

Abstract
Rationale: Pulmonary arterial hypertension (PAH) is a progressive disease characterized by elevated pulmonary artery pressure, vascular remodeling, and ultimately right ventricular heart failure. PAH can have a genetic component (heritable PAH), most often through mutations of bone morphogenetic protein receptor 2, and idiopathic and associated forms. Heritable PAH is not completely penetrant within families, with approximately 20% concurrence of inactivating bone morphogenetic protein receptor 2 mutations and delayed onset of PAH disease. Because one of the treatment options is using prostacyclin analogs, we hypothesized that prostacyclin synthase promoter sequence variants associated with increased mRNA expression may play a protective role in the bone morphogenetic protein receptor 2 unaffected carriers.
Objectives: To characterize the range of prostacyclin synthase promoter variants and assess their transcriptional activities in PAH-relevant cell types. To determine the distribution of prostacyclin synthase promoter variants in PAH, unaffected carriers in heritable PAH families, and control populations.
Methods: Polymerase chain reaction approaches were used to genotype prostacyclin synthase promoter variants in more than 300 individuals. Prostacyclin synthase promoter haplotypes’ transcriptional activities were determined with luciferase reporter assays.
Measurements and Main Results: We identified a comprehensive set of prostacyclin synthase promoter variants and tested their transcriptional activities in PAH-relevant cell types. We demonstrated differences of prostacyclin synthase promoter activities dependent on their haplotype.
Conclusions: Prostacyclin synthase promoter sequence variants exhibit a range of transcriptional activities. We discovered a significant bias for more active prostacyclin synthase promoter variants in unaffected carriers as compared with affected patients with PAH.
Keywords: lung diseases, pulmonary hypertension, genetic polymorphism, genetic predisposition to disease
At a Glance Commentary
Scientific Knowledge on the Subject
Heritable pulmonary arterial hypertension (HPAH) is often associated with mutations in BMPR2 gene and other members of the transforming growth factor-β pathway. HPAH is characterized by incomplete penetrance (∼20%), and during long-term follow-up unaffected carriers can be identified in family studies. The role of modifier genes influencing penetrance in HPAH has been proposed, although evidence supporting candidate genes is lacking.
What This Study Adds to the Field
Prostacyclin is a major regulator of vascular tone and proliferation of lung vascular endothelial and smooth muscle cells. Prostacyclin synthase (PGIS) is the key enzyme producing prostacyclin in these cell types, and its promoter has a wide variety of sequence polymorphisms. We demonstrate in this study that PGIS promoter sequence variants associated with increased transcriptional activity are over-represented in unaffected BMPR2 mutant carriers. We propose that increased expression of PGIS is protective in unaffected carriers and likely acts as a modifier gene influencing penetrance in HPAH.
Pulmonary arterial hypertension (PAH) is a rare disease, with annual incidence of 2–15 per million, with a female preponderance (≥2:1), characterized by elevations in mean pulmonary artery pressure (mPAP) of more than 25 mm Hg at rest, a pulmonary capillary wedge pressure of less than 15 mm Hg, vascular remodeling, and hyperproliferation of pulmonary artery endothelial and smooth muscle cells (1, 2). The incidence of new PAH cases is estimated to be approximately 1,000 per year in the United States (3). Presentation occurs most commonly in the third and fourth decades in women and men, respectively, although with treatment the 1-, 3-, and 5-year survival rates are 92, 75, and 66% (4). Currently, there is no cure for this disease, but targeted therapies that focus on altering vascular tone using prostacyclin analogs, dual endothelin antagonists, or phosphodiesterase-5 inhibitors have improved patient survival (1, 2, 4).
PAH is divided into three categories: (1) hereditary PAH (HPAH), characterized by inheritable disease; (2) idiopathic PAH, with no clear genetic or environmental cause in most cases; and (3) PAH associated with one of several other clinical factors (associated PAH). HPAH accounts for approximately 6% of PAH cases and has an autosomal-dominant mode of inheritance originally associated with mutations in bone morphogenetic protein receptor type 2 (BMPR2) gene (5, 6), although there are less frequent associations with germline mutations in activin receptor-like kinase type 1 (ACVRL1), endoglin (ENG, reviewed in [7]), the SMAD gene family of proteins (8), and recently in caveolin-1 (CAV1 [9]) and in the potassium channel subfamily K, member 3 (KCNK3 [10]). Idiopathic PAH includes cases with no clear cause, although the role for underlying mutations in BMPR2 in 5–25% of cases has been recently appreciated (7, 11). Associated PAH can occur as a result of congenital heart disease, human immunodeficiency virus, connective tissue disorders, and anorexigen or stimulant drug use.
Although BMPR2 mutations are often associated with HPAH (∼80% of the families), there is incomplete penetrance with approximately 20% of the BMPR2 mutant carriers presenting clinical manifestation of PAH. Unaffected carriers within HPAH families suggest the role of one or more additional genetic or mechanistic modifiers of the BMPR2 mutations and is an active area of investigation. The Vanderbilt Familial Pulmonary Hypertension Clinic has been following and characterizing families with BMPR2 mutations for more than three decades, long enough to accurately identify unaffected carriers at an advanced age that are unlikely to develop HPAH in their lifetime. Our investigation of the prostacyclin synthase (PGIS; official gene symbol PTGIS) DNA promoter sequence variants reveals a complexity of sequence polymorphisms within a short region (∼525 base pairs [bp]) encompassing the minimum promoter (231 bp, a CpG island), exon 1, and the 5′ side of intron 1 (12). By unraveling this sequence complexity, in conjunction with functional transcriptional studies, we find more active PGIS promoter variants are significantly associated with unaffected carriers in HPAH families, suggesting that an enhanced level of endogenous PGIS expression is a potential mechanism for protection from HPAH disease.
Methods
The patient and control populations were obtained through the Vanderbilt Familial PAH cohort (Vanderbilt study) or the Pulmonary Hypertension Breakthrough Initiative (PHBI) and are outlined in Table 1 (described in more detail in the online supplement and Table E1 in the online supplement). In short, the individuals were in three groups: (1) patients with PAH (n = 108; all forms of PAH), (2) unaffected BMPR2 mutation carriers (n = 24), and (3) control subjects (n = 101). Control subjects from Vanderbilt were either related HPAH family members who did not inherit BMPR2 mutations or unrelated married-in HPAH family members. The PHBI control subjects were individual organ donors whose lungs either did not match a recipient or could not be used in transplant (failed donors). As expected, there was a 2:1 female to male sex bias in the PAH group compared with control subjects (chi-square, 0.0002). All individuals were consented for genetic testing by their respective study group, and the samples were further deidentified from association to any individual’s personal information for the purpose of this study.
Table 1:
PAH Patient Population and Control Subjects
| N | |
|---|---|
| PHBI study | |
| Affected (associated PAH) | 20 |
| Affected (HPAH) | 6 |
| Affected (idiopathic PAH) | 38 |
| Affected (other) | 4 |
| Control subjects (failed donors) | 41 |
| Total | 109 |
| Vanderbilt study | |
| Affected (HPAH) | 40 |
| Unaffected carriers | 24 |
| Control subjects (noncarriers) | 16 |
| Control subjects (married-ins) | 44 |
| Total | 124 |
| Totals | |
| All PAH (affected) | 108 |
| Unaffected carriers | 24 |
| All control subjects | 101 |
| Total | 233 |
Definition of abbreviations: HPAH = hereditary pulmonary arterial hypertension; PAH = pulmonary arterial hypertension; PHBI = Pulmonary Hypertension Breakthrough Initiative.
Characterization of PGIS promoter sequences was completed using an initial polymerase chain reaction (PCR) product amplified from genomic DNA used in three subsequent different assays (12) (see online supplement and Figure E1). Each individual’s PGIS promoter genotype can be assembled by combining the three assay results at each of the four single-nucleotide polymorphisms (SNPs) and the repeat length of a variable number of tandem repeats (VNTRs) immediately 5′ of the PGIS protein initiation methionine (MET1). Linkage disequilibrium analysis of SNPs 1, 3, and 4 was performed using Haploview 4.2 (13).
All cloned PGIS promoter constructs, produced by PCR from genomic DNA, were made by fusing the 213 bp promoter variant in the native context of the MET1 of exon 1 of the PGIS as the MET1 of Luciferase (Luc) in pGL3-Basic (12). By constructing the PGIS promoter Luc plasmids in this way, each sequence polymorphism can be individually evaluated for promoter activity within a defined sequence context.
BEAS-2B normal lung epithelial cell line (ATCC, Manassas, VA) was grown in the recommended semidefined BEGM media (Lonza, Allendale, NJ). Primary cells (PromoCell GmbH, Heidelberg Germany) were grown in their recommended media. Cells were grown humidified at 37°C with 5% CO2. Chemical transfection conditions were optimized for each cell line or type in 12-well tissue culture plates by surveying a panel of six different reagents using pGL3-Control (Luciferase reporter) and pRL-CMV (Renilla reporter; Promega, Madison, WI) (see online supplement).
Total RNA and cDNA were prepared from PHBI lung tissue samples as described in the online supplement. Quantitative reverse-transcriptase PCR (qRT-PCR) was completed using 2 μl diluted cDNA in duplicate using ROX-containing FastStart Universal Probe Mastermix (Roche, Indianapolis, IN), gene-specific Taqman primer/probes (Applied Biosystems, Foster City, CA), and Bio-Rad (Hercules, CA) iCycler for 50 cycles. For normalizing each cDNA preparation, three endogenous control subjects (glyceraldehyde phosphate dehydrogenase, B2M, and TFRC) were run in duplicate on two different days, and the calculated Ct values were average across all three genes.
Results
The PGIS promoter region we focused on was determined to be a minimum promoter through directed deletion analysis (12). The PGIS promoter region is 231 bp proximal to the protein synthesis MET1 contained in exon 1 and is a CpG island. As we previously described, the PGIS promoter has approximately 85% guanine-cytosine content, two SNPs (green bars), a nine base VNTR (red arrows) with three to nine copies, and a SNP in the second repeat of the VNTR, counting 5′→3′ (Figure 1). The PGIS coding exon 1 is short encompassing the protein’s targeting sequence for lipid anchoring and asymmetric endoplasmic reticulum membrane insertion (14), followed by a long first intron (intron 1) of approximately 18 kilobase pairs. We expanded our investigation to include all of exon 1 and approximately 220 bp of the 5′ end of intron 1, and discovered a new SNP (SNP4, C325A) since our earlier report (12). With the completion of the 1,000 Genomes Project (15), SNP4 was identified (rs116939356) as C/A with a minor allele frequency (MAF) of 6% in whites.
Figure 1.

Genomic map of the prostacyclin synthase (PGIS) promoter. Diagram representing approximately 500 base pair (bp) of genomic DNA amplified by polymerase chain reaction containing the minimum PGIS promoter region, exon 1, and a small portion of the 5′ side of intron 1. Contained within this region are four single-nucleotide polymorphisms (SNPs) (SNP1, T22G [rs5580]; SNP2, G45A [rs5581]; SNP3, C185T [wt/M1] [rs5582]; and SNP4, C325A [rs116939356]; green bars) and a 9-base variable number of tandem repeats (VNTR) with three to nine copies of the tandem repeat (red arrows). SNPs 1, 2, and 3 along with the sequence numbering were previously described (9).
As an initial source of cloned PGIS promoter variants, we genotyped (see online supplement and Figure E1) 90 individuals from the Polymorphism Discovery Resource (PDR [16], Coriell Institute, Camden, NJ). The PDR was designed to capture the widest degree of genomic DNA sequence diversity in the human population. Table 2 summarizes the PGIS promoter genotypes found for the PDR90 set (complete genotypes for the PDR90 set are found in Table E2). Within the PDR90 set, the 6-repeat VNTR is the most common, with homozygous 6/6 representing approximately 54% of this diverse population. SNP1 T22G (rs5580) and SNP4 C325A allele frequencies match that currently reported in dbSNP (SNP1 G MAF = 14–42%; SNP4 A MAF = 6–12%, respectively). SNP1 T22G MAF shows a significant range depending on the composition of the population examined with a higher MAF in white populations (MAF = 42%) compared with the estimated Global MAF (14%) from the 1,000 Genomes Project (15). SNP2 G45A (rs5581) allele frequency is much lower than reported in a small Zimbabwe population (SNP2 A MAF = 0.5% vs. 5%). A Global MAF was not given probably because of difficulty sequencing this region of the genome. SNP3 C185T (rs5582) has MAF = 43%, although this was not previously described within its VNTR repeat context. The Global MAF for SNP3 is 12% but there are no specific data for whites. There was no evidence in the PDR population that these three SNPs deviated from expected Hardy-Weinberg equilibrium (17). Because the nucleotide position of SNP3 changes relative to PGIS MET1 and the VNTR repeat number, SNP3 is referred here as wt (C major allele) and M1 (T minor allele). From the PDR90 collection, the PAH study samples, and our previous work with lung cancer cell lines, we cloned into pGL-3 Basic and sequenced 20 different PGIS promoter haplotype variants from genomic DNA (see Table E3).
Table 2:
PGIS Promoter VNTR Repeat Length and SNP Genotype and Allele Frequencies
| Number | Frequency | |
|---|---|---|
| VNTR genotypes | ||
| 3/4 | 0 | 0.000 |
| 4/4 | 2 | 0.022 |
| 4/5 | 0 | 0.000 |
| 4/6 | 13 | 0.144 |
| 4/7 | 0 | 0.000 |
| 4/8 | 1 | 0.011 |
| 5/5 | 1 | 0.011 |
| 5/6 | 16 | 0.178 |
| 5/7 | 0 | 0.000 |
| 5/8 | 0 | 0.000 |
| 6/6 | 49 | 0.544 |
| 6/7 | 5 | 0.056 |
| 6/8 | 2 | 0.022 |
| 6/9 | 1 | 0.011 |
| Total | 90 | 1.000 |
| VNTR alleles | ||
| 3 | 0 | 0.000 |
| 4 | 18 | 0.100 |
| 5 | 18 | 0.100 |
| 6 | 135 | 0.750 |
| 7 | 5 | 0.028 |
| 8 | 3 | 0.017 |
| 9 | 1 | 0.006 |
| Total | 180 | 1.000 |
| SNP1 K22 genotypes | ||
| T/T | 26 | 0.289 |
| T/G | 51 | 0.567 |
| G/G | 13 | 0.144 |
| Total | 90 | 1.000 |
| SNP1 K22 alleles | ||
| T | 103 | 0.572 |
| G | 77 | 0.428 |
| Total | 180 | 1.000 |
| SNP3 Y185 genotypes | ||
| C/C | 27 | 0.300 |
| C/T | 48 | 0.533 |
| T/T | 15 | 0.167 |
| Total | 90 | 1.000 |
| SNP3 Y185 alleles | ||
| C | 102 | 0.567 |
| T | 78 | 0.433 |
| Total | 180 | 1.000 |
| SNP4 C325A genotypes | ||
| C/C | 75 | 0.833 |
| C/A | 14 | 0.156 |
| A/A | 1 | 0.011 |
| Total | 90 | 1.000 |
| SNP4 C325A alleles | ||
| C | 164 | 0.911 |
| A | 16 | 0.089 |
| Total | 180 | 1.000 |
Definition of abbreviations: SNP = single-nucleotide polymorphism; PGIS = prostacyclin synthase; VNTR = variable number of tandem repeats.
Given the diversity of variants within a short stretch of the genome, we completed PGIS promoter genotyping of the large CEPH Pedigree 1,463 (Coriell Institute) and found normal mendelian inheritance (see Figure E2). Linkage disequilibrium analysis was conducted on SNPs 1, 3, and 4 (see Figure E3; SNP2 was too rare to be included). These three SNPs are separated by only 304 bp but exhibit surprisingly weak linkage disequilibrium and low correlation values, making haplotype prediction less robust. A genome-wide examination of low-linkage disequilibrium genomic regions (potential recombination hotspots) identified a number of common sequence characteristics (18), many of which are found in the PGIS promoter region including high GC content, increased rate of sequence polymorphisms, repeat sequences (simple and total amount), genic-intronic bases, and transcription factor binding sites.
We investigated the transcriptional activities of a subset (Table 3) of this comprehensive collection of cloned PGIS promoter sequence variants by varying one parameter (a SNP or VNTR composition) at a time. The immortalized, normal lung epithelial cell line (BEAS-2B) was tested along with a variety of primary human cells: human umbilical vein endothelial cells, human umbilical artery endothelial cells (HUAEC), human pulmonary artery endothelial cells, human pulmonary artery smooth muscle cells (HPASMC), and human pulmonary microvascular endothelial cells. These cells were chosen to represent the major cell types found in the lung: epithelial, endothelial, and smooth muscle cells. Primary cell transcription reporter assays showed a range of standard deviation in the reporter assay measurements making some experimental comparison difficult (see Table E4A). The largest transcriptional activity effect was observed for the VNTR length series in BEAS-2B, human umbilical vein endothelial cells, and human pulmonary artery endothelial cells (Figure 2) suggesting an important role for VNTR length in both epithelial and endothelial cell types. There was no significant effect of VNTR length in HUAEC, or interestingly, HPASMC (see Figure E4), one of the expected sites of PGIS expression in the lung vasculature. However, the reporter responsiveness in HPASMC was muted compared with the other cells tested (ratio of pGL3-Basic to PGIS promoter constructs was ∼1.5x rather than the 5–10x typically). Human pulmonary microvascular endothelial cells could not be evaluated because they gave background levels of PGIS Luc activity even though the cells were transfected as exhibited by high Ren (from the CMV promoter). SNP1 (T22G; T/G) and SNP3 (C185T; wt/M1) were tested within the context of the 6-repeat VNTR structure (since all four possible haplotypes are found with the 6-repeat; see Table E3) but there were no statistically significant transcriptional effects (see Table E4B).
Table 3:
Cloned PGIS Promoter Haplotypes
| VNTR Repeat Length Transfection Study |
| T22_G45_3wt_LUC |
| T22_G45_4wt_LUC |
| T22_G45_5wt_LUC |
| T22_G45_6wt_LUC |
| T22_G45_7wt_LUC |
| T22_G45_8wt_LUC |
| SNP4 M325 Transfection Study |
| G22_G45_5wt_C325_LUC |
| G22_G45_5wt_A325_LUC |
Definition of abbreviations: PGIS = prostacyclin synthase; SNP = single-nucleotide polymorphism; VNTR = variable number of tandem repeats.
Figure 2.
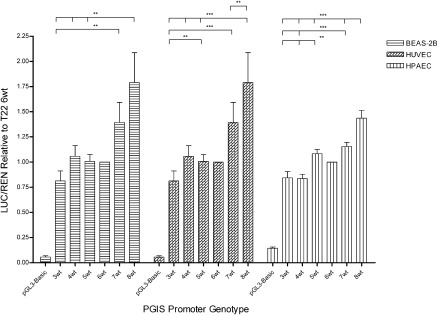
Transcriptional activity of prostacyclin synthase (PGIS) promoter variable number of tandem repeats (VNTR) variants. The transcriptional activity of the PGIS promoter VNTR variants was determined using the Luciferase reporter normalized to cotransfected Renilla reporter activity. Results from three different cell types in addition to normal are shown: immortalized human bronchial epithelial cell line (BEAS-2B), human umbilical vein endothelial cells (HUVEC), and human pulmonary artery endothelial cells (HPAEC). The VNTR repeats varied from three to eight copies keeping all the other single-nucleotide polymorphisms constant in each plasmid construct. The data were normalized to the activity of the most common PGIS promoter allele (T22_G45_6wt_C325). Error bars are the standard error of the mean and significance (**P = 0.005–0.05 or ***P < 0.005) was determined by two-tailed t tests.
In individuals where the PGIS promoter genotypes can be assigned to specific allelic haplotypes, the SNP4 A allele is found in association with the G22_G45_5wt_A325 or G22_G45_6wt_A325 haplotype. Because SNP4 occurs within the 5′ side of intron 1, a modified PGIS exon 1 was created by site-directed mutagenesis, converting MET1 to threonine with G22_G45_5wt making up the remainder of the promoter haplotype. This shifts the translational start from PGIS’s exon 1 to the MET1 of the Luciferase reporter. The PGIS promoter transcriptional activity of the SNP4 C325A alleles was measured by transfection studies (Figure 3) and the SNP4 A minor allele showed an increase of activity in BEAS-2B and HUAEC compared with SNP4 C major allele.
Figure 3.

Transcriptional activity of prostacyclin synthase (PGIS) promoter single-nucleotide polymorphism (SNP) 4 C325A variants. The transcriptional activity of the PGIS promoter SNP4 C325A variants was determined using the Luciferase reporter normalized to cotransfected Renilla reporter activity. Results from two different cell types in addition to normal are shown: immortalized human bronchial epithelial cell line (BEAS-2B) and human umbilical artery endothelial cells (HUAEC). The SNP4 C325A variants (C major allele, A minor allele) were tested in the context of the G22_G45_5wt PGIS promoter haplotype where the SNP4 A allele is most commonly associated. The data were normalized to the activity of the most common PGIS promoter allele (T22_G45_6wt_C325). Error bars are the standard error of the mean and significance (**P = 0.005–0.05) was determined by two-tailed t tests.
The clinical samples for this study were derived from two sources, the Vanderbilt study and the PHBI (Table 1) consisting of 108 patients with PAH (All PAH, includes all clinical definitions), 24 unaffected BMPR2 carriers, and 101 control subjects. The PGIS promoter genotypes for all 233 individuals were determined (see Table E2) and summarized in Table 4. Chi-square or Fisher exact tests were used to confirm statistically significant changes in the genotype distribution of each sample class (Table 5). We observed two general patterns of genotype distribution changes that are significant. First, the All PAH category had a VNTR composition shifted toward shorter repeat numbers compared with the control subjects and unaffected carriers (chi-square test, 0.028 and 0.053, respectively), with more SNP3 wt (C) alleles compared with control subjects (chi-square test, 0.025). Transfection data (Figure 2) suggest that shorter VNTRs are associated with lower PGIS transcriptional activity. Although SNP3 alleles did not show significant variation in transcriptional activity in the cells studied here, we previously found the wt (C) alleles had lower activity in lung cancer cell lines (12). The unaffected carriers had a significant bias for SNP4 minor allele (A) compared with all PAH and control groups (Fisher exact test, P = 0.019). When the SNP4 C325A SNP was tested by transfection reporter assays in PGIS G22_G45_5wt promoter haplotype (Figure 3), the SNP4 A allele (minor allele in the population/more common in unaffected carriers) was more active than the SNP4 C allele (major allele in the population/less common in unaffected carriers).
Table 4:
PGIS Promoter VNTR Repeat Length and SNP Genotype and Allele Frequencies
| All PAH |
Unaffected Carriers |
Control Subjects |
||||
|---|---|---|---|---|---|---|
| Number | Frequency | Number | Frequency | Number | Frequency | |
| VNTR genotypes | ||||||
| 3/4 | 1 | 0.009 | 0 | 0.000 | 0 | 0.000 |
| 4/4 | 3 | 0.028 | 1 | 0.042 | 1 | 0.010 |
| 4/5 | 3 | 0.028 | 0 | 0.000 | 2 | 0.020 |
| 4/6 | 24 | 0.222 | 5 | 0.208 | 24 | 0.238 |
| 4/7 | 3 | 0.028 | 0 | 0.000 | 0 | 0.000 |
| 4/8 | 0 | 0.000 | 0 | 0.000 | 0 | 0.000 |
| 5/5 | 0 | 0.000 | 1 | 0.042 | 0 | 0.000 |
| 5/6 | 9 | 0.083 | 6 | 0.250 | 9 | 0.089 |
| 5/7 | 3 | 0.028 | 1 | 0.042 | 1 | 0.010 |
| 5/8 | 0 | 0.000 | 1 | 0.042 | 0 | 0.000 |
| 6/6 | 56 | 0.519 | 7 | 0.292 | 60 | 0.594 |
| 6/7 | 5 | 0.046 | 2 | 0.083 | 4 | 0.040 |
| 6/8 | 1 | 0.009 | 0 | 0.000 | 0 | 0.000 |
| 6/9 | 0 | 0.000 | 0 | 0.000 | 0 | 0.000 |
| Total | 108 | 1.000 | 24 | 1.000 | 101 | 1.000 |
| VNTR alleles | ||||||
| 3 | 1 | 0.005 | 0 | 0.000 | 0 | 0.000 |
| 4 | 37 | 0.171 | 7 | 0.146 | 28 | 0.139 |
| 5 | 15 | 0.069 | 10 | 0.208 | 33 | 0.163 |
| 6 | 151 | 0.699 | 27 | 0.563 | 136 | 0.673 |
| 7 | 11 | 0.051 | 3 | 0.063 | 5 | 0.025 |
| 8 | 1 | 0.005 | 1 | 0.021 | 0 | 0.000 |
| 9 | 0 | 0.000 | 0 | 0.000 | 0 | 0.000 |
| Total | 216 | 1.000 | 48 | 1.000 | 202 | 1.000 |
| SNP1 K22 genotypes | ||||||
| T/T | 28 | 0.259 | 6 | 0.250 | 38 | 0.376 |
| T/G | 53 | 0.491 | 10 | 0.417 | 44 | 0.436 |
| G/G | 27 | 0.250 | 8 | 0.333 | 19 | 0.188 |
| Total | 108 | 1.000 | 24 | 1.000 | 101 | 1.000 |
| SNP1 K22 alleles | ||||||
| T | 109 | 0.505 | 22 | 0.458 | 120 | 0.594 |
| G | 107 | 0.495 | 26 | 0.542 | 82 | 0.406 |
| Total | 216 | 1.000 | 48 | 1.000 | 202 | 1.000 |
| SNP3 Y185 genotypes | ||||||
| C/C | 45 | 0.417 | 12 | 0.500 | 37 | 0.366 |
| C/T | 50 | 0.463 | 9 | 0.375 | 37 | 0.366 |
| T/T | 13 | 0.120 | 3 | 0.125 | 27 | 0.267 |
| Total | 108 | 1.000 | 24 | 1.000 | 101 | 1.000 |
| SNP3 Y185 alleles | ||||||
| C | 140 | 0.648 | 33 | 0.688 | 111 | 0.550 |
| T | 76 | 0.352 | 15 | 0.313 | 91 | 0.450 |
| Total | 216 | 1.000 | 48 | 1.000 | 202 | 1.000 |
| SNP4 C325A genotypes | ||||||
| C/C | 93 | 0.861 | 15 | 0.625 | 85 | 0.842 |
| C/A | 14 | 0.130 | 8 | 0.333 | 15 | 0.149 |
| A/A | 1 | 0.009 | 1 | 0.042 | 1 | 0.010 |
| Total | 108 | 1.000 | 24 | 1.000 | 101 | 1.000 |
| SNP4 C325A alleles | ||||||
| C | 200 | 0.926 | 38 | 0.792 | 185 | 0.916 |
| A | 16 | 0.074 | 10 | 0.208 | 17 | 0.084 |
| Total | 216 | 1.000 | 48 | 1.000 | 202 | 1.000 |
Definition of abbreviations: PAH = pulmonary arterial hypertension; PGIS = prostacyclin synthase; SNP = single-nucleotide polymorphism; VNTR = variable number of tandem repeats.
Table 5:
Statistical Analysis of Genotype Association with Clinical Category
| Unaffected Carriers | All Control Subjects | |
|---|---|---|
| VNTR alleles* | ||
| All PAH | 0.053 | 0.028 |
| Unaffected carriers | ns | |
| SNP3 Y185 genotypes* | ||
| All PAH | ns | 0.025 |
| Unaffected carriers | ns | |
| SNP4 C325A genotypes† | ||
| All PAH | 0.019 | ns |
| Unaffected carriers | 0.050 |
Definition of abbreviations: ns = not significant; PAH = pulmonary arterial hypertension; PGIS = prostacyclin synthase; SNP = single-nucleotide polymorphism; VNTR = variable number of tandem repeats.
Chi-square test.
Fisher exact test.
Two subgroup analyses were completed removing the noncarrier family members from the control subjects (16 individuals; see Tables E5A and E5B) and examining only patients with HPAH instead of all patients with PAH (62 patients with PAH; see Table E5C and E5D). As one would expect with large changes to the sample population being compared, there were changes in the level of significance of genotype comparisons shown in Table 5 with the different subgroup analyses (see Table E5). Generally, the pattern of results remained unchanged regardless of the sample population chosen.
There was no evidence in the genotypes of the clinical samples that SNPs 1, 3, and 4 deviated from expected Hardy-Weinberg equilibrium (17). Odds ratio analysis was conducted to examine the relationship of these genotypes to PAH risk (Table 6) (19, 20). Homozygous genotypes at SNP1 and SNP3 we hypothesized to increase PGIS gene expression were associated with decreased risk for PAH compared with control subjects. SNP1 T/T genotype showed a trend (odds ratio, 0.52; P = 0.090), whereas SNP3 T/T genotype had a significant risk reduction (odds ratio, 0.40; P = 0.020). SNP4 A containing genotypes (C/A + A/A) in unaffected carriers had a significant risk reduction compared with patients with PAH (odds ratio, 0.27; P = 0.007).
Table 6:
PGIS Promoter VNTR, SNP3, and SNP4 Genotype Association with PAH Risk
| Odds Ratio | 95% CI | Chi-Square Test | P Value | |
|---|---|---|---|---|
| SNP1 K22 Genotypes*† | ||||
| G/T vs. G/G | 0.85 | 0.42–1.72 | 0.21 | ns |
| T/T vs. G/G | 0.52 | 0.24–1.11 | 2.87 | 0.090 |
| SNP3 Y185 Genotypes*† | ||||
| C/T vs C/C | 1.11 | 0.60–2.04 | 0.12 | ns |
| T/T vs C/C | 0.40 | 0.18–0.87 | 5.40 | 0.020 |
| SNP4 C325A Genotypes*‡ | ||||
| C/A+A/A vs C/C | 0.27 | 0.10–0.72 | 7.36 | 0.007 |
Definition of abbreviations: CI = confidence interval; ns = not significant; PAH = pulmonary arterial hypertension; PGIS = prostacyclin synthase; SNP = single-nucleotide polymorphism; VNTR = variable number of tandem repeats.
Chi-square test.
Control subjects vs. all PAH.
Unaffected carriers vs. all PAH.
To support the transfection studies and assess the effects of several PGIS promoter polymorphisms in lung tissue, we measured the level of PGIS mRNA in lung tissue from the PHBI study samples by qRT-PCR (Figure 4; see online supplement). The first comparison looked at the effect of SNP3 C185T genotype on PGIS expression in lung tissue (Figure 4A). PGIS mRNA expression was increased in lung tissue when at least one PGIS promoter allele contains the SNP3 T allele. For SNP4 C325A, the minor allele A, which is overrepresented in the unaffected carriers, also had an increased level of PGIS mRNA in lung tissue (Figure 4B).
Figure 4.

Association of prostacyclin synthase (PGIS) mRNA expression and promoter variants in human lung tissue. Quantitative reverse-transcriptase polymerase chain reaction was used to quantitate the PGIS mRNA level in human lung tissue relative to three endogenous control genes. Error bars are the standard error of the mean and significance (*P = 0.05, **P = 0.025, or ns = not significant) was determined by two-tailed t tests. (A) Association of PGIS mRNA levels and PGIS promoter single-nucleotide polymorphism (SNP) 3 C185T alleles (wt [C] major allele, M1 [T] minor allele) occurring within the variable number of tandem repeats in human lung tissue. Each promoter variant category is grouped by SNP3 C185T genotype (wt/wt, n = 35; wt/M1, n = 32; M1/M1, n = 11). (B) Association of PGIS mRNA levels and PGIS promoter SNP4 C325A alleles (C major allele, A minor allele) in human lung tissue. Because the SNP4 C minor allele is linked with the G22_G45 composition at SNP1 and SNP2, individuals with heterozygous genotypes at SNP1 T22G (T major allele, G minor allele) in conjunction with C325/C325 (n = 33) or C325/A325 (n = 9) were used in this comparison.
There are several individuals with two of the more common PGIS promoter genotypes, allowing the direct comparison of VNTR structures of 4wt_6wt versus 6wt_6M1, keeping SNPs 1, 2, and 4 constant, combining the effects of VNTR repeat length and SNP3 C185T composition (Figure 5A). As anticipated, the longer VNTRs in association with the SNP3 T allele resulted in higher PGIS mRNA expression. Consistent with this finding, higher PGIS protein levels were detected in lung extracts from individuals with the 6wt_6M1 genotype compared with the 4wt_6wt genotype (sixfold increase relative to glyceraldehyde phosphate dehydrogenase; P = 0.01) (Figure 5B).
Figure 5.

Association of prostacyclin synthase (PGIS) mRNA and protein expression and promoter variants in human lung tissue quantitative reverse-transcriptase polymerase chain reaction was used to quantitate the PGIS mRNA level in human lung tissue relative to three endogenous control genes. (A) Association of PGIS mRNA levels and the two most common PGIS promoter genotypes in human lung tissue. The common PGIS promoter genotypes were compared; these vary in both variable number of tandem repeats (VNTR) length (4/6 to 6/6) and the presence of single-nucleotide polymorphism (SNP)3 C185T (wt/M1), keeping other SNP compositions constant (T22_G22/4wt_6wt, N = 12; T22_G22/6M1_6wt, N = 15). (B) Association of PGIS protein expression and the two most common PGIS promoter genotypes in human lung tissue. Western blot analysis (15 μg protein extract per lane) from human lung tissue is shown detecting endogenous levels of PGIS protein (50 kD) and two different endogenous control proteins (GAPDH and β-actin). Densitometry of PGIS protein expression relative to GAPDH showed a sixfold increase in PGIS level with the predicted more active PGIS promoter sequence (replicate blots, two-tailed t tests; P = 0.01). The common PGIS promoter genotypes were compared; these vary in both VNTR length (4/6 to 6/6) and the presence of SNP3 C185T (wt/M1), keeping other SNP compositions constant (T22_G22/4wt_6wt, N = 4; T22_G22/6M1_6wt, N = 4). Error bars are the standard error of the mean, and significance (**P = 0.025) was determined by two-tailed t tests.
We tested whether there was a correlation between PGIS expression levels and PAH lung inflammation index (see Figure E5) (21). We found there was an inverse correlation between PGIS expression and the lung inflammation index (r = −0.276; P = 0.038) indicating a lower inflammation index in PAH lungs expressing higher levels of PGIS mRNA. PGIS mRNA expression, however, showed a trend for correlation to mPAP (r = +0.240; P = 0.072) but was not correlated with pulmonary vascular resistance (PVR) (r = +0.172; P = 0.204; see Figure E6), even though mPAP and PVR are highly correlated to each other because of their derivation (r = +0.721; P = 3.8 × 10−10).
Discussion
Our study examined the diversity of sequence polymorphisms of the PGIS promoter and the relationship between promoter sequence variants and expression. We found that increasing VNTR length is correlated to promoter activity in both epithelial and endothelial cells. We also investigated the effect SNP4 genotype had on activity, because the minor allele (A) was significantly overrepresented in BMPR2 unaffected carriers. We found the minor allele to have increased transcriptional activity in both cell types. We expected similar trends in HPASMC, but did not observe this because of limited responsiveness of our promoter constructs in this assay. With the availability of lung tissue from the PHBI, we found similar relationships in endogenous PGIS mRNA and protein expression and promoter genotype.
PGIS mRNA expression showed a trend toward a positive correlation to mPAP but no correlation to PVR. This somewhat paradoxical result may be caused by several confounding factors. First, there was a significant time delay between the PAH patient’s last cardiac catheterization and lung transplantation (mean, 357 d; median, 138 d). Second, 93% of the patients with PAH received prostacyclin treatment making interpretation complicated. Finally, most patients with PAH are at or near end-stage disease (right ventricular heart failure) when transplantation occurs, perhaps making the correlation of PGIS mRNA levels in the lung tissue and previously collected hemodynamic measures unreliable.
Recently, Stacher and coworkers (21) published a semiquantitative, morphometric analysis of the vascular pathology in PAH using clinically and phenotypically annotated lung samples from patients and control subjects (failed donors) provided by the PHBI. Their results reinforced the importance of intima and media thickening observed in the PAH vasculature, and a correlation of perivascular inflammation with the remodeled vessels. Media fractional thickness and perivascular inflammation were significantly correlated with increased mPAP. Supporting an antiinflammatory role for prostacyclin, we found increased PGIS mRNA lung expression inversely correlated with inflammatory index from the same individuals.
Prostacyclin signaling is known to have strong antiinflammatory effects in various disease model systems (reviewed in [22]). In airway allergic inflammation, Th2 inflammatory cytokines are induced in prostacyclin receptor knockout mice (23–25). Exogenous treatment with prostacyclin decreased these effects. PGIS overexpression decreased nuclear factor-κB transcription factor activation driven by proinflammatory cytokines (26). Proinflammatory cytokines also seem to decrease the prostacyclin signaling in HPASMC (27). In the bleomycin model of pulmonary fibrosis, prostacyclin is protective by its antiproliferative action on fibroblasts and a reduction of inflammatory cells (28–30). The proposed model for prostacyclin’s effects included down-regulation of proinflammatory and profibrotic cytokines (31).
We hypothesized that increased PGIS expression in BMPR2 unaffected carriers is protective in preventing PAH. Increased lung PGIS expression predicted in unaffected carriers may be able to directly influence the pathologic vascular and inflammatory (22) changes by localized expression of prostacyclin at the appropriate levels, affecting the proper cell types. One limitation of this study has been the focus on patients with HPAH with BMPR2 mutations. Presumably, unaffected carriers also occur in the other known HPAH-based genetic loci. It would be interesting to test how generalizable our findings are in their disease progression.
The triple pharmacotherapy (endothelin receptor antagonists, phosphodiesterase 5 inhibitors, and prostanoids) seemed to produce little amelioration of the pathobiology observed in PAH lung tissue (21). Plexiform lesions were not correlated with either mPAP or PVR, perhaps contradicting the understanding that these vascular obstructions are a driver of PA hypertension. There was, however, a correlation of increased plexiform lesions in patients using prostanoids. We speculate that one explanation for little improvement in the lung vasculature of treated patients with PAH is that systemic administration of these drugs relieves the hypertensive aspects of the disease, while perhaps slowing but not preventing the progressive nature of the pathologic changes, including inflammation and vessel wall thickening. Part of end-stage disease may be contributed by vascular desensitization and tolerance of prostacyclin signaling as observed in pulmonary artery smooth muscle cell in vitro systems (27, 32). New therapeutics targeting perivascular inflammation and/or vessel wall thickening may be important future avenues of investigation. In conclusion, we found that unaffected carriers in HPAH families have a bias for PGIS promoter polymorphisms, which increase synthesis of PGIS, and more active PGIS promoter sequences were associated with a lower risk for developing PAH. Our findings support the hypothesis that PGIS promoter polymorphisms play a role as a genetic modifier in PAH disease.
Footnotes
Supported by NHLBI (R01 HL089508, P01 HL014985, R01 HL089508), and Cardiovascular Medical Research and Education Fund, IPAH Core Center for Genomics (M.W.G.); Parker B. Francis fellowship and NHLBI (K08HL105536) (B.B.G.); and University of Colorado Denver Aurora Lights Program, HRSA Health Career Opportunity Grant (M.J.D.R.).
Author Contributions: All authors meet the ICMJE criteria for authorship as defined as substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content; and final approval of the version to be published.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201309-1697OC on March 7, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Sitbon O, Morrell N. Pathways in pulmonary arterial hypertension: the future is here. Eur Respir Rev. 2012;21:321–327. doi: 10.1183/09059180.00004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waxman AB, Zamanian RT. Pulmonary arterial hypertension: new insights into the optimal role of current and emerging prostacyclin therapies. Am J Cardiol. 2013;111:1A–16A; quiz 17A–19A. doi: 10.1016/j.amjcard.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 3.U.S. National Library of Medicine. Genetics Home Reference [updated 2012 Sep, accessed 2013 Aug 15]. Available from. http://ghr.nlm.nih.gov/condition/pulmonary-arterial-hypertension
- 4.Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–1087. doi: 10.1183/09031936.00072709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm Circ. 2011;1:305–319. doi: 10.4103/2045-8932.87293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma L, Chung WK. The genetic basis of pulmonary arterial hypertension. Hum Genet. doi: 10.1007/s00439-014-1419-3. [DOI] [PubMed] [Google Scholar]
- 7.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(Suppl. 1):S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32:1385–1389. doi: 10.1002/humu.21605. [DOI] [PubMed] [Google Scholar]
- 9.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, III, Palomero T, Sumazin P, Kim HR, Talati MH, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Trégouët DA, Borczuk A, Rosenzweig EB, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–361. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jardim C, Hoette S, Souza R. Contemporary issues in pulmonary hypertension. Eur Respir Rev. 2010;19:266–271. doi: 10.1183/09059180.00008810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stearman RS, Grady MC, Nana-Sinkam P, Varella-Garcia M, Geraci MW. Genetic and epigenetic regulation of the human prostacyclin synthase promoter in lung cancer cell lines. Mol Cancer Res. 2007;5:295–308. doi: 10.1158/1541-7786.MCR-06-0221. [DOI] [PubMed] [Google Scholar]
- 13.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 14.Cathcart MC, Reynolds JV, O'Byrne KJ, Pidgeon GP. The role of prostacyclin synthase and thromboxane synthase signaling in the development and progression of cancer. Biochim Biophys Acta. 2010;1805:153–166. doi: 10.1016/j.bbcan.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez S, Gaunt TR, Day IN. Hardy-Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol. 2009;169:505–514. doi: 10.1093/aje/kwn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith AV, Thomas DJ, Munro HM, Abecasis GR. Sequence features in regions of weak and strong linkage disequilibrium. Genome Res. 2005;15:1519–1534. doi: 10.1101/gr.4421405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis CM, Knight J. Introduction to genetic association studies. Cold Spring Harbor Protocols. 2012;2012:297–306. doi: 10.1101/pdb.top068163. [DOI] [PubMed] [Google Scholar]
- 20.Solé X, Guinó E, Valls J, Iniesta R, Moreno V. SNPStats: a web tool for the analysis of association studies. Bioinformatics. 2006;22:1928–1929. doi: 10.1093/bioinformatics/btl268. [DOI] [PubMed] [Google Scholar]
- 21.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–272. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorris SL, Peebles RS., Jr PGI2 as a regulator of inflammatory diseases. Med Inflamm. 2012;2012:926–968. doi: 10.1155/2012/926968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaffar Z, Ferrini ME, Shaw PK, FitzGerald GA, Roberts K. Prostaglandin I₂promotes the development of IL-17-producing γδ T cells that associate with the epithelium during allergic lung inflammation. J Immunol. 2011;187:5380–5391. doi: 10.4049/jimmunol.1101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaffar Z, Wan KS, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J Immunol. 2002;169:5997–6004. doi: 10.4049/jimmunol.169.10.5997. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi Y, Tokuoka S, Masuda T, Hirano Y, Nagao M, Tanaka H, Inagaki N, Narumiya S, Nagai H. Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. Br J Pharmacol. 2002;137:315–322. doi: 10.1038/sj.bjp.0704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurgul-Convey E, Lenzen S. Protection against cytokine toxicity through endoplasmic reticulum and mitochondrial stress prevention by prostacyclin synthase overexpression in insulin-producing cells. J Biol Chem. 2010;285:11121–11128. doi: 10.1074/jbc.M109.054775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Haroun H, Clarke DL, Deacon K, Bradbury D, Clayton A, Sutcliffe A, Knox AJ. IL-1beta, BK, and TGF-beta1 attenuate PGI2-mediated cAMP formation in human pulmonary artery smooth muscle cells by multiple mechanisms involving p38 MAP kinase and PKA. Am J Physiol Lung Cell Mol Physiol. 2008;294:L553–L562. doi: 10.1152/ajplung.00044.2006. [DOI] [PubMed] [Google Scholar]
- 28.Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL, Koller BH. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2006;291:L144–L156. doi: 10.1152/ajplung.00492.2005. [DOI] [PubMed] [Google Scholar]
- 29.Zhou W, Dowell DR, Geraci MW, Blackwell TS, Collins RD, Polosukhin VV, Lawson WE, Wu P, Sussan T, Biswal S, et al. PGI synthase overexpression protects against bleomycin-induced mortality and is associated with increased NQO 1 expression. Am J Physiol Lung Cell Mol Physiol. 2011;301:L615–L622. doi: 10.1152/ajplung.00224.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, Xiao Y, Guo Z, Gao J. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir Res. 2010;11:34. doi: 10.1186/1465-9921-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou W, Hashimoto K, Goleniewska K, O’Neal JF, Ji S, Blackwell TS, Fitzgerald GA, Egan KM, Geraci MW, Peebles RS., Jr Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol. 2007;178:702–710. doi: 10.4049/jimmunol.178.2.702. [DOI] [PubMed] [Google Scholar]
- 32.Sobolewski A, Jourdan KB, Upton PD, Long L, Morrell NW. Mechanism of cicaprost-induced desensitization in rat pulmonary artery smooth muscle cells involves a PKA-mediated inhibition of adenylyl cyclase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L352–L359. doi: 10.1152/ajplung.00270.2003. [DOI] [PubMed] [Google Scholar]