

Abstract
We examined 1) whether global cerebral blood flow (CBF) would increase across a 6-h bout of normobaric poikilocapnic hypoxia and be mediated by a larger increase in blood flow in the vertebral artery (VA) than in the internal carotid artery (ICA); and 2) whether additional increases in global CBF would be evident following an α1-adrenergic blockade via further dilation of the ICA and VA. In 11 young normotensive individuals, ultrasound measures of ICA and VA flow were obtained in normoxia (baseline) and following 60, 210, and 330 min of hypoxia (FiO2 = 0.11). Ninety minutes prior to final assessment, participants received an α1-adrenoreceptor blocker (prazosin, 1 mg/20 kg body mass) or placebo. Compared with baseline, following 60, 220, and 330 min of hypoxia, global CBF [(ICAFlow + VAFlow) ∗ 2] increased by 160 ± 52 ml/min (+28%; P = 0.05), 134 ± 23 ml/min (+23%; P = 0.02), and 113 ± 51 (+19%; P = 0.27), respectively. Compared with baseline, ICAFlow increased by 23% following 60 min of hypoxia (P = 0.06), after which it progressively declined. The percentage increase in VA flow was consistently larger than ICA flow during hypoxia by ∼20% (P = 0.002). Compared with baseline, ICA and VA diameters increased during hypoxia by ∼9% and ∼12%, respectively (P ≤ 0.05), and were correlated with reductions in SaO2. Flow and diameters were unaltered following α1 blockade (P ≥ 0.10). In conclusion, elevations in global CBF during acute hypoxia are partly mediated via greater increases in VA flow compared with ICA flow; this regional difference was unaltered following α1 blockade, indicating that a heightened sympathetic nerve activity with hypoxia does not constrain further dilation of larger extracranial blood vessels.
Keywords: hypoxia, vasculature, α1-adrenoreceptor
the human brain is the most oxygen-dependent organ in the body. It has a limited capacity for substrate storage and a high metabolic rate, making the precise regulation of cerebral blood flow (CBF) critical for maintenance of constant nutrient and oxygen supply. The ability to preserve normal cerebral function in a hypoxic environment is dependent upon maintaining a normal oxygen delivery to the brain in the face of hypoxemia (48). To achieve this, compensatory elevations in CBF occur to offset the hypoxic-induced reductions in oxygen content. The traditional view regarding elevations in CBF, in response to a hypoxic stimuli, is that the decline in cerebral vascular resistance is mediated via dilation of the pial vessels (14, 28); however, recent findings in humans have revealed that hypoxic-induced dilation in other (larger) intracranial vessels [e.g., the middle cerebral artery (MCA) (46, 48); and extracranial blood vessels including the vertebral artery (VA), internal carotid artery (ICA), and common carotid artery (CCA) (33, 40, 46)]. Such findings are broadly consistent with animal studies that report that the larger cerebral arteries (including ICA and VA) are also sensitive to changes in blood gases (20, 24) and serve as a first-line defense in maintaining brain perfusion (20). Moreover, because of large-vessel dilation, these findings likely highlight the reason why blood velocity in the MCA [as assessed with transcranial Doppler (TCD) ultrasound] is either reduced (36) or unchanged (3, 5, 19, 41) during short-term exposure to poikilocapnic hypoxia. Under these conditions of hypoxia, velocity in the MCA might not reflect flow, and hence the assumption of constant diameter using TCD may not be valid. Although this does not necessarily invalidate all TCD measurements in hypoxia because, for example, once hypoxic dilation has occurred, the diameter of the MCA might remain constant and thus allow for certain TCD-based investigations during continued hypoxia. However, data to support or refute this possibility are needed.
During isocapnic hypoxia, blood flow to the brainstem increases more than it does to the anterior regions, as assessed by blood flow through the VA and ICA, respectively (33, 45). Consistently, positron emission tomography imaging data collected during isocapnic hypoxia reveal that cortical blood flow is less responsive to hypoxia than phylogenetically older areas of the brain (7). At least during slow (8–9 day) ascent to 5,050 m, we recently (46) reported no regional difference in ICA or VA blood flow. Conversely, upon rapid ascent to 5,300 m, Subudhi et al. (40) reported a greater cerebral oxygen delivery in the VA compared with the ICA. It is unknown whether such regional differences in blood flow are present over an acute bout of poikilocapnic hypoxia [e.g., the first 6 h, when initial ventilatory acclimatization is known to occur (1)].
Although experimental studies are minimal, several factors are believed to mediate cerebral vasodilation in hypoxia, including nitric oxide (6, 43), prostaglandins (18, 23), adenosine (13, 30), and sympathetic nerve activity (SNA) (2, 15). Hypoxia is a potent activator of the sympathetic nervous system (22, 37, 50); however, although the cerebral circulation is richly innervated with sympathetic nerve fibers (16, 32), the role of SNA on CBF regulation is likely insignificant in humans during normoxic and normotensive rest [reviewed in (47)]. Nevertheless, because the ICA and VA are well innervated with paravascular nerve bundles (9–11), it is plausible that marked elevations in SNA with hypoxia may constrain dilation of large cerebral vessels (2). For example, large increases in muscle SNA can decrease the diameter of conduit arteries even in the presence of elevated blood pressure (12, 34, 35), suggesting that SNA acts to regulate conduit artery tone. Whether this influence can be extrapolated to hypoxic-induced elevations in SNA to affecting large cerebral vessels has not been examined.
The aim of this study was to examine 1) changes in diameter, velocity, and blood flow in the CCA, ICA, and VA in response to an acute (6-h) exposure to normobaric poikilocapnic hypoxia (FiO2 = 0.11); and 2) the role of SNA in mediating changes in extracranial blood flow. We hypothesized 1) that global CBF would increase over time in acute hypoxia and be mediated via larger relative changes in blood flow in the VA compared with that in the ICA; and 2) that additional increases in global CBF will be evident following an α1-adrenergic blockade, mediated by further dilation of the ICA and VA.
METHODS
Participants.
Eleven healthy normotensive volunteers (7 men, 4 women; mean ± SD: age, 24 ± 3 yr; body mass, 75 ± 10 kg; height, 174 ± 6 cm; body mass index, 25 ± 3 kg/m2) participated in this randomized placebo-controlled experiment. This study was approved by the Human Ethics Committee of the University of British Columbia and conformed to the standards set by the Declaration of Helsinki. All volunteers provided written informed consent. Participants were nonsmokers, had no previous history of cardiovascular, cerebrovascular, or respiratory diseases, and were not taking any cardiovascular medications. All experimental testing took place at the University of British Columbia (altitude 344 m).
Study design and measures.
Each experimental session commenced between 8:00 and 9:00 A.M. Experimental testing followed a minimum of 12 h abstinence from alcohol, caffeine, and strenuous exercise. Due to the length of the experimental session, participants were asked to eat a small breakfast ∼2 hr before arrival to the laboratory. Subsequent to each measurement bout in the normobaric chamber, participants consumed a small snack (i.e., granola bar, ∼150 kcal) and ∼250 ml of water to maintain hunger comfort and hydration. The experimental sessions were separated by ≥7 days.
Measures (see below) were made in normoxia and during normobaric hypoxia (FiO2 = 0.11) at 60, 210, and 330 min, and then 60 min after returning to normobaric normoxia (Fig. 1). Participants ingested an α1-adrenoreceptor blocker (prazosin; 1 mg/20 kg body mass) or an identical placebo capsule 90 min before the last assessment in hypoxia. Prazosin has been shown to induce systemic peripheral arterial-dilation and venodilation via the removal of sympathetic nerve activity (4, 26). This acceptable clinical dose of prazosin in young normotensive individuals has been previously shown to have a functional block of ∼80%, and the 90- to 180-min postingestion period was appropriate for the peak activity of prazosin (27, 32). Additionally, the functional block of prazosin has been determined at high altitude (4,300 m) (29); compared with sea level, no decrement in the degree of blockade was evident with altitude exposure.
Fig. 1.

Measures were repeated in normoxia and following 60, 210, and 330 min of normobaric hypoxia (FiO2 = 0.11). Following 240 min of hypoxia [90 min prior to the last assessment in hypoxia (330 min)], participants received an oral dose of the α1-adrenoreceptor blocker prazosin (1 mg/20 kg body mass) or placebo capsule. All measures were repeated in normoxia 60 min following relief of the hypoxic exposure.
Following 20 min of supine rest, measurements of arterial oxygen saturation (SaO2) (Pulse Oximeter MD300K1; Vacumed, Ventura, CA), heart rate (HR; three-lead electrocardiogram, ML132; ADInstruments, Colorado Springs CO) and blood pressure (BP; manual auscultation) were obtained. In addition, continuous diameter, velocity, and blood flow recordings in the right CCA, right ICA, and right VA were obtained using a 10-MHz multifrequency linear array probe attached to a high-resolution ultrasound machine (Terason 3000; Teratech, Burlington, MA). One experienced sonographer conducted all measures, and measures were performed in the following order: CCA, ICA, VA.
The CCA and ICA were measured at least 1.5–2 cm from the carotid bifurcation while ensuring there was no evidence of turbulent or retrograde flow. The right VA was measured between the transverse process of C4 and the subclavian artery, but always at the same location within each subject. Average diameter and blood velocity recordings were made for ∼30 s (see below), and care was taken to ensure probe position was stable so that the angle of insonation did not vary from 60°. The sample volume was positioned in the center of the vessel and adjusted to cover the width of the vessel diameter. Measurement settings for each extracranial artery within each individual were standardized for all measurement sets.
All images were directly stored as an AVI file for off-line analysis. Custom-designed edge detection and wall-tracking software, which is largely independent of investigator bias, was utilized for the analysis of CCA, ICA, and VA diameter and velocity blood flow at 30 Hz [(8, 49) see Fig. 2]. This software provides continuous and simultaneous diameter and velocity measurements. Reproducibility of diameter measurements using this semiautomated software is significantly better than manual methods, significantly reduces observer error and bias, and possesses an intraobserver CV of 6.7% (49). Mean blood flow was determined as half the time-averaged maximum velocity (17) multiplied by the cross-sectional lumen area. This method has been adopted previously and is used instead of the intensity weighted mean because the latter is more susceptible to noise and other distorting influences (45, 46). Global CBF was estimated assuming symmetrical bilateral flow in the ICA and VA (33, 46) as global CBF = (ICAFlow + VAFlow) ∗ 2.
Fig. 2.

Effect of α1 blockade on common carotid artery (CCA), internal carotid artery (ICA; n = 9), vertebral artery (VA; n = 10) diameter, blood flow velocity, and blood flow. Measures were taken after 330 min of hypoxia.
Statistical analysis.
All data were analyzed using SPSS (version 21; Surrey, U.K.) and expressed as mean ± SD. Statistical significance was defined as P ≤ 0.05. To examine the interaction between any time course change and experimental condition (placebo vs. α1 blockade) a two-way repeated measures ANOVA was used; to further explore any significant main effects or interaction effects, a one-way repeated measures ANOVA and paired t-tests were used, respectively. We were unable to obtain sufficient ICA images for 2 of the 11 participants, and they are therefore not included in the ICA variable data analysis. Due to technical issues (i.e., loss of one recording), we were unable to gain a complete VA set for one participant (posthypoxic exposure); therefore, n = 10 was used for ANOVA analysis.
RESULTS
Effect of α1-adrenoreceptor blockade (following 330 min of hypoxia).
A significant interaction between the time course change and condition was evident for HR. For example, compared with the placebo trial, HR was 5 ± 1 beats/min higher in the α1 blockade trial (Table 1; P < 0.001). No other interaction effects were evident in any other variable (Table 1), including CCA, ICA, VA diameter, velocity, or flow (Fig. 2). Therefore, all the data from both trials were pooled for combined statistical analysis.
Table 1.
Time course effect of normobaric hypoxia on cardiovascular variables and affect of α1 blockade
| Condition | Normoxia | Hypoxia | Normoxia | |||
|---|---|---|---|---|---|---|
| Time, min | 0 | 60 | 220 | 330 | 60 | |
| SBP, mmHg | Placebo | 114 ± 11 | 121 ± 26 | 109 ± 9 | 109 ± 8 | 112 ± 13 |
| α1 Blockade | 109 ± 8 | 111 ± 8 | 116 ± 17 | 108 ± 12 | 113 ± 7 | |
| DBP, mmHg | Placebo | 76 ± 10 | 71 ± 10 | 70 ± 10 | 74 ± 9 | 74 ± 9 |
| α1 Blockade | 72 ± 8 | 71 ± 10 | 72 ± 12 | 65 ± 12 | 70 ± 10 | |
| MAP, mmHg | Placebo | 90 ± 9 | 88 ± 7 | 83 ± 8 | 86 ± 7 | 87 ± 9 |
| α1 Blockade | 83 ± 10‡ | 85 ± 9 | 79 ± 14 | 80 ± 8 | 84 ± 8 | |
| HR, beats/min | Placebo | 59 ± 10 | 68 ± 15 | 72 ± 15 | 72 ± 15*† | 60 ± 14 |
| α1 Blockade | 61 ± 9 | 66 ± 9 | 66 ± 8 | 77 ± 13‡ | 65 ± 10 | |
| SaO2, % | Placebo | 96 ± 2 | 72 ± 5*† | 76 ± 7*† | 80 ± 4*† | 97 ± 1 |
| α1 Blockade | 96 ± 1 | 79 ± 7 | 81 ± 6 | 79 ± 7 | 97 ± 1 |
Values expressed as mean ± SD.
DBP, diastolic blood pressure; HR, heart rate; MAP, mean arterial blood pressure; SaO2, arterial oxygen saturation; SBP, systolic blood pressure.
Significantly different from normoxia 0 min (baseline) in the placebo trial only.
Significantly different from normoxia 60 (recovery) in the placebo trial only (P ≤ 0.001).
Measurement time point in the α1 blockade trial significantly different from placebo trial (P ≤ 0.001).
Time-course changes with hypoxia.
During exposure to hypoxia at 60, 220, and 330 min, estimates of global CBF [(ICAFlow + VAFlow) ∗ 2] were increased by 160 ± 52 (+28%; P = 0.05), 134 ± 23 (+23%; P = 0.02), and 113 ± 51 ml/min (+19%; P = 0.27), respectively (Fig. 3). Following hypoxic exposure (60 min normoxia), global CBF was comparable to normoxic baseline (Fig. 3).
Fig. 3.

Changes in global cerebral blood flow (CBF; n = 9), VA blood flow and ICA blood flow during exposure to normobaric hypoxia. *Significantly different from normoxia baseline (0 min; P ≤ 0.05). †Significant difference from normoxic recovery (60 min; P = 0.03).
Compared with normoxic baseline at 60, 220, and 330 min of exposure to hypoxia, ICA blood flow increased by 108 ± 23 (+23%; P = 0.06), 82 ± −1 (+18%; P = 0.23), and 45 ± 306 ml/min (+10%; P = 1.0), respectively; VA blood flow increased by 43 ± 46 (+40%; P = 0.05), 35 ± 41 (+33%; P = 0.03), and 32 ± 38 ml/min (+30%; P = 0.04; Fig. 3). Although ICA blood flow increased with hypoxic exposure, the absolute and percent changes significantly decreased over the period of exposure (P = 0.05; Fig. 3). In contrast, the percent increase in VA blood flow remained elevated with hypoxic exposure (Fig. 4). Two-way ANOVA revealed that the percentage increase in VA blood flow was significantly larger than that of ICA blood flow at each measurement point by ∼20% (P = 0.002; Fig. 4). Following hypoxic exposure, blood flow in both the ICA and VA were comparable to normoxic baseline values (Fig. 3).
Fig. 4.

Percent change in VA and ICA (n =9) blood flow during exposure to normobaric hypoxia. *ICA flow was significantly reduced at each measurement point (P = 0.05). †VA significantly greater than ICA (P = 0.002).
Significant diameter changes in all three blood vessels were evident in response to hypoxic exposure (P < 0.001; Fig. 6). Compared with normoxic baseline, following 60, 220, and 330 min of exposure to hypoxia, CCA diameter increased by 0.05 ± 0.02 (+7%; P = 0.002), 0.05 ± 0.03 (+7%; P = 0.002), and 0.05 ± 0.03 cm (+7%; P = 0.002); ICA diameter increased by 0.05 ± 0.02 (+10%; P = 0.001), 0.05 ± 0.02 (+9%; P = 0.001), and 0.04 ± 0.03 cm (+8%; P = 0.05); and VA diameter increased by 0.04 ± 0.02 (+11%; P < 0.001), 0.05 ± 0.02 (+13%; P < 0.001), and 0.04 ± 0.02 cm (+12%; P = 0.001), respectively (Fig. 5). Although velocity in the VA increased by ∼10% following exposure to hypoxia (P = 0.05), it was unaltered in the CCA and ICA compared with normoxic baseline (P ≥ 0.08; Fig. 5). Following hypoxic exposure, CCA, ICA, and VA diameters were comparable to those at normoxic baseline (Fig. 5).
Fig. 6.

Correlation between percentage change from baseline in global CBF, extracranial arteries (CCA, ICA, and VA), and diameter with the absolute change in arterial oxygen saturation (SaO2) during normobaric hypoxia exposure. *Significance via Pearson's correlation; P ≤ 0.01. Global CBF and ICA (n = 9).
Fig. 5.
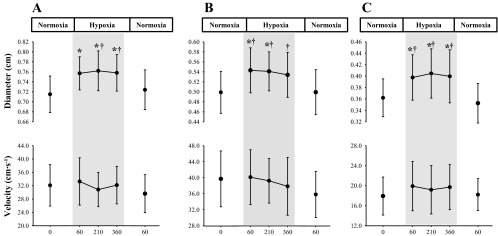
Absolute change in diameter and blood flow velocity in the CCA (A), ICA (B) (n = 9), and VA (C) (n = 10). Diameter changes significantly larger during hypoxia compared with normoxic baseline (*0 min; P < 0.01) and recovery (†60 min; P ≤ 0.04).
During hypoxia, an inverse relationship was evident between the change in SaO2 with the changes in global CBF (r = −0.72; P < 0.001), ICA blood flow (r = −0.62; P = 0.001), and VA blood flow (r = −0.73; P < 0.001; Fig. 6). Likewise, during hypoxia, a significant inverse relationship was evident between SaO2 with the relative changes (percentage change from baseline) in arterial diameter in the CCA (r = −0.44; P = 0.01), ICA (r = −0.48; P = 0.01), and VA (r = −0.51; P = 0.002; Fig. 6).
DISCUSSION
This study is the first to examine the short-term changes in extracranial vessel diameter, velocity, and flow in response to an acute (6-h) exposure to normobaric poikilocapnic hypoxia, and examine the role of SNA in mediating these responses. The main new findings were as follows: 1) elevations in global CBF over 6 h of hypoxia are determined more by an increase in arterial diameter rather than an increase in blood velocity; 2) elevations in global CBF over time are largely mediated via greater increases in blood flow in the VA compared with flow in the ICA; 3) changes in global CBF and regional blood flow in hypoxia are not altered via α1 blockade, indicating that heightened SNA with hypoxia does not constrain further dilation of the larger extracranial blood vessels; and 4) 60 min following the hypoxic stimulus, global CBF along with CCA, ICA, and VA blood flow, and arterial diameters were comparable to those at normoxic baseline.
Global increase in CBF.
In support of our hypothesis, global CBF increased by ∼28% within 60 min and remained elevated throughout the hypoxic stimulus. This finding, however, contrasts with other reports (over a comparable time frame) that have employed TCD and showed MCA velocity (as a surrogate of flow) is either reduced (36) or unchanged (3, 5, 19, 41) during short-term exposure to poikilocapnic hypoxia. The discrepancies between findings are likely explained via dilation of the MCA (46, 48). In support of the current study, Kety and Schmidt (28) [using an inert tracer (nitrous oxide) combined with arterial and jugular venous sampling] first reported a 35% increase in CBF following ∼20 min of breathing a gas mixture containing 10% oxygen. A number of experiments conducted at high altitude report an approximately 24–70% increase in global CBF following ∼2 hr to 2–3 days after exposure to high altitude (>3,600 m) (39, 40, 46).
In comparison with the current study, Subudhi et al. (40) reported a 70% increase in global CBF within 2–4 h following rapid ascent to 5,260 m while on supplemented oxygen; this increase is substantially larger than the increase observed in the current study despite similar levels of SaO2 (78% in this study vs. 76% in the Subudhi et al. study). Currently the effect of normobaria vs. hypobaria upon the hypoxic-induced increases in CBF are unknown, but this may have influenced the between-study differences in the increase in global CBF to ∼11% oxygen. Collectively, it seems likely that the time period of exposure to a level of hypoxia, the ascent profile to high altitude, the influence of barometric pressure, and methodological technique to measure CBF all potentially influence the increase in CBF. Further research is needed to unravel the influence of each of these factor on the increase in CBF with hypoxia.
Regional contribution to global flow with acute hypoxia.
In the current study, the increase in global CBF was influenced by an increase in blood flow in both the ICA (∼18%) and VA (∼37%); however, the percentage increase in VA blood flow was consistently higher than in the ICA by ∼20% (Fig. 4). These findings are consistent with previous reports during acute (10–15 min) isocapnic (45) and poikilocapnic hypoxia (33). Following the initial increase after 60 min of poikilocapnic hypoxia, we found that flow in the ICA declined ∼6% every ∼2 hr, and therefore, global CBF was maintained via consummate elevations in VA flow. These findings confirm our hypothesis that the overall increase in VA blood flow contributes more to the increase in global CBF than the ICA, and that this regional difference is present within 60 min of hypoxia and is maintained over at least 6 h of exposure. This favored distribution of CBF is believed to be advantageous for protecting regions of the brain with important and necessary homeostatic function during hypoxia, such as the respiratory and cardiovascular control centers (e.g., cerebellum, hypothalamus, thalamus, and brainstem). Although the mechanisms mediating this response are not known, it is noteworthy that the highest concentrations of the adenosine A2 receptor (which are believed to be involved in the regulation of vascular tone, albeit in rats) are located in the basal ganglia (25) and hence may explain the preferential distribution of flow to these regions.
Consistent dilation of extracranial blood vessels.
In contrast to our original hypothesis, for the first time we observed simultaneous increases in arterial diameter in the CCA, ICA, and VA in response to hypoxia. Furthermore, the ∼9% and 12% increase in ICA and VA diameters appeared to mediate the hypoxic increase in ICA and VA blood flow rather than an increase in velocity. Currently, there are inconsistencies between studies on what determines the increase in blood flow in the ICA and VA (i.e., increase in blood velocity vs. arterial dilation, or both). For example, with acute (15 min) isocapnic hypoxia (SaO2 ∼70%) dilation occurred selectively in the VA (45); however, no ICA or VA dilation was evident in another study during more modest levels of isocapnic hypoxia (SaO2 ∼93%) or poikilocapnic hypoxia (SaO2 ∼90%) (33). Studies at high altitude have reported an increase in blood velocity in the ICA and VA during exposure to 5,050 m following a 7- to 9-day ascent (46), or an increase in blood flow velocity in the ICA and dilation in the VA (40) during acute exposure to 5,260 m. Differences in experimental design and imaging methods likely explain these between-study differences. Nevertheless, under the hypoxic conditions of the current study, consistent arterial dilation in hypoxia challenges the concept that cerebrovascular resistance is solely modulated at the level of the pial vessels (14, 28) and supports the notion that arterial dilation occurs throughout the entire cerebrovascular arterial tress. These findings support the concept from animal studies that the larger extracranial vessels serve as a first-line defense in maintaining brain perfusion (20).
A significant inverse relationship between SaO2 and global CBF and arterial diameter was evident (Fig. 6). On the basis of the calculation of the coefficient of determination, the change in SaO2 accounted for 52%, 39%, and 53% of the increase in global CBF, and flow in the ICA and VA, respectively; the change in SaO2 also accounted for 23% and 26% of the increase in ICA and VA diameters, respectively, with hypoxia. These findings further extend the work of others (33, 40, 45) of a greater vasoreactivity to hypoxemia in the VA than in the ICA.
Mechanisms of hypoxic-induced vasodilation in large cerebral blood vessels.
In addition to the tone of the pial vessels (14, 28), the findings of the current study and others show that larger arteries in the neck (ICA, VA) and brain (MCA) also appear sensitive to hypoxemia (40, 45, 46, 48) and therefore contribute to an acute increase in global CBF with hypoxia. A myriad of neural, chemical, metabolic, and physical factors occur following hypoxic exposure and contribute to arterial dilation in acute hypoxia (2), yet little information is available on how these factors influence the dilation of the ICA and VA. Potential mechanism(s) include the following: 1) circulating factors such as adenosine, which is released during hypoxemia (30); in addition, intravenous infusion of aminophylline (an adenosine agonist) during isocapnic hypoxia (SaO2 of ∼80%) has previously been shown to reduce the hypoxic increase in CBF via vasodilation (13, 31); 2) endothelium-derived vasoactive factor such as nitric oxide has been shown to be important in regulating CBF during hypoxia (43), and use of phase-contrast magnetic resonance imaging has shown that the administration of the nitric oxide inhibitor N(G)-monomethyl-l-arginine prevented the hypoxia increase in CBF (43); 3) mechanical factors such as an increase in flow toward the brain, which ultimately results in an increase in vasculature wall shear stress, and subsequent release of vasodilatory compounds such as nitric oxide (21, 42). It is therefore possible that arterial dilation in the ICA and VA are influenced by a direct effect of circulating factors induced by hypoxia or indirectly by a shear stress–mediated increase in flow in the pial vessels, or a combination of both.
Effect of α1-adrenoreceptor blockade on hypoxic-induced increases in CBF.
To our knowledge this is the first study to examine the role of SNA on hypoxic-induced increase in CBF via large cerebral arteries. Hypoxia is a potent activator of the sympathetic nervous system (22, 37, 50). Because the ICA and VA are innervated with paravascular nerve bundles (9–11), we reasoned that SNA might constrain vasodilation and hence CBF. The finding of the current study rejects our hypothesis because the administration of the α1 blockade did not result in a further increase in global CBF; diameter, and blood flow velocity in the CCA, ICA and VA were unaltered. Because the degree of hypoxia in the current study stimulated arterial dilation, it is possible that the effect of hypoxemia out-competed any effect of elevations in SNA. Additionally, although the neurogenic control of the cerebral vasculature acts to buffer surges in perfusion pressure, it does not greatly regulate CBF during normotensive conditions [reviewed in (47)]. Therefore, because BP was relatively stable during hypoxic exposure, it would unlikely result in much engagement of cerebral SNA.
Recovery of global CBF following hypoxia.
In the current study, global CBF and flow in the ICA and VA (mediated by reductions in diameter) returned to their normoxic baseline levels following 60 min of normoxic recovery from the hypoxia challenge. A recent report has shown that CBF (as assessed by arterial spin labeling magnetic resonance imaging) is elevated (by ∼12%) above normoxic baseline 6–8 hr following the return to sea level from high altitude (3,800–4,350 m) (44). Notably, the inconsistency between reported studies likely is a reflection of the greater duration of hypoxic exposure in the altitude studies. Nevertheless, the current study suggests that the vascular responses within the large extracranial vessels to short-term hypoxia are fully reversible within 60 min of normoxic recovery.
Methodological limitations.
The current study was performed in a laboratory setting within a hypoxic chamber; as such, the findings of the current study must be interpreted with caution when comparing them with other research studies performed using a hypobaric chamber or field studies at high altitude. It is important to note that all ultrasound measures were performed on the right side of participants. Although there is no difference between blood flow in the right and left ICA, the blood flow in the right VA has been reported to be ∼20% lower compared with the left VA (38). We are unaware of any data that indicate whether there are bilateral differences in VA blood flow to hypoxia. Nevertheless, because our measurements were performed on the right side and the related significant findings, we feel this does not detract from our main study conclusions. If anything, we have likely underestimated the contribution of VA flow to global CBF. It is important to highlight that the measure of global CBF in the current study was estimated on the basis of blood flow in the ICA and VA as determined via ultrasound, and is therefore not an absolute direct measure of global CBF. It is well established that carbon dioxide is an important regulator of CBF and has a vital role in determining the increase in CBF observed with initial exposure to hypoxia (45).We did not measure or control changes in carbon dioxide during the hypoxic exposure, and therefore, the role of hypocapnia on the vascular response in the CCA, ICA, and VA was not quantified. Nevertheless, our aim in this study was the integrative physiological responses to short-term hypoxia over a period in which early ventilatory acclimation occurs (1) (as reflected in the progressive elevation in SaO2 over the 6 h). For example, although not significant, SaO2 slightly increased throughout the hypoxic exposure; these changes, likely coupled with reductions in arterial Pco2, would reduce the cerebral hypoxemic dilation and increase the vasoconstriction from hypocapnia.
Conclusion.
In summary, elevations in global CBF over 6 h of hypoxia are partly mediated via greater increases in VA flow and arterial dilation. The changes in global CBF and regional blood flow in hypoxia are not altered via α1 blockade, indicating that a heightened SNA with hypoxia does not constrain further dilation of the larger extracranial blood vessels. It appears the vascular responses in the large extracranial cerebral vessels to an acute hypoxic challenge are fully revisable with 60 min of normoxic recovery. The implications of these findings are to further our understanding of how hypoxemia influences regional CBF. Such patterns and mechanism(s) need to be understood to provide insight into how various pathologies associated with acute and chronic hypoxemia might affect the regulation of CBF.
GRANTS
Support for this study was provided by a Canada Research Chair in Cerebrovascular Physiology and Natural Sciences and Engineering Research Council of Canada Discovery Grant to P.N.A., by a postdoctoral fellowship from the Heart and Stroke Foundation of Canada to N.C.L., and by a Natural Sciences and Engineering Research Council of Canada Undergraduate Student Research Award to L.M.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.C.L., B.M., and P.N.A. conception and design of research; N.C.L. and L.M. performed experiments; N.C.L. and L.M. analyzed data; N.C.L. and P.N.A. interpreted results of experiments; N.C.L. prepared figures; N.C.L. drafted manuscript; N.C.L. and P.N.A. edited and revised manuscript; N.C.L., L.M., B.M., and P.N.A. approved final version of manuscript.
REFERENCES
- 1.Ainslie PN, Lucas SJ, Burgess KR. Breathing and sleep at high altitude. Respir Physiol Neurobiol 188: 233–256, 2013 [DOI] [PubMed] [Google Scholar]
- 2.Ainslie PN, Ogoh S. Regulation of cerebral blood flow in mammals during chronic hypoxia: a matter of balance. Exp Physiol 95: 251–262, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Ainslie PN, Poulin MJ. Ventilatory, cerebrovascular, and cardiovascular interactions in acute hypoxia: regulation by carbon dioxide. J Appl Physiol 97: 149–159, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Awan NA, Miller RR, Maxwell K, Mason DT. Effects of prazosin on forearm resistance and capacitance vessels. Clin Pharmacol Ther 22: 79–84, 1977 [DOI] [PubMed] [Google Scholar]
- 5.Bailey DM, Evans KA, James PE, McEneny J, Young IS, Fall L, Gutowski M, Kewley E, McCord JM, Moller K, Ainslie PN. Altered free radical metabolism in acute mountain sickness: implications for dynamic cerebral autoregulation and blood-brain barrier function. J Physiol 587: 73–85, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey DM, Taudorf S, Berg RM, Jensen LT, Lundby C, Evans KA, James PE, Pedersen BK, Moller K. Transcerebral exchange kinetics of nitrite and calcitonin gene-related peptide in acute mountain sickness: evidence against trigeminovascular activation? Stroke 40: 2205–2208, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Binks AP, Cunningham VJ, Adams L, Banzett RB. Gray matter blood flow change is unevenly distributed during moderate isocapnic hypoxia in humans. J Appl Physiol 104: 212–217, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Black MA, Cable NT, Thijssen DH, Green DJ. Importance of measuring the time course of flow-mediated dilatation in humans. Hypertension 51: 203–210, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Bleys RL, Cowen T, Groen GJ, Hillen B, Ibrahim NB. Perivascular nerves of the human basal cerebral arteries: I. Topographical distribution. J Cereb Blood Flow Metab 16: 1034–1047, 1996 [DOI] [PubMed] [Google Scholar]
- 10.Borodulya AV, Pletchkova EK. Cholinergic innervation of vessels of the base of the brain. Acta Anat (Basel) 96: 135–147, 1976 [DOI] [PubMed] [Google Scholar]
- 11.Borodulya AV, Pletchkova EK. Distribution of cholinergic and adrenergic nerves in the internal carotid artery. A histochemical study. Acta Anat (Basel) 86: 410–425, 1973 [DOI] [PubMed] [Google Scholar]
- 12.Boutouyrie P, Lacolley P, Girerd X, Beck L, Safar M, Laurent S. Sympathetic activation decreases medium-sized arterial compliance in humans. Am J Physiol Heart Circ Physiol 267: H1368–H1376, 1994 [DOI] [PubMed] [Google Scholar]
- 13.Bowton DL, Haddon WS, Prough DS, Adair N, Alford PT, Stump DA. Theophylline effect on the cerebral blood flow response to hypoxemia. Chest 94: 371–375, 1988 [DOI] [PubMed] [Google Scholar]
- 14.Cohen PJ, Alexander SC, Smith TC, Reivich M, Wollman H. Effects of hypoxia and normocarbia on cerebral blood flow and metabolism in conscious man. J Appl Physiol 23: 183–189, 1967 [DOI] [PubMed] [Google Scholar]
- 15.Curran-Everett DC, Meredith MP, Krasney JA. Acclimatization to hypoxia alters cerebral convective and diffusive O2 delivery. Respir Physiol 88: 355–371, 1992 [DOI] [PubMed] [Google Scholar]
- 16.Edvinsson L. Neurogenic mechanisms in the cerebrovascular bed. Autonomic nerves, amine receptors and their effects on cerebral blood flow. Acta Physiol Scand Suppl 427: 1–35, 1975 [PubMed] [Google Scholar]
- 17.Evans DH. On the measurement of the mean velocity of blood flow over the cardiac cycle using Doppler ultrasound. Ultrasound Med Biol 11: 735–741, 1985 [DOI] [PubMed] [Google Scholar]
- 18.Fan JL, Burgess KR, Thomas KN, Peebles KC, Lucas SJ, Lucas RA, Cotter JD, Ainslie PN. Influence of indomethacin on the ventilatory and cerebrovascular responsiveness to hypoxia. Eur J Appl Physiol 111: 601–610, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Fan JL, Subudhi AW, Evero O, Bourdillon N, Kayser B, Lovering AT, Roach RC. AltitudeOmics: enhanced cerebrovascular reactivity and ventilatory response to CO2 with high altitude acclimatization and reexposure. J Appl Physiol. First published December 19, 2013;. 10.1152/japplphysiol00704.2013. [DOI] [PubMed] [Google Scholar]
- 20.Faraci FM, Heistad DD, Mayhan WG. Role of large arteries in regulation of blood flow to brain stem in cats. J Physiol 387: 115–123, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green DJ, Bilsborough W, Naylor LH, Reed C, Wright J, O'Driscoll G, Walsh JH. Comparison of forearm blood flow responses to incremental handgrip and cycle ergometer exercise: relative contribution of nitric oxide. J Physiol 562: 617–628, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol 546: 921–929, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrell JW, Schrage WG. Cyclooxygenase-derived vasoconstriction restrains hypoxia-mediated cerebral vasodilation in young adults with metabolic syndrome. Am J Physiol Heart Circ Physiol 306: H261–H269, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heistad DD, Marcus ML, Abboud FM. Role of large arteries in regulation of cerebral blood flow in dogs. J Clin Invest 62: 761–768, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarvis MF, Williams M. Direct autoradiographic localization of adenosine A2 receptors in the rat brain using the A2-selective agonist, [3H]CGS 21680. Eur J Pharmacol 168: 243–246, 1989 [DOI] [PubMed] [Google Scholar]
- 26.Jauernig RA, Moulds RF, Shaw J. The action of prazosin in human vascular preparations. Arch Int Pharmacodyn Ther 231: 81–89, 1978 [PubMed] [Google Scholar]
- 27.Jones H, Lewis NC, Green DJ, Ainslie PN, Lucas SJ, Tzeng YC, Grant EJ, Atkinson G. α1-Adrenoreceptor activity does not explain lower morning endothelial-dependent, flow-mediated dilation in humans. Am J Physiol Regul Integr Comp Physiol 300: R1437–R1442, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest 27: 484–492, 1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazzeo RS, Dubay A, Kirsch J, Braun B, Butterfield GE, Rock PB, Wolfel EE, Zamudio S, Moore LG. Influence of alpha-adrenergic blockade on the catecholamine response to exercise at 4,300 meters. Metabolism 52: 1471–1477, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Meno JR, Ngai AC, Winn HR. Changes in pial arteriolar diameter and CSF adenosine concentrations during hypoxia. J Cereb Blood Flow Metab 13: 214–220, 1993 [DOI] [PubMed] [Google Scholar]
- 31.Nishimura M, Yoshioka A, Yamamoto M, Akiyama Y, Miyamoto K, Kawakami Y. Effect of theophylline on brain tissue oxygenation during normoxia and hypoxia in humans. J Appl Physiol 74: 2724–2728, 1993 [DOI] [PubMed] [Google Scholar]
- 32.Ogoh S, Brothers RM, Eubank WL, Raven PB. Autonomic neural control of the cerebral vasculature: acute hypotension. Stroke 39: 1979–1987, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Ogoh S, Sato K, Nakahara H, Okazaki K, Subudhi AW, Miyamoto T. Effect of acute hypoxia on blood flow in vertebral and internal carotid arteries. Exp Physiol 98: 692–698, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Padilla J, Young CN, Simmons GH, Deo SH, Newcomer SC, Sullivan JP, Laughlin MH, Fadel PJ. Increased muscle sympathetic nerve activity acutely alters conduit artery shear rate patterns. Am J Physiol Heart Circ Physiol 298: H1128–H1135, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perret F, Mooser V, Waeber B, Yanik T, Jean-Jacques M, Mooser E, Nussberger J, Brunner HR. Effect of cold pressor test on the internal diameter of the radial artery. Am J Hypertens 2: 727–728, 1989 [DOI] [PubMed] [Google Scholar]
- 36.Poulin MJ, Fatemian M, Tansley JG, O'Connor DF, Robbins PA. Changes in cerebral blood flow during and after 48 h of both isocapnic and poikilocapnic hypoxia in humans. Exp Physiol 87: 633–642, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Saito M, Mano T, Iwase S, Koga K, Abe H, Yamazaki Y. Responses in muscle sympathetic activity to acute hypoxia in humans. J Appl Physiol 65: 1548–1552, 1988 [DOI] [PubMed] [Google Scholar]
- 38.Schoning M, Walter J, Scheel P. Estimation of cerebral blood flow through color duplex sonography of the carotid and vertebral arteries in healthy adults. Stroke 25: 17–22, 1994 [DOI] [PubMed] [Google Scholar]
- 39.Severinghaus JW, Chiodi H, Eger EI, 2nd, Brandstater B, Hornbein TF. Cerebral blood flow in man at high altitude. Role of cerebrospinal fluid pH in normalization of flow in chronic hypocapnia. Circ Res 19: 274–282, 1966 [DOI] [PubMed] [Google Scholar]
- 40.Subudhi AW, Fan JL, Evero O, Bourdillon N, Kayser B, Julian CG, Lovering AT, Roach RC. AltitudeOmics: effect of ascent and acclimatization to 5,260 m on regional cerebral oxygen delivery. Exp Physiol. In press. First published June 27, 2013; 10.1113/expphysiol.2013.075184. [DOI] [PubMed] [Google Scholar]
- 41.Subudhi AW, Panerai RB, Roach RC. Effects of hypobaric hypoxia on cerebral autoregulation. Stroke 41: 641–646, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Toda N, Okamura T. Cerebral vasodilators. Jpn J Pharmacol 76: 349–367, 1998 [DOI] [PubMed] [Google Scholar]
- 43.Van Mil AH, Spilt A, Van Buchem MA, Bollen EL, Teppema L, Westendorp RG, Blauw GJ. Nitric oxide mediates hypoxia-induced cerebral vasodilation in humans. J Appl Physiol 92: 962–966, 2002 [DOI] [PubMed] [Google Scholar]
- 44.Villien M, Bouzat P, Rupp T, Robach P, Lamalle L, Tropres I, Esteve F, Krainik A, Levy P, Warnking JM, Verges S. Changes in cerebral blood flow and vasoreactivity to CO2 measured by arterial spin labeling after 6days at 4350m. Neuroimage 72: 272–279, 2013 [DOI] [PubMed] [Google Scholar]
- 45.Willie CK, Macleod DB, Shaw AD, Smith KJ, Tzeng YC, Eves ND, Ikeda K, Graham J, Lewis NC, Day TA, Ainslie PN. Regional brain blood flow in man during acute changes in arterial blood gases. J Physiol 590: 3261–3275, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willie CK, Smith KJ, Day TA, Ray LA, Lewis NC, Bakker A, Macleod DB, Ainslie PN. Regional cerebral blood flow in humans at high altitude: gradual ascent and two weeks at 5,050 m. J Appl Physiol First published June 27, 2013; 10.1152/japplphysiol.00594.2013. [DOI] [PubMed] [Google Scholar]
- 47.Willie CK, Tzeng YC, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol 592: 841–859, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson MH, Edsell ME, Davagnanam I, Hirani SP, Martin DS, Levett DZ, Thornton JS, Golay X, Strycharczuk L, Newman SP, Montgomery HE, Grocott MP, Imray CH. Cerebral artery dilatation maintains cerebral oxygenation at extreme altitude and in acute hypoxia–an ultrasound and MRI study. J Cereb Blood Flow Metab 31: 2019–2029, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woodman RJ, Playford DA, Watts GF, Cheetham C, Reed C, Taylor RR, Puddey IB, Beilin LJ, Burke V, Mori TA, Green D. Improved analysis of brachial artery ultrasound using a novel edge-detection software system. J Appl Physiol 91: 929–937, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Xie A, Skatrud JB, Puleo DS, Morgan BJ. Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol 91: 1555–1562, 2001 [DOI] [PubMed] [Google Scholar]