

Abstract
Nitric oxide (NO) has been implicated in the arterial vasodilation and associated vascular hyporesponsiveness to vasoconstrictors observed in liver cirrhosis. Bacteria, potent activators of NO and TNF-α synthesis, are found in the mesenteric lymph nodes (MLNs) of ascitic cirrhotic rats. Here, we investigated the impact of bacterial translocation (BT) to MLNs on TNF-α production, vascular NO release, and contractility in the mesenteric vasculature of ascitic cirrhotic rats. Vascular response to the α-adrenoagonist methoxamine, which is diminished in the superior mesenteric arterial beds of cirrhotic rats, is further blunted in the presence of BT. BT promoted vascular NO release in cirrhotic rats, an effect that depended on pressure-induced shear stress and was blocked by the NO inhibitor Nω-nitro-L-arginine. Removing the endothelium had the same effect. Endothelial NO synthase (eNOS), but not the inducible isoform (iNOS), was present in mesenteric vasculature of cirrhotic rats with and without BT, and its expression was enhanced compared with controls. TNF-α was induced in MLNs by BT and accumulated in parallel in the serum. This TNF-α production was associated with elevated levels of tetrahydrobiopterin (BH4), a TNF-α–stimulated cofactor and enhancer of eNOS-derived NO biosynthesis and NOS activity in mesenteric vasculature. These findings establish a link between BT to MLNs and increased TNF-α production and elevated BH4 levels enhancing eNOS-derived NO overproduction, further impairing contractility in the cirrhotic mesenteric vasculature.
Introduction
Arterial vasodilation, particularly that occurring in the splanchnic circulation, is a major causative factor in the pathogenesis of the hyperdynamic circulatory syndrome that is known to occur in cirrhosis and portal hypertension (1–3). A classic expression of these hemodynamic abnormalities is an impairment in vascular contractility, with resulting hyporeactivity to vasoconstrictors (4–9). This has been demonstrated to be caused largely by overproduction of endothelium-derived vasodilators, mainly nitric oxide (NO) (5–7, 10, 11). Recent investigations have shown that endothelial NO synthase (eNOS) is the major enzymatic source for this NO overproduction (12–17).
However, in various pathologic entities, proinflammatory cytokines, endotoxins, and bacterial infections have also been associated with enhanced production of NO, hyporesponsiveness to vasocon-strictors, and development of a hyperdynamic state (18–20). These physiological similarities, and the fact that endotoxemia is a common finding in cirrhosis (21–24), led to the hypothesis proposed by Vallance and Moncada that bacterial endotoxin, directly or involving cytokines, may account for hemodynamic changes in advanced cirrhosis (25) by stimulating the expression of inducible NO synthase (iNOS) and, thus, NO production.
The gut is a large reservoir of bacteria and endotoxins. Intestinal barrier failure can lead to passage of bacteria and microbial products through the epithelial mucosa to mesenteric lymph nodes (MLNs) and other organs, a process known as bacterial translocation (BT) (26, 27). In liver cirrhosis, intestinal bacterial overgrowth, impaired host defenses, and disruption of the gut mucosal barrier promote this process (28–32). In cirrhotic rats, BT has been reported to be increased after the development of ascites (33) and to occur with a frequency of 45–75% (34–36). Moreover, live bacteria have been isolated from MLNs in patients undergoing laparotomy — a situation associated with a significant increase in the development of postoperative sepsis (37). In experimental cirrhosis, the presence of BT has been shown to be directly correlated with the occurrence of severe bacterial infections, such as spontaneous bacteremia and spontaneous bacterial peritonitis (31, 38, 39).
TNF-α is a proinflammatory cytokine released by mononuclear cells in response to inflammatory stimuli (40). TNF-α blood concentration has been shown to be elevated in patients with chronic liver disease and in animals subjected to experimental portal hypertension (41–44). TNF-α is also known to be involved in the pathogenesis of the hyperdynamic circulatory syndrome in portal hypertension (42, 45). Finally, the gut and its associated lymphoid tissue, the largest immunologic organ of the body, has recently been shown to produce and release TNF-α in response to BT, even in the absence of portal or systemic spread of bacteria (46, 47).
We designed this study to determine (a) whether there is any relationship between BT to MLNs and vascular contractility in the superior mesenteric arterial bed in cirrhosis; (b) if so, whether this additional vascular impairment is also due to vascular NO release; and (c) if that is the case, what NOS isoform is responsible for this additional NO overproduction and by what mechanism this NOS isoform is upregulated; and (d) whether TNF-α is playing any role in this process.
Methods
All experimental procedures in this study were conducted according to the American Physiological Society principles for the care and use of laboratory animals.
Induction of liver cirrhosis in rats by CCl4.
Male Sprague-Dawley rats (Harlan Sprague Dawley Inc., Indianapolis, Indiana, USA), weighing 100–125 g, underwent inhalation exposure to CCl4 and had phenobarbital (0.35g/L) added to their drinking water, as described previously by ourselves and others (4, 5). This protocol produces a high yield of micronodular cirrhosis in about 12–16 weeks. Phenobarbital and CCl4 exposure were stopped at least 6 days before the perfusion experiments. Normal sex- and age-matched untreated rats were used as controls.
Assessment of BT.
On the study day, the animals were anesthetized with ketamine hydrochloride (100 mg/kg), and the abdominal skin was shaved and sterilized with an iodine solution. All of the following surgical procedures were performed under strict sterile conditions with sterile instruments. In cirrhotic animals, ascites was quantified. To exclude animals with systemic bacteremia for protocol 1 (see below), 3 cm3 of blood was withdrawn from the inferior vena cava and inoculated into aerobic and anaerobic Bactec culture bottles. The blood was incubated at 35°C, and the growth value (a measurement of CO2 production by bacteria) was continuously monitored for at least 7 days. None of these cultures showed bacterial growth, confirming that, in our laboratory, this model of CCl4-induced liver cirrhosis is free of systemic bacteremia (33). Therefore, we did not test for bacteremia in the following protocols. The caudal and cranial MLNs were removed and weighed with an E400D scale from Ohaus Corp. (Florham Park, New Jersey, USA), which is accurate to ±0.01 g. Tissues were then homogenized in a measured amount of saline, and 0.1-mL aliquots were plated onto blood, MacConkey, and phenylethyl alcohol blood agar plates (BBL Prepared Media; Becton Dickinson Microbiology Systems, Sparks, Maryland, USA). A swab of ascitic fluid from the peritoneal cavity was cultured. If this culture showed bacterial growth, the animal was excluded from the study. Spleen and liver were removed and weighed. Liver slices were fixed in 10% neutral buffered formaldehyde. Solid culture media were examined, and colonies were counted after 24 and 48 hours of aerobic incubation at 35°C. Any positive MLN cultures were considered indicative of BT from the intestinal lumen.
In vitro perfusion.
The in vitro perfusion system used was a partial modification of that originally described by McGregor (48); it has been used extensively in previous studies by our laboratory (4–7, 16, 17). Briefly, the superior mesenteric artery (SMA) was cannulated with a polyethylene PE-60 catheter, and then gently perfused with 15 mL of warm Krebs solution to eliminate blood. After isolating the SMA with its mesentery, the gut was cut off close to its mesenteric border. The arterial vasculature was then transferred to a 37°C water-jacketed container and perfused with oxygenated (95% O2, 5% CO2) 37°C Krebs solution, using a roller pump (Masterflex; Cole-Parmer Instrument Co., Barrington, Illinois, USA). The Krebs solution had the following composition (in mmol/L): NaCl, 118.0; KCl, 4.7; KH2PO4, 1.2; MgSO4, 1.2; CaCl2, 2.5; NaHCO3, 25.0; disodium EDTA, 0.026; glucose, 11.0 (pH was 7.4). The effluent of the perfused tissue was continuously removed from the perfusing chamber. The perfusion pressure was measured with a H-P pressure transducer (Hewlett Packard, Andover, Massachusetts, USA) mounted on a side arm just before the perfusing cannula. Pressure was recorded continuously on a 7D polygraph inscriber (Grass Instruments, Quincy, Massachusetts, USA).
Experimental protocols
All cirrhotic rats were studied prospectively, because at the time of experimental procedures, it was not known whether the cirrhotic animals were positive or negative for BT. A total of 78 cirrhotic rats with ascites and 26 age-matched control rats were included in this study. In all animals (except in protocol 4), before starting procedures to assess BT, the left femoral artery was exposed and cannulated with a PE-50 catheter. Blood (1.5 cm3) was drawn into a pyrogen-free Vacutainer (Becton Dickinson) and centrifuged at 5,000 rpm for 15 minutes. The separated serum was stored immediately at –70°C until analysis for NO metabolites (NOx) and TNF-α. Mean arterial pressure (MAP) was evaluated by connecting the catheter to the H-P pressure polygraph inscriber, and was recorded on a model 7D inscription recorder (Grass Instruments). Measurements of MAP were performed before blood was drawn (and after its replacement with 1.5 cm3 saline). In the animals used in protocols 2 and 3, additional MLN homogenate was stored at –70°C until measurement of NOx and TNF-α levels.
Protocol 1.
In 18 ascitic cirrhotic rats (with BT: n = 10; without BT: n = 8) and 10 control rats, in vitro perfusion of the mesenteric vessel bed was performed. Baseline perfusion at 4 mL/min was established for 60 minutes. At 30, 45, and 60 minutes, the perfusion solution from the mesenteric tissue was collected into glass tubes for 1 minute. Then 2 doses of methoxamine (MTx; 30 and 100 μM) were administered noncumulatively by continuous infusion for 2 minutes (first perfusion cycle), and the pressure response was documented. Three perfusate samples were collected for each pressure-response period, beginning with the increase of perfusion pressure. Each sample was collected for 1 minute, frozen immediately, and stored at –30°C until the NOx assay. After the first perfusion cycle was completed, each vessel preparation was perfused at the basal flow rate of 4 mL/min for 50 minutes (after that time, the perfusate NOx concentration has returned to baseline levels; data not shown).
Then the specific NOS inhibitor Nϖ-nitro-L-arginine (l-NNA; 10–4 M; Sigma Chemical Co., St. Louis, Missouri, USA) was added, and a second perfusion cycle was initiated after 20 minutes of incubation time. The NOS inhibitor was present at the same molar concentration in the perfusion system throughout the second perfusion cycle. Before initiation of the experiment, the Krebs solution that was circulated through the perfusion system (including the catheter, but not mesenteric tissue) was collected for use in determining background NOx levels. As shown previously, the perfusion system showed stable basal perfusion conditions and unchanged pressure response for the duration of this experiment (6, 7, 17).
Protocol 2.
Endothelial denudation of the mesenteric vasculature was performed in 16 cirrhotic ascitic rats (with BT: n = 8; without BT: n = 8) and in 6 control rats by a combined treatment of cholic acid (sodium salt) and distilled water (4). In brief, after cannulation of the SMA and gentle flushing with 10 mL of warmed Krebs solution to eliminate blood, perfusion with 1.5 mL of 0.5% cholic acid (for 10 seconds) followed by 15 mL of Krebs solution (to eliminate cholic acid) was performed. The preparation was then transferred to a 37°C water-jacketed container and perfused with oxygenated 37°C Krebs solution (4 mL/min for 10 minutes). After the mesenteric vasculature was relaxed, it was perfused with 37°C distilled water for 10 minutes. A period of 45 minutes was allowed before noncumulative concentration-response curves to MTx (1–100 μM) were derived. To assess whether the vessel was completely de-endothelialized and whether smooth muscle function was maintained, the mesenteric preparation was kept preconstricted with MTx (100 μM) at the end of each dose-response curve, while dose-dependent vasorelaxation response to the endothelium-dependent vasodilator acetylcholine (10–8 to 10–6 g; bolus of 0.1 mL) and the endothelium-independent vasodilator sodium nitroprusside (10–6 to 10–5 g; bolus of 0.1 mL) was tested.
Protocols 3 and 4.
In protocol 3, a total of 25 cirrhotic ascitic rats and 10 control rats were used for performance of blotting and tetrahydrobiopterin (BH4) measurements in the studied mesenteric vasculature. In protocol 4, a separate group of 19 cirrhotic ascitic animals were used for evaluation of NOS activity in mesenteric resistance vessels (MRVs).
Western blotting.
SMA vessels and MRVs were analyzed for the presence of eNOS and iNOS protein. SMA vessels were harvested after removal of MLNs and freeing the artery from surrounding tissue over a length of 3–4 cm, starting at its aortic origin. Tissue for Western blotting of MRVs was harvested from a portion of the capillary bed situated between the MLNs and the small intestine. The tissue was cut off from the SMA at the level of second-order arteries and dissected from the intestine as close as possible to the gut wall. Vessels were washed in PBS and homogenized in a lysis buffer described previously (17). Protein supernatants were quantitated using the Lowry assay, and equal amounts of protein from each sample were separated by SDS-PAGE and electroblotted onto nitrocellulose membranes. Membranes were probed with an mAb recognizing eNOS (Transduction Laboratories, Lexington, Kentucky, USA) and a polyclonal antibody recognizing iNOS (Affinity BioReagents Inc., Golden, Colorado, USA). The specificity of the eNOS and iNOS antibodies for rat tissue was established previously by using quiescent and activated rat sinusoidal endothelial cells, respectively, as positive controls. Enhanced chemiluminescence was used for protein detection.
BH4 assay.
Frozen mesenteric vasculature was weighed and homogenized in 3 volumes of 50 mM Tris-HCl (pH 7.4) containing 1 mM DTT and 1 mM EDTA. This mixture was centrifuged, and supernatant BH4 levels were measured by HPLC with fluorescence detection after oxidation, as described previously (49).
NOS activity assay.
NOS activity was measured by determining the conversion of 3H-labeled L-arginine to 3H-labeled L-citrulline, using a commercially available NOS assay kit (Calbiochem-Novabiochem Corp., San Diego, California, USA). Briefly, mesenteric tissue was harvested as described above, and then minced with fine scissors and homogenized in lysis buffer. Samples were incubated with a reaction buffer containing 1 mM NADPH, 3 μM BH4, 100 nM calmodulin, 2.5 mM CaCl2, 1 μM FAD, 1 μM FMN, and 1 μCi/μL of [3H]arginine or [14C]arginine. To determine NOS activity, duplicate samples were incubated in the presence and absence of Nϖ-nitro-L-arginine methylester (l-NAME; 1 mM) or vehicle. The reaction was continued for 1 hour at 35°C, and then stopped by the addition of 0.4 mL of cold stop buffer (50 mM HEPES and 5 mM EDTA; pH 5.5). After addition of 100 μL of equilibrated resin to each reaction mixture, samples were passed over prepared columns. The eluates were transferred into vials and analyzed using a liquid scintillation counter. Radiolabeled cpm of L-citrulline generation were measured.
Determination of NOx concentration.
The NOx concentration of each perfusate and serum sample was measured using the Nitric Oxide Analyzer from Sievers Instruments (Boulder, Colorado, USA), as described previously (16, 17). In brief, this assay is based upon spectrophotometric analysis after a chemiluminescent reaction between NO and ozone. Fifty microliters of each sample was placed into the purge vessel, which contained 3 mL of 0.1 M vanadium chloride in 1 M HCl at 95°C. The NO generated from nitrite and nitrate ions was carried into the analyzer by vacuum through a gas bubbler trap containing 5 mL of 1 M NaOH. This analyzer quantitates dissolved NO and NO2-derived NO that has been generated by acid and stripped from the solution by nitrogen gas. The NO then reacts with analyzer-generated ozone to form excited NO2, which releases light in the red and near-infrared regions of the spectrum and is detected by a thermoelectrically cooled, red-sensitive photomultiplier tube. The lower limit of sensitivity for this machine is below 2 picomoles of NO per second. Because of this high sensitivity, it is necessary to make sure that exogenous contamination does not affect the results. Therefore, we conducted control experiments in which the perfusate was circulated through the perfusion system without tissue for the duration of the experiment. Under these circumstances, NOx concentrations of the collected perfusate were low and practically the same at all timepoints. The displayed NOx concentrations and related NOx production rates were corrected for each experiment by subtracting background NOx concentrations. The net NOx release in response to MTx (Δ NOx concentration) is calculated as the difference between NOx concentration at baseline (without MTx) and after each pressure-response period. NOx production rate was obtained by multiplying the perfusate flow rate by the concentration of NOx in the perfusate, and is expressed in nanomoles per minute. For calculation of the regression coefficients between the amount of NOx production and shear stress, the average NOx concentration was calculated for each experimental period. Shear stress was calculated as described previously using the following equation (16, 17):
(Equation 1)
 |
1 |
where K = (η/2πL3)1/4 or K = 1/2(8η/πL3)1/4 = constant factor.
Assays.
Concentrations of TNF-α in serum and MLN homogenate were measured in duplicate with a commercially available rat-specific ELISA (sandwich ELISA) kit (Endogen Inc., Woburn, Massachusetts, USA) according to the protocol supplied by the manufacturer. The lower limit of sensitivity for TNF-α in this assay was 3 pmol/mL.
Statistical analysis.
Results were expressed as mean ± SEM. Statistical analysis was performed using 2-way, repeated-measures ANOVA and paired and unpaired Student’s t tests, with statistical significance set at P < 0.05. Each relationship of hemodynamic, perfusate, serum, and lymph node parameters was tested for regression by a simple regression analysis. To increase the sensitivity of the regression analysis, statistical significance for correlations was set at P < 0.01.
Results
All CCl4-treated rats (LC) used in these experiments showed macroscopically macro/micronodular cirrhosis of the liver. The weight of cirrhotic rats was found to be significantly lower than that of control rats (348.1 ± 6.0 g vs. 397.6 ± 10.3 g, respectively; P < 0.0001). This difference was more pronounced in animals that were also positive for BT (LC, BT+: 326.4 ± 7.1 g; LC, BT–: 370.9 ± 8.5 g; P < 0.01). The weights of the spleen and the mesenteric vasculature, expressed in grams per kilogram of body weight, were both significantly higher in cirrhotic rats than in control rats (LC: 3.70 ± 0.09 g/kg and 15.4 ± 0.5 g/kg; control: 1.94 ± 0.06 g/kg and 8.5 ± 0.5 g/kg, respectively; P < 0.0001). There were no significant differences between cirrhotic rats with or without BT. Similarly, no statistical difference in volume of ascites was found between cirrhotic rats with or without BT (LC, BT+: 19.6 ± 2.5 mL; LC, BT–: 17.4 ± 3.2 mL). MLN weight was not different among the study groups. BT to MLNs did not occur in any of the 26 normal rats. Among the 78 ascitic cirrhotic rats, MLN culture was positive for bacteria in 40 animals (51.3%).
Protocol 1
In vitro perfusion pressure in mesenteric arterial vessel bed.
Removal of MLNs from the mesenteric vasculature before performance of in vitro perfusion did not change unstimulated or MTx-induced perfusion pressures (data not shown). As shown before, basal perfusion pressures were significantly lower in vessel preparations of cirrhotic rats than in control rats (4, 5). The mesenteric arterial beds of cirrhotic rats showed a significantly lower pressure response to MTx than control rats did (P < 0.0001 by ANOVA). This vascular hyporesponsiveness to MTx was significantly more pronounced in cirrhotic rats with BT (P < 0.01 by ANOVA; Figure 1a).
Figure 1.
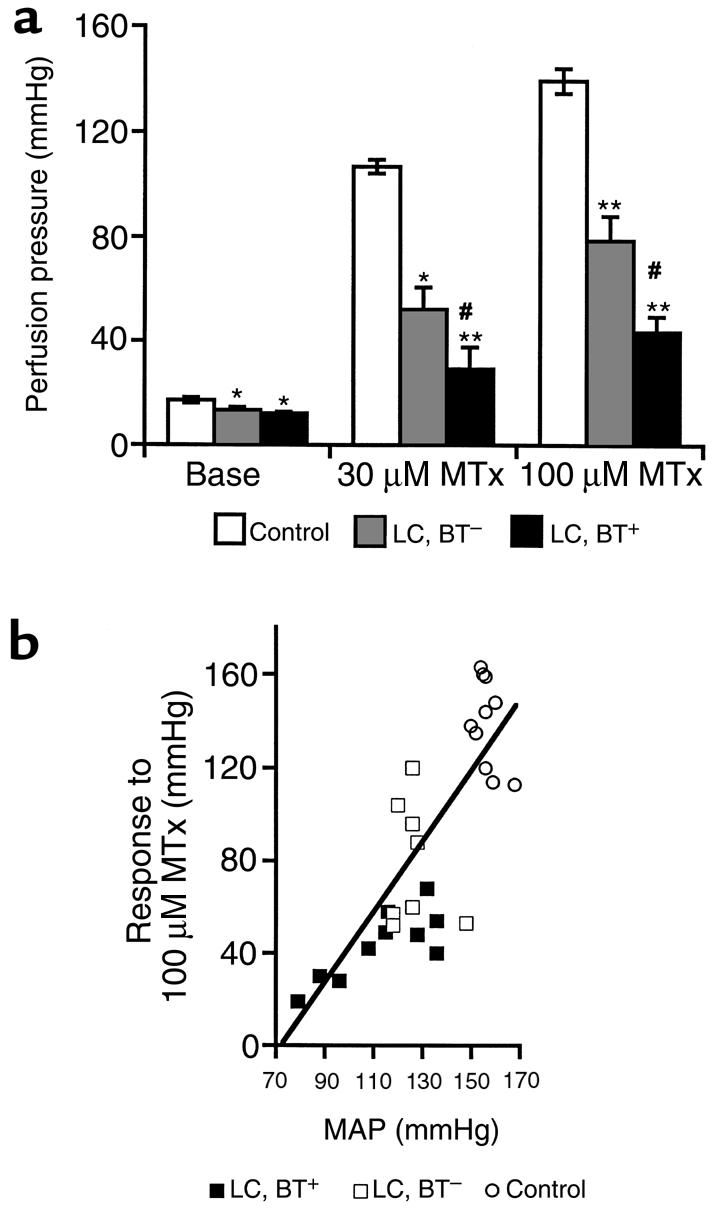
(a) Perfusion pressure under basal conditions (Base) and during the administration of 30 μM and 100 μM MTx in the in vitro–perfused superior mesenteric arterial bed. *P < 0.01 vs. control. **P < 0.0001 vs. control. #P < 0.01 vs. LC, BT–. (ANOVA for pressure-response periods: P < 0.0001 for control vs. LC, BT– or LC, BT+ rats; P < 0.01 for LC, BT+ vs. LC, BT– rats.) (b) Relationship between in vitro vascular responsiveness and systemic hemodynamic parameter MAP. A close correlation for in vitro pressure response to MTx (100 μM) and in vivo MAP was observed (r = 0.797, P < 0.0001).
Relationship between in vitro vascular contractility and in vivo MAP.
No difference in MAP obtained before and after blood drawing was observed. Cirrhotic rats showed significantly lower MAP than did control rats (LC, BT+: 117.9 ± 3.1 mmHg; LC, BT–: 133.1 ± 2.1 mmHg; control: 153.8 ± 1.6 mmHg; P < 0.0001). The presence of BT in cirrhotic rats was associated with a significant reduction in MAP (P < 0.01). Vascular responsiveness to MTx (100 μM) was found to be significantly correlated with MAP (r = 0.797, P < 0.0001; Figure 1b), demonstrating the hemodynamic importance of in vitro vascular reactivity as a classic manifestation of the vasodilatation that characterizes the hyperdynamic circulatory syndrome in portal hypertension. Additionally, when both cirrhotic rats and control rats were included in the analysis, a significant correlation was observed between in vitro baseline perfusion pressures and in vivo MAP (r = 0.537, P < 0.0001). There was a similar correlation between in vitro baseline perfusion pressures and pressure response to MTx (r = 0.656, P < 0.001).
Perfusate NOx concentrations.
In a preliminary study, we collected the perfusate from the mesenteric arterial beds of the animals in each of the different study groups under basal conditions at intervals of 15 minutes for 1.5 hours. In each study group, perfusate NOx concentration was found to remain unchanged during this time (data not shown). As shown in Figure 2a, basal perfusate NOx concentrations were significantly higher in cirrhotic rats with BT than in cirrhotic rats without BT and control rats. During MTx administration, NOx concentrations in perfusate increased in all study groups and were significantly elevated in cirrhotic rats compared with control rats (P < 0.0001 by ANOVA). Cirrhotic rats with BT showed significantly higher perfusate NOx concentrations than did cirrhotic rats without BT (P < 0.001 by ANOVA). A representative spectrophotometric tracing, used to determine NOx concentration in the perfusate from a typical experiment in each study group, is shown in Figure 2b. Regression analysis revealed that perfusate NOx concentrations correlated with the in vitro vascular pressure response (e.g., at 100 μM MTx; r = 0.507, P < 0.01). That is, the higher the NOx concentration, the lower the in vitro perfusion pressure. However, no correlation was found between basal perfusate NOx concentration and baseline perfusion pressure (for all: r = 0.16, P > 0.40). Net NOx release in response to MTx was significantly elevated in cirrhotic rats compared with control rats (P < 0.001 by ANOVA). In the presence of BT, an additional marked increase in NOx release was found (P < 0.01 by ANOVA). In each study group, a significant regression correlation between NOx production rate and shear stress index was found (LC, BT+: r = 0.72, P < 0.0001; LC, BT–: r = 0.65, P < 0.001; control: r = 0.69, P < 0.0001) (Figure 3). The slope of NOx production rate vs. shear stress index was significantly higher in each group of cirrhotic animals than in the control animals (LC, BT+: 1.36 ± 0.10; LC, BT–: 0.535 ± 0.049; controls: 0.233 ± 0.018; P < 0.0001). Cirrhotic rats with BT showed the highest slope (P < 0.0001), demonstrating significantly more pronounced shear stress–dependent NOx production by the mesenteric vascular endothelium in the presence of BT than in the absence of BT.
Figure 2.

(a) Absolute perfusate NOx concentrations under basal conditions and during pressure-response periods to MTx. *P < 0.01 vs. control. **P < 0.0001 vs. control. #P < 0.001 vs. LC, BT–. ##P < 0.0001 vs. LC, BT–. (ANOVA for pressure-response periods: P < 0.0001 for control vs. LC, BT– or LC, BT+ rats; P < 0.001 for LC, BT+ vs. LC, BT– rats.) (b) Representative NOx assays by chemiluminescence technique for rats of each study group. At baseline conditions and during pressure-response periods to 30 μM and 100 μM MTx, 3 perfusate samples (1, 2, 3) were collected, each for 1 minute. An increase in peak size (area under the curve) obtained in cirrhotic rats compared with that of controls and a further increase in peak size in readings from cirrhotic rats with BT is evident.
Figure 3.

Regression analysis for NOx production rate vs. applied pressure-induced shear stress index. A highly significant correlation was found for each experimental study group. LC, BT+: r = 0.72, P < 0.0001; LC, BT–: r = 0.65, P < 0.001; controls: r = 0.69, P < 0.0001. Note the markedly enhanced production of NOx at given shear stress levels in rats with BT compared with animals without BT.
Effect of inhibition of NO synthesis by l-NNA on the pressure response to MTx.
Incubation with L-NNA did not change baseline perfusion pressures. After inhibition of NO formation, no statistical difference in response to MTx could be observed between perfused mesenteric arterial beds of cirrhotic rats with and without BT. A significant hyporeactivity to MTx persisted in cirrhotic rats compared with control rats (P < 0.01; Figure 4a). Cirrhotic rats with BT had a significantly greater pressure response to L-NNA than did cirrhotic rats without BT (P < 0.01 by ANOVA). For example, pressure increases in response to treatment with 30 μM MTx were 236.5 ± 27.1% vs. 143.8 ± 27.4%, respectively (P < 0.01), demonstrating that NO synthesis blockade with L-NNA led to a significantly greater response in vessel preparations of animals with BT.
Figure 4.

(a) In vitro perfusion pressures in superior mesenteric vasculature after inhibition of NO synthesis by incubation with L-NNA. *P < 0.05 vs. control. **P < 0.001 vs. control. Blockade of NO formation corrected the hyporesponsiveness to MTx that is related to the presence of BT in cirrhotic rats. (b) Dose-response curves for MTx in de-endothelialized superior mesenteric vascular beds. *P < 0.05 vs. LC, BT+ and LC, BT–. **P < 0.001 vs. LC, BT+ and LC, BT–. P < 0.001 for control vs. LC, BT+ and LC, BT– (ANOVA). Note the lack of difference in vascular reactivity in mesenteric preparations from cirrhotic rats with and without BT.
Protocol 2
Effect of endothelium removal on the in vitro pressure response to MTx.
De-endothelialized mesenteric vasculature in all groups practically eliminated vasodilatory response to acetylcholine, indicating an appropriate removal of the endothelium (data not shown). Dose-dependent relaxation responses to sodium nitroprusside proved the viability of the smooth muscle, and were found to be significantly elevated in cirrhotic rats with BT compared with control rats (P < 0.05 by ANOVA). Removal of the endothelium significantly increased baseline perfusion pressures and response to MTx in all study groups (P < 0.01). However, a significantly lower baseline perfusion pressure, as well as hyporesponsiveness to MTx, persisted in cirrhotic rats compared with control rats (P < 0.05). Endothelial denudation completely abolished the difference in vascular responsiveness to MTx that was observed in intact mesenteric vasculature between cirrhotic rats with and without BT.
Protocol 3
Western blot analysis.
Protein expression of eNOS was evident in each vasculature examined. In cirrhotic rats without BT, eNOS protein levels in SMA and MRV tissue were markedly increased compared with those in control animals. In cirrhotic rats with BT, expression of eNOS protein in the studied vasculature appeared to be additionally enhanced. There was no detectable iNOS protein in SMA or MRVs from cirrhotic or control rats, whereas iNOS was detected in cell lysates from activated sinusoidal endothelial cells from rat liver and in MLNs from LPS-treated (6 mg/kg for 12 hours) rats, which served as positive controls (Figure 5).
Figure 5.

Expression of eNOS and iNOS protein in arterial vessels of control and cirrhotic ascitic rats with and without BT. The molecular weights of eNOS and iNOS are 140 and 135 kDa, respectively. Each lane was loaded with 100 mg of protein, and eNOS and iNOS protein was detected by enhanced chemiluminescence. (a) Western blotting for iNOS. No induction of iNOS was found in vessels from control or cirrhotic rats, including animals with BT. In iNOS-positive controls, strong iNOS bands detected with recombinant iNOS protein can be appreciated. (b) Western blotting for eNOS. For SMA, tissue was pooled from 3 animals per group; a representative blot from 3 independent experiments is shown. For MRVs, individual blots for each animal were performed, and a representative blot from 6 independent experiments is shown. Higher immunoreactivity in cirrhotic rats (LC) than in controls can be seen.
TNF-α, NOx, and BH4 levels.
TNF-α levels in MLN homogenate and serum were significantly elevated only in cirrhotic rats with BT (P < 0.0001; Figure 6, a and b). In cirrhotic rats without BT, slight (not significant) increases in TNF-α concentrations in serum and MLNs were noted. A significant correlation between TNF-α levels in serum and MLN homogenate was observed (r = 0.667, P < 0.0001; Figure 6c). NOx concentrations in serum were significantly elevated in cirrhotic rats compared with control rats (P < 0.0001; Figure 7a). Significantly higher serum NOx levels were detected in the presence of BT than in the absence of BT (P < 0.0001). Serum NOx concentrations were found to correlate significantly with levels of TNF-α in MLN homogenate (r = 0.561, P < 0.0001; Figure 7b) and serum (r = 0.529, P < 0.0001), suggesting a prominent role for TNF-α (particularly that of mesenteric origin) in NOx production. In MLN homogenate, NOx concentrations were significantly increased in cirrhotic rats with BT compared with cirrhotic rats without BT and control rats (P < 0.0001), and correlated significantly with local TNF-α concentrations (r = 0.450, P < 0.001). BH4 concentrations in the mesenteric vasculature were found to be significantly increased in cirrhotic rats with BT compared with cirrhotic rats without BT and controls (P < 0.05; Figure 7c); no difference in BH4 levels was observed between cirrhotic rats without BT and controls. BH4 levels in the mesenteric vascular bed were positively correlated with TNF-α concentrations in MLN homogenate (r = 0.524, P < 0.01; Figure 7d) and serum (r = 0.631, P < 0.001), suggesting the induction of BH4 synthesis by TNF-α.
Figure 6.

TNF-α concentrations in MLNs (a) and serum (b). *P < 0.0001 vs. control. #P < 0.001 vs. LC, BT–. (c) Observed correlation between TNF-α concentrations in MLNs and serum (r = 0.67, P < 0.0001).
Figure 7.

(a) Serum NOx concentrations. *P < 0.0001 vs. control. #P < 0.0001 vs. LC, BT–. (b) Serum NOx levels correlated significantly with TNF-α concentrations in MLNs (r = 0.561, P < 0.0001). (c) BH4 concentrations in mesenteric vasculature. *P < 0.05 vs. control. #P < 0.05 vs. LC, BT–. (d) Levels of BH4 in the studied vasculature were significantly correlated with TNF-α concentrations in MLNs (r = 0.524, P < 0.01), indicating an induction of BH4 synthesis by BT and an associated increase in TNF-α synthesis.
Relationship with hemodynamics.
MAP was found to be inversely correlated with serum levels of NOx and TNF-α (r = 0.675, P < 0.0001 and r = 0.575, P < 0.0001, respectively). TNF-α levels in MLN homogenate tended to correlate inversely with MAP (r = 0.36, P < 0.01). Moreover, BH4 levels in the studied mesenteric vasculature closely correlated inversely with MAP (r = 0.627, P < 0.01). The pressure response to MTx (100 μM) was inversely correlated with NOx concentrations in serum (r = 0.746, P < 0.0001), and tended to correlate inversely with serum TNF-α levels.
Protocol 4
NOS activity in mesenteric vasculature.
NOS catalytic activity (assessed as L-NAME–inhibitable conversion of 3H-labeled L-arginine to 3H-labeled L-citrulline) was significantly increased in mesenteric vasculature from cirrhotic rats with BT compared with animals without BT (1.4 ± 0.2 vs. 0.5 ± 0.2 pmol/mg protein, respectively; P < 0.01).
Discussion
This study provides direct evidence that there is a greater impairment in vascular reactivity in cirrhotic rats with BT to MLNs than in rats without BT, and that this additional impairment is largely mediated by endothelial overproduction of NO. We also demonstrate that, contrary to what may have been expected, this endothelial NO overproduction is shear stress–dependent and eNOS-derived and seems to be mediated by TNF-α through BH4.
NO is synthesized by different isoforms of NOS. Two such isoforms have been investigated extensively in the vasculature (50–52). NOS 2, or iNOS, is induced by LPS, endotoxins, and inflammatory cytokines, and produces large amounts of NO for extended periods of time. This enzyme is not stimulated by mechanical factors. NOS 3, or eNOS, releases NO for short periods, in response to physical stimuli such as flow- or pressure-induced shear stress — a specific and unique function of this enzyme (53, 54). In our model, we assessed endothelial NO release in response to MTx-induced vasoconstriction and the resulting increase in pressure and shear stress. In cirrhotic rats without BT, we found that in the mesenteric vessels NO was overproduced in response to vasoconstrictors, and that this vascular NO release was shear stress–dependent. This directly shows, in a functional manner, the involvement of eNOS-derived NO release in the observed vascular hyporesponsiveness found in cirrhotic rats without BT. Additionally, Western blotting with eNOS antibodies showed a consistent augmentation in the eNOS immunoreactivity in the mesenteric vasculature of animals without BT. These results are consistent with recent reports demonstrating NO-mediated vascular hyporesponsiveness (5–7, 10, 55) and eNOS upregulation (12–17) in portal hypertensive animals.
However, the novel finding of this study is that BT to MLNs was associated with an additional 45% decrease in pressure response to MTx in the superior mesenteric arterial bed. This vascular hyporeactivity in rats with BT was associated with significantly enhanced vascular NO release compared with that occurring in the absence of BT. Blockade of NO formation by L-NNA induced a significantly greater pressure response in cirrhotic rats with BT than in those without BT, thereby abrogating the difference in vascular responsiveness between the 2 study groups. This demonstrates that NO is the main causative vasodilator mediating the more pronounced impairment in vascular contractility in animals with BT. Additionally, de-endothelialization reversed the impairment in vascular reactivity observed in cirrhotic rats with BT, indicating that the vasoactive factor acting as the source of NO is the endothelium, not the vascular smooth muscle. The observation that a blunted response to MTx persisted after de-endothelialization or incubation with L-NNA has been made before (4, 56). This reduced response is probably due to structural changes in the vessel wall, which are known to be induced by the chronic high blood flow and plasma-volume expansion (57) conditions present in cirrhosis and portal hypertension. At basal conditions, the vasculature in our in vitro technique expresses no active vascular tone; therefore, basal perfusion pressure is determined solely by the flow resistance caused by the structure of the vessel wall (6). Basal perfusion pressure was significantly lower in cirrhotic rats than in controls, a phenomenon found to be independent of BT. The close correlation observed between basal perfusion pressures and pressure response to the highest MTx concentration used indicates that the vessel structure, at least in part, defines the maximum vasoconstrictive response to MTx that can be achieved in these vessels. Moreover, baseline pressures correlated significantly with MAP, suggesting that worsening of the hyperdynamic circulatory syndrome and subsequent increase in blood flow induces progressive vascular remodeling. Finally, the tissue weight of mesenteric vasculature was significantly higher in cirrhotic rats than in control rats, and there were no differences between animals with and without BT. This indicates vascular hypertrophy or angiogenic alterations in the mesenteric vasculature (or both), as has been recently reported in prehepatic portal hypertensive rats (58). Nonetheless, we cannot completely exclude the possibility that other vasoactive substances besides NO are playing a minor role in the observed vascular hyporeactivity in cirrhotic rats.
The additional enhancement in NO overproduction in conditions of BT was found to be clearly shear stress–dependent. This is shown in the significantly larger NO release in response to MTx-induced increase in perfusion pressure and in the highly significant regression coefficient for the relationship of NO release vs. shear stress in the animals (Figure 3). Additionally, in the studied vasculature, eNOS protein expression was slightly accentuated and enzyme activity was significantly enhanced in cirrhotic rats with BT compared with cirrhotic animals without BT. This marked upregulation of eNOS in the studied vessels probably explains the elevated perfusate NOx concentrations detected at basal conditions in cirrhotic rats with BT, because it leads to a higher sensitivity in mechanotransduction mechanisms that mediate shear stressinduced endothelial signaling (59). In contrast, expression of iNOS protein was not detectable in SMA or MRVs, indicating that the presence of bacteria and a local inflammatory process in the mesentery of rats with BT does not lead to a significant induction of iNOS and associated iNOS-derived NO synthesis in the splanchnic circulation of cirrhotic rats. In fact, NO production mediated by iNOS has been shown to be independent of physical factors such as flow and shear stress (50, 52, 60). Moreover, iNOS is known to be predominantly expressed in vascular smooth muscle (51), which is not the site of NO production in the studied arteries, as shown by the endothelium dependence of that production. This lack of induction of iNOS protein expression, even in rats with BT, further supports findings against a role for iNOS in the circulatory abnormalities in portal hypertension (55, 61–63). The explanation for this deficit in response to inflammatory stimuli remains to be elucidated. Taken together, the presence of BT is associated with significant enhancement of eNOS-derived NO overproduction in the mesenteric arterial bed in cirrhotic rats, aggravating vascular hyporesponsiveness to vasoconstrictors (Figure 1a) and arterial hypotension (Figure 1b). It is important to emphasize that the severity of arterial hypotension has been shown to be a strong independent predictor of survival in cirrhotic patients (64).
TNF-α is a multifunctional endogenous proinflammatory cytokine that has been demonstrated to be involved in the pathogenesis of the hyperdynamic circulatory syndrome in portal hypertension (antagonism of TNF-α with anti-TNF antibody or inhibition of TNF-α synthesis by thalidomide blunts the development of the hyperdynamic circulation in portal vein–ligated and cirrhotic rats; refs. 42, 45). Moreover, in patients with chronic liver disease and in animal models of portal hypertension, elevated blood concentrations of TNF-α have been reported, compared with normal controls (41–44). TNF-α is produced by mononuclear cells upon activation by infectious microorganisms or their cell-wall products, such as endotoxin (40). BT can be considered a primary event in the etiology of endotoxemia, as reflected by significant portal-to-peripheral venous gradients in endotoxin blood levels of patients with cirrhosis who are undergoing shunt surgery; such gradients demonstrate relatively high levels of endotoxin in the portal circulation (24). Moreover, it has been proposed that macrophages play a key role in the process of BT, as transporters of intestinal bacteria (65). The gut and its associated lymphoid tissue, the largest immunologic organ of the body, contains numerous mononuclear cells (66). Concordantly, we find increased levels of TNF-α in MLNs only in cirrhotic rats with BT. Local production of TNF-α correlated closely with corresponding serum levels that were also found to be significantly increased only in the presence of BT. This suggests that lymph node–derived TNF-α contributes to the associated systemic levels (e.g., by drainage into the systemic circulation). In fact, Mainous et al. demonstrated enhanced TNF-α bioactivity in efferent mesenteric lymph in conditions of zymosan-induced BT that appeared to correlate with the magnitude of inflammatory insult and was not different from systemic levels (46). Moreover, even in the absence of portal and systemic bacteremia, elevated portal concentrations of TNF-α were found in rats with shock-induced intestinal injury with BT (47). These findings demonstrate, in different models of BT, that the gut and its associated lymphoid tissue is capable of becoming a cytokine-releasing organ contributing to enhanced serum levels of TNF-α. This even occurs in conditions where BT is limited to MLNs (46, 47). Thus, our results suggest that the significantly increased systemic levels of TNF-α in cirrhotic rats with BT are, at least in part, due to enhanced mesenteric release of TNF-α.
The importance of this enhanced TNF-α production for NO synthesis in cirrhotic rats is reflected in the significant correlation observed between serum concentrations of TNF-α and NO. The significant elevation of serum NO levels in the presence of BT is specifically interesting in regard to results reported by Guarner et al. demonstrating a relationship between serum endotoxin levels and serum NO levels in patients with liver cirrhosis, particularly in those with ascites (21). That this increase in serum concentrations of TNF-α and NO in the presence of BT has a crucial impact on systemic hemodynamics and vascular contractility can be seen in the inverse correlations found between these two serum markers and in vivo MAP as well as in vitro pressure response to MTx.
Although TNF-α and endotoxins are widely known as stimulators of iNOS induction in vitro (67), this study failed to detect iNOS protein expression. BH4 is an endogenously synthesized essential cofactor for NO biosynthesis (68) and has been shown to be rate-limiting for in vivo NO activity (69). Moreover, BH4 can directly increase eNOS-derived NO bioavailability (69, 70). TNF-α and LPS have been demonstrated to stimulate gene expression and activity of the key enzyme for regulation of BH4 biosynthesis, GTP-cyclohydrolase I, in endothelial cells (71–74). Thus, TNF-α and LPS can directly increase the activity of eNOS in the absence of any induction of iNOS by upregulating production of BH4 (73, 75). BH4 was found to be increased in the studied vasculature only in cirrhotic rats with BT. The important role of BH4 for NO synthesis in conditions of BT and associated TNF-α production can be seen in the positive correlation between serum levels of NO and TNF-α. The close relationship between systemic NO levels and TNF-α concentrations in MLNs particularly emphasizes the importance of TNF-α of mesenteric origin. Furthermore, induction of BH4 by TNF-α is supported by significant correlations between TNF-α concentrations (in MLNs and serum) and levels of BH4 in mesenteric vasculature. Finally, the hemodynamic impact of this increase in splanchnic BH4 is apparent in its significant correlation with MAP, pointing out the special importance of the splanchnic bed for the circulatory abnormalities in liver cirrhosis, as has been suggested before (2).
In conclusion, BT to MLNs in cirrhotic rats is accompanied by a significant elevation in local and systemic concentrations of TNF-α. This is associated with increased BH4 levels and enhanced eNOS-derived NO overproduction by the mesenteric vascular endothelium, further impairing vascular contractility in this vascular bed. Our results indicate that BT may play a predominant role in the severity of arterial vasodilation observed in advanced liver cirrhosis by stimulating a novel pathway for eNOS upregulation mediated by TNF-α.
Acknowledgments
The authors gratefully acknowledge W.C. Sessa and V. Shah for very helpful discussion of the results, and Maryann Vergato for secretarial assistance and preparation of this manuscript. This work was supported by grants from the Veterans Administration Merit Review Fund and the B. Braun Foundation, Braun-Melsungen.
References
- 1.Colombato LA, Albillos A, Groszmann RJ. Temporal relationship of peripheral vasodilatation, plasma volume expansion and the hyperdynamic circulatory state in portal-hypertensive rats. Hepatology. 1992;15:323–328. doi: 10.1002/hep.1840150224. [DOI] [PubMed] [Google Scholar]
- 2.Groszmann RJ. Hyperdynamic circulation of liver disease 40 years later: pathophysiology and clinical consequences. Hepatology. 1994;20:1359–1363. [PubMed] [Google Scholar]
- 3.Schrier RW, et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151–1157. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 4.Atucha NM, Shah V, Garcia-Cardena G, Sessa WE, Groszmann RJ. Role of endothelium in the abnormal response of mesenteric vessels in rats with portal hypertension and liver cirrhosis. Gastroenterology. 1996;111:1627–1632. doi: 10.1016/s0016-5085(96)70026-4. [DOI] [PubMed] [Google Scholar]
- 5.Sieber CC, Lopez-Talavera JC, Groszmann RJ. Role of nitric oxide in the in vitro splanchnic vascular hyporeactivity in ascitic cirrhotic rats. Gastroenterology. 1993;104:1750–1754. doi: 10.1016/0016-5085(93)90655-v. [DOI] [PubMed] [Google Scholar]
- 6.Sieber CC, Groszmann RJ. In vitro hyporeactivity to methoxamine in portal hypertensive rats: reversal by nitric oxide blockade. Am J Physiol. 1992;262:G996–G1001. doi: 10.1152/ajpgi.1992.262.6.G996. [DOI] [PubMed] [Google Scholar]
- 7.Sieber CC, Groszmann RJ. Nitric oxide mediates hyporeactivity to vasopressors in mesenteric vessels of portal hypertensive rats. Gastroenterology. 1992;103:235–239. doi: 10.1016/0016-5085(92)91118-n. [DOI] [PubMed] [Google Scholar]
- 8.Murray BM, Paller MS. Decreased pressor reactivity to angiotensin II in cirrhotic rats. Evidence for a post-receptor defect in angiotensin action. Circ Res. 1985;57:424–431. doi: 10.1161/01.res.57.3.424. [DOI] [PubMed] [Google Scholar]
- 9.Kiel JW, Pitts V, Benoit JN, Granger DN, Shepherd AP. Reduced vascular sensitivity to norepinephrine in portal-hypertensive rats. Am J Physiol. 1985;248:G192–G195. doi: 10.1152/ajpgi.1985.248.2.G192. [DOI] [PubMed] [Google Scholar]
- 10.Lee FY, Albillos A, Colombato LA, Groszmann RJ. The role of nitric oxide in the vascular hyporesponsiveness to methoxamine in portal hypertensive rats. Hepatology. 1992;16:1043–1048. doi: 10.1002/hep.1840160430. [DOI] [PubMed] [Google Scholar]
- 11.Castro A, et al. Impaired responsiveness to angiotensin II in experimental cirrhosis: role of nitric oxide. Hepatology. 1993;18:367–372. [PubMed] [Google Scholar]
- 12.Cahill PA, Redmond EM, Hodges R, Zhang S, Sitzmann JV. Increased endothelial nitric oxide synthase activity in the hyperemic vessels of portal hypertensive rats. J Hepatol. 1996;25:370–378. doi: 10.1016/s0168-8278(96)80124-3. [DOI] [PubMed] [Google Scholar]
- 13.Gadano AC, et al. Endothelial calcium-calmodulin dependent nitric oxide synthase in the in vitro vascular hyporeactivity of portal hypertensive rats. J Hepatol. 1997;26:678–686. doi: 10.1016/s0168-8278(97)80435-7. [DOI] [PubMed] [Google Scholar]
- 14.Martin PY, et al. Upregulation of endothelial constitutive NOS: a major role in the increased NO production in cirrhotic rats. Am J Physiol. 1996;270:F494–F499. doi: 10.1152/ajprenal.1996.270.3.F494. [DOI] [PubMed] [Google Scholar]
- 15.Niederberger M, et al. Comparison of vascular nitric oxide production and systemic hemodynamics in cirrhosis vs. prehepatic portal hypertension in rats. Hepatology. 1996;24:947–951. doi: 10.1002/hep.510240432. [DOI] [PubMed] [Google Scholar]
- 16.Hori N, Wiest R, Groszmann RJ. Enhanced release of nitric oxide in response to changes in flow and shear stress in the superior mesenteric arteries of portal hypertensive rats. Hepatology. 1998;28:1467–1473. doi: 10.1002/hep.510280604. [DOI] [PubMed] [Google Scholar]
- 17.Wiest R, Shah V, Sessa WC, Groszmann RJ. Nitric oxide overproduction by eNOS precedes hyperdynamic splanchnic circulation in portal hypertensive rats. Am J Physiol. 1999;276:G1043–G1051. doi: 10.1152/ajpgi.1999.276.4.G1043. [DOI] [PubMed] [Google Scholar]
- 18.Szabo C, Mitchell JA, Thiemermann C, Vane JR. Nitric oxide-mediated hyporeactivity to noradrenaline precedes the induction of nitric oxide synthase in endotoxin shock. Br J Pharmacol. 1993;108:786–792. doi: 10.1111/j.1476-5381.1993.tb12879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer J, et al. Effects of nitric oxide synthesis inhibition in hyperdynamic endotoxemia. Crit Care Med. 1994;22:306–312. doi: 10.1097/00003246-199402000-00023. [DOI] [PubMed] [Google Scholar]
- 20.Tracey WR, Tse J, Carter G. Lipopolysaccharide-induced changes in plasma nitrite and nitrate concentrations in rats and mice: pharmacological evaluation of nitric oxide synthase inhibitors. J Pharmacol Exp Ther. 1995;272:1011–1015. [PubMed] [Google Scholar]
- 21.Guarner C, et al. Increased serum nitrite and nitrate levels in patients with cirrhosis: relationship to endotoxemia. Hepatology. 1993;18:1139–1143. [PubMed] [Google Scholar]
- 22.Lee FY, et al. Role of endotoxaemia in hyperdynamic circulation in rats with extrahepatic or intrahepatic portal hypertension. J Gastroenterol Hepatol. 1996;11:152–158. doi: 10.1111/j.1440-1746.1996.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 23.Lin RS, et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995;22:165–172. doi: 10.1016/0168-8278(95)80424-2. [DOI] [PubMed] [Google Scholar]
- 24.Lumsden AB, Henderson JM, Kutner MH. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology. 1988;8:232–236. doi: 10.1002/hep.1840080207. [DOI] [PubMed] [Google Scholar]
- 25.Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337:776–778. doi: 10.1016/0140-6736(91)91384-7. [DOI] [PubMed] [Google Scholar]
- 26.Berg, R.D. 1992. Translocation and the indigenous gut flora. In Probiotics. R. Fuller, editor. Chapman Hall. London, United Kingdom. 55–85.
- 27.Wells CL, Maddaus MA, Simmons RL. Proposed mechanisms for the translocation of intestinal bacteria. Rev Infect Dis. 1988;10:958–979. doi: 10.1093/clinids/10.5.958. [DOI] [PubMed] [Google Scholar]
- 28.Guarner C, Runyon BA, Young S, Heck M, Sheikh MY. Intestinal bacterial overgrowth and bacterial translocation in cirrhotic rats with ascites. J Hepatol. 1997;26:1372–1378. doi: 10.1016/s0168-8278(97)80474-6. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Tsao G, Albillos A, Barden GE, West AB. Bacterial translocation in acute and chronic portal hypertension. Hepatology. 1993;17:1081–1085. [PubMed] [Google Scholar]
- 30.Alexander JW, et al. The process of microbial translocation. Ann Surg. 1990;212:496–510. doi: 10.1097/00000658-199010000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyke RJ. Problems of bacterial infection in patients with liver disease. Gut. 1987;28:623–641. doi: 10.1136/gut.28.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popper H, Paronetto F. Problems in the immunology of hepatic diseases. Hepatogastroenterology. 1984;31:1–5. [PubMed] [Google Scholar]
- 33.Garcia-Tsao G, Lee FY, Barden GE, Cartun R, West AB. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology. 1995;108:1835–1841. doi: 10.1016/0016-5085(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 34.Runyon BA, Squier S, Borzio M. Translocation of gut bacteria in rats with cirrhosis to mesenteric lymph nodes partially explains the pathogenesis of spontaneous bacterial peritonitis. J Hepatol. 1994;21:792–796. doi: 10.1016/s0168-8278(94)80241-6. [DOI] [PubMed] [Google Scholar]
- 35.Llovet JM, et al. Bacterial translocation in cirrhotic rats. Its role in the development of spontaneous bacterial peritonitis. Gut. 1994;35:1648–1652. doi: 10.1136/gut.35.11.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casafont F, Sanchez E, Martin L, Aguero J, Romero FP. Influence of malnutrition on the prevalence of bacterial translocation and spontaneous bacterial peritonitis in experimental cirrhosis in rats. Hepatology. 1997;25:1334–1337. doi: 10.1002/hep.510250605. [DOI] [PubMed] [Google Scholar]
- 37.O’Boyle CJ, et al. Microbiology of bacterial translocation in humans. Gut. 1998;42:29–35. doi: 10.1136/gut.42.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Runyon BA, et al. Effect of selective bowel decontamination with norfloxacin on spontaneous bacterial peritonitis, translocation, and survival in an animal model of cirrhosis. Hepatology. 1995;21:1719–1724. [PubMed] [Google Scholar]
- 39.Llovet JM, et al. Translocated intestinal bacteria cause spontaneous bacterial peritonitis in cirrhotic rats: molecular epidemiologic evidence. J Hepatol. 1998;28:307–313. doi: 10.1016/0168-8278(88)80018-7. [DOI] [PubMed] [Google Scholar]
- 40.Old LJ. Tumor necrosis factor (TNF) Science. 1985;230:630–632. doi: 10.1126/science.2413547. [DOI] [PubMed] [Google Scholar]
- 41.Tilg H, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterology. 1992;103:264–274. doi: 10.1016/0016-5085(92)91122-k. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Talavera JC, Cadelina G, Olchowski J, Merrill W, Groszmann RJ. Thalidomide inhibits tumor necrosis factor alpha, decreases nitric oxide synthesis, and ameliorates the hyperdynamic circulatory syndrome in portal-hypertensive rats. Hepatology. 1996;23:1616–1621. doi: 10.1002/hep.510230644. [DOI] [PubMed] [Google Scholar]
- 43.Chu CJ, et al. Hyperdynamic circulation of cirrhotic rats with ascites: role of endotoxin, tumour necrosis factor and nitric oxide. Clin Sci (Colch) 1997;93:219–225. doi: 10.1042/cs0930219. [DOI] [PubMed] [Google Scholar]
- 44.Ohta M, et al. Does tumor necrosis factor alpha regulate nitric oxide synthase expression in the portal hypertensive gastric mucosa? Gastroenterology. 1998;112:A1351. doi: 10.1002/hep.510270403. (Abstr.) [DOI] [PubMed] [Google Scholar]
- 45.Lopez-Talavera JC, Merrill WW, Groszmann RJ. Tumor necrosis factor alpha: a major contributor to the hyperdynamic circulation in prehepatic portal-hypertensive rats. Gastroenterology. 1995;108:761–767. doi: 10.1016/0016-5085(95)90449-2. [DOI] [PubMed] [Google Scholar]
- 46.Mainous MR, Ertel W, Chaudry IH, Deitch EA. The gut: a cytokine-generating organ in systemic inflammation? Shock. 1995;4:193–199. [PubMed] [Google Scholar]
- 47.Deitch EA, Xu D, Franko L, Ayala A, Chaudry IH. Evidence favoring the role of the gut as a cytokine-generating organ in rats subjected to hemorrhagic shock. Shock. 1994;1:141–145. doi: 10.1097/00024382-199402000-00010. [DOI] [PubMed] [Google Scholar]
- 48.McGregor DD. The effect of sympathetic nerve stimulation on vasoconstrictor responses in perfused mesenteric vessels of the rat. J Physiol (Lond) 1965;177:21–30. doi: 10.1113/jphysiol.1965.sp007572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsutsui M, Milstien S, Katusic ZS. Effect of tetrahydrobiopterin on endothelial function in canine middle cerebral arteries. Circ Res. 1996;79:336–342. doi: 10.1161/01.res.79.2.336. [DOI] [PubMed] [Google Scholar]
- 50.Moncada S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann NY Acad Sci. 1997;811:60–67; discussion 67–69. doi: 10.1111/j.1749-6632.1997.tb51989.x. [DOI] [PubMed] [Google Scholar]
- 51.Sessa WC. The nitric oxide synthase family of proteins. J Vasc Res. 1994;31:131–143. doi: 10.1159/000159039. [DOI] [PubMed] [Google Scholar]
- 52.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 53.Buga GM, Gold ME, Fukuto JM, Ignarro LJ. Shear stress-induced release of nitric oxide from endothelial cells grown on beads. Hypertension. 1991;17:187–193. doi: 10.1161/01.hyp.17.2.187. [DOI] [PubMed] [Google Scholar]
- 54.Rubanyi GM, Romero JC, Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. Am J Physiol. 1986;250:H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- 55.Weigert AL, et al. Endothelium-dependent vascular hyporesponsiveness without detection of nitric oxide synthase induction in aortas of cirrhotic rats. Hepatology. 1995;22:1856–1862. [PubMed] [Google Scholar]
- 56.Ortiz MC, et al. Vascular hyporesponsiveness in aortic rings from cirrhotic rats: role of nitric oxide and endothelium. Clin Sci (Colch) 1996;91:733–738. doi: 10.1042/cs0910733. [DOI] [PubMed] [Google Scholar]
- 57.Tsoporis J, Fields N, Lee RM, Leenen FH. Arterial vasodilation and cardiovascular structural changes in normotensive rats. Am J Physiol. 1991;260:H1944–H1952. doi: 10.1152/ajpheart.1991.260.6.H1944. [DOI] [PubMed] [Google Scholar]
- 58.Sumanovski LT, Battegay E, Stumm M, Kooij M, Sieber CC. Increased mesenteric angiogenesis in portal hypertensive rats: role of nitric oxide. Hepatology. 1999;29:1044–1049. doi: 10.1002/hep.510290436. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi M, Ishida T, Traub O, Corson MA, Berk BC. Mechanotransduction in endothelial cells: temporal signaling events in response to shear stress. J Vasc Res. 1997;34:212–219. doi: 10.1159/000159225. [DOI] [PubMed] [Google Scholar]
- 60.Cooke JP, Rossitch E, Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinemann A, Stauber RE. The role of inducible nitric oxide synthase in vascular hyporeactivity of endotoxin-treated and portal hypertensive rats. Eur J Pharmacol. 1995;278:87–90. doi: 10.1016/0014-2999(95)00138-b. [DOI] [PubMed] [Google Scholar]
- 62.Sogni P, Smith AP, Gadano A, Lebrec D, Higenbottam TW. Induction of nitric oxide synthase II does not account for excess vascular nitric oxide production in experimental cirrhosis. J Hepatol. 1997;26:1120–1127. doi: 10.1016/s0168-8278(97)80121-3. [DOI] [PubMed] [Google Scholar]
- 63.Fernandez M, et al. Evidence against a role for inducible nitric oxide synthase in the hyperdynamic circulation of portal-hypertensive rats. Gastroenterology. 1995;108:1487–1495. doi: 10.1016/0016-5085(95)90698-3. [DOI] [PubMed] [Google Scholar]
- 64.Llach J, et al. Prognostic value of arterial pressure, endogenous vasoactive systems, and renal function in cirrhotic patients admitted to the hospital for the treatment of ascites. Gastroenterology. 1988;94:482–487. doi: 10.1016/0016-5085(88)90441-6. [DOI] [PubMed] [Google Scholar]
- 65.Wells CL, Maddaus MA, Simmons RL. Role of the macrophage in the translocation of intestinal bacteria. Arch Surg. 1987;122:48–53. doi: 10.1001/archsurg.1987.01400130054008. [DOI] [PubMed] [Google Scholar]
- 66.Aranow JS, Fink MP. Determinants of intestinal barrier failure in critical illness. Br J Anaesth. 1996;77:71–81. doi: 10.1093/bja/77.1.71. [DOI] [PubMed] [Google Scholar]
- 67.Gross SS, Jaffe EA, Levi R, Kilbourn RG. Cytokine-activated endothelial cells express an isotype of nitric oxide synthase that is tetrahydrobiopterin-dependent, calmodulin-independent and inhibited by arginine analogs with a rank-order of potency characteristic of activated macrophages. Biochem Biophys Res Commun. 1991;178:823–829. doi: 10.1016/0006-291x(91)90965-a. [DOI] [PubMed] [Google Scholar]
- 68.Werner ER, Werner-Felmayer G, Wachter H, Mayer B. Biosynthesis of nitric oxide: dependence on pteridine metabolism. Rev Physiol Biochem Pharmacol. 1996;127:97–135. doi: 10.1007/BFb0048266. [DOI] [PubMed] [Google Scholar]
- 69.Stroess E, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99:41–46. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wever RMF, Van Dam T, Van Rijn HJM, DeGroot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun. 1997;237:340–344. doi: 10.1006/bbrc.1997.7069. [DOI] [PubMed] [Google Scholar]
- 71.Werner ER, Werner-Felmayer G, Wachter H. Tetrahydrobiopterin and cytokines. Proc Soc Exp Biol Med. 1993;203:1–12. doi: 10.3181/00379727-203-43566a. [DOI] [PubMed] [Google Scholar]
- 72.Werner-Felmayer G, et al. Pteridine biosynthesis in human endothelial cells. Impact on nitric oxide-mediated formation of cyclic GMP. J Biol Chem. 1993;268:1842–1846. [PubMed] [Google Scholar]
- 73.Rosenkranz-Weiss P, et al. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J Clin Invest. 1994;93:2236–2243. doi: 10.1172/JCI117221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katusic ZS, Stelter A, Milstien S. Cytokines stimulate GTP cyclohydrolase I gene expression in cultured human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:27–32. doi: 10.1161/01.atv.18.1.27. [DOI] [PubMed] [Google Scholar]
- 75.Bhagat K, Vallance PJ. Induction of nitric oxide synthase in humans, in vivo: endothelial nitric oxide synthase masquerading as inducible nitric oxide synthase. Circulation. 1997;96:A4069. (Abstr.) [Google Scholar]