

Abstract
Systemic lupus erythematosus is characterized by the production of autoantibodies against nucleic acid-associated Ags. We previously found that Tlr7 was required for anti-Sm and Tlr9 for anti-chromatin autoantibodies. Yet, although Tlr7 deficiency ameliorated disease, Tlr9 deficiency exacerbated it. Despite the mechanistic and clinical implications of this finding, it has yet to be elucidated. In this study, we characterize MRL/lpr lupus-prone mice genetically deficient in Tlr7, Tlr9, both Tlr7 and Tlr9, or Myd88 to test whether Tlr7 and Tlr9 function independently or instead regulate each other. We find that disease that is regulated by Tlr9 (and hence is worse in its absence) depends on Tlr7 for its manifestation. In addition, although Tlr7 and Tlr9 act in parallel pathways on different subsets of autoantibodies, Tlr9 also suppresses the production of Tlr7-dependent RNA-associated autoantibodies, suggesting previously unrecognized cross-regulation of autoantibody production as well. By comparing disease in mice deficient for Tlr7 and/or Tlr9 to those lacking Myd88, we also identify aspects of disease that have Tlr- and Myd88-independent components. These results suggest new models for how Tlr9 regulates and Tlr7 enhances disease and provide insight into aspects of autoimmune disease that are, and are not, influenced by TLR signals.
The TLRs are a family of cell surface or endosomal pattern recognition receptors (1). Many of the known ligands for TLRs are microbial products such as bacterial LPS, but some TLR family members can be activated by nucleic acids that may be either pathogen or host derived (2, 3). TLR ligation results in the activation of transcription factors including NF-κB and IFN regulatory factor family members (4), which both activate the innate immune system and contribute to a robust adaptive immune response.
In addition to their role in the immune response to pathogens, TLRs have recently been implicated in autoimmunity (5, 6). Pioneering work demonstrated that B cells transgenically expressing a rheumatoid factor (RF) BCR can be activated ex vivo by immune complexes of anti-DNA or anti-RNA specificity (7, 8). This activation was dependent on Tlr9, the endosomal sensor for dsDNA, or Tlr7, an endosomal sensor for ssRNA; the signaling adaptor Myd88, which is downstream of both Tlr7 and Tlr9, was also required. Subsequent studies in RF BCR-transgenic animals confirmed and extended these findings in vivo (9). Other studies in anti-DNA and anti-RNA BCR-transgenic models demonstrated a similar requirement for Tlr9 or Tlr7, respectively, for activation of antinucleic acid B cells (10, 11). In contrast, the roles of TLRs in activating non-B cells, such as myeloid cells, in systemic autoimmunity are less clear. In vitro experiments implicate immune-complex, FcγR-dependent activation of dendritic cells (DCs) and plasmacytoid DCs, in promoting IFN-I and other cytokine secretion in a partially TLR-dependent fashion (12, 13). The roles of TLRs in DCs in vivo have not been specifically defined with respect to systemic autoimmunity.
Using the MRL/lpr murine model of systemic lupus erythematosus, we previously examined the role of Tlr9 and Tlr7 in spontaneous disease in vivo using mice that lacked one or the other of these genes in all tissues. Tlr9 was required for endogenous anti-DNA Ab production (14, 15), a finding that has since been reproduced in other murine models of lupus (10, 16, 17). Unexpectedly, in Tlr9-deficient animals, despite the absence of anti-DNA Abs—the clinical hallmark of lupus—several parameters of disease, including hypergammaglobulinemia, lymphocyte activation, and glomerulonephritis, were significantly worsened (14, 15). In contrast, Tlr7 deficiency had no effect on anti-DNA Abs but instead prevented the appearance of anti-Sm autoantibodies in the MRL/lpr model (15). Disease phenotypes were partially ameliorated in the absence of Tlr7 despite the continued presence of anti-DNA and antinucleosome Abs. The discovery of a paradoxical and unique regulatory role for Tlr9 in lupus was surprising and has important implications for the design of clinical strategies as well as for our understanding of the mechanisms of disease initiation and propagation. So far, there has been no additional insight into how Tlr9 might play a regulatory role. Indeed, this finding raised a major question. Because Tlr7 and Tlr9 are homologous receptors expressed in similar tissues with (as far as is known) identical signaling pathways, how is it that the lack of either of these two genes results in opposite effects on immune activation and global disease?
One hypothesis is that Tlr7 and Tlr9 work in parallel, independent pathways. This would be consistent with their apparent parallel effects on distinct subsets of autoantibodies (10, 11, 15). In this view, divergent disease outcomes might arise from unsuspected differences in signaling pathways and/or from expression in different tissues at different times coupled with differential exposure to ligands. In contrast, it is possible that Tlr7 and Tlr9 are operating in series, with signals mediated by one influencing the other either directly or indirectly. One model of Tlr9 and Tlr7 interdependency postulates that Tlr9-dependent anti-chromatin Abs are important in the clearance of cellular debris, which in turn contain the ligands for both Tlr9 (i.e., DNA-related) and Tlr7-dependent (i.e., RNA-related) activation (18–20). In the absence of Tlr9, such Abs would be missing, leading to overstimulation of Tlr7 as a result of excess ligand. A second model postulates a direct interaction, with Tlr9 regulating signals via Tlr7 within the same cell. This could occur whether there were competition for downstream signaling components and/or whether Tlr9 signals induced expression of genes that counterregulated both Tlr9 and Tlr7 signals (21). The first step in understanding the roles of Tlr7 and Tlr9 and their interactions is to determine, genetically, whether one acts upstream of the other or whether they operate in parallel. To investigate this, we generated cohorts of mice simultaneously deficient in both Tlr7 and Tlr9, and we report on their phenotype and the implications for how Tlr7 and Tlr9 regulate autoimmunity in divergent directions.
In addition, though the absence of Myd88 ameliorates disease and reduces autoantibodies (8, 22), whether in regulating lupus it acts only downstream of Tlr7 and Tlr9 or rather has additional upstream inputs, such as other Tlrs or IL-1/18, has not been addressed. Furthermore, it is not known whether all immune activation and disease in the MRL/lpr model is dependent on Myd88/Tlr signals or whether additional innate immune receptor families or other factors also contribute. To address these issues, we produced backcrossed mice deficient in the adaptor Myd88 and compared them to the mice doubly deficient for Tlr7 and Tlr9. In this aspect of the study, we found that although Tlr7 and Tlr9 are important upstream inputs for Myd88, the control of autoimmunity by innate immunity is more complex than anticipated, with evidence for Tlr7/9-independent components as well as Myd88-independent aspects of disease.
Materials and Methods
Mice
To generate a cohort including Tlr7y/−Tlr9−/− mice and littermates for comparison, previously described Tlr9−/− and Tlr7−/− mice on the lupusprone MRL/MpJlpr/lpr background (15) were intercrossed by standard breeding techniques. To simplify the breeding strategy, given that Tlr7 is located on the X chromosome, only male mice were included in this analysis, except where otherwise noted. Representative animals were genotyped using a 768 single nucleotide polymorphism Illumina mouse genotyping array and were determined to be >98% backcrossed to the MRL/MpJlpr/lpr background and were homozygous for MRL alleles at all known MRL lupus susceptibility loci (23–26). Mice were analyzed at 16 wk of age, except where indicated, because of the previously reported accelerated mortality of the Tlr9−/− group (15).
Myd88−/− mice on the MRL/MpJlpr/lpr background were generated by crossing Myd88−/− mice (mixed C57BL/6 and 129Sv background) with MRL/MpJlpr/lpr males for six generations. Mice heterozygous for the Myd88-deleted allele were intercrossed to generate Myd88+/+ and Myd88−/− littermates. These animals were aged to 16 or 24 wk for analysis.
All animal work was approved by the Yale Institutional Animal Care and Use Committee.
Measurement of serum autoantibodies and cytokines
HEp-2 immunofluorescence assays (Antibodies, Davis, CA) were performed as previously described (14) with serum diluted at 1/200 and were scored for relative fluorescence intensity of nuclear and cytoplasmic staining on a scale of 0–3 and for the presence or absence of mitotic chromatin by an observer blinded to the genotype of the mice.
For anti-nucleosome ELISA, polystyrene plates were coated with poly-L-lysine (Sigma-Aldrich, St. Louis, MO). Plates were then incubated with phenol-extracted and S1 nuclease-treated dsDNA from calf thymus (Sigma-Aldrich), followed by calf thymus histones type II-AS (Sigma-Aldrich). After blocking with 1% BSA in PBS, serial dilutions of serum from 1/200 to 1/5400 were added. Specific Abs were detected with alkaline phosphatase-conjugated goat anti-mouse IgG (Southern Bio-technology Associates, Birmingham, AL), and absorbance at 405/630 nm was compared with a purified PL2-3 monoclonal anti-nucleosome standard (27) for quantitation.
Anti-Sm ELISA was performed as previously described (14) with serial dilutions of serum from 1/200 to 1/5400. ELISAs for anti-ribosomal P Ag autoantibodies and anti-RNA Abs were described previously (28).
Anti-IgG2a RF titers were determined by ELISA essentially as described previously (29). Polystyrene plates were coated with purified mAb 23.3 (IgG2a,λ anti–4-hydroxy-3-nitro-phenylacetyl) overnight. After blocking with 1% BSA in PBS, serial dilutions of serum from 1/200 to 1/5400 were added. Specific Abs were detected with biotinylated anti-κ L chain (clone 187.1; BD Pharmingen, San Diego, CA), followed by alkaline-phosphatase–conjugated streptavidin (Invitrogen, Carlsbad, CA), and absorbance at 405/630 nm was compared with a 400tμ23 (IgM-RF) standard for quantitation.
Total serum IgG was determined by ELISA as described previously (14). Titers of individual IgG isotypes were measured by cytometric bead assay (Millipore, Bedford, MA) per the manufacturer’s protocols.
Serum IL-6, TNF-α, IL-12p40, IL-12p70, and IL-10 were measured by multiplex cytometric bead assay (Bio-Rad, Hercules, CA) per the manufacturer’s instructions. Serum IL-23(p19/p40) was measured by ELISA (eBioscience, San Diego, CA). Serum IFN-α was measured as previously described (14) with serum diluted 1/20.
Analysis of lymphocyte subsets
Spleen cells were isolated and stained as previously described (14) with the following Abs for FACS analysis: anti-CD90.2 (30H12), anti-CD45R (RA3-6B2, BD Biosciences), anti-CD4 (GK1.5; Biolegend, San Diego, CA), anti-CD44 (Pgp1), and anti-CD62L (Mel-14; eBioscience). Anti-CD90.2 and anti-CD44 were purified and fluorescence conjugated in our laboratory. Dead cells were excluded by staining with propidium iodide. For measurement of regulatory T cells, spleen and axillary lymph node cells were stained as above with anti-TCRβ (H57-597; BD Biosciences), anti-CD4, anti-CD25 (PC61; BD Biosciences), followed by fixation/permeabilization and anti-Foxp3 (FJK-16s; eBioscience) staining as recommended by the manufacturer. Dead cells were excluded with ethidium monoazide bromide.
Evaluation of clinical disease
For skin disease, mice were scored for the extent of dorsal lesion on a scale of 0–5, with up to one additional point for presence of ear dermatitis and facial rash or loss of whiskers as described previously (15). Proteinurea was measured using a colorimetric dipstick assay (Albustix; Bayer, Pittsburgh, PA). For kidney disease, formalin-fixed and paraffin-embedded sections stained with either H&E or periodic acid Schiff stain were scored for glomerular disease by an independent observer who was blinded to the genotype of the mice. For interstitial disease, H&E-stained slides were photographed, and the total area of infiltrates relative to the area of the kidney cross-section was determined using ImageJ software (National Institutes of Health, Bethesda, MD).
Results
Autoantibody regulation by Tlr7, Tlr9, and Myd88
Autoantibodies were measured in the serum of 16-wk-old Tlr7y/− Tlr9−/− and control mice by the HEp-2 anti-nuclear Ab (ANA) assay (Fig. 1A, 1B). As previously observed, the intensity of nuclear-staining HEp-2 patterns was significantly reduced in the absence of Tlr9 (Fig. 1B, □). Consistent with prior results (14, 15), the remaining nuclear staining observed in some Tlr9-deficient mice was restricted to a speckled staining pattern (data not shown), suggesting both a qualitative and a quantitative difference in the autoantibody repertoire. Importantly, mice lacking both Tlr9 and Tlr7 did not develop an ANA of either the homogenous or speckled pattern, reflecting that the speckled patterns in Tlr9-de-ficient mice were due to Tlr7-dependent autoantibodies. To confirm that the nuclear HEp-2 staining pattern is dependent on Tlr9 and Tlr7, we analyzed MRL/lpr mice deficient in Myd88; these mice also lacked an ANA at 16 and 24 wk of age (Fig. 1C, □), consistent with other reports by our group and others (8, 22). Hence, the classic ANA is entirely dependent on autoantibodies that require either Tlr9 or Tlr7, and Myd88, for their development.
FIGURE 1.

ANAs are absent in mice lacking Myd88 or both Tlr9 and Tlr7. A, Representative HEp-2 ANA staining from serum of 16-wk (Tlr) or 24-wk (Myd88) old MRL/lpr mice of the indicated genotypes. Arrows indicate mitotic chromatin staining. B and C, HEp-2 slides were scored for intensity of nuclear (□) and cytoplasmic (■) staining patterns. Data are represented as mean ± SEM. Statistics were calculated by two-tailed Mann-Whitney U test. *p < 0.05; **p < 0.01; ***p < 0.001. D and E, HEp-2 slides were scored for the presence or absence of mitotic chromatin staining.
Previously we observed an increased frequency of cytoplasm-staining autoantibodies when Tlr9 was absent (15). To evaluate this phenomenon, we scored HEp-2 slides for the intensity of cytoplasmic staining (Fig. 1B, ■). Cytoplasmic staining was increased in both frequency and intensity in mice lacking Tlr9. However, the cytoplasmic staining was reduced in both Tlr7y/−Tlr9+/− and Tlr7y/−Tlr9−/− mice, suggesting that anti-cytoplasmic autoantibodies are Tlr7 dependent regardless of the Tlr9 genotype. Cytoplasmic staining was more prevalent in 24-wk than 16-wk-old mice and was significantly reduced in the absence of Myd88 (Fig. 1C, ■). Thus, Tlr9 suppresses Tlr7-dependent and Myd88-dependent anti-cytoplasmic Abs.
The staining of mitotic chromosomes in the HEp-2 assay indicates the presence of anti-chromatin autoantibodies (Fig. 1A, arrows). In agreement with prior results (15), these autoantibodies were Tlr9 dependent but not Tlr7 dependent (Fig. 1D); they were also absent in Myd88-deficient mice (Fig. 1E). Overall, these data revealed that HEp-2 staining autoantibodies of all types in the serum of MRL/lpr mice were reduced to near-background levels when both Tlr7 and Tlr9, or their signaling adaptor Myd88, were absent.
Although it has the advantage of detecting autoantibodies binding to native structures, the HEp-2 assay provides limited qualitative information about the identity of these autoantigens. Therefore, we performed ELISAs to quantitate autoantibodies to defined autoantigens. Anti-nucleosome autoantibodies were significantly reduced in the absence of Tlr9 (Fig. 2A). The residual anti-nucleosome activity observed in Tlr9-deficient animals was further reduced in Tlr7y/−Tlr9−/− mice. Anti-nucleosome Abs were also significantly reduced in Myd88−/− mice (Fig. 2B). These data suggest that Tlr9 and Myd88 are required for the majority of spontaneous anti-nucleosome Ab formation in vivo, whereas intact Tlr7 permits a low level of anti-nucleosome Ab to develop.
FIGURE 2.

Tlr7, Tlr9, and Myd88 dependence of autoantibodies differs depending on the nature of the autoantigen. IgG serum autoantibodies were measured by ELISA in mice of the indicated genotypes and ages. A and B, Anti-nucleosome Abs measured in units relative to micrograms per milliliter PL2-3 binding. C and D, Anti-Sm Abs measured in units relative to micrograms per milliliter Y12 binding. E and F, Anti-riboP Abs expressed in OD units. G and H, Anti-RNA Abs measured in OD units. I and J, Total κ+ anti-IgG2a RF Abs measured relative to micrograms per milliliter 400tμ23 binding. Statistics were calculated by two-tailed Mann-Whitney U test. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we examined serum autoantibodies to RNA-associated proteins, such as Sm, a component of the nuclear pre-mRNA spliceosome (30, 31). At 16 wk of age, few animals had detectable anti-Sm Abs; however, the frequency and titer of anti-Sm were significantly elevated in mice lacking Tlr9 (Fig. 2C). By 24 wk of age, a greater proportion of wild-type MRL/lpr mice had developed anti-Sm, but mice lacking Myd88 did not develop significant anti-Sm titers (Fig. 2D). Analysis of an independent cohort of 24-wk-old Tlr7-deficient mice showed that anti-Sm Abs were dependent on Tlr7 (Supplemental Fig. 1), consistent with previous reports on a younger cohort (15). Similarly, Abs to ribosomal P Ag (32) were elevated in the absence of Tlr9 unless Tlr7 was also absent (Fig. 2E), implying that intact Tlr9 suppresses the development of Abs to these Tlr7-dependent RNA-associated proteins. Anti-ribosomal P Ag Abs were absent in 24-wk-old Myd88-deficient mice (Fig. 2F), although low titers were detectable in 16-wk-old Tlr7y/−Tlr9−/− mice (Fig. 2E), suggesting that Myd88-dependent pathways in addition to Tlr7 or Tlr9 may also contribute to the production of anti-ribosomal P Abs.
Autoantibodies reactive to RNA itself (28) were significantly reduced when Tlr7 was absent (Fig. 2G). These autoantibodies were further reduced when both Tlr7 and Tlr9 were absent; intriguingly, these data suggest that Tlr9 may be capable of activating anti-RNA B cells to some extent when Tlr7 is absent, particularly considering that Tlr7-dependent HEp-2 cytoplasmic staining was increased in Tlr9-deficient animals (Fig. 1B). Production of anti-RNA Abs required Myd88 (Fig. 2H), as expected.
Finally, we measured titers of anti-IgG2a RF autoantibodies (33). Interestingly, although absence of either Tlr7 or Tlr9 alone had no significant impact on RF titers, these titers were significantly reduced in Tlr7y/−Tlr9−/− (Fig. 2I) and Myd88-deficient (Fig. 2J) mice. These results suggest that nucleic acid-containing immune complexes are an important immunogen for RF B cells in vivo, as has been shown artificially in vitro (7, 8). However, both Tlr7y/−Tlr9−/− and Myd88−/− MRL/lpr mice had significantly greater levels of RF than did age-matched nonautoimmune BALB/c controls, suggesting that both Myd88-dependent and Myd88-independent pathways contribute to the production of these autoantibodies, presumably through immune complexes that do or do not contain nucleic acids and/or through the activation of MyD88-independent nucleic acid sensors (34).
Immune activation is regulated by TLR7, TLR9, and MyD88
Activated immune cells accumulate in MRL/lpr mice, resulting in increased serum Ig titers, splenomegaly, tissue damage, and lymphadenopathy (35). To assess global immune activation, we measured serum Ig by ELISA. As previously observed, total serum Ig was significantly elevated in mice deficient in Tlr9 compared with Tlr9+/+ MRL/lpr mice (Fig. 3A). Ig concentrations were lower in Tlr7y/−Tlr9−/− mice than in Tlr9−/−, indicating that the stimulatory effect of Tlr9 deficiency requires intact Tlr7. Similarly, mice deficient in Myd88 also had reduced serum Ig concentrations at 16 and 24 wk of age (Fig. 3B). Importantly, however, although Ig titers were reduced in Tlr7y/−Tlr9−/− and Myd88−/− mice, they remained significantly higher than in age-matched BALB/c controls. Moreover, the presence of high IgG titers in Tlr7y/−Tlr9−/− and Myd88−/− mice indicates that the markedly reduced amounts of selected serum autoantibodies in these mice (Figs. 1, 2) is not due to a nonspecific defect in B cell activation and Ig class switch.
FIGURE 3.

Hypergammaglobulinemia is Tlr and Myd88 dependent. A and B, Total serum IgG was measured by ELISA in mice of the indicated genotypes and ages. C–F, Total serum IgG2a and IgG3 were measured by cytometric bead assay. Statistics were calculated by two-tailed Mann-Whitney U test. *p < 0.05; **p < 0.01; ***p < 0.001.
Additionally, we measured concentrations of individual IgG isotypes. Serum IgG2a, IgG2b, and IgG3 were all elevated in Tlr9−/− mice (Fig. 3C, 3E, and Supplemental Fig. 2) as previously reported (15) but were reduced to concentrations below those of Tlr-intact MRL/lpr mice when Tlr7 was also absent. Hence, the hypergammaglobulinemia of Tlr9-deficient mice was again dependent on Tlr7. Amounts of each IgG isotype were also reduced in Myd88-deficient animals (Fig. 3D, 3F, and Supplemental Fig. 2). Nonetheless, titers of individual IgG isotypes remained markedly elevated in Tlr7y/−Tlr9−/− and Myd88−/− mice in comparison with BALB/c controls.
We also measured selected serum cytokines in a cohort of 13- to 17-wk old-animals (n ≥ 5, except n = 3 for Myd88−/−). There were no significant differences, as measured by cytometric bead assay, among the groups in the concentrations of IL-6, IL-10, IL-12, IL-23, and TNF-α (data not shown). A serum ELISA assay also did not reveal any differences in IFN-α concentrations in this cohort, although amounts in almost all mice were near the limits of detection (data not shown).
Tlr9−/− mice had significantly exacerbated splenomegaly and lymphadenopathy compared with Tlr9-intact littermates (Fig. 4A). However, this was ameliorated in mice also lacking Tlr7, indicating that Tlr7 is required for the increased spleen and lymph node weight observed in the Tlr9-deficient animals. Although spleen and lymph nodes were also similar in weight in both Myd88+/+ and Myd88−/− animals at 16 wk of age, by 24 wk their weights had increased significantly in Myd88+/+ but not Myd88−/−animals (Fig. 4B). Both Tlr7y/+Tlr9−/−and Myd88−/−MRL/lpr mice had larger spleens and lymph nodes than age-matched BALB/c controls.
FIGURE 4.

Splenomegaly and lymphadenopathy are Tlr and Myd88 dependent. Spleen (□) and axillary lymph nodes (■) from MRL/lpr mice of the indicated Tlr (A) or Myd88 (B) genotypes were weighed. Data are represented as mean ± SEM. Statistics were calculated by two-tailed Mann-Whitney U test. ***p < 0.001.
To determine the cellular composition of the spleens, we enumerated various cell populations by flow cytometry. In Myd88−/− mice, we found a 2- to 3-fold reduction in the number of T cells, plasmacytoid, and conventional DCs and CD11b+CD11c− macrophages (data not shown). Conversely, the increased spleen weight observed in Tlr7y/+Tlr9−/− mice compared with other groups was attributed to an expansion of T cells, especially CD45R+CD90+ cells, as well as an increase in CD11c+ cells, whereas B cell numbers increased only slightly (data not shown).
We evaluated CD4+ T cells for the expression of the activation markers CD44 and CD62L (Fig. 5A). Tlr7y/+Tlr9−/− mice had more CD44highCD62Lhigh activated and CD44highCD62Llow memory T cells compared with Tlr9-intact or Tlr9+/− groups (Fig. 5B). Again, when Tlr7 was also absent, the expansion of activated and memory T cells was not observed. Similarly, 16- and 24-wk-old Myd88+/− animals had an increase in the number of activated and memory CD4 T cells compared with Myd88-deficient animals (Fig. 5C). No differences were observed among any of the groups in Foxp3+ regulatory T cells as a proportion of CD4+ cells (data not shown).
FIGURE 5.

Activation of CD4 T cells. A, Representative FACS staining of CD45R−CD90.2+CD4+ T cells from 16-wk-old Myd88+/− and Myd88−/− MRL/lpr mice. B and C, Number of CD45R−CD90.2+CD4+ cells with naive (CD44lowCD62Lhigh, left), activated (CD44highCD62Lhigh, middle) and activated/memory (CD44highCD62Llow, right) phenotypes. Statistics were calculated by two-tailed Mann-Whitney U test. *p < 0.05; **p < 0.01.
Kidney disease is reduced in absence of Tlr7 and Tlr9 or Myd88
Aged MRL/lpr mice develop severe kidney disease characterized by glomerulonephritis and interstitial infiltrates composed of lymphocytes and monocytes, with resultant proteinuria (36). Proteinuria was significantly elevated in the Tlr9−/− mice but reduced in Tlr7y/−Tlr9+/− mice compared with the Tlr-intact controls (Fig. 6A). The elevated proteinuria in Tlr9−/− mice was dependent on Tlr7, because it was absent in the Tlr7y/−Tlr9−/− group. Proteinuria was also significantly reduced in 24 wk old Myd88−/− mice (Fig. 6B).
FIGURE 6.

Kidney disease is ameliorated in Tlr7/9 and Myd88-deficient mice. Disease parameters were assessed in mice of the indicated genotypes and ages. A and B, Proteinuria. C and D, Severity of glomerulonephritis. E and F, The area of interstitial and perivascular infiltrates expressed as a percentage of the total kidney area. Data are represented as mean ± SEM. Statistics were calculated by two-tailed Mann-Whitney U test. *p < 0.05; **p < 0.01; ***p < 0.001.
GlomerulonephritiswassignificantlymoresevereinTlr9−/−mice at 16 wk compared with controls (Fig. 6C). This exacerbated glomerulonephritis required Tlr7, because it was not observed in the Tlr7y/−Tlr9−/− group. Glomerulonephritis was also reduced in the absence of MyD88 (Fig. 6D). Notably, interstitial infiltrates were not significantly changed in the absence of either Tlr7 or Tlr9 alone but were significantly reduced when both Tlrs or Myd88 were absent (Fig. 6E, 6F), suggesting that either Tlr7 or Tlr9 is sufficient to promote interstitial disease, but that one or the other is required.
Dermatitis incidence, but not severity, is reduced in absence of Myd88
MRL/lpr mice develop dorsal skin lesions as they age, as well as a facial rash, loss of whiskers, and degradation of the soft tissue of the ears (37, 38). Because few mice of any genotype had developed dermatitis at 16 wk of age, a separate group of MRL/lpr, Tlr7y/−Tlr9−/− and Myd88−/− mice were aged to 24 wk to evaluate skin disease. Although we did not observe significant sex-dependent differences in other aspects of disease (data not shown), dermatitis incidence and severity were greater in female mice than male mice of each genotype (Fig. 7A, 7B), and so males and females were analyzed separately. The incidence of dermatitis (skin score > 1) was not different between MRL/lpr and Tlr7y/−Tlr9−/− MRL/lpr mice, indicating that skin disease does not require Tlr7 or Tlr9. However, dermatitis incidence was reduced in mice lacking Myd88 (Fig. 7A), suggesting that there may be a role for other TLR family members or signaling through the IL-1R family of cytokine receptors, which have been implicated in psoriasis and other dermatological diseases (39, 40). Notably, although the number of Myd88-deficient mice developing dermatitis by 24 wk was reduced, animals that did develop dermatitis had lesions of similar severity to Myd88-intact mice (Fig. 7B). The residual disease in Myd88-deficient animals indicates that MyD88 is not required for dermatitis, although it enhances its incidence.
FIGURE 7.
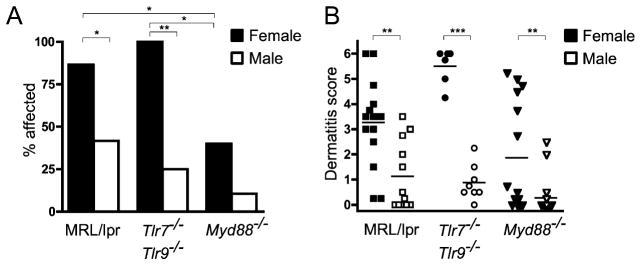
MRL/lpr mice deficient in Tlr7/9 or Myd88 develop dermatitis. A, Percentage of animals of the indicated genotypes and genders with a dorsal skin lesion of ≥5 mm diameter by 24 wk of age. Statistics were calculated by Fisher’s exact test. *p < 0.05; **p < 0.01. B, Dermatitis scores for female (solid symbols) or male (open symbols) MRL/lpr mice of the indicated genotypes at 24 wk of age. Scores were assigned as described in Materials and Methods.
Aged Tlr7y/−Tlr9−/− mice do not develop autoantibodies or lupus-like disease
To determine whether the mild disease and the absence of auto-antibodies observed in 16-wk-old Tlr7y/−Tlr9−/− MRL/lpr mice represented a delay in the progression of disease or an absolute requirement for Tlr-dependent signals, we aged a cohort of male and female Tlr7y/−Tlr9−/− MRL/lpr mice to 48 wk of age and evaluated serum autoantibodies and other disease parameters. Because all Tlr9−/− and most Tlr-intact MRL/lpr mice died before 48 wk, it was not possible to provide age-matched controls of these genotypes for direct comparison. Nonetheless, nine of nine Tlr7y/−Tlr9−/− mice were negative in the HEp-2 and anti-nucleosome ELISA assays as late as 48 wk of age (data not shown). Five of nine animals were negative in the anti-Sm ELISA, whereas the remaining animals had levels near the lower limit of detection in the assay (~4 μg/ml Y12 equivalents.) Serum IgG concentrations ranged from 4.9 to 29.4 mg/ml, similar to the amounts observed in the 16-wk cohort, indicating that hyper-gammaglobulinemia did not become more severe with increasing age. Kidneys were evaluated by histology; two of nine mice had moderate glomerulonephritis (GN score of 2), whereas none had significant interstitial disease (data not shown). Survival of mice deficient in either Tlr7 or both Tlr7 and Tlr9 was slightly improved compared with Tlr-intact mice, although this difference was not statistically significant, given the small cohort size, we were able to analyze at this advanced age (Supplemental Fig. 3). Thus, albeit in a limited study, the effects of lack of both Tlr7 and Tlr9 are persistent in substantially ameliorating serologic, histologic, and clinical signs of lupus.
Discussion
In previous studies, we described the phenotypes of MRL/lpr lupusprone mice lacking either Tlr7 or Tlr9. Tlr7 had no effect on ANA and anti-DNA Ab production, but disease was ameliorated when Tlr7 was absent (15). In contrast, Tlr9 was essential for anti-DNA Abs, the clinical hallmark of lupus, but disease was unexpectedly exacerbated in Tlr9-deficient animals (14, 15). To better understand the interaction of these genes in murine lupus and resolve the role of Tlr9 in disease, we generated and analyzed cohorts of mice simultaneously deficient in both Tlr genes, along with Tlr7-and Tlr9-deficient mice for direct comparison. Moreover, we analyzed animals deficient in the signaling adaptor Myd88, which is required for signaling through both Tlr7 and Tlr9, as well as other Tlr genes and the Il1r family of cytokine receptors.
Although the design of our current experiments was straightforward, we were able to reach a number of important conclusions that significantly advance our knowledge of how Tlr genes and Myd88 both promote and regulate autoimmunity, as well as the relationships among Tlr7, Tlr9, and Myd88. First, although we previously knew that Tlr7 and Tlr9 largely controlled independent sets of autoantibodies and were in this sense acting in parallel, we discovered that Tlr9 suppresses Tlr7-dependent anti-RNA associated autoantibodies. Thus, Tlr9 acts as an upstream regulator for this class of Ab. Second, for almost all elements of disease for which we previously ascribed a regulatory role for Tlr9, the exacerbated disease was no longer observed when Tlr7 was also absent. This implicates Tlr7 as functioning downstream of Tlr9 in promoting disease and again supports a model in which the Tlrs genetically interact. Taken together, these observations provide insight into the mechanism of the opposing effects of Tlr7 and Tlr9.
Furthermore, we demonstrated that Tlr7 and Tlr9 together were sufficient to account for the primary diagnostic characteristic of lupus, the ANA. The phenotypes of the Tlr7y/−Tlr9−/− mice were identical in this respect to the Myd88-deficient animals, establishing that no other Tlr, or other innate immune sensor, is involved in producing the ANA in MRL/lpr mice. A very recent report on the phenotype of Unc93b-deficient animals on the B6/lpr background similarly demonstrated reductions in antichromatin and RF, presumably due to effects of this gene on signaling from endosomal TLRs (41). Despite the exclusive influence of these receptors on autoantibodies, we discovered that there remained substantial aspects of disease that were independent of both Tlr and Myd88. Thus, some aspects of systemic autoimmunity either depend on other innate immune receptors yet to be identified or, in contrast to current theory (42), lack any need for innate stimulation. These findings support the need for further investigation to determine the roles of other innate receptors in systemic autoimmunity, as might be suggested by reports of TLR-independent DNA sensing pathways that lead to autoimmunity and IFN-I production (43, 44), including the pathway downstream of Trex1 (45).
On the basis of our findings, we propose a model by which the loss of Tlr9 exacerbates Tlr7-dependent disease. This might occur in cis, in trans, or both. In a cis-interaction model, the presence of Tlr9 directly inhibits the activity of Tlr7 within a single cell. This might occur through competition for rate-limiting downstream signaling components, such as MyD88 and others, so that (for example) a B cell with anti-RNA specificity could be more responsive to Tlr7 signals when Tlr9 is absent. A recent report (46) on cell lines suggested that a truncated form of the Unc93b gene product could differentially interact with TLR7 and TLR9, providing a potential mechanism by which TLR9 could inhibit TLR7. However, the authors could not find a naturally occurring truncated protein. Alternatively, Tlr9 signaling may be constitutively active to some degree because of both the abundance and stability of DNA ligands and the steady expression of the receptor and may tonically induce counterregulatory molecules, which in turn would inhibit subsequent signals via Tlr7. When Tlr9 is not expressed, such inhibitors would be absent, leading to greater stimulation via Tlr7. In support of this model, there is at least one report that overexpression of TLR9 can inhibit TLR7 signals in cell lines (47).
In a fundamentally different way, the interactions between Tlr7 and Tlr9 might occur in trans, through the effects of Tlr-dependent cytokine production, the activities of the different subsets of autoantibodies themselves or competition for niches for antibody-forming cells (48). In a limited survey of inflammatory cytokines, however, we did not observe differences in serum concentrations among the groups. In one particular model of trans-regulation, anti-DNA Abs controlled by Tlr9 are important in the clearance of pathogenic cellular debris, which contain ligands for both Tlr9 and Tlr7 (18–20, 49, 50). In the absence of Tlr9, Tlr7 might be over-stimulated because of increased availability of its ligands. To distinguish among these cis and trans models will require lineage-specific deletion of the Tlr genes, especially Tlr9, so that the role of Tlr in generating autoantibodies can be distinguished from effects in other cells and on end-organ damage. The key observation made in this study that Tlr9 regulates Tlr7-dependent disease provides a framework and strong rationale for such future studies.
Although we found that autoantibody production was mediated by parallel Tlr-dependent pathways, our findings in the doubly-deficient mice also revealed a previously unappreciated degree of overlap between the DNA- and RNA-associated specificities. This in turn has implications for how Tlrs recognize self-Ags in vivo. It is likely that the in vivo autoantigens, which include dead and dying cells and cellular debris, are heterogeneous mixtures containing both Tlr9- and Tlr7-activating DNA and RNA components (18, 20, 49, 50). If that is the case, then the low level of antinucleosome autoantibody in the single Tlr9 knockouts and the residual anti-RNA activity in Tlr7 knockouts may be explained by activation of the opposite specificity of Tlr by the mixed Ag; in the absence of both Tlr7 and Tlr9, or the common adaptor Myd88, neither type of autoantibody is generated. A second possibility is that the paradigm of Tlr7 recognizing exclusively RNA and Tlr9 recognizing exclusively DNA may be incomplete; indeed, TLR7 can bind to DNA (51). Either of these possibilities would be consistent with our data, as well as with findings in transgenic BCR systems of autoimmune activation (11). For example, antinucleosome immune complex activation of RF B cells was shown to be only partially dependent on either Tlr9 or Tlr7 alone, although it was abrogated in the absence of both Tlrs or Myd88 (9).
Mice lacking Myd88 were equivalent to the Tlr7/9-deficient animals in many respects, having nearly undetectable autoantibody titers and little disease, suggesting that Tlr7 and Tlr9 are required for the majority of disease in the MRL/lpr model and that other Myd88-dependent pathways are of less import. However, despite the lack of HEp-2–reactive autoantibodies in both Tlr7/9- and Myd88-deficient animals, both groups of mice developed dermatitis. The incidence of dermatitis was diminished in Myd88-deficient mice compared with Tlr7/9-deficient animals, suggesting that there may be a contribution of other Myd88-dependent pathways to development of dermatitis. In addition to its role in Tlr signaling, Myd88 is required for signal transduction downstream of the Il1r family of cytokine receptors, which have been implicated in psoriasis and other dermatological conditions (39, 40). Nonetheless, the dermatitis phenotype had a substantial Myd88-independent component. Increased spleen and lymph node weights and hypergammaglobulinemia were also still present in both Tlr7/9 or Myd88-deficient animals compared with a nonautoimmune strain. This could in part be caused by the lpr mutation of Fas, which prevents Fas/Fas ligand-mediated apoptosis (52). Importantly, however, the lpr mutation was not sufficient to provoke development of specific lupus autoantibodies or significant kidney disease in the absence of Tlr7/9 and Myd88-mediated signaling, consistent with the notion that the lpr mutation is an accelerator of disease mediated by other lupus-susceptibility loci in the MRL background. This role of Fas-deficiency has been confirmed in numerous studies of MRL mice deficient in key genes or cell types in the immune system (53–57). Taken together, these findings do suggest that Myd88-dependent innate signaling is not sufficient to account for all aspects of systemic autoimmune disease. Indeed, such results point to the possibility of other innate signaling mechanisms in stimulating certain types of autoimmunity.
Given the importance of nucleic acid recognition in a variety of systemic autoimmune syndromes, other recently-described pattern recognition receptors that recognize nucleic acids—including DAI, RIG-I, Mda5, and AIM2 (58–67)—might be playing unsuspected roles, either accounting for Myd88-independent aspects of disease or making contributions downstream of autoantibody production, which was largely dependent upon Tlr7 and Tlr9. However, it is important to realize that even if Tlr7 and/or Tlr9 are essential for some aspects of disease and autoantibody production, this does not rule out an additional essential role for other nucleic-acid sensing pathways. Alternatively or in addition, residual aspects of disease in the Tlr7y/−Tlr9−/− and Myd88−/− groups may depend upon protein autoantigens, which are not nucleic acid dependent. Nonetheless, it is clear that Tlr7 and Tlr9 play very essential roles in most aspects of disease. Perhaps most importantly, our results demonstrate that the regulatory role of Tlr9 is best understood in terms of regulating Tlr7-dependent signals. Work is underway to distinguish among the several possible ways in which this could occur; resolution will require the development of conditional alleles for Tlr9 and Tlr7 and their deployment in autoimmune models. Biochemical approaches will also be useful in testing some aspects of these models.
For the present, these findings have important therapeutic implications (68), as they suggest that inhibitors that target Tlr7 should be beneficial, whereas those that target Tlr9 alone may paradoxically exacerbate disease; however, inhibitors that target both may have even more efficacy. Indeed, studies of a synthetic inhibitor of both Tlrs in the MRL/lpr (69) and (NZB × NZW)F1 (70) models of lupus found significantly reduced lymphadenopathy and glomerulonephritis, as well as improved survival in treated animals.
Acknowledgments
We thank Yale Animal Resources Center for their outstanding animal husbandry and Ann Marshak-Rothstein and Eicke Latz for stimulating discussions.
This work was supported by National Institutes of Health Grant P01-AR050256. K.M.N. was supported by 1F32-AR055472 and a grant from the S.L.E. Lupus Foundation.
Abbreviations used in this paper
- ANA
anti-nuclear Ab
- DC
dendritic cell
- RF
rheumatoid factor
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007;220:251–269. doi: 10.1111/j.1600-065X.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 3.Krug A. Nucleic acid recognition receptors in autoimmunity. Handb Exp Pharmacol. 2008;38:129–151. doi: 10.1007/978-3-540-72167-3_7. [DOI] [PubMed] [Google Scholar]
- 4.Kaisho T, Tanaka T. Turning NF-κB and IRFs on and off in DC. Trends Immunol. 2008;29:329–336. doi: 10.1016/j.it.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Fischer M, Ehlers M. Toll-like receptors in autoimmunity. Ann N Y Acad Sci. 2008;1143:21–34. doi: 10.1196/annals.1443.012. [DOI] [PubMed] [Google Scholar]
- 6.Christensen SR, Shlomchik MJ. Regulation of lupus-related autoantibody production and clinical disease by Toll-like receptors. Semin Immunol. 2007;19:11–23. doi: 10.1016/j.smim.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leadbetter EA, I, Rifkin R, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 8.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, Imanishi-Kari T. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–440. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Lövgren T, Eloranta ML, Ba°ve U, Alm GV, Rönnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 13.Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, Rifkin IR. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178:6876–6885. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 14.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Lartigue A, Courville P, Auquit I, François A, Arnoult C, Tron F, Gilbert D, Musette P. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. 2006;177:1349–1354. doi: 10.4049/jimmunol.177.2.1349. [DOI] [PubMed] [Google Scholar]
- 17.Yu P, Wellmann U, Kunder S, Quintanilla-Martinez L, Jennen L, Dear N, Amann K, Bauer S, Winkler TH, Wagner H. Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int Immunol. 2006;18:1211–1219. doi: 10.1093/intimm/dxl067. [DOI] [PubMed] [Google Scholar]
- 18.Cocca BA, Cline AM, Radic MZ. Blebs and apoptotic bodies are B cell autoantigens. J Immunol. 2002;169:159–166. doi: 10.4049/jimmunol.169.1.159. [DOI] [PubMed] [Google Scholar]
- 19.Peng Y, Martin DA, Kenkel J, Zhang K, Ogden CA, Elkon KB. Innate andadaptiveimmune responseto apoptoticcells. J Autoimmun. 2007;29:303–309. doi: 10.1016/j.jaut.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172:6692–6700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- 21.O’Neill LA. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, Igawa T, Sugiyama N, Niiro H, Harada M. Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum. 2007;56:1618–1628. doi: 10.1002/art.22571. [DOI] [PubMed] [Google Scholar]
- 23.Watson ML, Rao JK, Gilkeson GS, Ruiz P, Eicher EM, Pisetsky DS, Matsuzawa A, Rochelle JM, Seldin MF. Genetic analysis of MRL-lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med. 1992;176:1645–1656. doi: 10.1084/jem.176.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Nose M, Kamoto T, Nishimura M, Hiai H. Host modifier genes affect mouse autoimmunity induced by the lpr gene. Am J Pathol. 1997;151:1791–1798. [PMC free article] [PubMed] [Google Scholar]
- 25.Qu WM, Miyazaki T, Terada M, Lu LM, Nishihara M, Yamada A, Mori S, Nakamura Y, Ogasawara H, Yazawa C, et al. Genetic dissection of vasculitis in MRL/lpr lupus mice: a novel susceptibility locus involving the CD72c allele. Eur J Immunol. 2000;30:2027–2037. doi: 10.1002/1521-4141(200007)30:7<2027::AID-IMMU2027>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 26.Vidal S, Kono DH, Theofilopoulos AN. Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/6-Faslpr mice. J Clin Invest. 1998;101:696–702. doi: 10.1172/JCI1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monestier M, Novick KE. Specificities and genetic characteristics of nucleosome-reactive antibodies from autoimmune mice. Mol Immunol. 1996;33:89–99. doi: 10.1016/0161-5890(95)00115-8. [DOI] [PubMed] [Google Scholar]
- 28.Blanco F, Kalsi J, Isenberg DA. Analysis of antibodies to RNA in patients with systemic lupus erythematosus and other autoimmune rheumatic diseases. Clin Exp Immunol. 1991;86:66–70. doi: 10.1111/j.1365-2249.1991.tb05775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shlomchik MJ, Zharhary D, Saunders T, Camper SA, Weigert MG. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- 30.Bloom DD, Davignon JL, Retter MW, Shlomchik MJ, Pisetsky DS, Cohen PL, Eisenberg RA, Clarke SH. V region gene analysis of anti-Sm hybridomas from MRL/Mp-lpr/lpr mice. J Immunol. 1993;150:1591–1610. [PubMed] [Google Scholar]
- 31.Pettersson I, Hinterberger M, Mimori T, Gottlieb E, Steitz JA. The structure of mammalian small nuclear ribonucleoproteins: Identification of multiple protein components reactive with anti-(U1)ribonucleoprotein and anti-Sm autoantibodies. J Biol Chem. 1984;259:5907–5914. [PubMed] [Google Scholar]
- 32.Bonfa E, Marshak-Rothstein A, Weissbach H, Brot N, Elkon K. Frequency and epitope recognition of anti-ribosome P antibodies from humans with systemic lupus erythematosus and MRL/lpr mice are similar. J Immunol. 1988;140:3434–3437. [PubMed] [Google Scholar]
- 33.Wolfowicz CB, Sakorafas P, Rothstein TL, Marshak-Rothstein A. Oligoclonality of rheumatoid factors arising spontaneously in lpr/lpr mice. Clin Immunol Immunopathol. 1988;46:382–395. doi: 10.1016/0090-1229(88)90057-8. [DOI] [PubMed] [Google Scholar]
- 34.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 35.Dixon FJ, Andrews BS, Eisenberg RA, McConahey PJ, Theofilopoulos AN, Wilson CB. Etiology and pathogenesis of a spontaneous lupus-like syndrome in mice. Arthritis Rheum. 1978;21(Suppl 5):S64–S67. doi: 10.1002/art.1780210909. [DOI] [PubMed] [Google Scholar]
- 36.Kelley VE, Roths JB. Interaction of mutant lpr gene with background strain influences renal disease. Clin Immunol Immunopathol. 1985;37:220–229. doi: 10.1016/0090-1229(85)90153-9. [DOI] [PubMed] [Google Scholar]
- 37.Sanchez R, Jonsson R, Tarkowski A. Phenotypes of immunocompetent cells and Ia antigen expression in oral mucosa and skin of autoimmune mouse strains. Autoimmunity. 1988;1:243–252. doi: 10.3109/08916938809010678. [DOI] [PubMed] [Google Scholar]
- 38.Furukawa F, Tanaka H, Sekita K, Nakamura T, Horiguchi Y, Hamashima Y. Dermatopathological studies on skin lesions of MRL mice. Arch Dermatol Res. 1984;276:186–194. doi: 10.1007/BF00414018. [DOI] [PubMed] [Google Scholar]
- 39.Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 40.Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, Kanaly ST, Towne JE, Willis CR, Kuechle MK, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204:2603–2614. doi: 10.1084/jem.20070157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, Theofilopoulos AN. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci USA. 2009;106:12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med. 2005;202:1333–1339. doi: 10.1084/jem.20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 45.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukui R, Saitoh S, Matsumoto F, Kozuka-Hata H, Oyama M, Tabeta K, Beutler B, Miyake K. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206:1339–1350. doi: 10.1084/jem.20082316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Shao Y, Bennett TA, Shankar RA, Wightman PD, Reddy LG. The functional effects of physical interactions among Toll-like receptors 7, 8, and 9. J Biol Chem. 2006;281:37427–37434. doi: 10.1074/jbc.M605311200. [DOI] [PubMed] [Google Scholar]
- 48.Manz RA, Radbruch A. Plasma cells for a lifetime? Eur J Immunol. 2002;32:923–927. doi: 10.1002/1521-4141(200204)32:4<923::AID-IMMU923>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 49.Rosen A, Casciola-Rosen L, Ahearn J. Novel packages of viral and self-antigens are generated during apoptosis. J Exp Med. 1995;181:1557–1561. doi: 10.1084/jem.181.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haas T, Metzger J, Schmitz F, Heit A, Müller T, Latz E, Wagner H. The DNA sugar backbone 2′ deoxyribose determines toll-like receptor 9 activation. Immunity. 2008;28:315–323. doi: 10.1016/j.immuni.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 53.Peng SL, Madaio MP, Hayday AC, Craft J. Propagation and regulation of systemic autoimmunity by gammadelta T cells. J Immunol. 1996;157:5689–5698. [PubMed] [Google Scholar]
- 54.Peng SL, Madaio MP, Hughes DP, Crispe IN, Owen MJ, Wen L, Hayday AC, Craft J. Murine lupus in the absence of α β T cells. J Immunol. 1996;156:4041–4049. [PubMed] [Google Scholar]
- 55.Peng SL, Cappadona J, McNiff JM, Madaio MP, Owen MJ, Hayday AC, Craft J. Pathogenesis of autoimmunity in αβ T cell-deficient lupus-prone mice. Clin Exp Immunol. 1998;111:107–116. doi: 10.1046/j.1365-2249.1998.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peng SL, Moslehi J, Robert ME, Craft J. Perforin protects against autoimmunity in lupus-prone mice. J Immunol. 1998;160:652–660. [PubMed] [Google Scholar]
- 57.Chan OT, Madaio MP, Shlomchik MJ. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J Immunol. 1999;163:3592–3596. [PubMed] [Google Scholar]
- 58.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 59.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, Tamura T, Takaoka A, Nishikura K, Taniguchi T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA. 2008;105:5477–5482. doi: 10.1073/pnas.0801295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 62.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 65.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vilaysane A, Muruve DA. The innate immune response to DNA. Semin Immunol. 2009;21:208–214. doi: 10.1016/j.smim.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 68.Barrat FJ, Coffman RL. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev. 2008;223:271–283. doi: 10.1111/j.1600-065X.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 69.Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–1731. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- 70.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]