

Abstract
Adduction of a nitric oxide (NO) moiety (NO•) to cysteines termed as S-nitrosylation (SNO) has emerged as a crucial mechanism for NO signaling crucial for mediating the vascular effects of estrogens. Mitochondrion is a known vascular risk factor; however, the effects of estrogens on mitochondrial SNO are incompletely understood. In this study we determined the effects of estradiol-17β (E2β) on mitochondrial protein SNO in primary human umbilical vein endothelial cells and compared the mitochondrial nitroso-proteomes in E2β- and a NO donor S-nitrosoglutathione (GSNO)-treated cells using a proteomics approach. Treatment with 10 nM E2β and 1 mM GSNO for 30 minutes significantly increased the levels of mitochondrial SNO-proteins. Subcellular localization of SNO-proteins showed mitochondria as the major cellular organelle for protein SNO in response to E2β and GSNO. E2β stimulated mitochondrial endothelial nitric oxide synthase (eNOS) phosphorylation and mitochondrial protein SNO that was enhanced by overexpression of mitochondrion or Golgi, but not membrane targeting eNOS constructs. We identified 11, 32, and 54 SNO-proteins in the mitochondria from the untreated, E2β-, and GSNO-treated human umbilical vein endothelial cells, respectively. Comparisons of the nitroso-proteomes revealed that common and different mitochondrial SNO-proteins were affected by endogenous NO on E2β stimulation and exogenous NO from donor. These SNO-proteins were associated with various mitochondrial functions, including energy and redox regulation, transport, iron homeostasis, translation, mitochondrial morphology, and apoptosis, etc. Collectively, we conclude that estrogens rapidly stimulate protein SNO in endothelial mitochondria via mitochondrial eNOS, providing a mechanism for mediating the vascular effects of estrogens.
Endothelial cells, lining the inner surface of all blood vessels throughout the body, play a key role in vascular health largely by stimulating nitric oxide (NO) production via endothelial NO synthase (eNOS) because NO possesses potent antiinflammatory, antiapoptotic, antithrombotic, and antioxidant effects (1–3). Formation of cyclic guanosine monophosphate (cGMP) well known to mediate many biological functions of NO, including vascular remodeling, vessel relaxation, platelet aggregation, etc. (4); however, many biological activities of NO are cGMP independent (5). Once synthesized, NO can be rapidly converted to other reactive nitrogen species (RNS) such as nitrogen trioxide (N2O3), peroxinitrite (ONOO−), and nitrosoglutathione (GSNO) (6). These RNS can donate a NO moiety (NO•) to cysteines in peptides or proteins, a process called S-nitrosylation (SNO) that results in the formation of nitrothiols. This rapid, reversible, and redox-sensitive posttranslational modification has emerged as a crucial cGMP-independent signaling pathway for NO (7), which modulates the functions of a plethora of proteins and participates in the regulation of nearly all major biological pathways (8).
NO signaling plays a key role in mediating the so-called “protective” effects of estrogens in the vasculature. Estrogens stimulate endothelial NO production via rapid eNOS activation and increased eNOS expression via specific estrogen receptor (eg, ERα and ERβ)-dependent mechanisms (9–12). Of note, the current understanding of estrogen action on endothelial cells is predominantly formulated from studies of NO biosynthesis via eNOS; however, the targets and functional sequelae of enhanced NO production by estrogen stimulation remain largely unknown. We have recently shown that estradiol-17β (E2β) rapidly stimulates protein SNO via specific ER- and eNOS-dependent mechanisms in primary endothelial cells (13, 14). These findings suggest that SNO serves as a novel mechanism in mediating the vascular effects of estrogens. By using biotin switch-based proteomics approach, we have obtained a partial estrogen-responsive endothelial nitroso-proteome in which many SNO-proteins are found to be mitochondrial proteins (13, 14). A few studies have also recently shown that SNO is important for the cardioprotective effects of estrogens in ischemic heart because ischemic preconditioning results in increased SNO of a variety of proteins that are mostly mitochondrial proteins (15, 16). These findings suggest that mitochondrion may be the major organelle that is affected by estrogens via SNO.
Mitochondrion is the organelle for synthesizing ATP and energy production; with reactive oxygen species (ROS) as major byproducts it also plays a crucial role in various key cellular functions such as oxidative phosphorylation, oxidative stress, and apoptosis (17). Mitochondrion is the primary organelle that NO targets because NO exposure results in substantial changes in mitochondrial respiration (18). Numerous studies have previously shown that estrogens have profound effects on mitochondrial functions in endothelial cells, including mitochondria biogenesis (19), energy generation (20), aging (21, 22), etc. It has been demonstrated that NO derived from mitochondrial eNOS plays a major role in mediating the mitochondrial effects of estrogens in endothelial cells (20, 23, 24); however, the mechanisms post-NO biosynthesis such as SNO, by which estrogens regulate mitochondrial functions, are incompletely understood. Although a few mitochondrial SNO-proteins have been identified (13, 14, 25–27), a large-scale analysis of estrogen-responsive endothelial mitochondrial nitroso-proteome has not been performed to date. Therefore, this study was performed to develop a comprehensive proteomics approach for analyzing global changes in the estrogen-responsive mitochondrial SNO-proteins in endothelial cells.
Materials and Methods
Chemicals and antibodies
Estradiol-17β (E2β), sodium ascorbate, HEPES, diethylene triamine pentaacetic acid, neocuproine, copper chloride, fatty acid-free BSA, methanol, N-ethylmeleimide methyl methanethiosulfonate, 3-(3-Cholamidopropyl)dimethylammonio-1-propanesulfonate, sodium dodecyl sulfate (SDS), dimethyl sulfoxide, and all other chemicals unless specified, were from Sigma. N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio) propionamide (biotin-HPDP) was from Thermo Scientific. S-Nitrosoglutathione (GSNO) was from BioVision. Trypsin Gold (mass spectrometry grade) was from Promega Corp. Antibiotin antibody was from Cell Signaling Technology. 2-(6-biotinoyl-amino-hexanoyl amino) ethylmethanethiosulfonate-Texas red was from Toronto Research Chemicals. Anti-β-actin monoclonal antibody was from Ambion. Antibodies against eNOS and glyceraldehyde 3-phosphate dehydrogenase and ser1177 phosphorylated eNOS were from Santa Cruz Biotechnology, Inc. Antibodies against lamin B and cytochrome c oxidase IV were from Abcam. Oregon-Green wheat germ agglutinin, alexa488-Con A, MitoTracker-Green FM, prolong Gold antifade reagent, MCDB131, and M199 were from Invitrogen.
Cell culture and treatment
Human umbilical cord vein endothelial cells (HUVECs) were isolated by collagenase digestion as described previously (13). Umbilical cords were collected from the University of California Irvine Medical Center Hospital (Orange, CA) and approved by the Institutional Review Boards. Cells were cultured in endothelial cell medium (purchased from ScienCell) containing 5% fetal bovine serum (ScienCell) and supplemented with 1% antibiotics and 1% endothelial cell growth supplement and used within 5 passages. Cells at approximately 70% confluence were cultured in serum-free M-199 (phenol red-free M-199 containing 0.1% fatty acid-free BSA, 1% fetal calf serum, and 25 mM HEPES) for 16 hours. After 1-hour equilibration with fresh M199–0.1% BSA-25 mM HEPES, the cells were treated with E2β for 20–30 minutes. Ethanol (final concentration <0.01%) was used to dissolve E2β, which did not alter cellular responses. GSNO was used as a NO donor for a positive control.
Biotin-switch (BST), SDS-PAGE, and immunoblotting
Biotin switch was performed as previously described (13). Briefly, HUVECs (∼4 × 106 cells) or the purified mitochondrial proteins were lysed in blocking buffer (25 mM HEPES, pH 7.7; 1 mM diethylene triamine pentaacetic acid; 0.1 mM neocuproine; 50 mM N-ethylmeleimide; and 2.5% SDS). The protein content of the samples was determined and was adjusted to 0.6 mg/mL. The samples (50 μg/group) were transferred to 1.5-mL amber Eppendorf tubes and were incubated at 50°C for 30 minutes in the dark, followed by precipitation with cooled (−20°C) acetone (1:3, vol/vol) and incubation at −20°C for 2 hours. Following centrifugation (12 000 × g, 10 minutes), the acetone-precipitated proteins were washed once with 75% cold acetone, and then resuspended in labeling buffer (25 mM HEPES, pH 7.7; 30 mM sodium ascorbate; 0.1 μM CuCl2; 0.4 mM N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio) propionamide; and 1% SDS). After adjusting the protein content to 0.6 mg/mL, the samples were incubated at 37°C for 1 hour in the dark with occasional agitation. Finally, the samples were mixed with SDS sample buffer (without dithiothreitol or 2-mercaptoethanol) for SDS-PAGE and immunoblotting analyses. Total SNO-proteins of each sample were detected by immunoblotting with antibiotin antibody. All proteins in a lane were summed as the level of SNO-proteins of the sample.
Subcellular localization of SNO-proteins by fluorescence microscopy
SNO-proteins were detected in intact cells by a modified BST protocol as previously described (13, 28). Briefly, HUVECs were cultured on glass coverslips and then cultured in serum-free M-199 when reaching 70% confluence and treated with E2β or GSNO for 20–30 minutes. The cells were washed with cold PBS and fixed with methanol at −20°C for 15 minutes. Free thiols were blocked with 0.2 M methyl methanethiosulfonate in HEN/methanol buffer (80% methanol; 100 mM HEPES, pH 7.7; 1 mM EDTA; and 0.1 mM neocuproine) at 50°C for 30 minutes in darkness. After 3 washes with 100 mM HEPES/80% methanol, the cells were incubated with 0.2 M ascorbate and 0.2 μM 2-(6-biotinoyl-amino-hexanoyl amino) ethylmethanethiosulfonate-Texas Red in 100 mM HEPES/80% methanol at 37°C for 1 hour in the dark. After extensive washing with methanol, the cells were washed with PBS and then incubated with 1% BSA in PBS for 10 minutes. After a brief washing with PBS, the cells were incubated with Oregon-Green-wheat germ agglutinin, Alexa488-concanavalin A, or MitoTracker-Green FM for 50 minutes at room temperature to label Golgi apparatus, endoplasmic reticulum, and mitochondria, respectively. After washing with PBS, the cells were mounted with Prolong Gold antifade reagent and examined under a fluorescence microscope; fluorescence images were captured by a Hamamatsu charge-coupled device camera using SimplePCI software (Compix).
Mitochondrion extraction
Mitochondrion extraction was performed by using the Mitochondrion Isolation Kit (Miltenyi Biotec) with the manufacturer's instructions. Briefly, HUVECs (∼1 × 107 cells) were quickly washed and trypsinized at 37°C for 3 minutes. The cells were resuspended in 1 mL ice-cold Lysis Buffer and were homogenized in a Dounce homogenizer (80 strokes). The lysates were transferred into a 15-mL tube with 9 mL of ice-cold Separation Buffer. After incubation with 50 μL microbeads for 1 hour at 4°C, which were precoated with monoclonal antibody against the translocase of outer mitochondrial membrane 22, the microbead-bound mitochondria were purified on the LS column using the magnetic MidiMACS Separation Unit. The purified mitochondria were pelleted by centrifugation (13 000 × g, 2 minutes) and resuspended in 100 μL of Storage Buffer and stored at −80°C for further analysis. Protein content was determined by the Piece BCA protein assay kit.
Mitochondrion and plasma membrane eNOS targeting and cell transfection
The pCDNA3 plasmids carrying the wild-type (WT), plasma membrane targeting, and mitochondrion targeting eNOS cDNAs were kindly provided by Dr. Stephen Felton (Medical College of Georgia). The plasma membrane targeting eNOS (CAAX-eNOS) was reconstructed by fusion of the 15-amino acid membrane targeting sequence from K-ras (CAAX) in-frame to the C terminus of the cytosolic myristoylation site mutant eNOS (G2A-eNOS) that is catalytically identical to WT-eNOS as previously described (29). The mitochondrion-targeting eNOS (Mito-eNOS) was constructed by fusion of a targeting sequence derived from human cytochrome-c oxidase subunit VIII to G2A-eNOS as previously described (30). HUVECs were seeded at a density of 1.5 × 105 cells per well in a 6-well plate and transfected on the next day with the plasmids carrying cDNAs for WT-, Mito-, and CAAX-eNOS using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's instructions.
To assess the activity of the overexpressed eNOS on S-nitrosylation, the ratio of SNO-proteins in organelle-eNOS-transfected and total SNO-proteins was calculated by the following equation: . Basal eNOS is endogenous eNOS in nontransfected cells; basal SNO is constitutively SNO-proteins; [Total eNOS − Basal eNOS] stands for the overexpressed organelle-specific eNOS; [Total SNO − Basal SNO] stands for SNO mediated by overexpressed eNOS; was used to normalize the data.
Trypsin digestion, avidin capture of SNO-peptides, and liquid chromatography (LC) mass spectrometry (MS)n analysis
Protein digestion and avidin capture were performed as previously described (31). Briefly, the biotin-labeled protein samples (1 mg) by BST were precipitated by acetone and resuspended in 200 μL of 50 mM ammonium bicarbonate/1 M urea. The samples were incubated with trypsin (20 μg) at 37°C for 18 hours, followed by incubation with NeutrAvidin beads (25 μL) prewashed in 50 mM ammonium bicarbonate at room temperature for 1 hour. The beads were washed 3 times with 1 M ammonium bicarbonate, followed by 3× washes with H2O. Peptides were eluted from the beads with 100 μL of 0.4% trifluoroacetic acid/30% acetonitrile at room temperature for 30 minutes and then eluted one more time with 50 μL of the solution. The samples were dried using a SpeedVac and resuspended in 4% acetonitrile/0.1% formic acid for mass spectrometric analysis. Complete drying was avoided to diminish sample loss and thiol oxidation.
LC multistage tandem MS (MS/MS) analysis of cross-linked peptides was carried out using an LTQ-Orbitrap XL MS (Thermo Scientific) coupled with an Eksigent NanoLC system (Eksigent). Briefly, the LC analysis was performed using a capillary column (100 μm i.d. × 150 mm long) packed with C18 resins (GL Sciences) and the peptides were eluted using a linear gradient of 2%–40% B in 35 minutes; (Solvent A: 100% H2O/0.1% formic acid; Solvent B: 100% acetonitrile /0.1% formic acid). For LC MS/MS analysis, a cycle of one full Fourier transform ion cyclotron resonance mass spectrometry scan mass spectrum (350–1800 mass to charge ratio [m/z], resolution of 60 000 at m/z 400) was followed by 10 data-dependent MS/MS acquired in the linear ion trap with normalized collision energy (setting of 35%). Target ions selected for MS/MS were dynamically excluded for 30 seconds.
The MS data was extracted and analyzed as previously described (32). Monoisotopic masses of parent ions and corresponding fragment ions, parent ion charge states, and ion intensities from LC-MS/MS spectra were extracted using in-house software based on Raw_Extract script from Xcalibur v2.4. Following automated data extraction, the resultant peak lists for each LC-MS/MS experiment were submitted to the development version (v5.8.0) of Protein Prospector (University of California, San Francisco) for database searching using a concatenated SwissProt database composed of a SwissProt database and its randomized version (SwissProt.2010.08.10.random.concat). Homo sapiens was selected as the restricted species. Trypsin was set as the enzyme with a maximum of 2 missed cleavage sites. The mass tolerances for parent and fragment ions were set as 20 ppm and 0.8 Da, respectively. Chemical modifications such as protein N-terminal acetylation, methionine oxidation, N-terminal pyroglutamine, and deamidation of asparagine were selected as variable modifications. Maximal modifications on a given peptide were set as 3. The Search Compare program in Protein Prospector was used for summarization, validation, and comparison of results. To determine the expectation value cutoff that corresponds to a percent false positive (% FP) rate, the plot of the expectation values vs % FP rate for each search result was automatically obtained using the Search Compare Program. Based on these results, we chose an expectation value cutoff for all peptides corresponding to ≤1% FP.
Protein identification and canonical pathway analysis
Protein identification was based on at least 2 SNO-peptides identified by the LC- multistage tandem mass spectrometry. The biological functions of the identified proteins were analyzed using Ingenuity Pathways Analysis (version 7.1; http://www.ingenuity.com). Based on the local networks by computational algorithms, identified proteins were connected with hub proteins, forming a functional protein cross talk.
Experimental replication and statistical analysis
All experiments were repeated at least 3 times using cells from different fetuses. Data were presented as means ± SEM, and analyzed by one-way ANOVA, followed by Bonferroni test for multiple comparisons using SigmaStat 3.5 (Systat Software, Inc). Student's t test was used for comparison of data between the 2 groups. Significance was defined as P < .05.
Results
E2β stimulation of protein SNO mainly occurs in the mitochondria in endothelial cells
We presented data affirming the stimulatory effects of E2β on protein SNO in endothelial cells as we recently reported (13, 14). Consistently, we detected various proteins that are constitutively nitrosylated in untreated control HUVECs, and treatment with E2β (10 nM) or GSNO (1 mM) for 30 minutes significantly increased the levels of SNO-proteins in HUVEC (Figure 1). To determine the subcellular location of E2β-induced SNO-proteins in endothelial cells, we detected SNO-proteins in HUVECs in situ. After treatment with or without E2β (10 nM) or GSNO (1 mM) for 30 minutes, the cells were fixed; SNO-proteins were labeled in red fluorescence by BST, and the cellular organelles, including Golgi, endoplasmic reticulum, and mitochondria were labeled in green fluorescence by using specific organelle trackers. As shown in Figure 2, in untreated control cells, constitutively nitrosylated proteins were mainly detected in the mitochondria, with weak signals in the Golgi complex and endoplasmic reticulum. Treatment with E2β or GSNO dramatically increased the red fluorescence intensities of SNO-proteins in the cytosol and nucleus of HUVECs. The E2β- and GSNO-induced SNO-proteins in the cytosol were mainly detected in the mitochondria similar to untreated cells; however, E2β and GSNO seemed to stimulate protein SNO in the endoplasmic reticulum but not in the Golgi complex. Interestingly, increased SNO-proteins were also detected in the nucleus of E2β- and GSNO-treated cells, implicating a role of SNO in the regulation of nuclear signaling of estrogens.
Figure 1.
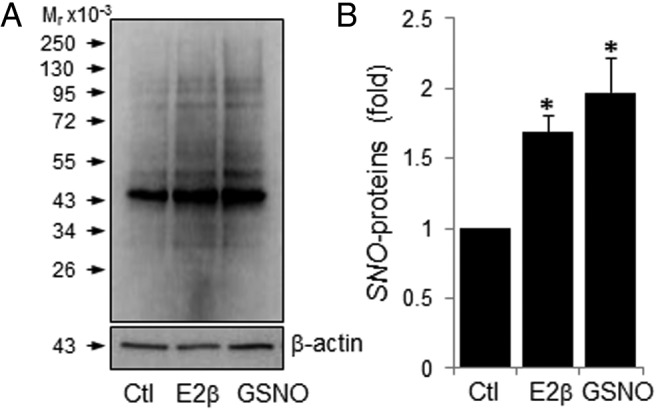
E2β and GSNO stimulate protein S-nitrosylation in HUVECs. HUVECs were treated with 10 nM E2β or 1 mM GSNO for 30 minutes. Total protein extracts were harvested and subjected to biotin-switch. The biotin-labeled SNO-proteins were analyzed by immunoblotting with antibiotin antibody. β-Actin was used as a loading control. A, Image of a representative experiment is shown; B, Plot summarized data (mean ± SEM) from 3 independent experiments. *, P < .05. Ctl, control.
Figure 2.
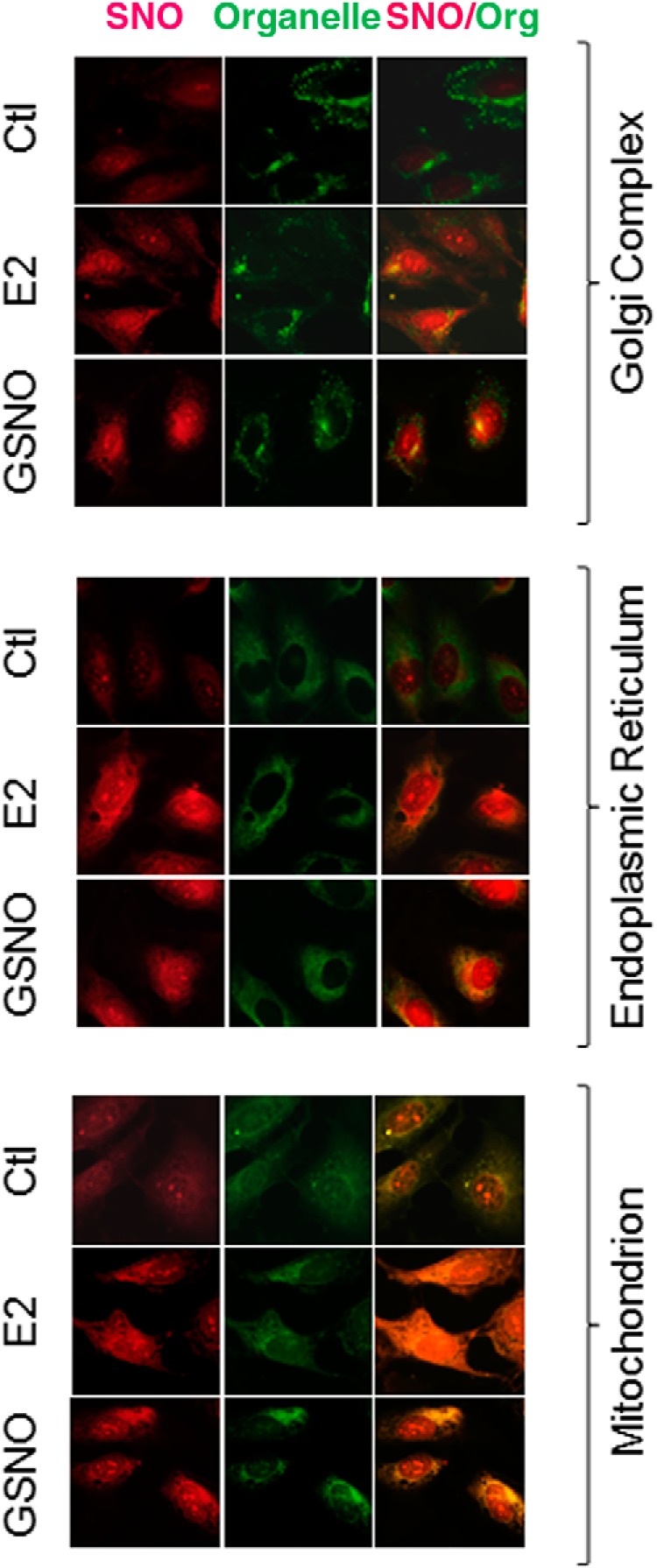
In situ detection of SNO-proteins in E2β- and GSNO-treated HUVECs. HUVECs were treated with 10 nM E2β or 1 mM GSNO for 30 minutes, followed by double immune fluorescence labeling. SNO-proteins were labeled by BST; Oregon-Green-wheat germ agglutinin (WGA), Alexa488-concanavalin A, or MitoTracker-Green FM, were used to label Golgi apparatus, endoplasmic reticulum, and mitochondria, respectively. Ctl, control.
E2β stimulates mitochondrial protein SNO in endothelial cells
To further demonstrate the effects of estrogens on mitochondrial protein SNO, we purified mitochondria from HUVECs by a novel method for mitochondria isolation using magnetic microbeads coated with anti-translocase of outer mitochondrial membrane 22 antibody (33). The mitochondrial preparations obtained were positive with cytochrome c oxidase IV, but not GAPDH, which are markers for mitochondrion and cytosol, respectively. The mitochondrial proteins purified from control, E2β- or GSNO- treated cells were subjected to BST for labeling SNO-proteins, and levels of mitochondrial SNO-proteins were determined by immunoblotting with antibiotin antibody. Constitutively nitrosylated proteins were readily detectable in the mitochondria of untreated control cells. Treatment with E2β or GSNO significantly increased total levels of mitochondrial SNO-proteins (Figure 3). In the mitochondrial preparations, immunoreactive eNOS protein was detected. Moreover, treatment with E2β stimulated eNOS phosphorylation in the mitochondria preparations in a time-dependent manner, similarly to that in whole-cell extracts (Figure 4).
Figure 3.

E2β and GSNO stimulate mitochondrial protein S-nitrosylation in HUVECs. HUVECs were treated with 10 nM E2β or 1 mM GSNO for 30 minutes. a, Mitochondrial proteins were purified and subjected to biotin-switch. The biotin-labeled mitochondrial SNO-proteins were analyzed by immunoblotting with antibiotin antibody. Cytochrome c oxidase IV (COX IV) was used as a loading control for mitochondrion. b, Data (mean ± SEM) were summarized from 3 independent experiments. *, P < .05. c, Validation of mitochondria isolation from HUVECs by immunoblotting with antibodies against COX4, a mitochondrial marker. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a protein marker for cytosol.
Figure 4.
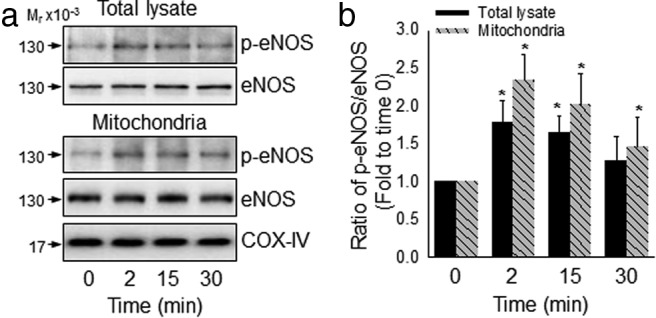
E2 activates mitochondrial eNOS in HUVECs. HUVECs were treated with or without 10 nM E2β for different times. Activation of eNOS in the purified mitochondria and whole-cell extracts was measured by immunoblotting with specific antibody against ser1177 phosphorylated eNOS. Images (a) show blots of a typical experiment and graph (b) summarized data (mean ± SEM) from 3 independent experiments. *, P < .05.
Overexpression of mitochondrion- but not membrane-targeted-eNOS enhanced E2β-stimulated protein SNO in HUVECs
When the plasmids of Mito-eNOS or CAAX-eNOS (membrane targeting) were transfected in HUVECs, overexpressed eNOS was shown to be targeted into mitochondria and plasma membrane, respectively, as previously described (29, 30). Transfections with the WT-, Mito-, and CAAX-eNOS all resulted in increased eNOS expression; however, E2β-stimulated protein SNO was enhanced by overexpression of the WT- and Mito-eNOS, but not CAAX-eNOS in HUVECs (Figure 5).
Figure 5.

E2-stimulated protein SNO was enhanced by overexpression of mitochondrial or Golgi, but not membrane- targeted eNOS in HUVECs. Mitochondrial eNOS (Mito-eNOS), CAAX-eNOS, and wt-eNOS were overexpressed in HUVECs, respectively. After treatment with or without 10 nM E2β, cells were lysed and subjected to biotin-switch. The biotin-labeled SNO-proteins were analyzed by immunoblotting with antibiotin antibody. Expression levels of eNOS were determined with anti-eNOS antibody. Graph summarized data (mean ± SEM) from 3 independent experiments. Bars with different letters differ significantly (P < .05).Ctl, control.
Identification of mitochondrial SNO-proteins in HUVEC by LC-MS/MS analysis
We labeled total mitochondrial SNO-proteins from untreated, E2β-, and GSNO-treated cells. Following trypsin digestion, the biotin-labeled SNO-peptides were purified for identification of mitochondrial SNO-proteins by mass spectrometry. The SNO-peptides identified were summarized in Table 1. A total of 90 SNO-peptides were identified in the purified endothelial mitochondria, including 14 from the untreated and, 41 and 83 from the E2β- and GSNO- treated HUVECs, respectively. These SNO-peptides had overlaps among the groups (Figure 6a). Eight SNO-peptides were identified in both the untreated and E2β-treated HUVECs: 10 in the untreated and GSNO-treated HUVECs and 36 in the E2β- and GSNO-treated HUVECs. Of note, 6 SNO-peptides were found in all 3 groups. Mass spectrometric analyses of these identified SNO-peptides suggest a total of 57 SNO-proteins in the mitochondrial preparations, including 11, 32, and 53 SNO-proteins in the untreated, E2β-, and GSNO-treated cells, respectively. Similarly, the SNO-proteins had overlap among the 3 groups (Figure 6b). Nine were found in both the untreated and E2β-treated HUVECs; 10 in the untreated and the GSNO-treated HUVECs; 28 in the E2β- and GSNO-treated HUVECs. Additionally, the 6 constitutive SNO-peptides with other specific peptides identified in either control, or E2, or GSNO-treated cells led to 8 possible constitutive mitochondrial SNO-proteins.
Table 1.
Mitochondrial nitroso-Proteome in HUVECs
| Function | UniProt Identification | Protein Name | SNO-Peptide | Best Expected Value |
||
|---|---|---|---|---|---|---|
| Ctl | E2β | GSNO | ||||
| Fatty acid oxidation | P49748 | Acyl-CoA dehydrogenase | ELGAFGLQVPSELGGVGLC*NTQYAR | ND | ND | 1.4E-05 |
| Q13011 | δ (3,5)-δ (2,4)-dienoyl-CoA isomerase | YC*AQDAFFQVK | ND | ND | 4.0E-04 | |
| P40939 | Trifunctional enzyme subunit α | TGIEQGSDAGYLC*ESQK | ND | 6.9E-06 | 1.1E-07 | |
| EVEAVIPDHC*IFASNTSALPISEIAAVSK | ND | ND | 1.1E-07 | |||
| C*LAPMMSEVIR | ND | 6.9E-06 | 1.1E-07 | |||
| Q16698 | 2,4-dienoyl-CoA reductase | VHAIQC*DVR | ND | ND | 1.1E-03 | |
| Q9H845 | Acyl-CoA dehydrogenase family member 9 | GSNTC*EVHFENTK | ND | ND | 4.9E-07 | |
| P50416 | Carnitine O-palmitoyltransferase 1, liver isoform | TLETANC*MSSQTK | ND | ND | 3.2E-05 | |
| SCTTESC*DFVR | ND | 3.2E-05 | ||||
| P13804 | Electron transfer flavoprotein subunit α | TIYAGNALC*TVK | ND | ND | 8.3E-05 | |
| LGGEVSC*LVAGTK | ND | 7.5E-04 | 8.3E-05 | |||
| VAQDLC*K | ND | ND | 8.3E-05 | |||
| P24752 | Acetyl-CoA acetyltransferase | IHMGSC*AENTAK | ND | 7.2E-06 | 1.9E-06 | |
| O14561 | Acyl carrier protein | LMC*PQEIVDYIADK | ND | ND | 1.0E-03 | |
| TCA cycle | O75390 | Citrate synthase | LPC*VAAK | 6.2E-06 | ND | 1.0E-05 |
| Q99798 | Aconitase | VGLIGSC*TNSSYEDMGR | ND | 1.8E-06 | 5.5E-06 | |
| P50213 | Isocitrate dehydrogenase subunit α | IEAAC*FATIK | ND | ND | 2.7E-03 | |
| P48735 | Isocitrate dehydrogenase2 (NADP+), mitochondrial | C*ATITPDEAR | ND | 3.5E-03 | 2.6E-03 | |
| Q02218 | 2-oxoglutarate dehydrogenase | IC*EEAFAR | ND | 1.1E-03 | ND | |
| P40926 | Malate dehydrogenase | TIIPLISQC*TPK | ND | ND | 4.0E-09 | |
| SQETEC*TYFSTPLLLGK | ND | 6.8E-09 | 4.0E-09 | |||
| EGVVEC*SFVK | ND | ND | 4.0E-09 | |||
| GC*DVVVIPAGVPR | ND | 6.8E-09 | 4.0E-09 | |||
| GYLGPEQLPDC*LK | ND | ND | 4.0E-09 | |||
| P31040 | Succinate dehydrogenase flavoprotein subunit | VGSVLQEGC*GK | 5.6E-06 | 2.8E-05 | 1.5E-07 | |
| AAFGLSEAGFNTAC*VTK | ND | ND | 1.5E-07 | |||
| TYFSC*TSAHTSTGDGTAMITR | 5.6E-06 | ND | ND | |||
| P21912 | Succinate dehydrogenase iron-sulfur subunit | CHTIMNC*TR | ND | 3.7E-05 | ND | |
| P53007 | Tricarboxylate transport protein, mitochondrial | GIGDC*VR | ND | ND | 5.1E-03 | |
| Electron transport chains | O95299 | NADH dehydrogenase 1 α subcomplex subunit 10 | VITVDGNIC*TGK | ND | 1.8E-03 | 3.6E-03 |
| C*EVLQYSAR | ND | 8.6E-04 | 3.7E-04 | |||
| O75380 | NADH dehydrogenase iron-sulfur protein 6 | VIAC*DGGGGALGHPK | ND | 8.6E-04 | 3.7E-04 | |
| P28331 | NADH-ubiquinone oxidoreductase 75 kDa | VLFLLGADGGC*ITR | ND | ND | 1.8E-04 | |
| P31930 | Cytochrome b-c1 complex subunit 1 | YIYDQC*PAVAGYGPIEQLPDYNR | ND | 1.7E-05 | 1.9E-05 | |
| LC*TSATESEVAR | 8.3E-04 | 1.7E-05 | 1.9E-05 | |||
| NALVSHLDGTTPVC*EDIGR | ND | 1.7E-05 | 1.9E-05 | |||
| P22695 | Cytochrome b-c1 complex subunit 2 | NALANPLYC*PDYR | 8.6E-05 | 2.6E-06 | 1.8E-07 | |
| ENMAYTVEC*LR | 8.6E-05 | ND | 1.8E-07 | |||
| P53701 | Cytochrome c-type heme lyase | AYEYVEC*PIR | ND | 5.9E-04 | 2.5E-03 | |
| O75947 | ATP synthase subunit δ | SC*AEWVSLSK | ND | 3.4E-04 | 3.9E-04 | |
| P56381 | ATP synthase subunit ϵ | YSQIC*AK | ND | 2.6E-03 | 2.9E-03 | |
| P36542 | ATP synthase subunit γ | GLC*GAIHSSIAK | ND | 7.2E-05 | 3.7E-05 | |
| P48047 | ATP synthase subunit O | GEVPC*TVTSASPLEEATLSELK | ND | ND | 5.5E-08 | |
| One carbon metabolism | Q6UB35 | Monofunctional C1-tetrahydrofolate synthase | AEIDLVC*ELAK | ND | ND | 6.8E-05 |
| P13995 | Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase | IC*NAVSPDKDVDGFHVINVGR | ND | ND | 6.9E-03 | |
| P34897 | Serine hydroxymethyltransferase | AALEALGSC*LNNK | ND | ND | 5.0E-05 | |
| NTC*PGDR | ND | ND | 5.0E-05 | |||
| GLELIASENFC*SR | ND | 3.3E-04 | 5.0E-05 | |||
| Other metabolism | P19367 | Hexokinase-1 | ATDC*VGHDVVTLLR | ND | 2.1E-04 | 1.4E-05 |
| TVC*GVVSR | ND | ND | 1.4E-05 | |||
| P00367 | Glutamate dehydrogenase 1 | C*AVVDVPFGGAK | ND | ND | 7.4E-05 | |
| P43304 | Glycerol-3-phosphate dehydrogenase | NYLSC*DVEVR | ND | ND | 3.4E-03 | |
| Q5JRX3 | Presequence protease | C*SVNATPQQMPQTEK | ND | ND | 2.2E-06 | |
| P32322 | Pyrroline-5-carboxylate reductase 1 | C*MTNTPVVVR | ND | ND | 2.5E-05 | |
| VGAFTMVC*K | ND | 2.5E-07 | 4.1E-07 | |||
| P00505 | Aspartate aminotransferase | TC*GFDFTGAVEDISK | 2.5E-07 | 4.1E-07 | ||
| NLDKEYLPIGGLAEFC*K | ND | ND | 4.1E-07 | |||
| P05091 | Aldehyde dehydrogenase | LLC*GGGIAADR | ND | ND | 6.8E-05 | |
| Transport | P10809 | 60 kDa heat shock protein | AAVEEGIVLGGGC*ALLR | 2.4E-05 | 8.2E-07 | 1.1E-05 |
| C*EFQDAYVLLSEK | 2.4E-05 | 8.2E-07 | 1.1E-05 | |||
| C*IPALDSLTPANEDQK | 2.4E-05 | ND | 1.1E-05 | |||
| Q12931 | Heat shock protein 75 kDa | NIYYLC*APNR | ND | ND | 5.8E-04 | |
| P38646 | Stress-70 protein | MEEFKDQLPADEC*NK | 7.5E-04 | 1.0E-05 | 6.6E-06 | |
| Q3ZCQ8 | Mitochondrial import inner membrane translocase subunit TIM50 | VVVVDC*K | ND | 1.6E-04 | ND | |
| P12235 | ADP/ATP translocase 1 | GADIMYTGTVDC*WR | ND | ND | 6.4E-09 | |
| YFAGNLASGGAAGATSLC*FVYPLDFAR | ND | ND | ||||
| P05141 | ADP/ATP translocase 2 | GLGDC*LVK160 | ND | 4.6E-10 | 6.4E-09 | |
| KGTDIMYTGTLDC*WR257 | 4.8E-07 | ND | 6.4E-09 | |||
| YFAGNLASGGAAGATSLC*FVYPLDFAR129 | 4.8E-07 | ND | ND | |||
| P12236 | ADP/ATP translocase 3 | GLGDC*LVK | ND | 4.6E-10 | 6.4E-09 | |
| GADIMYTGTVDC*WR | ND | 6.4E-09 | ||||
| YFAGNLASGGAAGATSLC*FVYPLDFAR | 4.8E-07 | ND | ND | |||
| Q9Y512 | Sorting and assembly machinery component 50 homolog | IC*DGVQFGAGIR | ND | ND | 7.6E-04 | |
| Q00325 | Phosphate carrier protein | FAC*FER | 5.0E-05 | 2.4E-05 | ND | |
| Q9Y6C9 | Mitochondrial carrier homolog 2 | QVC*QLPGLFSYAQHIASIDGR | ND | ND | 6.6E-06 | |
| P21796 | Voltage-dependent anion-selective channel protein 1 | YQIDPDAC*FSAK | ND | 3.2E-08 | 1.0E-04 | |
| EHINLGC*DMDFDIAGPSIR | ND | ND | 1.0E-04 | |||
| P45880 | Voltage-dependent anion-selective channel protein 2 | SC*SGVEFSTSGSSNTDTGK | ND | ND | 3.2E-07 | |
| WC*EYGLTFTEK | ND | 1.9E-04 | 3.2E-07 | |||
| WNTDNTLGTEIAIEDQIC*QGLK | ND | 1.9E-04 | 3.2E-07 | |||
| Q9Y277 | Voltage-dependent anion-selective channel protein 3 | MC*NTPTYCDLGK | ND | ND | 1.0E-04 | |
| VC*NYGLTFTQK | ND | 5.5E-07 | 1.0E-04 | |||
| SC*SGVEFSTSGHAYTDTGK | ND | 5.5E-07 | 1.0E-04 | |||
| Transcription | Q99714 | 3-Hydroxyacyl-CoA dehydrogenase type-2 | VDVAVNC*AGIAVASK | ND | ND | 4.5E-06 |
| VC*NFLASQVPFPSR | ND | ND | 4.5E-06 | |||
| P42704 | Leucine-rich pentatricopeptide-repeat motif-containing protein, mitochondrial | LIASYC*NVGDIEGASK | ND | ND | 7.2E-08 | |
| VFNDTC*R | ND | 9.9E-09 | 7.2E-08 | |||
| IHDVLC*K | ND | ND | 7.2E-08 | |||
| C*VANNQVETLEK | ND | ND | 7.2E-08 | |||
| DILIAC*R | 6.5E-08 | 9.9E-09 | ND | |||
| P49411 | Elongation factor Tu | KGDEC*ELLGHSK | ND | 4.8E-06 | 8.2E-04 | |
| Fusion | O95140 | Mitofusin-2 | EQQVYC*EEMR | ND | ND | 1.4E-03 |
| Apoptosis | O60313 | Dynamin-like 120-kDa protein | C*NEEHPAYLASDEITTVR | ND | ND | 1.7E-04 |
| Q07021 | Complement component 1 Q subcomponent-binding protein | ALVLDC*HYPEDEVGQEDEAESDIFSIR | ND | 2.0E-04 | 3.9E-03 | |
, N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio) propionamide (Biotin-HPDP)-labeled SNO-cysteines; CoA, coenzyme A; NADH, reduced nicotinamide adenine dinucleotide; ND, not detected; TCA, trichloroacetic acid.
Figure 6.

Mitochondrial nitroso-proteomes in HUVECs. Overlaps of the mitochondrial SNO-peptides (a) and SNO-proteins (b) identified from resting, and E2β- or GSNO-treated HUVECs. The mitochondrial E2β-responsive SNO-protein network (c) was generated by ingenuity pathway analysis. Proteins were represented as nodes; the biological relationship between 2 nodes is represented as a line. All lines are supported by at least one published reference. Solid lines represent a direct relationship, and dashed lines represent an indirect relationship. The red node color represents E2β stimulation. The shape of each node represents a functional class of proteins.
Pathway analysis
Ingenuity pathway analysis was used to explore the potential biological functions of the identified mitochondrial SNO-proteins in control and E2β-treated HUVECs. Function analysis suggested that the SNO-proteins are associated with various mitochondrial functions, including nucleic acid metabolism, small molecule biochemistry, cellular assembly and organization, molecular transport, and cell morphology. Signaling pathway analysis suggested that the mitochondrial SNO-proteins identified are linked to various fundamental cellular physiologies, including mitochondrial dysfunction, citrate cycle, oxidative phosphorylation, glyoxylate and dicarboxylate metabolism, and butanoate metabolism (Table 2). Of note, many mitochondrial SNO-proteins identified form a network for regulating cell functions (Figure 6c). According to the mitochondrial SNO-proteins identified in the resting and E2β-treated HUVECs, SNO apparently plays a critical role in the maintenance of normal mitochondrial physiology and is also important for the cell to respond to extracellular stimuli.
Table 2.
Ingenuity Pathway Analysis of HUVEC Mitochondrial nitroso-Proteome: Signaling Pathways and Molecular Functions
| Name | List of Molecules | n | Ratio | P Value |
|---|---|---|---|---|
| Mitochondrial dysfunction | ATP5C1, CPT1A, GPD2, HSD17B10, NDUFA10,NDUFAB1, NDUFS1, NDUFS6,OGDH, SDHA, SDHB, UQCRC1, UQCRC2 | 13 | 13/174 (0.075) | 1.72E-15 |
| Citrate cycle | ACO2, CS, IDH2, IDH3A, MDH2, OGDH, SDHA, SDHB | 8 | 8/57 (0.14) | 9.48E-14 |
| Oxidative phosphorylation | ATP5C1, ATP5E, ATP5H, ATP5O, NDUFA10, NDUFS1, NDUFAB1, NDUFS6, SDHA, SDHB, UQCRC1, UQCRC2 | 12 | 12/159 (0.075) | 1.49E-13 |
| Glyoxylate and dicarboxylate metabolism | ACO2, CS, MDH2, MTHFD, MTHFD1 liter | 5 | 5/112 (0.045) | 1.21E-08 |
| Butanoate metabolism | ACAT1, ALDH2, HADHA, HSD17B10, SDHA, SDHB | 6 | 6/128 (0.047) | 8.21E-08 |
| Nucleic acid metabolism | ALDH2, ATP5C1, ATP5E, ATP5H,ATP5O, CS, GPD2, HSPD1, IDH3A, MDH2, MFN2, NDUFS, OGDH, OPA1, SLC25A5,VDAC1 | 16 | ND | 1.26E-10– 6.43E-09 |
| Small molecule biochemistry | ALDH2, ATP5C1, ATP5E, ATP5H,ATP5O, GPD2, HSPD1, IDH3A, MDH2, MFN2, NDUFS1,OGDH, OPA1, SLC25A5, VDAC1 | 15 | ND | 1.26E-10– 6.43E-09 |
| Cellular assembly and organization | MFN2, OPA1, SLC25A5, SLC25A6, TIMM50, VDAC1 | 6 | ND | 2.00E-07– 4.14E-06 |
| Molecular transport | SLC25A4, SLC25A5, VDAC3 | 3 | ND | 2.00E-07 |
| Cell morphology | GPD2, HSPD1, MFN2, NDUFAB1, NDUFS1, OPA1, SLC25A6, VDAC1 | 8 | ND | 6.38E-07 |
ND, undetermined.
Discussion
In the endothelial mitochondrial nitroso-proteomes identified herein, we have found that 25 are E2β-responsive SNO-proteins, including 23 nitrosylated and 2 de-nitrosylated ones. Functional analysis classified these SNO-proteins as energy and redox metabolic enzymes responsible for carbohydrate, fatty acid oxidation, ATP synthesis, and oxidative phosphorylation. Six of them are involved in citric acid cycle, including citrate synthase, aconitase, isocitrate dehydrogenase 2, 2-oxoglutarate dehydrogenase, malate dehydrogenase, and succinate dehydrogenase iron-sulfur subunit. In addition, hexokinase-1, a glycolysis enzyme with docking sites to the outer membrane in mitochondria, is also identified as an E2β-responsive SNO-protein in the endothelial mitochondria. The identification of these mitochondrial SNO-proteins suggests that SNO is one of the mechanisms mediating the effects of estrogens in mitochondrial energy production. Notably, comparisons between the GSNO-and E2β-responsive endothelial nitroso-proteomes have identified common and different nitroso-proteins (Figure 6). These findings show that exogenous NO from donor such as GSNO exerts different cellular responses than endogenous NO from eNOS activation upon E2 stimulation via SNO-dependent mechanisms in endothelial cells.
The respiratory chain composed of 4 electron transport chain complexes (I–IV) and the ATP synthase (complex V) are the major enzymes of mitochondria for ATP synthesis (34). NO has long been known to inhibit complex I (35), presumptively protection against mitochondrial reactive oxygen species (ROS) production and calcium overloading upon ischemic preconditioning (36). Our current study has identified the ATP synthase subunit δ, ϵ, and γ, as well as 2 of the complex I reduced nicotinamide adenine dinucleotide dehydrogenase 1 α subcomplex subunit 10 and reduced nicotinamide adenine dinucleotide dehydrogenase iron-sulfur protein 6, as E2β-responsive SNO-targets. Thus, the estrogen-responsive endothelial mitochondrial SNO-proteins identified herein have further shown a critical role of SNO in energy generation upon estrogen stimulation.
Estrogens increase the efficiency of energy production and decrease ROS-mediated damage of mitochondria (20). ROS, as byproducts of the process of ATP synthesis, oxidize NO to form RNS that can further induce protein SNO (37). From this standpoint, SNO may provide an important mechanism for estrogens to protect oxidative stress-mediated damage of the mitochondria.
We also have shown that E2β stimulates SNO of 3 fatty acid oxidation related proteins, including trifunctional enzyme subunit α, electron transfer flavoprotein subunit α, and acetyl-CoA dehydrogenase in the mitochondria. The trifunctional enzymes catalyze the initial oxidation step and subsequent hydration of unsaturated carbons in the β-oxidation pathway (38); electron-transferring flavoprotein transfers electrons derived from the oxidation of fatty acids to ubiquinone (39); acetyl-CoA acetyltransferase plays a major role in ketone body metabolism (40). In addition, serine hydroxylmethyltransferase is also an estrogen-responsive mitochondrial SNO-protein the function of which is to support the flow of one-carbon units into the methyl cycle in embryonic mitochondria (41). Thus, these findings show that SNO targets numerous mitochondrial metabolic enzymes in endothelial cells. Consistently, 8 enzymes responsive for carbohydrate and fatty acid metabolism in rat heart mitochondria have been previously reported to be regulated by SNO (42).
Other E2β-responsive mitochondrial SNO-proteins identified in this study include 4 cellular transportation proteins (ie, mitochondrial import inner membrane translocase subunit TIM50 and voltage-dependent anion-selective channel protein 1, 2, and 3) and elongation factor-τ. These channels allow the diffusion of small hydrophilic molecules (43). Elongation factor-τ promotes the GTP-dependent binding of aminoacyl-tRNA to the A-site of ribosomes during protein biosynthesis (44). Therefore, identification of these SNO-targets underscores the significance of SNO in mediating mitochondrial protein synthesis and transport upon estrogen stimulation. Aspartate aminotransferase is another estrogen-response mitochondrial SNO-protein that catalyzes reversible transfer of α-amino group between aspartate and glutamate during protein synthesis (45). Physiological levels of NO are also known to promote cell survival and are antiapoptotic in endothelial cells (46). Accordingly, we have identified complement component 1 Q subcomponent-binding protein, a component of the complement system that is involved in the clearance of apoptotic cells and binds to surface blebs of apoptotic cells, followed by subsequent phagocytosis (47).
Although the major cellular source of NO is thought to be via eNOS localized at the plasma membrane (29), previous studies also have shown that mitochondria are able to synthesize NO via eNOS at the outer membrane of mitochondria (48, 49). We also have shown that E2β stimulates eNOS ser1177 phosphorylation in the mitochondria in a timely dependent manner similar to whole-cell extracts. By overexpressing organelle-targeting eNOS constructs (29, 30, 50), we have observed that mitochondrion-targeting eNOS significantly enhanced E2β stimulation of protein SNO in the mitochondria, consistent with a previous report (51). In addition, we have found that mitochondria are the major organelle of SNO-proteins in E2β-treated cells. These data thus suggest that local NO production via mitochondrial eNOS activation might be a major mechanism for E2β stimulation of mitochondrial protein SNO.
Levels of 2 SNO-proteins, ie, citrate synthase and ADP/ATP translocase 1, are decreased in E2β-treated cells, indicative of de-nitrosylation; however, levels of both are increased in GSNO-treated cells. These data suggest that endogenous NO generated by eNOS activation upon E2β stimulation and exogenous NO from donors like GSNO lead to opposite changes in some SNO-proteins, although E2β and GSNO stimulate SNO of many common mitochondrial proteins. De-nitrosylation of mitochondrial proteins upon E2β stimulation is unexpected, but not a surprise, as we have reported protein de-nitrosylation by E2β in intact endothelial cells (13, 14). However, the exact mechanisms by which E2β results in mitochondrial protein de-nitrosylation are not known. It is suspicious that some SNO-proteins can donate their NO groups to trans-nitrosylate other proteins (8). It is also possible that estrogens may activate one or more of the enzymes such as GSNO reductase (52, 53) and thioredoxin reductase (54), which in turn regulate dynamic protein SNO. However, these ideas need to be further investigated. There are many SNO-proteins that were stimulated only by GSNO, possibly due to the relative high concentration of GSNO used in comparison to the physiological levels of endogenous NO upon E2β stimulation (55). However, 3 E2β-stimulated mitochondrial SNO-proteins, including 2-oxoglutarate dehydrogenase, succinate dehydrogenase iron-sulfur subunit, and mitochondrial import inner membrane translocase subunit TIM50, are not detected in the GSNO-treated cells. Regardless, comparison of the nitroso-proteomes between the E2β- and GSNO-treated HUVECs provides insights regarding the mitochondrial SNO targets that are affected by endogenous NO produced by E2β stimulation and exogenous NO from the NO donor GSNO.
Interestingly, the specific SNO-sites of the constitutive SNO-proteins can be altered in response to stimulation. For example, there are 3 different SNO-peptides in ADP/ATP translocase 2 identified, implicating 3 SNO-sites (Cys129/160/257) in this constitutive SNO-protein. Among them, Cys129 (YFAGNLASGGAAGATSLC*FVYPLDFAR) and Cys-257 (KGTDIMY TGTLDC*WR) were identified as the SNO-sites in the untreated HUVECs. However, these SNO-peptides were not found in the E2β-treated HUVECs, but rather a different Cys160 (GLGDC*LVK) was identified. Moreover, GSNO stimulated SNO on Cys160 and results in de-nitrosylation on Cys129. These data suggest that SNO-regulated protein function may be even more complicated because this posttranslational modification may alter constitutive SNO-proteins via alterations on the SNO-sites in response to different stimuli.
There are a few limitations in our current BST-based proteomics approach for analyzing S-nitrosylation, although in theory this powerful method has the potential of extracting all SNO-proteins from a proteome with the identification of specific SNO-sites simultaneously. However, the accuracy of protein identification is expected to be reduced because only SNO-peptides were purified and sequenced by LC-MS/MS analysis; this leads to less peptide sequence(s) obtained for protein speculation. In addition, this method can only determine the relative changes of SNO-proteins among treatments. An unbiased quantization of changes in SNO-proteins would require input of internal standards or use stable isotope labeling of amino acids in cell culture (SILAC)-based quantitative proteomics approach (56).
Altogether, the present study has confirmed our recent studies (13, 14) showing that E2β stimulates protein SNO in endothelial cells and identified mitochondrion as a major cellular organelle in which protein SNO occurs in endothelial cells upon E2β stimulation. We have further analyzed, for the first time, the E2β- and GSNO-responsive mitochondrial nitroso-proteomes in endothelial cells. Function analysis of the mitochondrial nitroso-proteomes has revealed a network of SNO-proteins that are linked to diverse mitochondrial functions. We also have shown that forced overexpression of mitochondrion-targeting eNOS increases the E2β-stimulated SNO response. In keeping with our recent studies showing that estrogen stimulation of endothelial protein SNO is mediated by eNOS-derived NO via a specific ER-dependent mechanism (13, 14), the biological functions of the mitochondrial SNO-proteins revealed by informatics analysis suggest mitochondrial SNO as an important mechanism for mediating the endothelial effects of estrogens after biosynthesis of NO. Future studies of analyzing the functional sequelae of the estrogen-responsive mitochondrial SNO-targets are warranted and are expected to pave a new avenue for the understanding of the cardiovascular effects of estrogens.
Acknowledgments
We thank Dr David Fulton, Georgia Reagents University, for kindly donating the organelle targeting eNOS constructs.
This work was supported by National Institutes of Health grants RO1 HL70562 and R21 HL98746 (to D.B.C.). H.H.Z. was supported by an American Heart Association Postdoctoral Fellowship (AHA 11Post7610115).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BST
- biotin switch technique
- cGMP
- cyclic guanosine monophosphate
- eNOS
- endothelial nitric oxide synthase
- E2β
- estradiol-17β
- FP
- false positive
- GSNO
- S-nitrosoglutathione
- HUVECs
- human umbilical vein endothelial cells
- LC
- liquid chromatography
- Mito-eNOS
- mitochondrion-targeting eNOS
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- NO
- nitric oxide
- RNS
- reactive nitrogen species
- ROS
- reactive oxygen species
- SNO
- S-nitrosylation
- WT
- wild type.
References
- 1. Praticò D. Antioxidants and endothelium protection. Atherosclerosis. 2005;181:215–224 [DOI] [PubMed] [Google Scholar]
- 2. Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O, Sica V, Ignarro LJ. Nitric oxide and atherosclerosis: an update. Nitric Oxide. 2006;15:265–279 [DOI] [PubMed] [Google Scholar]
- 3. Taylor EL, Megson IL, Haslett C, Rossi AG. Nitric oxide: a key regulator of myeloid inflammatory cell apoptosis. Cell Death Differ. 2003;10:418–430 [DOI] [PubMed] [Google Scholar]
- 4. Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wanstall JC, Homer KL, Doggrell SA. Evidence for, and importance of, cGMP-independent mechanisms with NO and NO donors on blood vessels and platelets. Curr Vasc Pharmacol. 2005;3:41–53 [DOI] [PubMed] [Google Scholar]
- 6. Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med. 2001;30:463–488 [DOI] [PubMed] [Google Scholar]
- 7. Lane P, Hao G, Gross SS. S-Nitrosylation is emerging as a specific and fundamental posttranslational protein modification: head-to-head comparison with O-phosphorylation. Sci STKE. 2001;(86):re1. [DOI] [PubMed] [Google Scholar]
- 8. Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166 [DOI] [PubMed] [Google Scholar]
- 9. Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology. 2004;145:113–125 [DOI] [PubMed] [Google Scholar]
- 10. Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenfeld CR, Chen C, Roy T, Liu X. Estrogen selectively up-regulates eNOS and nNOS in reproductive arteries by transcriptional mechanisms. J Soc Gynecol Investig. 2003;10:205–215 [DOI] [PubMed] [Google Scholar]
- 12. MacRitchie AN, Jun SS, Chen Z, et al. Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res. 1997;81:355–362 [DOI] [PubMed] [Google Scholar]
- 13. Zhang HH, Feng L, Wang W, Magness RR, Chen DB. Estrogen-responsive nitroso-proteome in uterine artery endothelial cells: role of endothelial nitric oxide synthase and estrogen receptor-β. J Cell Physiol. 2012;227:146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang HH, Feng L, Livnat I, et al. Estradiol-17β stimulates specific receptor and endogenous nitric oxide-dependent dynamic endothelial protein S-nitrosylation: analysis of endothelial nitrosyl-proteome. Endocrinology. 2010;151:3874–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun J, Aponte AM, Kohr MJ, Tong G, Steenbergen C, Murphy E. Essential role of nitric oxide in acute ischemic preconditioning: S-nitros(yl)ation versus sGC/cGMP/PKG signaling? Free Radic Biol Med. 2013;54:105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kohr MJ, Sun J, Aponte A, et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296 [DOI] [PubMed] [Google Scholar]
- 19. Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–965 [DOI] [PubMed] [Google Scholar]
- 21. Duckles SP, Krause DN, Stirone C, Procaccio V. Estrogen and mitochondria: a new paradigm for vascular protection? Mol Interv. 2006;6:26–35 [DOI] [PubMed] [Google Scholar]
- 22. Viña J, Borrás C, Gambini J, Sastre J, Pallardó FV. Why females live longer than males? Importance of the upregulation of longevity-associated genes by oestrogenic compounds. FEBS Lett. 2005;579:2541–2545 [DOI] [PubMed] [Google Scholar]
- 23. Siow RC, Li FY, Rowlands DJ, de Winter P, Mann GE. Cardiovascular targets for estrogens and phytoestrogens: transcriptional regulation of nitric oxide synthase and antioxidant defense genes. Free Radic Biol Med. 2007;42:909–925 [DOI] [PubMed] [Google Scholar]
- 24. Nazarewicz RR, Zenebe WJ, Parihar A, et al. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007;67:1282–1290 [DOI] [PubMed] [Google Scholar]
- 25. Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163 [DOI] [PubMed] [Google Scholar]
- 26. Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chouchani ET, Hurd TR, Nadtochiy SM, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430:49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang Y, Loscalzo J. S-nitrosoprotein formation and localization in endothelial cells. Proc Natl Acad Sci USA. 2005;102:117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fulton D, Babbitt R, Zoellner S, et al. Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt- versus calcium-dependent mechanisms for nitric oxide release. J Biol Chem. 2004;279:30349–30357 [DOI] [PubMed] [Google Scholar]
- 30. Jagnandan D, Sessa WC, Fulton D. Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol. 2005;289:C1024–C1033 [DOI] [PubMed] [Google Scholar]
- 31. Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103:1012–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaake RM, Milenković T, Przulj N, Kaiser P, Huang L. Characterization of cell cycle specific protein interaction networks of the yeast 26S proteasome complex by the QTAX strategy. J Proteome Res. 2010;9:2016–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hornig-Do HT, Günther G, Bust M, Lehnartz P, Bosio A, Wiesner RJ. Isolation of functional pure mitochondria by superparamagnetic microbeads. Anal Biochem. 2009;389:1–5 [DOI] [PubMed] [Google Scholar]
- 34. Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54:1015–1069 [DOI] [PubMed] [Google Scholar]
- 35. Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139 [DOI] [PubMed] [Google Scholar]
- 36. Prime TA, Blaikie FH, Evans C, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2009;106:10764–10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252 [DOI] [PubMed] [Google Scholar]
- 38. Bartlett K, Eaton S. Mitochondrial beta-oxidation. Eur J Biochem. 2004;271:462–469 [DOI] [PubMed] [Google Scholar]
- 39. Watmough NJ, Frerman FE. The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim Biophys Acta. 2010;1797:1910–1916 [DOI] [PubMed] [Google Scholar]
- 40. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF., Jr Ketone bodies, potential therapeutic uses. IUBMB Life. 2001;51:241–247 [DOI] [PubMed] [Google Scholar]
- 41. Pike ST, Rajendra R, Artzt K, Appling DR. Mitochondrial C1-tetrahydrofolate synthase (MTHFD1L) supports the flow of mitochondrial one-carbon units into the methyl cycle in embryos. J Biol Chem. 2010;285:4612–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zalman LS, Nikaido H, Kagawa Y. Mitochondrial outer membrane contains a protein producing nonspecific diffusion channels. J Biol Chem. 1980;255:1771–1774 [PubMed] [Google Scholar]
- 44. Pape T, Wintermeyer W, Rodnina MV. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. EMBO J. 1998;17:7490–7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kochhar S, Christen P. Mechanism of racemization of amino acids by aspartate aminotransferase. Eur J Biochem. 1992;203:563–569 [DOI] [PubMed] [Google Scholar]
- 46. Shen YH, Wang XL, Wilcken DE. Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett. 1998;433:125–131 [DOI] [PubMed] [Google Scholar]
- 47. Taylor PR, Carugati A, Fadok VA, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–195 [DOI] [PubMed] [Google Scholar]
- 49. Giulivi C. Functional implications of nitric oxide produced by mitochondria in mitochondrial metabolism. Biochem J. 1998;332 (Pt 3):673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. García-Cardeña G, OH P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci U S A. 1996;93:6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iwakiri Y, Satoh A, Chatterjee S, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci USA. 2006;103:19777–19782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lima B, Lam GK, Xie L, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci USA. 2009;106:6297–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494 [DOI] [PubMed] [Google Scholar]
- 54. Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem. 2004;279:25891–25897 [DOI] [PubMed] [Google Scholar]
- 56. Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958 [DOI] [PubMed] [Google Scholar]