

Abstract
Large-conductance voltage- and Ca2+-activated K+ (BK) channels are critical regulators of detrusor smooth muscle (DSM) excitability and contractility. PKC modulates the contraction of DSM and BK channel activity in non-DSM cells; however, the cellular mechanism regulating the PKC-BK channel interaction in DSM remains unknown. We provide a novel mechanistic insight into BK channel regulation by PKC in DSM. We used patch-clamp electrophysiology, live-cell Ca2+ imaging, and functional studies of DSM contractility to elucidate BK channel regulation by PKC at cellular and tissue levels. Voltage-clamp experiments showed that pharmacological activation of PKC with PMA inhibited the spontaneous transient BK currents in native freshly isolated guinea pig DSM cells. Current-clamp recordings revealed that PMA significantly depolarized DSM membrane potential and inhibited the spontaneous transient hyperpolarizations in DSM cells. The PMA inhibitory effects on DSM membrane potential were completely abolished by the selective BK channel inhibitor paxilline. Activation of PKC with PMA did not affect the amplitude of the voltage-step-induced whole cell steady-state BK current or the single BK channel open probability (recorded in cell-attached mode) upon inhibition of all major Ca2+ sources for BK channel activation with thapsigargin, ryanodine, and nifedipine. PKC activation with PMA elevated intracellular Ca2+ levels in DSM cells and increased spontaneous phasic and nerve-evoked contractions of DSM isolated strips. Our results support the concept that PKC activation leads to a reduction of BK channel activity in DSM via a Ca2+-dependent mechanism, thus increasing DSM contractility.
Keywords: detrusor, phorbol 12-myristate 13-acetate, ryanodine receptors
detrusor smooth muscle (DSM) contractility is regulated by a complex integration of neurogenic and myogenic mechanisms (1). Alteration of these mechanisms may result in DSM dysfunction and development of bladder disorders, such as overactive bladder (OAB). DSM contraction during the urinary bladder voiding phase is predominantly initiated by activation of muscarinic, particularly M3, receptors (1, 10). The principal downstream effect of the M3 receptor signaling pathway is activation of phospholipase C via the α-subunits of Gq/11 proteins, subsequently leading to the formation of inositol trisphosphate (IP3) and diacylglycerol (DAG) (10, 36). IP3 activates IP3 receptors in the sarcoplasmic reticulum (SR) to promote SR Ca2+ release, whereas DAG activates PKC. PKC is a key enzyme involved in regulation of DSM contractility. A number of downstream targets of PKC, including phosphorylation of the CPI-17 regulatory protein (6, 35) and direct interactions with L-type voltage-gated Ca2+ (CaV) channels (19), promote bladder contraction. However, the molecular targets, especially ion channels, for the PKC signaling pathway in DSM are not completely understood.
DSM expresses several types of K+ channels, including large-conductance voltage- and Ca2+-activated K+ (BK) channels, small-conductance Ca2+-activated K+ channels, voltage-gated K+ channels, and two-pore domain K+ channels (33). The BK channel is the most physiologically relevant K+ channel involved in regulation of DSM function due to its large single-channel conductance and its unique properties controlled by voltage and Ca2+ (12, 14, 16, 17, 24, 33, 34). In some non-DSM tissues, BK channels can be activated by stretch (35, 44); however, the direct effect of stretch on BK channel activity in DSM remains to be investigated. BK channels play a significant role in the pathology of certain forms of OAB, such as neurogenic detrusor overactivity (NDO) and OAB associated with bladder obstruction due to benign prostatic hyperplasia (7, 13, 31). Furthermore, the BK channel has been identified as a target for pharmacological and genetic control of OAB (7, 8, 14, 17, 21, 25, 26, 31, 34, 40). Despite the key importance of the BK channel in DSM, many aspects concerning regulatory mechanisms are not completely understood. In DSM, localized Ca2+ releases from ryanodine receptors (RyRs), known as “Ca2+ sparks,” activate adjacent BK channels, inducing transient BK currents (TBKCs), also known as spontaneous transient outward currents (12, 14, 16, 17, 34, 45–47).
In addition to Ca2+ and voltage, protein kinases, such as PKA (16, 45–47) and PKC (20), also modulate BK channel activity in DSM. PKA activation increases BK channel activity in DSM cells (5, 16, 34, 45–47); however, the mechanism of the PKC-BK channel interaction in DSM is less clear. A recent study demonstrated a functional link between M3 receptors and BK channels in DSM, where pharmacological activation of M3 receptors with carbachol has been shown to inhibit BK channel activity (32). It has been suggested that this process may be mediated by PKC. However, the involvement of PKC signaling pathways in M3 receptor-induced inhibition of BK channels in DSM remains unexplored. Another study using the PKC inhibitor bisindolylmaleimide (Bim-1) and the PKC activator phorbol 12,13-dibutyrate (PDBu), as well as the BK channel activators NS-1619 and isopimaric acid, suggests that PKC may play an important role in DSM contractility by affecting BK channels (20). Despite these initial observations, the cellular and molecular mechanisms by which PKC interacts with BK channels have not been elucidated in DSM.
In arterial smooth muscle, PKC activation usually leads to inhibition of BK channel activity (3, 4, 27, 38, 42, 49). However, in Sprague-Dawley rat pulmonary arteries, stimulation of PKC increases BK channel activity (2, 50). This variation of PKC-induced effects may be attributed to experimental conditions based on species, strains, and tissue/organ differences. Furthermore, functional association of BK channels with different PKC isoenzymes, splice variants of the α- or β-subunit composition of the BK channel, as well as regulatory input from distinct signaling pathways, may also play a role.
Another important issue is how PKC interacts with the BK channels at the cellular level. In different cell types, the molecular mechanism of PKC-BK channel functional coupling can involve a direct phosphorylation of the BK channel α-subunit (37, 43, 49) or an indirect interaction (37). The indirect interactions could involve modulation of BK channel activity via intracellular Ca2+ (4, 37). In cerebral arteries, activation of PKC with PMA decreases Ca2+ spark and TBKC frequency (4).
The aim of the present study is to provide a novel mechanistic insight into BK channel regulation by PKC in DSM and to elucidate the physiological significance of these interactions at the cellular and tissue levels. We used a combined experimental approach, including patch-clamp electrophysiology, live-cell Ca2+ imaging, recordings of isometric tension in guinea pig isolated DSM strips, and pharmacological protocols utilizing the PKC activator PMA. The results are consistent with the idea that, in DSM cells, PKC inhibits BK channel activity indirectly via a Ca2+-dependent mechanism involving attenuation of Ca2+ release through RyRs while increasing the global intracellular Ca2+ levels necessary to activate DSM contraction.
MATERIALS AND METHODS
Tissue preparation.
Sixty-nine male Harley albino guinea pigs (Charles River Laboratories, Raleigh, NC) were used in this study (434.4 ± 8.1 g body wt). The animals were euthanized with CO2 and then subjected to a thoracotomy according to Animal Use Protocol 1747, reviewed and approved by the Institutional Animal Care and Use Committee of the University of South Carolina. DSM tissue dissection and preparation of DSM strips were performed as previously described (15, 39, 45, 47).
DSM cell isolation.
Guinea pig DSM single cells were enzymatically isolated using papain and collagenase, as previously described (15, 39, 45, 47). Single DSM cells were used for patch-clamp and live-cell Ca2+-imaging studies within 12 h after isolation.
Perforated patch-clamp recordings.
A few drops of the DSM cell suspension were placed into a glass bottom of a recording chamber. The cells were allowed to settle for ≥20 min and then washed with an extracellular bath solution. The amphotericin B-perforated whole cell patch-clamp technique was used to record TBKCs, membrane potential, and voltage-step depolarization-induced whole cell BK current. TBKCs and voltage-step depolarization-induced whole cell BK current were recorded in voltage-clamp mode, whereas membrane potential and spontaneous transient hyperpolarization (STH) were measured in current-clamp mode. An Axopatch 200B amplifier (Digidata 1322A) and pCLAMP version 10.2 software (Molecular Devices, Union City, CA) were used, and the currents were filtered using an eight-pole Bessel filter (model 900CT/9L8L, Frequency Devices, Ottawa, IL). The patch-clamp pipettes were made from borosilicate glass (Sutter Instruments, Novato, CA) and pulled using a vertical puller (model PP-830, Narishige Group, Tokyo, Japan). The pipettes were then polished with a fire polisher (Micro Forge MF-830, Narishige Group). Pipette resistance was 4–6 MΩ. All patch-clamp experiments were conducted at room temperature (22–23°C).
Single-channel recordings.
Single BK channel recordings were performed using the cell-attached patch-clamp technique. Single-channel recordings were conducted in the presence of thapsigargin (100 nM), a blocker of SR Ca2+-ATPase, ryanodine (30 μM), a blocker of RyRs, and nifedipine (1 μM), a blocker of CaV channels, to eliminate all major cellular sources of Ca2+ for BK channel activation. We applied command voltage of −60 mV to determine the effect of PMA on BK channel open probability (NPo). This approximation corresponds to cell membrane potential of +60 mV, if it is assumed that the cell membrane potential is 0 mV under the recording conditions of high K+ for bath and pipette solutions. All single-channel recordings were conducted at room temperature (22–23°C).
Ca2+-imaging experiments.
Intracellular Ca2+-imaging experiments were performed as previously described (15, 39, 47).
Isometric DSM tension recordings.
Isometric DSM tension recordings were conducted using mucosa-free DSM strips (∼2–3 mm wide × 5–6 mm long), as previously described (15, 39, 45, 47). In the first set of experimental series, the DSM strips exhibiting spontaneous phasic contractions were allowed to equilibrate for ≥30 min. Thereafter, increasing concentrations of PMA (1 nM–30 μM) or its inactive analog 4α-PMA (1 nM–30 μM) were applied at 10-min intervals. In a separate set of experimental series, paxilline (1 μM), a BK channel blocker, was applied; then the DSM contractions were allowed to reach a stable level before cumulative application of PMA (1 nM–30 μM) or 4α-PMA (1 nM–30 μM). To minimize potential effects caused by neurotransmitter release, the experiments on DSM strips exhibiting spontaneous phasic contractions were performed in the presence of 1 μM tetrodotoxin (TTX), a selective blocker of neuronal voltage-gated Na+ channels.
In the second experimental series, nerve-evoked DSM contractions were induced by electrical field stimulation (EFS) using a pair of platinum electrodes mounted in the tissue bath parallel to the DSM strip. The EFS pulses were generated using a PHM-152I stimulator (Med Associates, Georgia, VT). The EFS pulse parameters were as follows: 0.75-ms pulse width, 20-V pulse amplitude, and 3-s stimulus duration, with polarity reversed for alternating pulses. For EFS studies, after the equilibration period, DSM strips were subjected to continuous stimulation with a 20-Hz stimulation frequency at 1-min intervals.
Solutions and drugs.
The Ca2+-free dissection solution, the extracellular bath solution used in the perforated patch-clamp and Ca2+-imaging experiments, and the patch-clamp pipette solution for perforated patch-clamp experiments were prepared as previously described (14, 15). Pipette solutions in perforated patch-clamp experiments were supplemented with freshly dissolved (every 1–2 h) 200 μg/ml amphotericin B. The patch-pipette solution for single BK channel recordings contained (in mM): 140 KCl, 1.08 MgCl2, 5 EGTA, 1 HEPES, and 3.16 CaCl2 [free Ca2+ concentration was calculated at ∼300 nM with WEBMAXC Standard (http://www.stanford.edu/∼cpatton/webmaxcS.htm, Chris Patton)]. The pH of the solution was adjusted to 7.2 with NaOH. Extracellular (bath) solution used for the single-channel recordings contained (in mM): 140 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, and 4 NaOH (pH 7.35). For DSM contraction studies, physiological saline solution (in mM: 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgSO4, and 11 glucose, aerated with 95% O2-5% CO2 to obtain pH 7.4) was prepared daily. BSA and amphotericin B were obtained from Thermo Fisher Scientific, ryanodine was obtained from Enzo Life Sciences (Farmingdale, NY), and all other drugs were obtained from Sigma-Aldrich (St. Louis, MO). Stock solutions of TTX and nifedipine were prepared in citrate buffer and ethanol, respectively. PMA and 4α-PMA were dissolved in DMSO, and the final DMSO concentration in the bath did not exceed 0.1%.
Statistical analysis.
MiniAnalysis software (Synaptosoft, Decatur, GA) was used to analyze the amplitude and frequency of TBKCs and STHs and DSM contraction parameters. Whole cell steady-state BK current and membrane potential were analyzed by Clampfit 10.2 software (Molecular Devices). To evaluate the effect of PMA on voltage-step-induced whole cell current, the mean value of the last 50 ms of the 200-ms pulse before and after application of PMA (100 nM) was calculated. The effects of PMA on the amplitude and frequency of TBKCs or STHs were normalized to control values and are expressed in percentages. The values for single-channel total NPo were obtained using a built-in algorithm in Clampfit, which calculates as follows: NPo = TO/(TO + TC), where TO and TC correspond to total open time and total closed time during the recording interval, respectively. Single-channel events were analyzed over 10-min intervals prior to and after addition of PMA (100 nM). DSM contractile activity was quantified by measuring average phasic contraction amplitude (the difference between the force-time baseline curve and the maximum peak of the contractions), frequency (contractions/min), muscle force integral (calculated by integrating the area under the force-time baseline curve), phasic contraction duration (defined as width of the individual phasic contraction at 50% of the amplitude), and DSM tone (the difference between the zero line and the force-time baseline curve). To compare the contraction parameters for spontaneous, paxilline-induced, or EFS-induced contractions, data were normalized to the last 5 min of the contractions before addition of the first concentration of PMA or 4α-PMA in the presence or absence of paxilline (taken to be 100% for all configuration parameters) and are expressed as percentages. Statistical analyses were performed with GraphPad Prism version 4.3 (GraphPad Software, La Jolla, CA), and CorelDraw Graphic Suite X3 software (Corel) was used to illustrate the data. EC50 values were obtained using a sigmoidal fitting function in the GraphPad Prism software and are reported as means (95% confidence interval). Values are means ± SE, where n is the number of strips or cells and N is the number of guinea pigs. Statistical significance was tested using two-way ANOVA or paired Student's t-test; P < 0.05 was considered significant.
RESULTS
Pharmacological activation of PKC with PMA decreases the amplitude and frequency of TBKCs in freshly isolated DSM cells.
The average DSM cell capacitance of all cells used in perforated patch-clamp experiments was 17.8 ± 1.3 pF (n = 40, N = 29). TBKCs were recorded in voltage-clamp mode, and DSM cells were held −20 mV. Pharmacological activation of PKC with PMA (100 nM) significantly inhibited TBKC amplitude and frequency by 65.4 ± 16.6% and 93.9 ± 2.7%, respectively, in DSM cells (n = 7, N = 7, P < 0.05; Fig. 1, A and B).
Fig. 1.
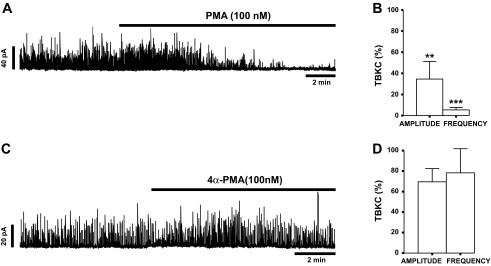
Pharmacological activation of PKC with PMA leads to inhibition of spontaneous transient large-conductance voltage- and Ca2+-activated K+ (BK) currents (TBKCs) in guinea pig detrusor smooth muscle (DSM) cells. A: original recording illustrating the inhibitory effect of PMA (100 nM) on TBKC activity in a guinea pig DSM cell. B: summary data depicting the inhibitory effect of 100 nM PMA on amplitude and frequency of TBKCs (n = 7, N = 7). **P < 0.01, ***P < 0.001. C: original recording illustrating lack of an effect of 100 nM 4α-PMA on TBKC amplitude and frequency in a DSM cell. D: summary data showing lack of a significant effect of 4α-PMA (100 nM) on amplitude and frequency of TBKCs (n = 7, N = 5, P > 0.05).
To confirm that the inhibitory effects on TBKCs were due to PKC activation, rather than some other effect of PMA, we performed additional experiments with the inactive PMA analog 4α-PMA, which does not activate PKC. As illustrated in Fig. 1, C and D, 4α-PMA (100 nM) did not significantly change the amplitude or frequency of TBKCs (n = 7, N = 5, P > 0.05). Taken together, the data indicate that PKC is a critical regulator of TBKCs in DSM cells.
PKC activation with PMA leads to reductions in STH amplitude and frequency and depolarizes the membrane potential in freshly isolated DSM cells.
In this experimental series, the effect of PMA on membrane potential in DSM cells was investigated using the current-clamp mode of the perforated patch-clamp technique. Activation of PKC with PMA (100 nM) depolarized the membrane potential by 3.9 ± 1.3 mV (n = 16, N = 13, P < 0.05; Fig. 2, A and B). In current-clamp mode, DSM cells exhibit STHs, which correspond to TBKCs recorded in voltage-clamp mode and are caused by Ca2+ sparks from RyRs (12, 47). PKC activation with PMA (100 nM) caused a significant inhibition of STH amplitude and frequency by 42.3 ± 15.3% and 64.6 ± 16.9%, respectively (n = 5, N = 5, P < 0.05; Fig. 2C).
Fig. 2.

PKC activation with PMA leads to inhibition of spontaneous transient hyperpolarizations (STHs) and depolarizes the membrane potential (MP) in freshly isolated DSM cells. A: original recording illustrating the effect of 100 nM PMA on STHs and membrane potential recorded in current-clamp mode in a DSM cell. B: summary data illustrating the depolarizing effect of PMA (100 nM) on membrane potential (MP) in guinea pig DSM cells (n = 16, N = 13; *P < 0.05). C: summary data depicting the inhibitory effect of PMA (100 nM) on amplitude and frequency of STHs in DSM cells (n = 5, N = 5; *P < 0.05). D: original recording in current-clamp mode illustrating lack of an effect of PMA (100 nM) on membrane potential in the presence of paxilline (1 μM). E: summary data illustrating lack of an effect of 100 nM PMA on DSM cell membrane potential in the presence of 1 μM paxilline (n = 5, N = 5, P > 0.05).
To investigate whether the inhibitory effects of PMA were due to BK channel activity, we performed membrane potential recordings in the presence of the selective BK channel blocker paxilline. In the presence of paxilline (1 μM), DSM cells did not exhibit STHs, confirming that STHs are caused by BK channel activity. The results indicate that PMA (100 nM) did not affect the membrane potential of DSM cells after inhibition of the BK channels with 1 μM paxilline (n = 5, N = 5, P > 0.05; Fig. 2, D and E). These results suggest a key physiological role for the BK channels in mediating the inhibitory effects of PMA and support the concept that PKC activation increases DSM cell excitability in a BK channel-dependent manner.
The PKC activator PMA has no effect on the voltage-step-induced whole cell steady-state BK current in DSM cells after inhibition of all major Ca2+ sources for BK channel activation.
In the next experimental series, DSM cells were held at −70 mV, and brief voltage-step depolarizations (200 ms) from −40 to +80 mV in 20-mV intervals were applied. To remove all major Ca2+ sources for BK channel activation, nifedipine (1 μM), a selective CaV channel inhibitor, thapsigargin (100 nM), a selective SR Ca2+ pump inhibitor, and ryanodine (30 μM), a selective RyR inhibitor, were applied in the bath solution. Under these experimental conditions, DSM cells responded to each depolarizing voltage step by gradually increasing outward BK currents (Fig. 3A). As illustrated in Fig. 3, activation of PKC with PMA (100 nM) did not significantly affect the whole cell steady-state BK current density at all recording voltages (n = 6, N = 4, P > 0.05; Fig. 3B). At +80 mV, current density was 11.7 ± 1.3 pA/pF for controls and 12.2 ± 1.6 pA/pF for cells activated with 100 nM PMA (n = 6, N = 4, P > 0.05; Fig. 3B). These results indicate that, when all major Ca2+ sources for BK channel activation are inhibited, PKC activation did not affect the voltage-step-induced whole cell steady-state BK current in guinea pig DSM cells.
Fig. 3.

PKC activation with PMA has no effect on voltage-step-induced whole cell steady-state BK currents after inhibition of all known major sources of Ca2+ for BK channel activation. A: original recording illustrating voltage-step-induced whole cell steady-state BK current prior to (control) and after addition of 100 nM PMA. B: current-voltage relationship illustrating lack of an effect of PMA (100 nM) on voltage-step-induced whole cell steady-state BK current in DSM cells (n = 6, N = 4, P > 0.05). All recordings were performed with thapsigargin (100 nM), ryanodine (30 μM), and nifedipine (1 μM) in the bath solution.
PMA does not change the single BK channel activity in the presence of thapsigargin, ryanodine, and nifedipine.
To examine whether PMA exerts a direct effect on single BK channel activity, we conducted single BK channel recordings using the cell-attached configuration of the single-channel patch-clamp technique. These recordings were conducted under conditions of pharmacological inhibition of major cellular sources of Ca2+ for BK channel activation with thapsigargin (100 nM), ryanodine (30 μM), and nifedipine (1 μM). In the absence of PMA, NPo was 0.009 ± 0.007 at a membrane potential of +60 mV (n = 5, N = 5; Fig. 4). PMA (100 nM) did not significantly alter the single BK channel NPo (n = 5, N = 5, P > 0.05; Fig. 4) or the unitary current amplitudes (n = 5, N = 5, P > 0.05; Fig. 4). These experiments indicate that the PKC activator PMA did not directly alter the activity of single BK channels when all major sources of Ca2+ for BK channel activation were inhibited.
Fig. 4.

Lack of an inhibitory effect of PMA on single BK channels recorded with cell-attached configuration in freshly isolated DSM cells under conditions of pharmacological inhibition of all major cellular sources of Ca2+ for BK channel activation. A: representative recordings of single BK channel currents measured at membrane potential of +60 mV prior to (control) and after addition of 100 nM PMA. C, closed channel state. B: single BK channel open probability (NPo) values over a 10-min interval before and after addition of 100 nM PMA for patch-clamp recording in A. C: summary data illustrating lack of an inhibitory effect of PMA (100 nM) on mean single BK channel NPo (n = 5, N = 5, P > 0.05). D: summary data demonstrating no change in mean single BK channel current amplitude in the presence and absence of 100 nM PMA (n = 5, N = 5, P > 0.05). All single-channel recordings were performed with thapsigargin (100 nM), ryanodine (30 μM), and nifedipine (1 μM) in the bath solution.
Pharmacological activation of PKC with PMA increases intracellular Ca2+ levels in freshly isolated DSM cells.
To determine the effect of PKC activation on intracellular Ca2+ levels, live-cell real-time Ca2+ imaging was performed with fura 2. As shown in Fig. 5, intracellular Ca2+ levels were significantly increased following application of PMA (100 nM). The fluorescence intensity ratio (ratio of fluorescence at 340 nm to fluorescence at 380 nm) was 0.79 ± 0.03 under control conditions and, in the presence of 100 nM PMA, increased to 1.03 ± 0.03 (n = 16, N = 6, P < 0.05; Fig. 5). These results revealed that pharmacological activation of PKC with PMA increases intracellular Ca2+ levels in freshly isolated DSM cells.
Fig. 5.

Pharmacological activation of PKC with PMA increases intracellular Ca2+ in freshly isolated DSM cells. A: original recording representing an increase in intracellular Ca2+ following application of 100 nM PMA. F340/F380, ratio of fluorescence intensity at 340 nm to fluorescence intensity at 380 nm. B: summary data illustrating a significant increase in intracellular Ca2+ after application of 100 nM PMA (n = 16, N = 6). *P < 0.05.
PKC activation with PMA increases spontaneous phasic contractions in DSM isolated strips.
Since our electrophysiological experiments revealed that PKC activation caused an inhibition of the TBKC activity in single DSM cells, our next step was to study the functional link between PKC and BK channel activity in DSM isolated strips. We tested the effect of PKC activation with PMA on DSM spontaneous phasic contractions. DSM isolated strips exhibiting spontaneous phasic contractions were exposed to increasing concentrations of PMA (1 nM–30 μM; Fig. 6A). PMA induced a concentration-dependent increase in spontaneous phasic contraction amplitude, duration, muscle force integral, and muscle tone in DSM isolated strips (n = 10, N = 6, P < 0.05; Fig. 6, A and C). PMA (30 μM) increased DSM spontaneous phasic contraction amplitude to 186 ± 26.1%, contraction duration to 132 ± 11.4%, muscle force integral to 257 ± 4.6%, and muscle tone to 168 ± 25.8% (n = 10, N = 6, P < 0.05 for all parameters; Fig. 6C). EC50 values for PMA-induced spontaneous phasic contraction parameters were as follows: phasic contraction amplitude, 0.18 (0.06–0.53, 95% confidence interval) μM; muscle force integral, 0.65 (0.17–2.53) μM; contraction duration, 6.18 (0.70–55.2) μM; tone, 0.83 (0.32–2.13) μM. These results indicate a key role for PKC in the regulation of DSM spontaneous phasic contractions.
Fig. 6.

Pharmacological activation of PKC with PMA leads to a significant increase in spontaneous phasic contractions of DSM isolated strips. A: original myograph recording showing contractile effects of PMA (1 nM–30 μM) on spontaneous phasic contractions in a DSM isolated strip. B: original myograph recording showing effects of 1 nM–30 μM PMA on spontaneous phasic contractions in a DSM isolated strip in the presence of paxilline (1 μM). C: cumulative PMA concentration-response (spontaneous phasic contraction amplitude, muscle force integral, duration, and muscle tone) curves of DSM strips in the presence (n = 12, N = 9) and absence (n = 10, N = 6) of 1 μM paxilline (Pax). *P < 0.05, **P < 0.01.
To evaluate the role of BK channels in PMA-induced spontaneous contractions, we examined the effects of PMA (1 nM–30 μM) in the presence of the BK channel selective inhibitor paxilline (1 μM). PMA slightly increased the spontaneous phasic contraction amplitude in the presence of 1 μM paxilline, but the effect was significantly less pronounced than in the absence of paxilline (n = 12, N = 9; Fig. 6). Furthermore, in the presence of 1 μM paxilline, PMA did not significantly affect phasic contraction duration, muscle force integral, or muscle tone. In the presence of paxilline, PMA (30 μM) changed the spontaneous DSM contraction amplitude to 119 ± 9.0%, contraction duration to 89 ± 4.9%, muscle force integral to 109 ± 10%, and muscle tone to 111 ± 10.5% (P > 0.05 for all parameters except amplitude, for which P < 0.05; Fig. 6C). Concentration-response curves for the effects of PMA (1 nM–30 μM) on all parameters of the spontaneous phasic contractions, in the absence or presence of 1 μM paxilline, are illustrated in Fig. 6C. These results suggest that the effects of PKC on DSM myogenic spontaneous phasic contractions are mediated by BK channels. To further confirm that the PMA effects were due to PKC activation, we performed experiments with the inactive PMA analog 4α-PMA. The results revealed that 4α-PMA (1 nM–30 μM) did not affect the spontaneous phasic contraction parameters of DSM isolated strips (n = 14, N = 9, P > 0.05).
PKC activation with PMA increases EFS-induced contractions in DSM isolated strips.
Next, we investigated the effect of PKC activation with PMA (1 nM–30 μM) on EFS-induced DSM contractions. In the absence of TTX, EFS-induced contractions were evoked by repetitive EFS (20 Hz each minute; Fig. 7). PKC activation with PMA (1 nM–30 μM) increased the amplitude and muscle force integral of EFS-induced contractions in a concentration-dependent manner (Fig. 7, A and C). The effects of PMA on EFS-induced contractions reached a maximum at 30 μM. PMA (30 μM) increased the amplitude of the EFS-induced contractions to 133 ± 9.4% and muscle force integral to 135 ± 9.8% (n = 8, N = 7, P < 0.05 for both parameters; Fig. 7A). EC50 values were as follows: contraction amplitude, 2.03 (0.95–4.33) μM; muscle force integral, 1.53 (0.71–3.29) μM.
Fig. 7.

PKC activation with PMA leads to a significant increase in continuous 20-Hz electrical field stimulation (EFS)-induced contractions of DSM isolated strips. A: original myograph recording showing contractile effect of PMA (1 nM–30 μM) on 20-Hz EFS-induced contractions of DSM isolated strips. B: original myograph recording showing effect of PMA (1 nM–30 μM) on 20-Hz EFS-induced contractions in the presence of paxilline (1 μM) in a DSM isolated strip. C: cumulative PMA concentration-response (20-Hz EFS-induced contraction amplitude and muscle force integral) curves of DSM strips in the presence (n = 9, N = 8) and absence (n = 8, N = 7) of 1 μM paxilline. *P < 0.05, **P < 0.01, ***P < 0.005.
In the next experimental series, we tested the effects of PKC activation with PMA on EFS-induced contractions in DSM isolated strips in the presence of the BK channel selective inhibitor paxilline. In the presence of 1 μM paxilline, the effects of PMA (1 nM–30 μM) on EFS-induced contractions were significantly reduced (n = 9, N = 8, P < 0.05; Fig. 7B). These experiments demonstrated that the effects of PMA on EFS-induced contractions in DSM are mediated by BK channels. 4α-PMA (1 nM–30 μM) did not alter the amplitude or muscle force of 20-Hz EFS-induced contractions of guinea pig DSM isolated strips (n = 8, N = 8, P > 0.05). The results from these experiments indicate that PKC activation plays an essential role in nerve-evoked contractions in DSM and that these effects are mediated by BK channels.
DISCUSSION
This study investigated the functional link between PKC and BK channels in DSM at the cellular and tissue levels. We reveal that pharmacological activation of PKC with PMA inhibits the amplitude and frequency of BK channel-mediated TBKCs and STHs and depolarizes the membrane potential in freshly isolated guinea pig DSM cells. Furthermore, activation of PKC with PMA increases intracellular Ca2+ levels in DSM cells and enhances the spontaneous phasic and nerve-evoked contractions in DSM isolated strips. The experiments with the selective BK channel inhibitor paxilline indicate that the effects of PMA on DSM excitability and contractility are mediated by the BK channel. Most importantly, we provide experimental evidence that the mechanism of BK channel inhibition by PKC in DSM is Ca2+-dependent.
The existence of a functional link between the BK channel and PKC in DSM has been recently suggested (20, 32), but the cellular mechanism of this interaction has not been investigated. In non-DSM, PKC can interact with BK channels through various signaling pathways, either directly or indirectly (4, 37, 43, 49). To study the cellular mechanism of PKC-BK channel interaction in DSM, we performed perforated patch-clamp experiments on freshly isolated DSM cells. This method was selected, because the perforated patch-clamp mode is less invasive than the conventional patch-clamp mode, and it preserves the cellular environment, allowing investigation of BK channels under native physiological conditions with all intracellular signaling pathways intact. The results from our perforated patch-clamp experiments reveal that pharmacological activation of PKC with PMA significantly reduces TBKC activity in DSM cells (Fig. 1). The current-clamp recordings show that PMA decreases the amplitude and frequency of STHs and depolarizes DSM cell membrane potential (Fig. 2). Since TBKCs and STHs in DSM are mediated by BK channel activity (14, 46), we hypothesized that the effects of PMA on DSM excitability involve the BK channels. This assumption is supported by the fact that the depolarizing effect of PMA on DSM membrane potential was not observed following inhibition of the BK channels with paxilline (Fig. 2). This finding is in line with recent reports indicating that the BK channels and PKC may interact at functional and cellular levels in DSM (20, 32).
In some vascular smooth muscle cells, PKC can inhibit BK channels directly via phosphorylation of the BK channel α-subunit (37, 43, 49), whereas in cerebral arteries, PKC inhibits BK channels via attenuation of Ca2+ release from the SR (4). Our results demonstrate that, following pharmacological inhibition of all major sources of Ca2+ for BK channel activation, PMA does not significantly change the voltage-step-induced whole cell steady-state BK current in DSM cells (Fig. 3). This observation indicates that intracellular Ca2+ is of key importance for the inhibitory effect of PMA on BK channel activity. To confirm this hypothesis, we performed additional experiments to measure single BK channel activity in the cell-attached mode of the patch-clamp technique in the presence of nifedipine, thapsigargin, and ryanodine. Activation of PKC with PMA did not significantly change single BK channel NPo in DSM cells after inhibition of all major sources of Ca2+ for BK channel activation (Fig. 4). This finding confirms our initial observation that a Ca2+-dependent mechanism mediates PKC-induced inhibition of BK channel activity in DSM.
RyRs are potential targets for PKC phosphorylation (5, 22). PKC phosphorylation of the RyRs has been suggested to be the primary mechanism for PKC-induced inhibition of the Ca2+ sparks in cerebral artery myocytes (4). In airway smooth muscle cells, PKC decreases Ca2+ sparks and contractile activity via a direct interaction with RyRs (22). On the basis of our electrophysiological experiments, it is reasonable to expect that a similar mechanism of PKC-induced phosphorylation of RyRs operates in DSM, but the regulatory protein of SR Ca2+-ATPase activity, phospholamban (PLB), is another plausible target for PKC phosphorylation. The latter hypothesis is based on an observation that PKC modulates the PLB activity in cardiac myocytes and, thus, regulates SR Ca2+ release (48). Although we cannot completely rule out the possibility of a direct PKC-BK channel interaction in DSM, our results clearly support the concept that PKC activation suppresses BK channel activity in DSM via an indirect mechanism involving SR Ca2+ stores.
Pharmacological inhibition of the BK channel with selective blockers, including peptides, such as iberiotoxin, or small molecules, such as paxilline, significantly increases DSM contractions (9, 11, 41). Enhancement of DSM contractility is a result of membrane depolarization, activation of CaV channels, and increase in global intracellular Ca2+ levels. Therefore, we expected and observed experimentally that inhibition of BK channel activity with PMA would increase intracellular Ca2+ levels in DSM cells (Fig. 5).
Furthermore, the results from live-cell Ca2+-imaging experiments confirm that PMA significantly increases intracellular Ca2+ levels in freshly isolated single DSM cells (Fig. 5), which supports the concept that PKC activity regulates intracellular Ca2+ dynamics in DSM. Elevation of intracellular Ca2+ by PKC activation leads to an increase in DSM contractions.
Our functional studies of DSM contractility show that pharmacological activation of PKC with PMA causes concentration-dependent increases in the parameters of spontaneous phasic contractions in DSM isolated strips (Fig. 6). The inactive 4α-PMA analog did not significantly change the parameters of spontaneous phasic DSM contractions, suggesting that stimulatory effects of PMA on DSM contractility are due to PKC activation. The PMA stimulatory effects on DSM contractility were significantly less pronounced in the presence of paxilline (Fig. 6), revealing involvement of BK channels in PMA-induced activation of DSM phasic contractions. These results support the concept that PKC increases DSM contractility by inhibition of BK channel activity. Our results are in line with the recent observation that suggests functional interactions between PKC and BK channels in DSM (20).
DSM contractility is regulated by autonomic neuronal pathways via release of excitatory neurotransmitters, such as acetylcholine. We recently showed that cholinergic stimulation of M3 receptors leads to BK channel inhibition (32). Furthermore, abnormal neuronal activity of elevated acetylcholine release could excessively increase DSM contractility, causing NDO (1). Recently, our research group revealed that BK channel expression and function are reduced in NDO (13). Here we show that pharmacological activation of PKC with PMA increases EFS-induced contractions in DSM isolated strips and that the effect is significantly less pronounced in the presence of paxilline (Fig. 7). The inactive analog 4α-PMA did not significantly change the parameters of EFS-induced contraction in DSM, supporting the concept that the contractile effects are due to PKC activation. Taken together, the results support the idea that PKC regulates nerve-evoked DSM contractions in a BK channel-dependent manner. Data from the EFS-induced contraction experiments (Fig. 7) suggest that PKC-BK channel interaction could play an important role in bladder contraction during the voiding phase. However, we cannot exclude the possibility that PKC has a regulatory role in maintaining bladder force during the storage phase.
Studies with another PKC activator (PDBu) showed a biphasic effect on DSM contractility in rabbits. Specifically, it reduced amplitude and increased frequency of DSM contractions at a lower concentration (10 nM) and increased muscle force at a higher concentration (1 μM) (20). The discrepancy between this observation and our findings is likely related to species differences in DSM physiology between guinea pigs and rabbits. Another plausible explanation is that the aforementioned study used PDBu, whereas we utilized the PKC activator PMA. PMA and PDBu are phorbol esters that mimic DAG, the natural activator of PKC; however, differences in pharmacological properties of PDBu and PMA were reported, and the results showed that PMA is a more potent PKC activator (30). We should also consider that there are 11 different PKC isoforms, classified into 3 major groups (18, 23, 28, 29). PKC isoforms differ markedly in expression and prevalence in different cell lines, tissues, and species (23). PKCα is suggested to be the major isoform in rabbit DSM (6); however, the isoform-specific downstream effects of PKC activation in DSM remain to be elucidated in future studies.
On the basis of the data obtained in this study, a proposed mechanism by which PKC regulates BK channel function in DSM cells is illustrated in Fig. 8. Activated PKC phosphorylates proteins, such as RyRs or PLB, in the SR (Fig. 8). This leads to inhibition of Ca2+ release from the SR (Ca2+ sparks) and subsequent inhibition of TBKCs. BK channel inhibition causes DSM cell membrane potential depolarization, activation of the CaV channels, increase in global intracellular Ca2+ concentration, and activation of DSM contraction (Fig. 8).
Fig. 8.

Schematic diagram illustrating proposed cellular mechanisms by which PKC regulates BK channel function in DSM. PMA activates PKC, which leads to inhibition of ryanodine receptors (RyRs) or sarcoplasmic reticulum (SR) Ca2+ pump. This causes a decrease in Ca2+ spark and TBKC activity, leading to DSM cell membrane depolarization, L-type voltage-dependent Ca2+ (CaV) channel activation, an increase in intracellular Ca2+, and DSM contraction. Pharmacological tools used in this study to inhibit cellular sources of Ca2+ are indicated. PLB, phospholamban.
In conclusion, we found that PKC and BK channels are functionally linked in DSM. Our patch-clamp studies reveal that pharmacological activation of PKC with PMA significantly decreases the amplitude and frequency of TBKCs and STHs and depolarizes the DSM cell membrane potential. In the absence of all major cellular sources of Ca2+ for BK channel activation, PKC activation with PMA did not change voltage-step-induced whole cell BK current or the single BK channel NPo recorded in the cell-attached mode. These findings support the concept that PKC inhibits BK channel activity indirectly by suppressing intracellular Ca2+ release through SR RyRs. Therefore, this study provides novel mechanistic insight into the regulation of DSM excitability. Our Ca2+-imaging experiments reveal that PKC activation with PMA elevates intracellular Ca2+ levels in DSM cells. Our functional studies on DSM contractility reveal that pharmacological activation of PKC with PMA significantly increases the parameters of spontaneous phasic and nerve-evoked DSM contractions and that these effects are mediated by the BK channel.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-084284 to G. V. Petkov.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.L.H., A.C.S., S.P.P., J.M., and G.V.P. performed the experiments; K.L.H., A.C.S., S.P.P., J.M., and G.V.P. analyzed the data; K.L.H., A.C.S., S.P.P., J.M., and G.V.P. interpreted the results of the experiments; K.L.H., A.C.S., S.P.P., J.M., and G.V.P. prepared the figures; K.L.H., A.C.S., and G.V.P. drafted the manuscript; K.L.H., A.C.S., S.P.P., J.M., and G.V.P. edited and revised the manuscript; K.L.H., A.C.S., S.P.P., J.M., and G.V.P. approved the final version of the manuscript; G.V.P. is responsible for conception and design of the research.
ACKNOWLEDGMENTS
We thank Dr. Wenkuan Xin, Aaron Provence, and Vítor Fernandes for critical evaluation of the manuscript.
REFERENCES
- 1.Andersson KE, Arner A. Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol Rev 84: 935–986, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Barman SA, Zhu S, White RE. PKC activates BKCa channels in rat pulmonary arterial smooth muscle via cGMP-dependent protein kinase. Am J Physiol Lung Cell Mol Physiol 286: L1275–L1281, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Barman SA, Zhu S, White RE. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 286: L149–L155, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bonev AD, Jaggar JH, Rubart M, Nelson MT. Activators of protein kinase C decrease Ca2+ spark frequency in smooth muscle cells from cerebral arteries. Am J Physiol Cell Physiol 273: C2090–C2095, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Brown SM, Bentcheva-Petkova LM, Liu L, Hristov KL, Chen M, Kellett WF, Meredith AL, Aldrich RW, Nelson MT, Petkov GV. β-Adrenergic relaxation of mouse urinary bladder smooth muscle in the absence of large-conductance Ca2+-activated K+ channel. Am J Physiol Renal Physiol 295: F1149–F1157, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang S, Hypolite JA, Mohanan S, Zderic SA, Wein AJ, Chacko S. Alteration of the PKC-mediated signaling pathway for smooth muscle contraction in obstruction-induced hypertrophy of the urinary bladder. Lab Invest 89: 823–832, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang SH, Gomes CM, Hypolite JA, Marx J, Alanzi J, Zderic SA, Malkowicz B, Wein AJ, Chacko S. Detrusor overactivity is associated with downregulation of large-conductance calcium- and voltage-activated potassium channel protein. Am J Physiol Renal Physiol 298: F1416–F1423, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christ GJ, Day NS, Day M, Santizo C, Zhao W, Sclafani T, Zinman J, Hsieh K, Venkateswarlu K, Valcic M, Melman A. Bladder injection of “naked” hSlo/pcDNA3 ameliorates detrusor hyperactivity in obstructed rats in vivo. Am J Physiol Regul Integr Comp Physiol 281: R1699–R1709, 2001 [DOI] [PubMed] [Google Scholar]
- 9.DeFarias FP, Carvalho MF, Lee SH, Kaczorowski GJ, Suarez-Kurtz G. Effects of the K+ channel blockers paspalitrem-C and paxilline on mammalian smooth muscle. Eur J Pharmacol 314: 123–128, 1996 [DOI] [PubMed] [Google Scholar]
- 10.Frazier EP, Peters SL, Braverman AS, Ruggieri MR, Sr, Michel MC. Signal transduction underlying the control of urinary bladder smooth muscle tone by muscarinic receptors and β-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol 377: 449–462, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrera GM, Heppner TJ, Nelson MT. Regulation of urinary bladder smooth muscle contractions by ryanodine receptors and BK and SK channels. Am J Physiol Regul Integr Comp Physiol 279: R60–R68, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Herrera GM, Heppner TJ, Nelson MT. Voltage dependence of the coupling of Ca2+ sparks to BKCa channels in urinary bladder smooth muscle. Am J Physiol Cell Physiol 280: C481–C490, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Hristov KL, Afeli SA, Parajuli SP, Cheng Q, Rovner ES, Petkov GV. Neurogenic detrusor overactivity is associated with decreased expression and function of the large conductance voltage- and Ca2+-activated K+ channels. PLos One e68052: 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hristov KL, Chen M, Kellett WF, Rovner ES, Petkov GV. Large-conductance voltage- and Ca2+-activated K+ channels regulate human detrusor smooth muscle function. Am J Physiol Cell Physiol 301: C903–C912, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hristov KL, Chen M, Soder RP, Parajuli SP, Cheng Q, Kellett WF, Petkov GV. KV2.1 and electrically silent KV channel subunits control excitability and contractility of guinea pig detrusor smooth muscle. Am J Physiol Cell Physiol 302: C360–C372, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hristov KL, Cui X, Brown SM, Liu L, Kellett WF, Petkov GV. Stimulation of β3-adrenoceptors relaxes rat urinary bladder smooth muscle via activation of the large-conductance Ca2+-activated K+ channels. Am J Physiol Cell Physiol 295: C1344–C1353, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hristov KL, Parajuli SP, Soder RP, Cheng Q, Rovner ES, Petkov GV. Suppression of human detrusor smooth muscle excitability and contractility via pharmacological activation of large conductance Ca2+-activated K+ channels. Am J Physiol Cell Physiol 302: C1632–C1641, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hug H, Sarre TF. Protein kinase C isoenzymes: divergence in signal transduction? Biochem J 291: 329–343, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huster M, Frei E, Hofmann F, Wegener JW. A complex of CaV1.2/PKC is involved in muscarinic signaling in smooth muscle. FASEB J 24: 2651–2659, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Hypolite JA, Lei Q, Chang S, Zderic SA, Butler S, Wein AJ, Malykhina AP, Chacko S. Spontaneous and evoked contractions are regulated by PKC-mediated signaling in detrusor smooth muscle: involvement of BK channels. Am J Physiol Renal Physiol 304: F451–F462, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Layne JJ, Nausch B, Olesen SP, Nelson MT. BK channel activation by NS11021 decreases excitability and contractility of urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol 298: R378–R384, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu QH, Zheng YM, Korde AS, Li XQ, Ma J, Takeshima H, Wang YX. Protein kinase C-ε regulates local calcium signaling in airway smooth muscle cells. Am J Respir Cell Mol Biol 40: 663–671, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal 10: 529–542, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Malysz J, Rovner ES, Petkov GV. Single-channel biophysical and pharmacological characterizations of native human large-conductance calcium-activated potassium channels in freshly isolated detrusor smooth muscle cells. Pflügers Arch 465: 965–975, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melman A, Bar-Chama N, McCullough A, Davies K, Christ G. Plasmid-based gene transfer for treatment of erectile dysfunction and overactive bladder: results of a phase I trial. Isr Med Assoc J 9: 143–146, 2007 [PubMed] [Google Scholar]
- 26.Melman A, Biggs G, Davies K, Zhao W, Tar MT, Christ GJ. Gene transfer with a vector expressing Maxi-K from a smooth muscle-specific promoter restores erectile function in the aging rat. Gene Ther 15: 364–370, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Minami K, Hirata Y, Tokumura A, Nakaya Y, Fukuzawa K. Protein kinase C-independent inhibition of the Ca2+-activated K+ channel by angiotensin II and endothelin-1. Biochem Pharmacol 49: 1051–1056, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258: 607–614, 1992 [DOI] [PubMed] [Google Scholar]
- 29.Nishizuka Y. The molecular heterogeneity of protein kinase-C and its implications for cellular regulation. Nature 334: 661–665, 1988 [DOI] [PubMed] [Google Scholar]
- 30.Nourshargh S, Hoult JR. Divergent effects of co-carcinogenic phorbol esters and a synthetic diacylglycerol on human neutrophil chemokinesis and granular enzyme secretion. Br J Pharmacol 91: 557–568, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oger S, Behr-Roussel D, Gorny D, Bernabe J, Comperat E, Chartier-Kastler E, Denys P, Giuliano F. Effects of potassium channel modulators on myogenic spontaneous phasic contractile activity in human detrusor from neurogenic patients. BJU Int 108: 604–611, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Parajuli SP, Petkov GV. Activation of muscarinic M3 receptors inhibits large-conductance voltage- and Ca2+-activated K+ channels in rat urinary bladder smooth muscle cells. Am J Physiol Cell Physiol 305: C207–C214, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petkov GV. Role of potassium ion channels in detrusor smooth muscle function and dysfunction. Nat Rev Urol 9: 30–40, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petkov GV, Nelson MT. Differential regulation of Ca2+-activated K+ channels by β-adrenoceptors in guinea pig urinary bladder smooth muscle. Am J Physiol Cell Physiol 288: C1255–C1263, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Qi Z, Chi S, Su X, Naruse K, Sokabe M. Activation of a mechanosensitive BK channel by membrane stress created with amphipaths. Mol Membr Biol 22: 519–527, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Schneider T, Fetscher C, Krege S, Michel MC. Signal transduction underlying carbachol-induced contraction of human urinary bladder. J Pharmacol Exp Ther 309: 1148–1153, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Schubert R, Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol Sci 22: 505–512, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Schubert R, Noack T, Serebryakov VN. Protein kinase C reduces the KCa current of rat tail artery smooth muscle cells. Am J Physiol Cell Physiol 276: C648–C658, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Smith AC, Hristov KL, Cheng Q, Xin W, Parajuli SP, Earley S, Malysz J, Petkov GV. Novel role for the transient potential receptor melastatin 4 channel in guinea pig detrusor smooth muscle physiology. Am J Physiol Cell Physiol 304: C467–C477, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soder RP, Petkov GV. Large conductance Ca2+-activated K+ channel activation with NS1619 decreases myogenic and neurogenic contractions of rat detrusor smooth muscle. Eur J Pharmacol 670: 252–259, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suarez-Kurtz G, Garcia ML, Kaczorowski GJ. Effects of charybdotoxin and iberiotoxin on the spontaneous motility and tonus of different guinea pig smooth muscle tissues. J Pharmacol Exp Ther 259: 439–443, 1991 [PubMed] [Google Scholar]
- 42.Taguchi K, Kaneko K, Kubo T. Protein kinase C modulates Ca2+-activated K+ channels in cultured rat mesenteric artery smooth muscle cells. Biol Pharm Bull 23: 1450–1454, 2000 [DOI] [PubMed] [Google Scholar]
- 43.Toro L, Wallner M, Meera P, Tanaka Y. Maxi-KCa, a unique member of the voltage-gated K channel superfamily. News Physiol Sci 13: 112–117, 1998 [DOI] [PubMed] [Google Scholar]
- 44.Wang W, Huang H, Hou D, Liu P, Wei H, Fu X, Niu W. Mechanosensitivity of STREX-lacking BKCa channels in the colonic smooth muscle of the mouse. Am J Physiol Gastrointest Liver Physiol 299: G1231–G1240, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Xin W, Cheng Q, Soder RP, Petkov GV. Inhibition of phosphodiesterases relaxes detrusor smooth muscle via activation of the large-conductance voltage- and Ca2+-activated K+ channel. Am J Physiol Cell Physiol 302: C1361–C1370, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xin W, Cheng Q, Soder RP, Rovner ES, Petkov GV. Constitutively active phosphodiesterase activity regulates urinary bladder smooth muscle function: critical role of KCa1.1 channel. Am J Physiol Renal Physiol 303: F1300–F1306, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xin W, Soder RP, Cheng Q, Rovner ES, Petkov GV. Selective inhibition of phosphodiesterase 1 relaxes urinary bladder smooth muscle: role for ryanodine receptor-mediated BK channel activation. Am J Physiol Cell Physiol 303: C1079–C1089, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamura K, Steenbergen C, Murphy E. Protein kinase C and preconditioning: role of the sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol 289: H2484–H2490, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Zhou XB, Wulfsen I, Utku E, Sausbier U, Sausbier M, Wieland T, Ruth P, Korth M. Dual role of protein kinase C on BK channel regulation. Proc Natl Acad Sci USA 107: 8005–8010, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu S, White RE, Barman SA. Effect of PKC isozyme inhibition on forskolin-induced activation of BKCa channels in rat pulmonary arterial smooth muscle. Lung 184: 89–97, 2006 [DOI] [PubMed] [Google Scholar]