel to the chondrocyte columns. We assessed growth plate height and column density in 3 areas of each growth plate and averaged the results. The number of resting cells was calculated as the number of resting chondrocytes per 1-mm growth plate width. The number of proliferative and hypertrophic cells was counted in approximately 15 intact columns per growth plate, and the counts for each cell type were averaged for individual growth plates. For this analysis, cells with a cell height of < 10 μm were counted as proliferative chondrocytes, whereas hypertrophic chondrocytes were defined by a height ≥ 10 μm (20). The terminal hypertrophic chondrocyte was defined as the cell in the last lacuna that was not invaded by metaphyseal blood vessels. The height of this cell was measured in 25 different columns per growth plate and averaged.
Statistics
All data were expressed as mean ± SEM. The overall effects of time and treatment in each growth plate location were evaluated by two-way ANOVA, followed by pair-wise comparisons of treatment effects at the 5- and 10-week time points using Student's t test. P values were corrected for multiple comparisons using the Holm correction. The effects of time on each growth plate location in vehicle-treated animals were evaluated by one-way ANOVA. The effect of estrogen on growth plate fusion was assessed using log-rank Kaplan-Meier survival analysis.
Results
Estrogen irreversibly advanced structural growth plate senescence and hastened epiphyseal fusion
In the vehicle-treated rabbits, with time, there was a significant decline in the total growth plate height of the PT and the distal radius (DR)(PT, P < .001; DR, P < .001; Figures 1, 2A, 2B, and Supplemental Figure 1), the number of proliferative chondrocytes per column (PT, P < .001; DR, P < .001; Figures 2C and D), and the number of hypertrophic chondrocytes per column (PT, P < .001; DR, P < .001; Figures 2E and F). During estrogen treatment, the decline in all of these structural growth plate parameters was accelerated in estrogen-treated compared with vehicle-treated animals, including the total growth plate height (PT, P < .05; DR, P < .05; Figures 1, 2A, 2B, and Supplemental Figure 1), the number of proliferative zone chondrocytes per column (PT, P < .001; DR, P < .001; Figures 2C and D), and the number of hypertrophic chondrocytes per column (PT, P = .07; DR, P < .001; Figures 2E and F).
Figure 1.
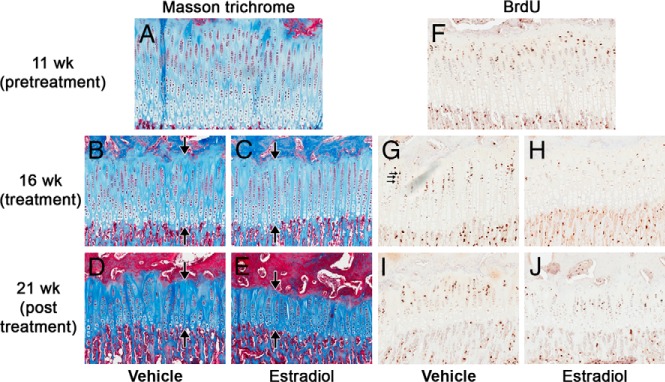
PT growth plate morphology and proliferation before (11 weeks), at the end of treatment (16 weeks), and 5 weeks after the estrogen treatment was stopped (21 weeks). Representative sections of PT growth plates from 11-(A and F), 16- (B, C, G, and H), and 21-week-old (D, E, I, and J) control (A, B, D, F. G, and I) and estrogen-treated (C, E, H, and J) animals stained with Masson Trichrome (A–E) or by BrdU immunohistochemistry (F–J). Estrogen treatment (16-week time point) affected the structure of the growth plate, decreasing the overall height as well as the number of cells in each zone. Interestingly, 5 weeks after the estrogen treatment was stopped (21-week time point) these structural changes remained advanced. Estrogen treatment also decreased the number of BrdU-labeled cells in estrogen treated animals (H) compared with controls (G). However, 5 weeks after the estrogen treatment was stopped (21 weeks) the number of BrdU-labeled cells in estrogen- treated animals (J) was similar to that of controls (I). Large arrows indicate the height of the growth plate, whereas small arrows indicate BrdU-labeled cells.
Figure 2.

Growth plate structure and epiphyseal fusion. Ovariectomized rabbits were treated with estradiol cypionate (closed symbols) or vehicle (open symbols) for 5 weeks starting at 11 weeks of age (denoted by boxes within each graph). Quantitative histology (mean ± SEM) was performed on Masson Trichrome-stained sections of the PT and distal radial growth plates by an observer blinded to treatment and age. In vehicle-treated animals, all structural parameters declined significantly with age (P < .001 by ANOVA) in both PT (A, C, E, and G) and distal radial (B, D, F, and H) growth plates. Estrogen treatment (16-week time point) accelerated the decline in growth plate height (A and B) and in the number of proliferative (C and D) hypertropic (E and F), and resting zone chondrocytes (G and H). Five weeks after the treatment was stopped (21-week time point), all structural growth plate parameters including the number of resting-zone chondrocytes remained advanced in previously estrogen-treated animals compared with controls. The percent of TUNEL (terminal deoxynucleotide transferase mediated dUTP nick end labeling)-positive resting zone chondrocytes was assessed in estrogen- and vehicle-treated animals (16-week time point; I). Estrogen treatment significantly hastened epiphyseal fusion in distal tibia (J). *, P < .05; **, P < .01; ***, P < .001 estradiol vs vehicle.
Interestingly, after the estrogen treatment had been discontinued, the decline of these structural parameters did not normalize, but instead remained advanced. Thus, at 21 weeks of age, which was 5 weeks after the estrogen treatment had been discontinued, most structural parameters remained significantly different in animals that had previously been treated with estrogen vs those previously treated with vehicle: the total growth plate height (PT, P = .12; DR, P < .01; Figures 1, 2A, 2B, and Supplemental Figure 1), the number of proliferative zone chondrocytes per column (PT, P < .001; DR, P < .001; Figures 2C and D) and the number of hypertrophic chondrocytes per column (PT, P < .05; DR, P < .01; Figures 2E and F).
Estrogen also hastened growth plate fusion as assessed at the distal tibial growth plate (Figure 2J). The distal femur, distal radial, and PT growth plates all remained open at the end of the study (data not shown). Lack of fusion in these other growth plates was expected because they normally undergo epiphyseal fusion substantially later than the distal tibia, at approximately 22–26 weeks of age in estrogen-treated animals and even later than that in ovariectomized animals (20).
Estrogen treatment reversibly decreased tibial growth rate, chondrocyte proliferation, and hypertrophic cell size
In vehicle-treated animals, tibial growth rate declined with age (P < .001, Figure 3B). Estrogen treatment decreased the PT growth rate (P < .05, Figure 3B). Interestingly, after the estrogen treatment was stopped, the PT growth rate of estrogen-treated animals returned to normal or close to normal (P = .76, Figure 3B).
Figure 3.

Growth plate function and body weight gain. Ovariectomized rabbits were treated with estradiol cypionate (closed symbols) or vehicle (open symbols) for 5 weeks starting at 11 weeks of age (denoted by boxes within each graph). Tibial growth rate was assessed from serial radiographs obtained every 2.5 weeks, and the calculated growth rates were plotted at the midpoint between the 2 time points. Proliferation rate was defined as the number of BrdU-labeled cells per cell column, assessed by immunohistochemistry. Hypertrophic cell size was defined as the height of the terminal hypertrophic cells, measured on Masson Trichrome-stained sections. Measurements (mean ± SEM) were made by an observer blinded to treatment and age. Estrogen-treated animals gained more weight during treatment, but their weights normalized after the treatment was stopped. In vehicle-treated animals, PT growth rate, proliferation rate, and hypertrophic cell size all declined with age. This decline was accelerated by estrogen (16-week time point) but, after estrogen treatment ended, these functional parameters normalized (21-week time point). *, P < .05; **, P < .01; ***, P < .001.
The number of BrdU-labeled cells per column and thus the rate of chondrocyte proliferation decreased with age in the proliferative zone of vehicle-treated animals (PT, P < .001, Figures 1 and 3C; DR, P < .001, Figure 3D and Supplemental Figure 1). Estrogen treatment further decreased chondrocyte proliferation (PT, P < .01, Figures 1H and 3C; DR, P < .001, Figure 3D and Supplemental Figure 1H). However, once the estrogen treatment was stopped, the chondrocyte proliferation rate in previously estrogen-treated animals did not remain suppressed but returned to a level similar to that of the vehicle-treated animals (PT, P = .52, Figures 1J and 3C; DR, P = .16, Figure 3D and Supplemental Figure 1J). The same pattern was seen for the size of the terminal hypertrophic chondrocytes, which is another important determinate of growth rate. Estrogen treatment caused a small but significant decrease in the size of the hypertrophic chondrocytes (PT, P < .01, Figure 3E; DR, P < .001, Figure 3F), but when the treatment was discontinued, the size of the terminal hypertrophic cells returned to a size similar to that of vehicle-treated animals (Figure 3, E and F).
Estrogen-treated animals also gained more weight during treatment (P < .05), but their weights normalized after the treatment was stopped (Figure 3A).
Estrogen treatment irreversibly advanced the loss of resting zone chondrocytes
With increasing age, there was a significant decline in the number of resting zone chondrocytes per mm growth plate width (PT, P < .001; DR, P < .001; Figure 2, G and H). Estrogen treatment accelerated this loss of resting zone chondrocytes (PT, P < .01; DR, P < .001; Figure 2, G and H). This loss of resting zone progenitor cells does not appear to be explained by increased apoptosis because the percent of TUNEL (terminal deoxynucleotide transferase mediated dUTP nick end labeling)-positive resting zone cells was similar in estrogen- (4.6 ± 0.6%) and vehicle- (4.4 ± 1.0%) treated animals (P = .87, Figure 2I). Interestingly, 5 weeks after the estrogen treatment was discontinued, the decline in number of resting zone chondrocytes per mm growth plate width remained advanced in animals previously treated with estrogen compared with vehicle-treated animals (PT, P = .07; DR, P < .01; Figure 2, G and H). This finding, in particular, may indicate a mechanism by which estrogen permanently advances structural growth plate senescence.
Discussion
We previously showed evidence that estrogen acts to accelerate the program of growth plate senescence and thus hastens growth cessation and epiphyseal fusion (20). Consistent with these prior findings, we here found that a transient exposure to estrogen accelerates the decline in growth plate height and other structural parameters of the growth plate senescence program and that these structural parameters remain advanced after the estrogen treatment is discontinued. Transient estrogen exposure also hastened epiphyseal fusion.
Also consistent with previous findings, we found that estrogen accelerated the decline in all studied functional parameters of growth plate senescence, including the growth rate, chondrocyte proliferation rate, and hypertrophic cell size. However, in contrast to the structural parameters, once the estrogen treatment was discontinued, the functional parameters (growth rate, chondrocyte proliferation rate, and hypertrophic cell size), did not remain low compared with controls, but instead normalized. This unexpected finding suggests that estrogen has a reversible suppressive effect on these functional parameters, rather than an irreversible effect related to advancing senescence. However, it is also important to note that, after estrogen was stopped, the growth rate, proliferation rate, and hypertrophic size returned to normal levels but did not rebound above normal. Thus, no catch-up growth occurred after estrogen treatment was discontinued. Consequently, the decreased tibial length of estrogen-treated animals was at least as pronounced 5 weeks after the estrogen treatment was stopped as it was at the end of the treatment. This pattern differs from that of other growth-inhibiting conditions, including hypothyroidism, glucocorticoid excess, and malnutrition, all of which are followed by increased chondrocyte proliferation and catch-up growth (7, 10, 11, 16). We have previously shown evidence that this catch-up growth occurs because growth inhibition slows growth plate senescence (7, 10, 11). Unlike other growth-inhibiting conditions, estrogen does not slow functional senescence, which may explain the lack of catch-up growth after growth inhibition by estrogen.
In humans, indirect evidence suggests that estrogen does accelerate functional growth plate senescence. In children with precocious puberty, the growth plates are exposed to increased estrogen levels. When treated with a GnRH analog, estrogen concentrations return to prepubertal levels and the growth rates are subsequently below normal for age and correlate with the advancement in bone age, suggesting that, in humans, estrogen irreversibly accelerates functional growth plate senescence (23).
In the current study, we hypothesized that estrogen accelerates senescence by accelerating the depletion of resting zone progenitor cells. Consistent with this hypothesis, we found that the loss of resting zone chondrocytes occurred more rapidly in estrogen-treated animals than in vehicle-treated animals. Furthermore, this decrease in resting zone cell number was maintained after the estrogen treatment stopped, suggesting that it represents an irreversible depletion, rather than a transient suppression of cell numbers. Thus estrogen accelerates the depletion of resting zone chondrocytes and accelerates growth plate senescence. In contrast, glucocorticoid excess and hypothyroidism slow resting zone chondrocyte depletion and slow growth plate senescence (7, 11, 13), further supporting the concept that loss of resting zone cells is an important determinant of growth plate senescence.
The mechanism for this proposed effect of resting zone chondrocyte depletion on acceleration of growth plate senescence and epiphyseal fusion is not known. There is evidence that resting zone chondrocytes act as progenitor cells to produce new clones of chondrocytes in the proliferative zone (1). These clones, which are aligned in columns, are important for bone elongation. Therefore declining numbers of resting zone chondrocytes might lead to senescence because, with time, there are fewer progenitor cells to replace exhausted proliferative columns (8). Eventually, no new columns can be formed and as the old columns stop proliferating, growth stops and epiphyseal fusion occurs. A second possible mechanism by which the number of resting zone chondrocytes might drive senescence is through paracrine factors. Resting zone chondrocytes produce parathyroid hormone-related protein (PTHrP) (21, 24), which, together with Indian hedgehog, forms a feed-back loop that controls the length of the proliferative columns (25–27). Loss of resting cell chondrocytes may therefore result in reduced PTHrP production and therefore shorter proliferative columns and possibly other structural changes. Resting zone chondrocytes also secrete other factors including bone morphogenetic protein inhibitors gremlin, chordin, and Wnt-inhibitor sfrp5 (28, 29), which may inhibit the differentiation program of growth plate chondrocytes (30–33). Loss of resting zone chondrocytes may therefore result in a decline in the production of PTHrP and other factors that inhibit the hypertrophic differentiation program and thus contribute to the functional and structural changes as well as to hastened epiphyseal fusion (34).
The mechanisms by which estrogen accelerates the depletion of resting zone chondrocytes are also not well understood. We did not find evidence that increased apoptosis contributes to the accelerated loss of resting-zone progenitor cells. However, we previously found that estrogen treatment decreases proliferation, and thus self-renewal, of resting zone chondrocytes (13), which could contribute to depletion. Interestingly, dexamethasone treatment also slows chondrocyte proliferation in the resting zone, but, unlike estrogen, actually conserves the number of resting zone chondrocytes (13), suggesting that dexamethasone may additionally inhibit the differentiation of resting to proliferative chondrocytes. Therefore, taken together, the findings suggest that estrogen accelerates the loss of progenitor cells in the resting zone by decreasing proliferation of resting zone chondrocytes without delaying or possibly accelerating their differentiation to proliferative zone chondrocytes.
The finding that estrogen accelerates the loss of resting-zone chondrocytes and simultaneously inhibits proliferation of proliferative chondrocytes suggests that, during estrogen exposure, there are fewer proliferative chondrocyte replications per lost resting zone chondrocyte, thus providing a cellular explanation for the clinical observations that premature estrogen exposure results in a gradual loss of growth potential proportional to the time of exposure (14, 23). In humans, this loss of growth potential may be partially offset by the stimulatory effect of estrogen on GH secretion.
After the estrogen treatment stopped, the proliferation rate, hypertrophic cell size, and growth rate of the experimental group and the control group converged. We attribute this convergence to recovery from the estrogen treatment. Such convergence could also occur simply because the decline in the control group must eventually level off. However, the data indicate that none of these parameters leveled off during the duration of the experiment. Similarly, in a prior study (20) leveling off of these parameters did not occur in the proximal tibia even up to 24 weeks of age in control rabbits of the same strain, housed and fed under the same conditions.
In summary, we found that estrogen has 2 distinct effects on the growth plate. First, estrogen reversibly suppresses growth plate function, including proliferation in the proliferative zone, hypertrophic cell size, and consequently the rate of longitudinal bone growth. Second, estrogen irreversibly advances structural senescence of the growth plate and hastens epiphyseal fusion. In addition, we found that estrogen irreversibly accelerates depletion of progenitor cells in the resting zone, providing a potential mechanism underlying the acceleration in growth plate senescence. Thus, these findings provide a possible cellular mechanism for the well-documented clinical observations that premature estrogen exposure in children leads to premature growth cessation and early epiphyseal fusion whereas delayed exposure to estrogen leads to delayed growth cessation and late fusion.
Acknowledgments
The work of O.N., M.W., E.B.M.L., and J.B. was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health. O.N. was supported by an ESPE Research Fellowship grant, supported by Novo Nordisk, and grants from the Swedish Research Council (K2012–99X-21998–01-3), the Swedish Society of Medicine, Her Royal Highness Crown Princess Lovisa's Foundation for Pediatric Care, Wera Ekstrom's Foundation for Pediatric Research, Märta och Gunnar V Philipson's Foundation, Sällskapet Barnavård, Stiftelsen Frimurare Barnhuset i Stockholm, and Karolinska Institutet.
Disclosure Summary: M.W., J.M., K.B., and J.B. have nothing to declare.
Footnotes
- BrdU
- bromodeoxyuridine
- DR
- distal radius
- PT
- proximal tibia.
References
- 1. Abad V, Meyers JL, Weise M, et al. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143(5):1851–1857 [DOI] [PubMed] [Google Scholar]
- 2. Hunziker EB. Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc Res Tech. 1994;28(6):505–519 [DOI] [PubMed] [Google Scholar]
- 3. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–336 [DOI] [PubMed] [Google Scholar]
- 4. Walker KV, Kember NF. Cell kinetics of growth cartilage in the rat tibia. I. Measurements in young male rats. Cell Tissue Kinet. 1972;5(5):401–408 [DOI] [PubMed] [Google Scholar]
- 5. Baron J, Klein KO, Yanovski JA, et al. Induction of growth plate cartilage ossification by basic fibroblast growth factor. Endocrinology. 1994;135(6):2790–2793 [DOI] [PubMed] [Google Scholar]
- 6. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5(6):623–628 [DOI] [PubMed] [Google Scholar]
- 7. Marino R, Hegde A, Barnes KM, et al. Catch-up growth after hypothyroidism is caused by delayed growth plate senescence. Endocrinology. 2008;149(4):1820–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nilsson O, Baron J. Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion. Trends Endocrinol Metab. 2004;15(8):370–374 [DOI] [PubMed] [Google Scholar]
- 9. Walker KV, Kember NF. Cell kinetics of growth cartilage in the rat tibia. II. Measurements during ageing. Cell Tissue Kinet. 1972;5(5):409–419 [DOI] [PubMed] [Google Scholar]
- 10. Forcinito P, Andrade AC, Finkielstain GP, Baron J, Nilsson O, Lui JC. Growth-inhibiting conditions slow growth plate senescence. J Endocrinol. 2011;208(1):59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gafni RI, Weise M, Robrecht DT, et al. Catch-up growth is associated with delayed senescence of the growth plate in rabbits. Pediatr Res. 2001;50(5):618–623 [DOI] [PubMed] [Google Scholar]
- 12. Hunziker EB, Schenk RK. Physiological mechanisms adopted by chondrocytes in regulating longitudinal bone growth in rats. J Physiol. 1989;414:55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schrier L, Ferns SP, Barnes KM, et al. Depletion of resting zone chondrocytes during growth plate senescence. J Endocrinol. 2006;189(1):27–36 [DOI] [PubMed] [Google Scholar]
- 14. Carel JC, Lahlou N, Roger M, Chaussain JL. Precocious puberty and statural growth. Hum Reprod Update. 2004;10(2):135–147 [DOI] [PubMed] [Google Scholar]
- 15. Uriarte MM, Baron J, Garcia HB, Barnes KM, Loriaux DL, Cutler GB., Jr The effect of pubertal delay on adult height in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1992;74(2):436–440 [DOI] [PubMed] [Google Scholar]
- 16. Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80(12):3689–3698 [DOI] [PubMed] [Google Scholar]
- 17. Quaynor SD, Stradtman EW, Jr, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med. 2013;369(2):164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331(16):1056–1061 [DOI] [PubMed] [Google Scholar]
- 19. Börjesson AE, Windahl SH, Karimian E, et al. The role of estrogen receptor-α and its activation function-1 for growth plate closure in female mice. Am J Physiol Endocrinol Metab. 2012;302(11):E1381–E1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weise M, De-Levi S, Barnes KM, Gafni RI, Abad V, Baron J. Effects of estrogen on growth plate senescence and epiphyseal fusion. Proc Natl Acad Sci USA. 2001;98(12):6871–6876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koziel L, Wuelling M, Schneider S, Vortkamp A. Gli3 acts as a repressor downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development. 2005;132(23):5249–5260 [DOI] [PubMed] [Google Scholar]
- 22. Heinrichs C, Yanovski JA, Roth AH, et al. Dexamethasone increases growth hormone receptor messenger ribonucleic acid levels in liver and growth plate. Endocrinology. 1994;135(3):1113–1118 [DOI] [PubMed] [Google Scholar]
- 23. Weise M, Flor A, Barnes KM, Cutler GB, Jr, Baron J. Determinants of growth during gonadotropin-releasing hormone analog therapy for precocious puberty. J Clin Endocrinol Metab. 2004;89(1):103–107 [DOI] [PubMed] [Google Scholar]
- 24. Chau M, Forcinito P, Andrade AC, et al. Organization of the Indian hedgehog–parathyroid hormone-related protein system in the postnatal growth plate. J Mol Endocrinol. 2011;47(1):99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung UI, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest. 2001;107(3):295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lanske B, Karaplis AC, Lee K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273(5275):663–666 [DOI] [PubMed] [Google Scholar]
- 27. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273(5275):613–622 [DOI] [PubMed] [Google Scholar]
- 28. Lui JC, Andrade AC, Forcinito P, et al. Spatial and temporal regulation of gene expression in the mammalian growth plate. Bone. 2010;46(5):1380–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nilsson O, Parker EA, Hegde A, Chau M, Barnes KM, Baron J. Gradients in bone morphogenetic protein-related gene expression across the growth plate. J Endocrinol. 2007;193(1):75–84 [DOI] [PubMed] [Google Scholar]
- 30. Dao DY, Jonason JH, Zhang Y, et al. Cartilage-specific β-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J Bone Miner Res. 2012;27(8):1680–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Luca F, Barnes KM, Uyeda JA, et al. Regulation of growth plate chondrogenesis by bone morphogenetic protein-2. Endocrinology. 2001;142(1):430–436 [DOI] [PubMed] [Google Scholar]
- 32. Shu B, Zhang M, Xie R, et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci. 2011;124(Pt 20):3428–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102(14):5062–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirai T, Chagin AS, Kobayashi T, Mackem S, Kronenberg HM. Parathyroid hormone/parathyroid hormone-related protein receptor signaling is required for maintenance of the growth plate in postnatal life. Proc Natl Acad Sci USA. 2011;108(1):191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]