

Abstract
Ischemia, lack of blood flow, and reperfusion, return of blood flow, is a common phenomenon affecting millions of Americans each year. Roughly 30,000 Americans per year experience intestinal ischemia-reperfusion (IR), which is associated with a high mortality rate. Previous studies of the intestine established a role for neutrophils, eicosanoids, the complement system and naturally occurring antibodies in IR-induced pathology. Furthermore, data indicate involvement of a lipid or lipid-like moiety in mediating IR-induced damage. It has been proposed that exposure of neo-antigens are recognized by antibodies, triggering action of the complement cascade. While it is evident that the pathophysiology of IR-induced injury is complex and multi-factorial, we focus this review on the involvement of eicosanoids, phospholipids and neo-antigens in the early pathogenesis. Lipid changes occurring in response to IR, neo-antigens exposed and the role of a phospholipid transporter, phospholipid scramblase 1 will be discussed.
Keywords: ischemia, reperfusion, lipids, neo-antigen
Ischemia/Reperfusion
Ischemia, the lack of sufficient blood supply to tissues, results in cellular dysfunction and eventually necrosis. However, the return of blood flow, termed reperfusion, exacerbates the damage begun during the ischemic period. Ischemia and reperfusion (IR) events occur in numerous organs as a result of various insults, such as trauma, shock, routine surgery and organ transplantation. The intestinal mucosa is among the organs most sensitive to IR (reviewed in [1]) with acute mesenteric arterial or venous thrombosis, embolism, and obstruction being common causes of intestinal IR (reviewed in [2, 3]).
Cases of acute mesenteric ischemia are classified by cause. In order of incidence, they are arterial embolism, arterial thrombus, non-occlusive and venous thrombosis (reviewed in [2]). Symptoms are non-specific and can be subtle, resulting in a delay of correct diagnosis which decreases survival probability. Furthermore, common sequelae include acute lung injury and multiple organ failure. Bowel resection is indicated in most cases which leaves surviving patients with bowel problems, such as short bowel syndrome, for the remainder of their lives. Despite decades of improved imaging devices and medical advances, the mortality rate associated with intestinal IR remains at 50 to 80% (reviewed in [4]).
The mesenteric arteries can maintain adequate perfusion of the intestine over a broad range of blood pressures, however below 40 to 45 mm Hg, perfusion is compromised (reviewed in [3]). The gastrointestinal tract has a very high capillary density, many collateral vessels and receives approximately 25% of total cardiac output at rest (reviewed in [3]). Cellular injury in humans is detectable by 20 minutes of total ischemia and within 60 minutes in the case of partial ischemia (reviewed in [3]).
Rodents are commonly used as research models for intestinal IR and have provided a large knowledge base regarding associated physiology and pathogenesis. Studies in mice have shown changes in the internal pH of enterocytes, abruptly increasing from 6.8 to 7.1 at the initiation of ischemia, and rapidly stabilizing at a pH of 6.3 by three minutes into ischemia [1]. Upon reperfusion, cascades of cellular events lead to eicosanoid production, signaling molecules derived from 20 carbon fatty acids via oxidation, formation of reactive oxygen species, secretion of cytokines and activation of the innate immune response. The barrier between luminal contents and intestinal mucosa is compromised as enterocytes are shed, by necrosis and apoptosis, and capillary permeability increases [5–7] (Fig. 1). The duration of ischemia influences the severity of damage and mortality. A recent study with mice demonstrated increasing severity of tissue damage, as assessed by histology, with increasing duration of ischemia [8]. Furthermore, mice subjected to 30 and 35 minutes of ischemia survived a minimum of 18 hours while mice subjected to 40 and 45 minutes of ischemia all succumbed within six hours post ischemia [8]. In contrast, experimental models of intestinal IR in horses frequently utilize two hours of ischemia and horses survive for 18 hours of reperfusion [9–11]. In fact, 10 of 11 horses subjected to two hours of jejunal ischemia survived 10 days, to the study’s end-point [12]. Thus, the duration of ischemia tolerated varies between species.
Figure 1. Factors contributing to IR-induced tissue damage.
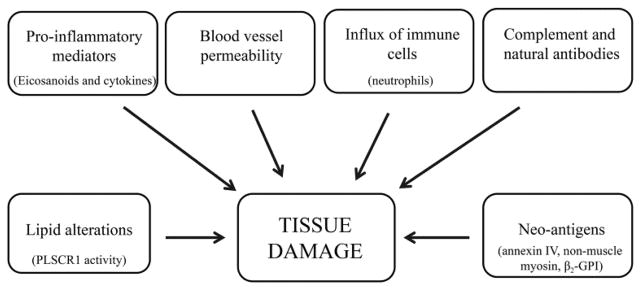
The pathogenesis of IR-induced injury is complex with many contributing factors. Shown are a few of the factors involved.
Several factors contributing to IR-induced pathology will be discussed briefly; however, the majority of this review will focus on lipids, neo-antigens and phosopholipid scramblase 1 as they relate to IR-induced pathology. Eicosanoids, derivatives of arachidonic acid, are rapidly produced during the reperfusion period. Prostaglandin E2 (PGE2) was found to be required but not sufficient for tissue damage in a mouse model of intestinal IR [13]. The conversion of arachidonic acid (AA) to prostaglandins requires the cyclooxygenase (Cox) enzymes. It is well established that the expression of Cox 2, the inducible isoform, is elevated in the intestinal tissue following ischemia. Several animal models have demonstrated an increase in Cox 2 transcription and translation in the intestine following varying lengths of ischemia and reperfusion [13–18]. The use of Cox inhibitors, NS-398 and FK3311, has further demonstrated the importance of this pathway in reperfusion injury in some but not all animal models [14, 19]. It is possible that structural differences in the inhibitors tested or species differences account for the discrepancy in results.
The role of Cox 1, the constitutive isoform, in the post-ischemia production of prostaglandins is debated in the literature. The expression of Cox 1 has primarily been reported in equine studies. In the equine jejunum, Cox 1 protein is constitutively expressed and is increased, as is Cox 2, at 18 hours reperfusion following a two hour ischemic period [10, 20]. Transcription of Cox 1 also increased, although not significantly, in response to 75 minutes of reduced blood flow to the jejunum [15]. Cox 1 is constitutively expressed in the lamina propria of equine colon and IR significantly increases protein levels in the epithelial cells [9, 11]. Our in vitro model using murine endothelial cells results in significant increases of both Cox 1 and Cox 2, with a greater percent increase over control levels for Cox 1 (unpublished data). The role of Cox 1 in IR-induced pathology may be underappreciated.
The Cox enzymes catalyze the formation of prostaglandins, which, as stated above, are necessary but not sufficient for intestinal IR damage, at least in mice [13]. Prostaglandins are potent signaling molecules primarily derived from AA (reviewed in [21]) that are synthesized upon stimulation rather than being stored by the cell (reviewed in [22]). PGE2 is often associated with inflammation due to its vasodilatory effect and enhancement of vascular permeability ([23], reviewed in [24]). Several in vivo studies have documented increased PGE2 production in response to intestinal IR [13, 25–29]. While increased transcription of the Cox enzymes begins during the ischemic period, reperfusion is necessary for PGE2 production [27]. Endothelial cells produce prostaglandins (reviewed in [22]); however, not all endothelial cells generate the same prostaglandin profile. PGI2 is the primary prostaglandin produced from endothelial cells of the large vessels, but microvessel endothelial cells, like those found in the mesenteric vasculature, synthesize more PGE2 and PGF2α [30]. It has long been postulated that PGE2 may paradoxically contribute to tissue healing by promoting angiogenesis and epithelial cell migration (reviewed in [31]). Higher doses of Cox inhibitors which more profoundly suppress PGE2 production are inhibitory for mucosal repair following ischemia in a porcine model [17]. An ex vivo study of porcine ileum showed that PGE2 application could increase intracellular cyclic adenosine monophosphate (cAMP) levels and contribute to closure of leaky tight junctions [23]. It has been proposed that PGE2 may act to downregulate nuclear factor kappa light chain enhancer of activated B cells (NFκB) activity in a negative feedback fashion (reviewed in [32]). While the precise mechanisms by which PGE2 exerts opposing effects are not well understood, concentration, timing and ligation of differing receptors are likely possibilities (reviewed in [24]).
Another eicosanoid derived from AA in response to intestinal IR is leukotriene B4 (LTB4). LTB4, produced by endothelial cells, is chemotactic for neutrophils and facilitates adherence and degranulation of neutrophils (reviewed in [33]). The chemotactic property of LTB4 has been well documented, both in vitro and in vivo. Reperfusion greatly increases the number of neutrophils in the intestinal tissue ([34], reviewed in [35]). Increased neutrophil adherence to endothelial cells was observed via intravital microscopy and in ex vivo assays on tissues subjected to intestinal IR [36, 37]. Activated neutrophils produce reactive oxygen compounds which contribute to the reperfusion damage (reviewed in [38]). Accordingly, IR induces release of myeloperoxidase, an enzyme of neutrophils [39]. LTB4 receptor antagonists, LTB4-DMA and LY-255283, have been shown to attenuate the effects of hypoxia, increased survival and decrease intestinal myeloperoxidase activity, while application of LTB4 enhanced leukocyte adhesion in a dose-dependent manner [36, 40, 41]. Similarly, LTB4 production and intestinal myeloperoxidase activity was reduced in dogs subjected to three hours of ischemia and one hour of reperfusion when treated with the 5-lipoxygenase inhibitor A-64077 [42]. Furthermore, 5-Lox−/− mice sustained less intestinal damage, had decreased intestinal myeloperoxidase activity and improved survival compared to wildtype mice following intestinal IR [43].
The Cox enzymes also mediate the production of thromboxanes which promote coagulation and are increased by intestinal IR, similar to prostaglandins and leukotrienes. In both rats and dogs, thromboxane B2 (TxB2) increased significantly over baseline during the 60 minute reperfusion period [29, 44]. An equine study also revealed increased TxB2, but only during the first of three hours of reperfusion [45]. Thromboxanes contribute to IR-associated systemic effects, such as pulmonary compromise. The release of TxB2 from the lungs and pulmonary permeability of rats subjected to intestinal IR was significantly greater when compared to Sham treated rats [46]. Furthermore, the pulmonary permeability was attenuated with thromboxane inhibitors [46]. It is clear that the synthesis of eicosanoids from AA potentiate IR-induced injury.
Antibodies and Neo-antigens
The complement system, comprised of over 30 proteins, plays a significant role in the pathology resulting from IR. Traditionally, the complement system is described as having three different methods of activation, converging at a common endpoint. Several approaches were taken to delineate the contribution of each activation pathway due to the significant overlap and crosstalk between them. The involvement of both the classical and alternative pathways in IR injury was demonstrated in the early 1990s. Administration of soluble complement receptor type 1 attenuated intestinal damage and neutrophil infiltration in a rat model [47]. Later studies using this same complement inhibitor confirmed that both the classical and alternative pathways of complement activation contributed to IR injury [48]. The generation of factor D−/− mice allowed for investigation of the alternative pathway. Mice deficient in factor D experienced attenuated intestinal injury and neutrophil infiltration following IR [49]. The lectin pathway was subsequently shown to contribute to IR pathology (reviewed in [50]). A supporting study demonstrated the requirement for MBL in IR injury as MBL−/− mice were protected from IR damage [51, 52] but susceptible after reconstitution with MBL [51]. Two native inhibitors of the complement pathway, complement C1 inhibitor (classical and lectin) and Crry (classical and alternative), attenuate tissue injury and reduce neutrophil infiltration [53, 54].
In accordance with the involvement of the classical complement pathway, antibodies are essential for IR injury. Rag-1−/− mice do not produce antibodies and do not sustain IR damage; however, administration of pooled wildtype antibodies as well as the IgM fraction alone results in intestinal damage similar to that seen in wildtype animals [27, 55]. It was later shown that human IgM can also elicit injury and complement deposition in Rag-1−/− and Rag -2−/− mice [56, 57]. Further support for the involvement of antibodies came out of studies investigating complement receptor 2 (CR2), a B cell membrane protein. CR2−/− mice produce antibodies but have defects in the generation of the normal antibody repertoire [58, 59]. CR2−/− mice are protected from IR injury; and, like Rag-1−/− mice, administration of wildtype IgM results in intestinal injury and complement deposition [59, 60]. Additional studies with CR2−/− mice identified specific antibodies capable of inducing IR damage. An anti-phospholipid antibody and anti-β2-glycoprotein I (β2-GPI) antibody were each able to induce IR injury and complement deposition in CR2−/− mice; however both antibodies were required in Rag-1−/− mice [61].
IR-Induced Neo-Antigens
β 2-GPI is one of three recently identified proteinacoeus neo-antigens involved in IR injury. This 54 kDa protein, originally named apolipoprotein H, is one of the most abundant human plasma proteins with an average concentration of 200 μg per ml (reviewed in [62]). Five short consensus repeats comprise the 326 amino acid protein categorizing it as a member of the complement control superfamily (reviewed in [62]). β2-GPI primarily circulates unaccompanied but can be found bound to circulating lipid (reviewed in [62]). A stretch of lysine residues (amino acids 282–287) in Domain V allows for binding to anionic phospholipids of cellular membranes which can activate the cells and promote apoptosis of the bound cell ([63], reviewed in [62]). The results of the aforementioned study strongly suggested the involvement of β2-GPI [61] and further studies have demonstrated the efficacy of peptides derived from Domain V, the lipid binding domain, in protecting wildtype mice from IR damage [64]. Additionally, treatment with the β2-GPI -derived peptides minimized the intestinal injury, complement deposition and eicosanoid production resulting from administration of wildtype antibodies to Rag-1−/− mice [64]. Interestingly, administration of purified human β2-GPI to wildtype mice prior to intestinal IR attenuated injury, complement deposition, and PGE2 production [65]. The authors speculate that the anti-β2-GPI antibodies produced by the mice bind the human β2-GPI, thus reducing the titer of anti-β2-GPI antibodies available for binding the mouse protein [65]. This hypothesis is supported by further data demonstrating attenuation of IR injury and sequelae when Rag-1−/− mice were administered pooled wildtype antibodies that had specifically been depleted of anti-β 2-GPI [64].
The other two identified neo-antigens induced by IR are the intracellular proteins non-muscle myosin heavy chain II and annexin IV. Screening experiments probing ischemic tissue with several monoclonal antibodies identified monoclonal antibodies against non-muscle myosin heavy chain II isoforms A and C or annexin IV that rendered Rag-1−/− mice susceptible to IR injury complement deposition, neutrophil infiltration or eicosanoid production [66–68]. Importantly, the anti-annexin IV monoclonal antibody does not recognize non-muscle myosin or phospholipids [69]. A peptide based on non-muscle myosin heavy chain II provides additional evidence that this protein serves as a neo-antigen following IR [70]. Non-muscle myosin heavy chain II may be involved in other models of IR injury as this same peptide has conferred protection in hind-limb and myocardial IR models [71, 72]. In wildtype mice, recombinant annexin IV reduces tissue injury as well as neutrophil infiltration and eicosanoid production [67, 69]. The authors propose that the recombinant annexin IV binds the circulating anti-annexin IV antibodies, greatly reducing the likelihood of antibody binding annexin IV expressed on damaged tissue [67, 69]. Although multiple neo-antigens have been identified, it is unknown how these proteinaceous neo-antigens interact.
The Contribution of Lipids to Cellular Signaling and Processes
The cellular membrane consists of a bilayer of phospholipids, which under normal conditions, exists in an asymmetric distribution with neutral phospholipids such as phosphatidylcholine (PC) residing in the outer leaflet of the bilayer and anionic aminophospholipids including phosphatidylserine (PS) remaining in the inner leaflet. Despite advances in “lipidomics”, or mass spectrometry-based lipid analysis, only a few studies have applied this technology to investigate intestinal lipid composition.
PC, PS, phosphatidylinositol (PI), phosphatidylethanolamine (PE) and sphingomyelin were the main lipid classes present in both wildtype and Rag-1−/− mice after Sham treatment [27]. Similar results were obtained by ESI-MS/MS analysis of untreated mouse and rat intestines [73, 74]. The prominent PC peaks in the rat intestine were composed of 16:0–18:2 PC species [73]. These data correlate with that of Sparkes et al., in which the most prominent PC species was 34:2 [27]. A study by Braun et al. also showed that normal mouse jejunum expressed a 496 Da and 524 Da lysoPC in the highest concentrations [75]. These masses correlate with the predominant 16:0 and 18:0 lysoPC species found in Sham-treated animals [27]. This data is consistent with formation of lysophospholipids by a lipase acting at the sn-2 position since mammalian lipids typically are enriched in saturated fatty acids at the 1-position and polyunsaturated fatty acids at the 2-position.
Phospholipases catalyze phospholipid hydrolysis and are classified by their cleavage sites. Phospholipase A cleaves fatty acyl chains from the glycerol backbone of a phospholipid. Three classes of PLA2, secretory, calcium-independent, and cytosolic, cleave the fatty acyl chain from the second carbon of the glycerol backbone (sn-2) (reviewed in [76]). The ubiquitous cytosolic PLA2 (cPLA2) has an affinity for phospholipids containing polyunsaturated fatty acids, particularly AA, at the sn-2 position (reviewed in [76–78]). While no preference for a phospholipid head group has been identified [79], AA is most often associated with phospholipids containing choline, ethanolamine or inositol as the head group [79]. The activity of cPLA2 is calcium-dependent and is synergistically increased by mitogen activated protein kinase mediated phosphorylation [80, 81]. Calcium is required for the translocation of cPLA2 from the cytosol to the plasma and intracellular membranes and for binding to a phospholipid substrate (reviewed in [76, 78]). Although the majority of AA is released from PC by the action of PLA2 [82], the activity of phospholipase C and D can indirectly contribute to the AA pool (reviewed in [83]).
PLA2 activity not only increases the level of free AA, but also lysophospholipids (Fig. 2). Several lysophospholipids are biologically active and circulate through the vasculature (reviewed in [84]). The term “lyso” was applied to these lipids because they lyse red blood cells (reviewed in [84]); structurally lysophospholipids are those with a single acyl chain. Just as PC is the most abundant phospholipid of cellular membranes, lysoPC is the most abundant lysophospholipid in serum (reviewed in [84]). Beyond their contribution to cellular structure, phospholipids are biologically active and participate in intracellular signaling.
Figure 2. Representative diagram of lipid metabolism.

This basic schematic demonstrates the cleavage of a fatty acyl chain from PC by PLA2 resulting in a lysoPC and free fatty acid, in this case AA. Free AA can then be metabolized by Cox and Lox enzymes to form eicosanoids.
PA and lysoPA influence several aspects of immune function. Many cells of the immune system express one or more of the nine known G protein coupled receptors for lysoPA (reviewed in [85]). GPR92, the lysoPA receptor mentioned above, is expressed by the resident lymphocytes of the intestine, including those in the epithelial layer, lamina propria, Peyer’s patches and mesenteric lymph nodes [86]. Stimulation of the immune system can increase the concentration of lysoPA, which is chemotactic for leukocytes, elevates intracellular calcium concentrations and compromises the endothelial barrier ([87], reviewed in [85, 88]).
High concentrations (75 μmol/L) of lysoPC are toxic to cells. The addition of exogenous lysoPC (16:0) to cultures of human aortic endothelial cells reduced mitochondrial respiration, as assessed by MTT assay, promoted detachment of cells from the matrix and increased the number of apoptotic cells in a time- and dose- dependent manner [89]. Uptake of extracellular calcium is promoted by lysoPC, increasing the intracellular calcium concentration, which appears necessary for lysoPC-mediated apoptosis [89, 90].
A hallmark of apoptosis is externalization of PS (reviewed in [91]). As the process of apoptosis begins, cardiolipin, the major constituent of mitochondrial membranes, is oxidized [92]. Cytochrome c is then released into the cytoplasm where it can oxidize PS residing in the inner leaflet of the plasma membrane [92]. Subsequently, oxidized PS is flipped to the outer leaflet where it is recognized as an apoptotic marker by the macrophage membrane protein CD36, promoting phagocytosis [92, 93]. Although transient exposure of PS occurs often, such as during cellular activation or fusion of membranes [94], the phospholipid is rapidly re-internalized under physiological conditions. LysoPS can also be flipped to the outer leaflet; however, lysoPS is not re-internalized, thus serving as a marker for phagocytosis (reviewed in [84]). PS is also involved in the clotting of blood when externalized by platelets and acts as a cofactor for maximal activity of protein kinase C and sodium/potassium ATPase [95].
Hypoxia activated PLA2 in a study exposing primary human umbilical vein endothelial cells (HUVEC) to zero percent O2 for two hours [96]. However, in the context of IR, reperfusion seems essential for stimulating PLA2 activity and elevating lysophospholipid content [97]. A decrease in total phospholipids is observed after intestinal IR and IR studies in rats indicated that administration of quinacrine, a non-specific inhibitor of PLA2 enzymes, reduced intestinal permeability and lowered the ratio of lysoPC to PC suggesting a role for PLA2 in IR injury [97, 98]. Quinacrine also attenuated the loss of total phospholipids following IR in a porcine heart model [99]. The total phospholipid content per gram of tissue following ischemia of the heart decreased in an investigation of the metabolic effects due to ischemia in cardiac tissue [99, 100].
An ex vivo study of myocardial cells indicated loss of PC and PE with a concomitant increase of lysoPC and lysoPE in response to four hours of zero percent O2 exposure [101]. An increase in fatty acids including AA was detected in the culture supernatant [101]. PC and PE were similarly decreased and lysophospholipids increased in a rat model of cerebral IR [102]. Two independent studies found a decrease in total phospholipids, up to 20%, when primary porcine pulmonary artery endothelial cells were exposed to zero percent O2 for 24 to 48 hours [103, 104]. In accordance with the myocardial cell study, culture supernatants contained elevated levels of AA and other free fatty acids [103, 104]. Primary HUVECs exposed to zero percent O2 for two hours followed by 45 minutes re-oxygenation released approximately 15% more AA than the controls [96]. Accordingly, eicosanoid production was augmented when HUVECs and bovine aortic endothelial cells were enriched with fatty acids in a hypoxia/re-oxygenation (HR) study [105]. Thus, several in vitro and ex vivo studies suggest a role for phospholipids and lysolipids in IR-induced damage.
The limited investigation into the composition of the healthy lipid profile extends to diseased tissue. A small number of studies have used ESI-MS/MS to study lipids in intestinal disease with variable results depending on the disease model. IR increased lysoPC with a concomitant decrease in PC [27]. Furthermore, phospholipase A2 (PLA2) activity and the subsequent lysoPC:PC ratio increased in response to IR [97]. These data correlate with previous studies indicating that lysoPC increases the intestinal permeability and PLA2 activity as a result of intestinal IR [106]. In addition to increased lysoPC levels following IR treatment, intestinal levels of free AA increased.
The altered phospholipid composition resulting from hypoxia increases the fluidity of the cellular membrane, an effect that is reversible with a sufficient re-oxygenation period [103]. In vivo studies demonstrated the effect of lysophospholipids on endothelial permeability. Addition of lysoPC to the lumen of the ileum resulted in increased permeability to molecules as large as 70 kDa [107]. LysoPC also enhanced the permeability that ischemia alone causes [108].
Inflamed tissue exhibits some of the same lipid changes found in HR studies. Colon biopsies from inflammatory bowel disease patients and inflamed intestinal mucosal samples contained significantly more AA than biopsies and mucosa from healthy control patients [109, 110]. LysoPE in the inflamed intestinal mucosal samples was also elevated in comparison to healthy control samples [110].
Evidence of lipid oxidation is found in both in vitro HR and in vivo IR studies. An increase in malondialdehyde occurred in primary pulmonary artery endothelial cells after a hypoxic period of eight hours [103]. However, evidence of free radical production and lipid oxidation was detected with a 45 minute hypoxia, 15 minute re-oxygenation treatment of aortic endothelial cells in a separate study [111]. A rat model of intestinal IR showed that reperfusion was necessary for an increase in malondialdehyde [97]. Interestingly, a model of oxidative tissue damage in which mice are exposed to γ-irradiation revealed an increase in oxidation of cardiolipin and PS but no other phospholipids, suggesting an increased susceptibility of cardiolipin and PS to oxidative stress [74]. Together, these studies suggest that the lipid profile of a tissue can change and that lysophospholipids and oxidized lipids are important components of intestinal reperfusion injury.
The Response of Phospholipid Scramblase 1 to Hypoxia
The cellular membrane consists of a bilayer of phospholipids which form a hydrophobic barrier between the cellular constituents and the outside environment. This bilayer is not a passive barrier however; the composition and distribution of membrane phospholipids are highly regulated. Under normal conditions, the membrane exists in an asymmetric distribution, with neutral phospholipids residing in the outer leaflet of the bilayer and anionic aminophospholipids remaining in the inner leaflet. This asymmetric distribution of phospholipids is not present in all membranes that fuse with the cellular membrane, such as during endocytosis. Thus, transmembrane proteins shuttle phospholipids from one leaflet to the other as a means of maintaining the asymmetric distribution of phospholipids (Fig. 3).
Figure 3. Schematic of membrane proteins involved in regulating phospholipid asymmetry.
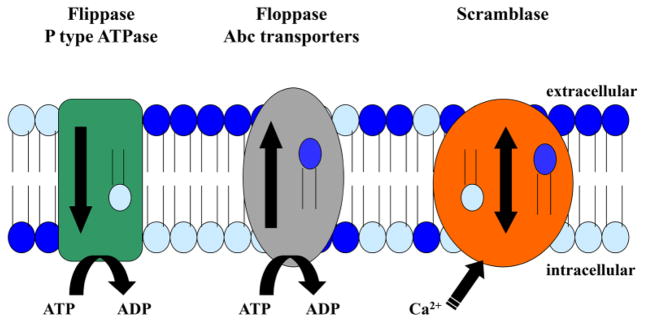
The three main classes of proteins involved in regulating the phospholipid bilayer are illustrated. Flippases and floppases are ATP-dependent and move specific phospholipids in one direction. Scramblases are activated by calcium and transport many different phospholipids in both directions.
Phospholipids move between membranes by several mechanisms. Phospholipids can be transferred by vesicles and proteins (reviewed in [112]). Proteins specific for the transport of phospholipids between organelles reside in the cytoplasm (reviewed in [113]). Additionally, contact between membranes allows for lateral diffusion of phospholipids whereby relocation can be achieved (reviewed in [112]).
The first of these lipid transporting proteins to be characterized was aminophospholipid translocase [114]. Use of spin-labeled analogs with bovine serum albumin back-extraction confirmed an affinity for PS and PE and demonstrated more rapid internalization of PS compared to PE [115, 116]. This aminophospholipid translocase was found to be ATP-dependent with a stoichiometry of one ATP per PS or PE transported [114, 117]. There is some evidence that basic fibroblast growth factor signaling regulates activity as incubation with an anti-basic fibroblast growth factor immunoglobulin inhibited aminophospholipid translocase activity in cultured bovine aortic endothelial cells [118].
A counterpart to the aminophospholipid translocase, which transports aminophospholipids from the outer leaflet to the inner leaflet of the bilayer, was proposed for the cell to maintain shape and function. Early evidence for such a counterpart was provided in studies with red blood cells [119]. Subsequently, this activity was also shown to be ATP-dependent, although independent of the aminophospholipid translocase [116]. This putative transporter was called flippase; for consistency and simplicity, the aminophospholipid translocase became known as floppase.
A third class of transmembrane proteins involved in regulation of membrane asymmetry were discovered nearly a decade later and named scramblases. Four members of this protein class have been identified in human ([120, 121], reviewed in [122]) and in mouse (reviewed in [122]). The properties of the scramblases are in stark contrast to those of the floppases and flippases. While floppases and flippases transport phospholipids in one direction, from inner leaflet to outer leaflet or vice versa, respectively, scramblases are bi-directional transporters, capable of moving phospholipids between leaflets in both directions (reviewed in [122]). Additionally, scramblases show very little specificity for phospholipid head-groups, transporting both neutral and aminophospholipids at a similar rate (reviewed in [123, 124]). Thus, scramblase activity serves to disrupt and reduce the membrane asymmetry ([125, 126], reviewed in [122]). The exposure of aminophospholipids, particularly PS, is at times desired, such as for initiation of the coagulation cascade and clearance of apoptotic cells (reviewed in [127]). Conceptually, scramblase activity should be tightly regulated even in cases of physiologic benefit. Experimentally, activation of scramblase corresponds with very specific intracellular conditions. Scramblase is activated by high concentrations of intracellular calcium [125, 126, 128–130] and acidic pH [130]. In contrast to the constitutive activity of floppases and flippases, scramblase activity is highly regulated and ATP-independent.
The scramblases are a conserved family, with orthologs found in several diverse organisms including the model organisms Mus musculus, Drosophila melanogaster, Danio rerio and Saccharomyces cerevisae (reviewed in [122]). The four human homologs are similar in sequence with PLSCR2 – 4 exhibiting 46 to 59% protein identity with PLSCR1 [120]. Each of the four known scramblases appears to have distinct localizations and functions. The localization of each murine protein has been investigated and all findings regarding the localization of human homologs are consistent with the murine data. Phospholipid scramblase 1 (PLSCR1), the first to be characterized [128, 129], primarily localizes to the cellular membrane [131–134] while PLSCR3 resides in the outer mitochondrial membrane [135]. Expression of PLSCR2 is only detected in the testes [120] and PLSCR4 appears to distribute both to the cellular membrane and the nucleus [136], though the functions of these two family members remain unidentified.
Activation of PLSCR1 involves the binding of calcium or potentially other divalent cations [128, 137] to a predicted single cation binding site [137] in the cytoplasmic portion. Several studies have demonstrated an increase in phospholipid scrambling in the presence of calcium [128, 129, 138]. Recent work confirms the importance of the C-terminal alpha helix for both calcium binding and scrambling activity [121, 139]. It is hypothesized that calcium binding is followed by a conformational change and perhaps self-aggregation [131, 137, 138]. In the absence of calcium, acidic conditions (pH < 6.0) activate PLSCR1 in erythrocyte-derived inside out vesicles [130]. Data suggest that protein kinase Cδ phosphorylates PLSCR1 at the threonine residue at position 161 following calcium binding [140]. This phosphorylation appears to be required for PLSCR1 activity; as either specific inhibition of protein kinase Cδ or transfection of PLSCR1 alone in cells intrinsically lacking both protein kinase Cδ and PLSCR1 resulted in loss of phospholipid scrambling [140]. Additionally, PLSCR1 and epidermal growth factor receptor are primarily localized to lipid rafts. There is evidence that PLSCR1 is a component of the epidermal growth factor receptor complex as epidermal growth factor stimulation allows for the co-immunoprecipitation of phosphorylated PLSCR1, epidermal growth factor receptor and the adaptor protein Shc [133]. Furthermore, data suggest that signaling via epidermal growth factor receptor activates synthesis of PLSCR1 [133].
Normally, PLSCR1 is found in the cellular membrane and to a lesser extent membranes of secretory vesicles, often associated with lipid rafts [131, 133, 134, 141]. The protein contains 18 cysteine residues [132], each of which could potentially serve as a site of palmitoylation. Using a mutated PLSCR1 that cannot be palmitoylated or the native structure with inhibition of palmitoylation revealed that palmitoylation is required for the protein’s association with the membrane [132]. Site directed mutagenesis studies in which alanine residues were systematically substituted for cysteine residues provided insight into which cysteine residues were important for palmitoylation. Only when all five of the cysteine residues between amino acids 184 and 189 were changed to alanine residues was palmitoylation lost [132]. Additionally, the binding affinity for calcium and activity of PLSCR1 is greatly decreased in the absence of palmitoylation [142].
In the absence of palmitoylation, PLSCR1 is found diffusely in the nucleus [132]. While PLSCR1 does not contain a classical nuclear localization signal [143], the amino acid sequence from residues 257 to 266 is necessary for active import into the nucleus by importin α and β [143, 144]. However, the full significance of PLSCR1’s function in the nucleus is still unclear.
A study by Rami et al. performed immunohistochemical staining for PLSCR1 on the hippocampal region of human brain samples from patients who had experienced and temporarily recovered from an ischemic insult and found an increase in PLSCR1 protein versus control samples in which IR did not occur [145]. Our unpublished data indicate a decrease in endothelial cell protein levels during a period of acute hypoxia but a significant increase in transcription within 15 minutes of re-oxygenation. While it is important to point out the different cell types under investigation, neurons in the Rami study and endothelial cells in ours, both suggest an increase in PLSCR1 shortly after reperfusion is established. As for the decrease in PLSCR1 protein during the hypoxic period that we observed, the potential for PLSCR1 to be released from the cell membrane must be considered and may be due to shedding of the protein into the culture media as was recently described for PLSCR3 [146].
Summary
Although the phenomenon of intestinal IR has been recognized for centuries and medical knowledge and technology have progressed immensely, a high mortality rate remains for those afflicted. The pathogenesis is multi-factorial and the numerous molecular and cellular interactions contribute to the difficulty of designing effective therapeutics. While several aspects of intestinal IR-induced pathogenesis have been identified, the initial molecular response remains unknown. Figure 4 represents one possibility for initiation of the cascade.
Figure 4. Proposed model of early events in the pathogenesis of IR-induced injury.

During normoxia, the asymmetry of the bilayer is maintained by flippases, floppases and scramblases. The neo-antigen β2-GPI circulates in a closed conformation and annexin IV and non-muscle myosin, additional neo-antigens, remain intracellular. Upon oxygen deprivation (ischemia or hypoxia) PLSCR1 is activated and the bilayer asymmetry disrupted with translocation of anionic phospholipids such as PS to the outer leaflet. Upon binding anionic phospholipids, β2-GPI adopts an open conformation exposing an epitope for antibody binding which can serve as a trigger for initiation of the complement cascade and inflammatory responses. The intracellular neo-antigens may also be exposed to the extracellular environment as a result of PLSCR1 activation.
Recent studies have identified neo-antigens as potential triggers, but their roles remain undefined [64, 66, 68]. The involvement of a lipid moiety is suggested by data in the literature. Antibodies are known to be one of the required components for IR-induced tissue damage as antibody-deficient Rag-1−/− mice are protected and reconstitution of Rag-1−/− mice with antibodies from wildtype mice prior to intestinal IR renders the Rag-1−/− mice susceptible to IR-induced injury [27]. Likewise, reconstitution with a monoclonal anti-phospholipid antibody and anti-phospholipid binding protein, β2-GPI, (both found in wildtype sera) produces IR-induced pathology in the otherwise protected Rag-1−/− mice [147].
Mass spectrometry has been used to compare the intestinal lipid profiles of polar lipids and free fatty acids between different strains of mice, C57Bl6, Rag-1−/−, TLR9−/−, TLR2−/− and CR2−/− ([26, 27] and unpublished data). IR treatment results in considerable changes in the lipid composition; however these alterations are largely independent of genetic background. Importantly, the levels of lysolipids increased, especially lysoPC and lysoPE, as did the levels of AA. Release of AA through phospholipase activity is essential for production of eicosanoids. These data show that cellular lipids are altered in response to intestinal IR and provide support for the hypothesis that a lipid moiety participates in the pathogenesis of IR-induced injury.
As the lipid profiles from various strains of mice prior to and following IR are quite similar, this may be the inciting stimulus for the pathogenic cascade leading to tissue damage. Ischemia activates PLSCR1 resulting in disruption of the phospholipid bilayer (Fig. 4). Following exposure of negatively charged phospholipids, such as PS, circulating β2-GPI is able to deposit on the cellular membranes of endothelial cells. The conformational change that occurs in β2-GPI upon binding allows for antibody recognition of the now exposed neo-antigens. The complement cascade can then be activated and an inflammatory response initiated.
Future Directions
In addition to their role in activating the complement cascade, antibodies are required for the up-regulation of Cox 2 transcription. In the absence of antibodies, Cox 2 transcription is not up-regulated and the production of PGE2, another required component for IR-induced damage, is not increased [27]. The mechanism by which antibodies influence Cox 2 transcription remains unidentified. Future studies addressing the relationship between antibodies and Cox 2 transcription will be important not only for the development of therapeutics for IR-induced injury but also for other conditions in which a Cox 2-mediated inflammatory response is detrimental. The contribution of the Cox 3 isoform to IR-induced injury is another area of study that may be of therapeutic value.
The identification of IR-induced neo-antigens is an important step in further dissecting the early molecular events involved in the pathogenesis. The mechanism by which these neo-antigens, being large intracellular or serum proteins, are exposed to the extracellular milieu remains a mystery. It seems likely that facilitated transport of some sort, either direct or indirect, must occur to allow for the intracellular neo-antigens to traverse the membrane. Likewise, it is probable that some change at the membrane triggers the deposition of the serum neo-antigen, β2-GPI.
PLSCR1 is sensitive to changes in O2 tension. Transcription and activity of PLSCR1 are promoted by hypoxia. The activity of PLSCR1 involves disruption of the cellular membrane, with phospholipids flipping between the inner and outer leaflets of the membrane. To successfully disrupt the membrane, this action is fairly non-specific and affects a relatively large area of the membrane. Accordingly, PLSCR1 activity could provide a means for externalization of intracellular neo-antigens. Going forward, with the knowledge that PLSCR1 is activated under hypoxic conditions (unpublished data), it will be important to assess the relationship between PLSCR1 activation and neo-antigen externalization. In addition to determining if the intracellular neo-antigens are exposed to the extracellular environment via PLSCR1 activity, studies examining the binding of β2-GPI to endothelial cells following hypoxia in the absence of PLSCR1 will further clarify the potential value in pursuing PLSCR1 as a therapeutic target. Hypothesized to be one of the first cellular responses to hypoxia, PLSCR1 may become an important target for the development of IR-related therapeutics.
Highlights.
Membrane lipids are altered during Intestinal ischemia/reperfusion.
Eicosanoids are required but not sufficient for intestinal ischemia/reperfusion-induced injury.
Membrane lipid alterations result in neoantigen exposure to initiate injury in response to intestinal ischemia/reperfusion
Phospholipid scramblase 1 appears to induce intestinal lipid alterations during ischemia/reperfusion.
Acknowledgments
This work was supported by grants from National Institutes of Health [R01 AI061691 to S.D.F, P20 GM103418, RR016475]; the American Heart Association [to S.D.F.]; the Kansas State University National Science Foundation GK-12 program [to E.A.S] and Kansas State University. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Institute of Health or National Science Foundation or Kansas State University.
Abbreviations
- AA
arachidonic acid
- ATP
adenosine tri-phosphate
- β2-GPI
beta2-glycoprotein 1
- cAMP
cyclic adenosine mono-phosphate
- Cox
cyclooxygenase
- cPLA2
cytosolic phospholipase A2
- CR2
complement receptor 2
- ESI-MS/MS
electrospray ionization-tandem mass spectrometry
- HR
hypoxia re-oxygenation
- HUVEC
human umbilical vein endothelial cells
- Ig
immunoglobulin
- IR
ischemia reperfusion
- Lox
lipoxygenase
- LTB4
leukotriene B4
- MBL
mannose binding lectin
- miRNA
micro ribonucleic acid
- mTOR
mammalian target of rapamycin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PGE2
prostaglandin E2
- PGF2α
prostaglandin F2 alpha
- PGI2
prostaglandin I2
- PI
phosphatidylinositol
- PLA2
phospholipase A2
- PLSCR
phospholipid scramblase
- PS
phosphatidylserine
- Rag
recombination activating gene
- sCR1
soluble complement receptor type 1
- TxB2
thromboxane B2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Emily Archer Slone, Email: eslone@ksu.edu.
Sherry D. Fleming, Email: sdflemin@ksu.edu.
References
- 1.Guan Y, Worrell RT, Pritts TA, Montrose MH. Intestinal ischemia-reperfusion injury: reversible and irreversible damage imaged in vivo. Am J Physiol Gastrointest Liver Physiol. 2009;297:G187–196. doi: 10.1152/ajpgi.90595.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oldenburg WA, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD. Acute mesenteric ischemia: a clinical review. Archives of Internal Medicine. 2004;164:1054–1062. doi: 10.1001/archinte.164.10.1054. [DOI] [PubMed] [Google Scholar]
- 3.Haglund U, Bergqvist D. Intestinal ischemia -- the basics. Lang Arch Surg. 1999;384:233–238. doi: 10.1007/s004230050197. [DOI] [PubMed] [Google Scholar]
- 4.Stamatakos M, Stefanaki C, Mastrokalos D, Arampatzi H, Safioleas P, Chatziconstantinou C, Xiromeritis C, Safioleas M. Mesenteric ischemia: still a deadly puzzle for the medical community. Tohoku J Exp Med. 2008;216:197–204. doi: 10.1620/tjem.216.197. [DOI] [PubMed] [Google Scholar]
- 5.Noda T, Iwakiri R, Fujimoto K, Matsuo S, Aw TY. Programmed cell death induced by ischemia-reperfusion in rat intestinal mucosa. Am J Physiol. 1998;274:G270–276. doi: 10.1152/ajpgi.1998.274.2.G270. [DOI] [PubMed] [Google Scholar]
- 6.Haglund U. Gut ischaemia. Gut. 1994;35:S73–S76. doi: 10.1136/gut.35.1_suppl.s73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Droy-Lefaix MT, Drouet Y, Geraud G, Hosford D, Braquet P. Superoxide dismutase (SOD) and the PAF-antagonist (BN 52021) reduce small intestinal damage induced by ischemia-reperfusion. Free radical research communications. 1991;12–13(Pt 2):725–735. doi: 10.3109/10715769109145852. [DOI] [PubMed] [Google Scholar]
- 8.Stringa P, Lausada N, Romanin D, Machuca M, Cabanne A, Rumbo M, Gondolesi G. Defining the nonreturn time for intestinal ischemia reperfusion injury in mice. Transplant Proc. 2012;44:1214–1217. doi: 10.1016/j.transproceed.2011.11.066. [DOI] [PubMed] [Google Scholar]
- 9.Matyjaszek SA, Morton AJ, Freeman DE, Grosche A, Polyak MM, Kuck H. Effects of flunixin meglumine on recovery of colonic mucosa from ischemia in horses. American journal of veterinary research. 2009;70:236–246. doi: 10.2460/ajvr.70.2.236. [DOI] [PubMed] [Google Scholar]
- 10.Cook VL, Jones Shults J, McDowell MR, Campbell NB, Davis JL, Marshall JF, Blikslager AT. Anti-inflammatory effects of intravenously administered lidocaine hydrochloride on ischemia-injured jejunum in horses. American journal of veterinary research. 2009;70:1259–1268. doi: 10.2460/ajvr.70.10.1259. [DOI] [PubMed] [Google Scholar]
- 11.Morton AJ, Grosche A, Rotting AK, Matyjaszek SA, Blikslager AT, Freeman DE. Expression of cyclooxygenase-1 and -2 in the left dorsal colon after different durations of ischemia and reperfusion in horses. American journal of veterinary research. 2009;70:1536–1544. doi: 10.2460/ajvr.70.12.1536. [DOI] [PubMed] [Google Scholar]
- 12.Dabareiner RM, White NA, 2nd, Donaldson L. Evaluation of Carolina Rinse solution as a treatment for ischaemia reperfusion of the equine jejunum. Equine veterinary journal. 2003;35:642–646. doi: 10.2746/042516403775696302. [DOI] [PubMed] [Google Scholar]
- 13.Moses T, Wagner L, Fleming SD. TLR4-mediated Cox-2 expression increases intestinal ischemia/reperfusion-induced damage. J Leukoc Biol. 2009;86:971–980. doi: 10.1189/jlb.0708396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato N, Kozar RA, Zou L, Weatherall JM, Attuwaybi B, Moore-Olufemi SD, Weisbrodt NW, Moore FA. Peroxisome proliferator-activated receptor gamma mediates protection against cyclooxygenase-2-induced gut dysfunction in a rodent model of mesenteric ischemia/reperfusion. Shock. 2005;24:462–469. doi: 10.1097/01.shk.0000183483.76972.ae. [DOI] [PubMed] [Google Scholar]
- 15.Hilton H, Nieto JE, Moore PF, Harmon FA, Naydan DK, Snyder JR. Expression of cyclooxygenase genes in the jejunum of horses during low-flow ischemia and reperfusion. American journal of veterinary research. 2011;72:681–686. doi: 10.2460/ajvr.72.5.681. [DOI] [PubMed] [Google Scholar]
- 16.Marshall JF, Blikslager AT. The effect of nonsteroidal anti-inflammatory drugs on the equine intestine. Equine Vet J. 2011;43(Suppl 39):140–144. doi: 10.1111/j.2042-3306.2011.00398.x. [DOI] [PubMed] [Google Scholar]
- 17.Blikslager AT, Zimmel DN, Young KM, Campbell NB, Little D, Argenzio RA. Recovery of ischaemic injured porcine ileum: evidence for a contributory role of COX-1 and COX-2. Gut. 2002;50:615–623. doi: 10.1136/gut.50.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe T, Kobata A, Tanigawa T, Nadatani Y, Yamagami H, Watanabe K, Tominaga K, Fujiwara Y, Takeuchi K, Arakawa T. Activation of the MyD88 signaling pathway inhibits ischemia-reperfusion injury in the small intestine. Am J Physiol. 2012;303:G324–334. doi: 10.1152/ajpgi.00075.2012. [DOI] [PubMed] [Google Scholar]
- 19.Kawata K, Takeyoshi I, Iwanami K, Sunose Y, Tsutsumi H, Ohwada S, Matsumoto K, Morishita Y. The effects of a selective cyclooxygenase-2 inhibitor on small bowel ischemia-reperfusion injury. Hepato-gastroenterology. 2003;50:1970–1974. [PubMed] [Google Scholar]
- 20.Cook VL, Meyer CT, Campbell NB, Blikslager AT. Effect of firocoxib or flunixin meglumine on recovery of ischemic-injured equine jejunum. American journal of veterinary research. 2009;70:992–1000. doi: 10.2460/ajvr.70.8.992. [DOI] [PubMed] [Google Scholar]
- 21.Stuart MJ. Prostaglandins and hemostasis: an overview. Advances in pediatrics. 1983;30:321–364. [PubMed] [Google Scholar]
- 22.Schror K. Prostaglandins, other eicosanoids and endothelial cells. Basic Res Cardiol. 1985;80:502–514. doi: 10.1007/BF01907914. [DOI] [PubMed] [Google Scholar]
- 23.Blikslager AT, Roberts MC, Rhoads JM, Argenzio RA. Prostaglandins I2 and E2 have a synergistic role in rescuing epithelial barrier function in porcine ileum. J Clin Invest. 1997;100:1928–1933. doi: 10.1172/JCI119723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egan K, FitzGerald GA. Eicosanoids and the vascular endothelium. Handbook of experimental pharmacology. 2006:189–211. doi: 10.1007/3-540-32967-6_6. [DOI] [PubMed] [Google Scholar]
- 25.Pope MR, Hoffman SM, Tomlinson S, Fleming SD. Complement regulates TLR4-mediated inflammatory responses during intestinal ischemia reperfusion. Mol Immunol. 2010;48:356–364. doi: 10.1016/j.molimm.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slone EA, Pope MR, Roth M, Welti R, Fleming SD. TLR9 is dispensable for intestinal ischemia/reperfusion-induced tissue damage. Am J Clin Exp Immunol. 2012;1:124–135. [PMC free article] [PubMed] [Google Scholar]
- 27.Sparkes BL, Slone EE, Roth M, Welti R, Fleming SD. Intestinal lipid alterations occur prior to antibody-induced prostaglandin E2 production in a mouse model of ischemia/reperfusion. Biochim Biophys Acta. 2010;1801:517–525. doi: 10.1016/j.bbalip.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turnage RH, Kadesky KM, Bartula L, Guice KS, Oldham KT, Myers SI. Splanchnic PGI2 release and “no reflow” following intestinal reperfusion. J Surg Res. 1995;58:558–564. doi: 10.1006/jsre.1995.1088. [DOI] [PubMed] [Google Scholar]
- 29.Mangino MJ, Anderson CB, Murphy MK, Brunt E, Turk J. Mucosal arachidonate metabolism and intestinal ischemia-reperfusion injury. Am J Physiol. 1989;257:G299–307. doi: 10.1152/ajpgi.1989.257.2.G299. [DOI] [PubMed] [Google Scholar]
- 30.Charo IF, Shak S, Karasek MA, Davison PM, Goldstein IM. Prostaglandin I2 is not a major metabolite of arachidonic acid in cultured endothelial cells from human foreskin microvessels. J Clin Invest. 1984;74:914–919. doi: 10.1172/JCI111509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawkey CJ, Rampton DS. Prostaglandins and the gastrointestinal mucosa: are they important in its function, disease, or treatment? Gastroenterology. 1985;89:1162–1188. doi: 10.1016/0016-5085(85)90225-2. [DOI] [PubMed] [Google Scholar]
- 32.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. Journal of investigative medicine : the official publication of the American Federation for Clinical Research. 2009;57:703–708. doi: 10.2310/JIM.0b013e31819aaa76. [DOI] [PubMed] [Google Scholar]
- 33.Bray MA. Leukotriene B4: an inflammatory mediator with vascular actions in vivo. Agents and actions Supplements. 1982;11:51–61. [PubMed] [Google Scholar]
- 34.Grosche A, Morton AJ, Graham AS, Valentine JF, Abbott JR, Polyak MM, Freeman DE. Mucosal injury and inflammatory cells in response to brief ischaemia and reperfusion in the equine large colon. Equine veterinary journal. Supplement. 2011:16–25. doi: 10.1111/j.2042-3306.2011.00415.x. [DOI] [PubMed] [Google Scholar]
- 35.Gayle JM, Blikslager AT, Jones SL. Role of neutrophils in intestinal mucosal injury. Journal of the American Veterinary Medical Association. 2000;217:498–500. doi: 10.2460/javma.2000.217.498. [DOI] [PubMed] [Google Scholar]
- 36.Karasawa A, Guo JP, Ma XL, Tsao PS, Lefer AM. Protective actions of a leukotriene B4 antagonist in splanchnic ischemia and reperfusion in rats. Am J Physiol. 1991;261:G191–198. doi: 10.1152/ajpgi.1991.261.2.G191. [DOI] [PubMed] [Google Scholar]
- 37.Oliver MG, Specian RD, Perry MA, Granger DN. Morphologic assessment of leukocyte-endothelial cell interactions in mesenteric venules subjected to ischemia and reperfusion. Inflammation. 1991;15:331–346. doi: 10.1007/BF00917350. [DOI] [PubMed] [Google Scholar]
- 38.Grisham MB, Granger DN. Neutrophil-mediated mucosal injury. Role of reactive oxygen metabolites. Dig Dis Sci. 1988;33:6S–15S. doi: 10.1007/BF01538126. [DOI] [PubMed] [Google Scholar]
- 39.Schmeling DJ, Caty MG, Oldham KT, Guice KS. Cytoprotection by diclofenac sodium after intestinal ischemia/reperfusion injury. Journal of pediatric surgery. 1994;29:1044–1048. doi: 10.1016/0022-3468(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 40.Steiner DR, Gonzalez NC, Wood JG. Leukotriene B(4) promotes reactive oxidant generation and leukocyte adherence during acute hypoxia. Journal of applied physiology. 2001;91:1160–1167. doi: 10.1152/jappl.2001.91.3.1160. [DOI] [PubMed] [Google Scholar]
- 41.Casillan AJ, Gonzalez NC, Johnson JS, Steiner DR, Wood JG. Mesenteric microvascular inflammatory responses to systemic hypoxia are mediated by PAF and LTB4. J Appl Physiol. 2003;94:2313–2322. doi: 10.1152/japplphysiol.00047.2002. [DOI] [PubMed] [Google Scholar]
- 42.Mangino MJ, Murphy MK, Anderson CB. Effects of the arachidonate 5-lipoxygenase synthesis inhibitor A-64077 in intestinal ischemia-reperfusion injury. J Pharmacol Exp Therap. 1994;269:75–81. [PubMed] [Google Scholar]
- 43.Cuzzocrea S, Rossi A, Serraino I, Di Paola R, Dugo L, Genovese T, Caputi AP, Sautebin L. 5-lipoxygenase knockout mice exhibit a resistance to splanchnic artery occlusion shock. Shock. 2003;20:230–236. doi: 10.1097/00024382-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Murthy S, Hui-Qi Q, Sakai T, Depace DE, Fondacaro JD. Ischemia/reperfusion injury in the rat colon. Inflammation. 1997;21:173–190. doi: 10.1023/a:1027318203971. [DOI] [PubMed] [Google Scholar]
- 45.Moore RM, Muir WW, Bertone AL, Oliver JL. Effect of platelet-activating factor antagonist L-691,880 on low-flow ischemia-reperfusion injury of the large colon in horses. Veterinary surgery : VS. 1998;27:37–48. doi: 10.1111/j.1532-950x.1998.tb00096.x. [DOI] [PubMed] [Google Scholar]
- 46.Turnage RH, LaNoue JL, Kadesky KM, Meng Y, Myers SI. Thromboxane A2 mediates increased pulmonary microvascular permeability after intestinal reperfusion. Journal of applied physiology. 1997;82:592–598. doi: 10.1152/jappl.1997.82.2.592. [DOI] [PubMed] [Google Scholar]
- 47.Hill J, Lindsay TF, Ortiz F, Yeh CG, Hechtman HB, Moore FD. Soluble complement receptor type 1 ameliorates the local and remote organ injury after intestinal ischemia-reperfusion in the rat. J Immunol. 1992;149:1723–1728. [PubMed] [Google Scholar]
- 48.Eror AT, Stojadinovic A, Starnes BW, Makrides SC, Tsokos GC, Shea-Donohue T. Anti-inflammatory effects of soluble complement receptor type 1 promote rapid recovery of ischemia/reperfusion injury in rat small intestine. Clin Immunol. 1999;90:266–275. doi: 10.1006/clim.1998.4635. [DOI] [PubMed] [Google Scholar]
- 49.Stahl GL, Xu Y, Hao L, Miller M, Buras JA, Fung M, Zhao H. Role for the alternate complement pathway in ischemia/reperfusion injury. Am J Pathol. 2003;162:449–455. doi: 10.1016/S0002-9440(10)63839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arumugam TV, Magnus T, Woodruff TM, Proctor LM, Shiels IA, Taylor SM. Complement mediators in ischemia-reperfusion injury. Clinica Chimica Acta. 2006 doi: 10.1016/j.cca.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Hart ML, Ceonzo KA, Shaffer LA, Takahashi K, Rother RP, Reenstra WR, Buras JA, Stahl GL. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol. 2005;174:6373–6380. doi: 10.4049/jimmunol.174.10.6373. [DOI] [PubMed] [Google Scholar]
- 52.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177:4727–4734. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 53.Karpel-Massler G, Fleming SD, Kirschfink M, Tsokos GC. Human C1 esterase inhibitor attenuates murine mesenteric ischemia/reperfusion induced local organ injury. J Surg Res. 2003;115:247–256. doi: 10.1016/s0022-4804(03)00192-6. [DOI] [PubMed] [Google Scholar]
- 54.Rehrig S, Fleming SD, Anderson J, Guthridge JM, Rakstang J, McQueen CE, Holers VM, Tsokos GC, Shea-Donohue T. Complement inhibitor, complement receptor 1-related gene/protein y-Ig attenuates intestinal damage after the onset of mesenteric ischemia/reperfusion injury in mice. J Immunol. 2001;167:5921–5927. doi: 10.4049/jimmunol.167.10.5921. [DOI] [PubMed] [Google Scholar]
- 55.Williams JP, Pechet TTV, Weiser MR, Reid R, Kobzik L, Moore FD, Carroll MC, Hechtman HB. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol. 1999;86:938–942. doi: 10.1152/jappl.1999.86.3.938. [DOI] [PubMed] [Google Scholar]
- 56.Zhang M, Alicot EM, Carroll MC. Human natural IgM can induce ischemia/reperfusion injury in a murine intestinal model. Mol Immunol. 2008;45:4036–4039. doi: 10.1016/j.molimm.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiser MR, Williams JP, Moore FD, Kobzik L, Ma M, Hechtman HB, Carroll MC. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183:2343–2348. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fleming SD, Shea-Donohue T, Guthridge JM, Kulik L, Waldschmidt TJ, Gipson MG, Tsokos GC, Holers VM. Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J Immunol. 2002;169:2126–2133. doi: 10.4049/jimmunol.169.4.2126. [DOI] [PubMed] [Google Scholar]
- 59.Reid RR, Woodcock S, Shimabukuro-Vornhagen A, Austen WG, Kobzik L, Zhang M, Hechtman HB, Moore FD, Carroll MC. Functional activity of natural antibody is altered in Cr2-deficient mice. J Immunol. 2002;169:5433–5400. doi: 10.4049/jimmunol.169.10.5433. [DOI] [PubMed] [Google Scholar]
- 60.Fleming SD, Shea-Donohue T, Guthridge JM, Kulik L, Waldschmidt TJ, Gipson MG, Tsokos GC, Holers VM. Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J Immunol. 2002;169:2126–2133. doi: 10.4049/jimmunol.169.4.2126. [DOI] [PubMed] [Google Scholar]
- 61.Fleming SD, Egan RP, Chai C, Girardi G, Holers VM, Salmon J, Monestier M, Tsokos GC. Anti-phospholipid antibodies restore mesenteric ischemia/reperfusion-induced injury in complement receptor 2/complement receptor 1-deficient mice. J Immunol. 2004;173:7055–7061. doi: 10.4049/jimmunol.173.11.7055. [DOI] [PubMed] [Google Scholar]
- 62.Miyakis S, Giannakopoulos B, Krilis SA. Beta 2 glycoprotein I--function in health and disease. Thromosis Research. 2004;114:335–346. doi: 10.1016/j.thromres.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 63.Meroni PL, Raschi E, Testoni C, Parisio A, Borghi MO. Innate immunity in the antiphospholipid syndrome: role of toll-like receptors in endothelial cell activation by antiphospholipid antibodies. Autoimmun Rev. 2004;3:510–515. doi: 10.1016/j.autrev.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 64.Fleming SD, Pope MR, Hoffman SM, Moses T, Bukovnik U, Tomich JM, Wagner LM, Woods KM. Domain V peptides inhibit beta2-glycoprotein I-mediated mesenteric ischemia/reperfusion-induced tissue damage and inflammation. J Immunol. 2010;185:6168–6178. doi: 10.4049/jimmunol.1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tomasi M, Hiromasa Y, Pope MR, Gudlar S, Tomich JM, Fleming SD. Human B2-glycoprotein I attenuates mouse intestinal ischemia/reperfusion induced injury and inflammation. Mol Immunol. 2012;52:207–216. doi: 10.1016/j.molimm.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, Andrews KA, Markaryan A, Quigg RJ, Silverman GJ, Tsokos GC, Holers VM. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol. 2009;182:5363–5373. doi: 10.4049/jimmunol.0803980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elvington A, Atkinson C, Kulik L, Zhu H, Yu J, Kindy MS, Holers VM, Tomlinson S. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. J Immunol. 2012;188:1460–1468. doi: 10.4049/jimmunol.1102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler B, Shimaoka M, Chan R, Friend D, Mahmood U, Weissleder R, Moore FD, Carroll MC. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–152. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, Andrews KA, Markaryan A, Quigg RJ, Silverman GJ, Tsokos GC, Holers VM. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol. 2009;182:5363–5373. doi: 10.4049/jimmunol.0803980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler B, Shimaoka M, Chan R, Friend D, Mahmood U, Weissleder R, Moore FD, Carroll MC. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–152. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan RK, Verna N, Afnan J, Zhang M, Ibrahim S, Carroll MC, Moore FD., Jr Attenuation of skeletal muscle reperfusion injury with intravenous 12 amino acid peptides that bind to pathogenic IgM. Surgery. 2006;139:236–243. doi: 10.1016/j.surg.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 72.Haas MS, Alicot EM, Schuerpf F, Chiu I, Li J, Moore FD, Carroll MC. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc Res. 2010;87:618–627. doi: 10.1093/cvr/cvq141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hicks AM, DeLong CJ, Thomas MJ, Samuel M, Cui Z. Unique molecular signatures of glycerophospholipid species in different rat tissues analyzed by tandem mass spectrometry. Biochim Biophys Acta. 2006;1761:1022–1029. doi: 10.1016/j.bbalip.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 74.Tyurina YY, Tyurin VA, Epperly MW, Greenberger JS, Kagan VE. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44:299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 75.Braun A, Treede I, Gotthardt D, Tietje A, Zahn A, Ruhwald R, Schoenfeld U, Welsch T, Kienle P, Erben G, Lehmann WD, Fuellekrug J, Stremmel W, Ehehalt R. Alterations of phospholipid concentration and species composition of the intestinal mucus barrier in ulcerative colitis: a clue to pathogenesis. Inflamm Bowel Dis. 2009;15:1705–1720. doi: 10.1002/ibd.20993. [DOI] [PubMed] [Google Scholar]
- 76.Leslie CC. Properties and regulation of cytosolic phospholipase A2. J Biol Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 77.Anderson BO, Moore EE, Banerjee A. Phospholipase A2 regulates critical inflammatory mediators of multiple organ failure. J Surg Res. 1994;56:199–205. doi: 10.1006/jsre.1994.1032. [DOI] [PubMed] [Google Scholar]
- 78.Clark JD, Schievella AR, Nalefski EA, Lin LL. Cytosolic phospholipase A2. Journal of lipid mediators and cell signalling. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- 79.Lister MD, Deems RA, Watanabe Y, Ulevitch RJ, Dennis EA. Kinetic analysis of the Ca2+-dependent, membrane-bound, macrophage phospholipase A2 and the effects of arachidonic acid. J Biol Chem. 1988;263:7506–7513. [PubMed] [Google Scholar]
- 80.Lin LL, Lin AY, Knopf JL. Cytosolic phospholipase A2 is coupled to hormonally regulated release of arachidonic acid. Proc Natl Acad Sci U S A. 1992;89:6147–6151. doi: 10.1073/pnas.89.13.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 82.Schoonderwoerd K, Stam H. Lipid metabolism of myocardial endothelial cells. Mol Cell Biochem. 1992;116:171–179. doi: 10.1007/BF01270585. [DOI] [PubMed] [Google Scholar]
- 83.Rink C, Khanna S. Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid Redox Signal. 2011;14:1889–1903. doi: 10.1089/ars.2010.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frasch SC, Bratton DL. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog Lipid Res. 2012;51:199–207. doi: 10.1016/j.plipres.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goetzl EJ, Wang W, McGiffert C, Huang MC, Graler MH. Sphingosine 1-phosphate and its G protein-coupled receptors constitute a multifunctional immunoregulatory system. J Cell Biochem. 2004;92:1104–1114. doi: 10.1002/jcb.20053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kotarsky K, Boketoft A, Bristulf J, Nilsson NE, Norberg A, Hansson S, Owman C, Sillard R, Leeb-Lundberg LM, Olde B. Lysophosphatidic acid binds to and activates GPR92, a G protein-coupled receptor highly expressed in gastrointestinal lymphocytes. J Pharmacol Exp Ther. 2006;318:619–628. doi: 10.1124/jpet.105.098848. [DOI] [PubMed] [Google Scholar]
- 87.Hines OJ, Ryder N, Chu J, McFadden D. Lysophosphatidic acid stimulates intestinal restitution via cytoskeletal activation and remodeling. J Surg Res. 2000;92:23–28. doi: 10.1006/jsre.2000.5941. [DOI] [PubMed] [Google Scholar]
- 88.Lum H. Lysophospholipids in the regulation of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1335–1336. doi: 10.1152/ajplung.2001.281.6.L1335. [DOI] [PubMed] [Google Scholar]
- 89.Matsubara M, Hasegawa K. Benidipine, a dihydropyridine-calcium channel blocker, prevents lysophosphatidylcholine-induced injury and reactive oxygen species production in human aortic endothelial cells. Atherosclerosis. 2005;178:57–66. doi: 10.1016/j.atherosclerosis.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 90.Chaudhuri P, Colles SM, Damron DS, Graham LM. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol. 2003;23:218–223. doi: 10.1161/01.atv.0000052673.77316.01. [DOI] [PubMed] [Google Scholar]
- 91.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 92.Tyurin VA, Tyurina YY, Kochanek PM, Hamilton R, DeKosky ST, Greenberger JS, Bayir H, Kagan VE. Oxidative lipidomics of programmed cell death. Methods Enzymol. 2008;442:375–393. doi: 10.1016/S0076-6879(08)01419-5. [DOI] [PubMed] [Google Scholar]
- 93.Fadeel B, Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit Rev Biochem Mol Biol. 2009;44:264–277. doi: 10.1080/10409230903193307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smrz D, Lebduska P, Draberova L, Korb J, Draber P. Engagement of phospholipid scramblase 1 in activated cells: implication for phosphatidylserine externalization and exocytosis. J Biol Chem. 2008;283:10904–10918. doi: 10.1074/jbc.M710386200. [DOI] [PubMed] [Google Scholar]
- 95.Yamaji-Hasegawa A, Tsujimoto M. Asymmetric distribution of phospholipids in biomembranes. Biol Pharm Bull. 2006;29:1547–1553. doi: 10.1248/bpb.29.1547. [DOI] [PubMed] [Google Scholar]
- 96.Michiels C, Arnould T, Knott I, Dieu M, Remacle J. Stimulation of prostaglandin synthesis by human endothelial cells exposed to hypoxia. Am J Physiol. 1993;264:C866–874. doi: 10.1152/ajpcell.1993.264.4.C866. [DOI] [PubMed] [Google Scholar]
- 97.Otamiri T, Franzen L, Lindmark D, Tagesson C. Increased phospholipase A2 and decreased lysophospholipase activity in the small intestinal mucosa after ischaemia and revascularisation. Gut. 1987;28:1445–1453. doi: 10.1136/gut.28.11.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Otamiri T, Tagesson C. Role of phospholipase A2 and oxygenated free radicals in mucosal damage after small intestinal ischemia and reperfusion. Am J Surg. 1989;157:562–565. doi: 10.1016/0002-9610(89)90699-5. discussion 566. [DOI] [PubMed] [Google Scholar]
- 99.Das DK, Engelman RM, Otani H, Rousou JA, Breyer RH, Lemeshow S. Effect of superoxide dismutase and catalase on myocardial energy metabolism during ischemia and reperfusion. Clin Physiol Biochem. 1986;4:187–198. [PubMed] [Google Scholar]
- 100.Shaikh NA, Downar E. Time course of changes in porcine myocardial phospholipid levels during ischemia. A reassessment of the lysolipid hypothesis. Circ Res. 1981;49:316–325. doi: 10.1161/01.res.49.2.316. [DOI] [PubMed] [Google Scholar]
- 101.Kawaguchi H, Shoki M, Iizuka K, Sano H, Sakata Y, Yasuda H. Phospholipid metabolism and prostacyclin synthesis in hypoxic myocytes. Biochim Biophys Acta. 1991;1094:161–167. doi: 10.1016/0167-4889(91)90004-h. [DOI] [PubMed] [Google Scholar]
- 102.Drgova A, Likavcanova K, Dobrota D. Changes of phospholipid composition and superoxide dismutase activity during global brain ischemia and reperfusion in rats. Gen Physiol Biophys. 2004;23:337–346. [PubMed] [Google Scholar]
- 103.Block ER, Patel JM, Edwards D. Mechanism of hypoxic injury to pulmonary artery endothelial cell plasma membranes. Am J Physiol. 1989;257:C223–231. doi: 10.1152/ajpcell.1989.257.2.C223. [DOI] [PubMed] [Google Scholar]
- 104.Bhat GB, Block ER. Effect of hypoxia on phospholipid metabolism in porcine pulmonary artery endothelial cells. Am J Physiol. 1992;262:L606–613. doi: 10.1152/ajplung.1992.262.5.L606. [DOI] [PubMed] [Google Scholar]
- 105.Oudot F, Cordelet C, Sergiel JP, Grynberg A. Polyunsaturated fatty acids influence prostanoid synthesis in vascular endothelial cells under hypoxia and reoxygenation. Int J Vitam Nutr Res. 1998;68:263–271. [PubMed] [Google Scholar]
- 106.Cunningham TJ, Yao L, Lucena A. Product inhibition of secreted phospholipase A2 may explain lysophosphatidylcholines’ unexpected therapeutic properties. J Inflamm (Lond) 2008;5:17. doi: 10.1186/1476-9255-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tagesson C, Franzen L, Dahl G, Westrom B. Lysophosphatidylcholine increases rat ileal permeability to macromolecules. Gut. 1985;26:369–377. doi: 10.1136/gut.26.4.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Otamiri T, Sjodahl R, Tagesson C. Lysophosphatidylcholine potentiates the increase in mucosal permeability after small-intestinal ischaemia. Scand J Gastroenterol. 1986;21:1131–1136. doi: 10.3109/00365528608996433. [DOI] [PubMed] [Google Scholar]
- 109.Pacheco S, Hillier K, Smith C. Increased arachidonic acid levels in phospholipids of human colonic mucosa in inflammatory bowel disease. Clin Sci (Lond) 1987;73:361–364. doi: 10.1042/cs0730361. [DOI] [PubMed] [Google Scholar]
- 110.Morita H, Nakanishi K, Dohi T, Yasugi E, Oshima M. Phospholipid turnover in the inflamed intestinal mucosa: arachidonic acid-rich phosphatidyl/plasmenyl-ethanolamine in the mucosa in inflammatory bowel disease. J Gastroenterol. 1999;34:46–53. doi: 10.1007/s005350050215. [DOI] [PubMed] [Google Scholar]
- 111.Kramer JH, Dickens BF, Misik V, Weglicki WB. Phospholipid hydroperoxides are precursors of lipid alkoxyl radicals produced from anoxia/reoxygenated endothelial cells. J Mol Cell Cardiol. 1995;27:371–381. doi: 10.1016/s0022-2828(08)80034-x. [DOI] [PubMed] [Google Scholar]
- 112.Bishop WR, Bell RM. Assembly of phospholipids into cellular membranes: biosynthesis, transmembrane movement and intracellular translocation. Annual review of cell biology. 1988;4:579–610. doi: 10.1146/annurev.cb.04.110188.003051. [DOI] [PubMed] [Google Scholar]
- 113.Baranska J. Biosynthesis and transport of phosphatidylserine in the cell. Advances in lipid research. 1982;19:163–184. doi: 10.1016/b978-0-12-024919-0.50011-4. [DOI] [PubMed] [Google Scholar]
- 114.Seigneuret M, Devaux PF. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc Natl Acad Sci U S A. 1984;81:3751–3755. doi: 10.1073/pnas.81.12.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morrot G, Herve P, Zachowski A, Fellmann P, Devaux PF. Aminophospholipid translocase of human erythrocytes: phospholipid substrate specificity and effect of cholesterol. Biochemistry. 1989;28:3456–3462. doi: 10.1021/bi00434a046. [DOI] [PubMed] [Google Scholar]
- 116.Connor J, Pak CH, Zwaal RF, Schroit AJ. Bidirectional transbilayer movement of phospholipid analogs in human red blood cells. Evidence for an ATP-dependent and protein-mediated process. J Biol Chem. 1992;267:19412–19417. [PubMed] [Google Scholar]
- 117.Beleznay Z, Zachowski A, Devaux PF, Navazo MP, Ott P. ATP-dependent aminophospholipid translocation in erythrocyte vesicles: stoichiometry of transport. Biochemistry. 1993;32:3146–3152. doi: 10.1021/bi00063a029. [DOI] [PubMed] [Google Scholar]
- 118.Julien M, Tournier JF, Tocanne JF. Basic fibroblast growth factor modulates the aminophospholipid translocase activity present in the plasma membrane of bovine aortic endothelial cells. Eur J Biochem. 1995;230:287–297. [PubMed] [Google Scholar]
- 119.Andrick C, Broring K, Deuticke B, Haest CW. Fast translocation of phosphatidylcholine to the outer membrane leaflet after its synthesis at the inner membrane surface in human erythrocytes. Biochim Biophys Acta. 1991;1064:235–241. doi: 10.1016/0005-2736(91)90307-t. [DOI] [PubMed] [Google Scholar]
- 120.Wiedmer T, Zhou Q, Kwoh DY, Sims PJ. Identification of three new members of the phospholipid scramblase gene family. Biochim Biophys Acta. 2000;1467:244–253. doi: 10.1016/s0005-2736(00)00236-4. [DOI] [PubMed] [Google Scholar]
- 121.Francis VG, Mohammed AM, Aradhyam GK, Gummadi SN. The single C-terminal helix of human phospholipid scramblase 1 is required for membrane insertion and scrambling activity. FEBS J. 2013;280:2855–2869. doi: 10.1111/febs.12289. [DOI] [PubMed] [Google Scholar]
- 122.Sahu SK, Gummadi SN, Manoj N, Aradhyam GK. Phospholipid scramblases: an overview. Arch Biochem Biophys. 2007;462:103–114. doi: 10.1016/j.abb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 123.Sims PJ, Wiedmer T. Unraveling the mysteries of phospholipid scrambling. Thromb Haemost. 2001;86:266–275. [PubMed] [Google Scholar]
- 124.Bevers E, Comfurius P, Zwaal RFA. Regulatory mechanisms in maintenance and modulation of transmembraane lipid asymmetry: pathophysiologic implications. Lupus. 1996;5:480–487. doi: 10.1177/096120339600500531. [DOI] [PubMed] [Google Scholar]
- 125.Williamson P, Kulick A, Zachowski A, Schlegel RA, Devaux PF. Ca2+ induces transbilayer redistribution of all major phospholipids in human erythrocytes. Biochemistry. 1992;31:6355–6360. doi: 10.1021/bi00142a027. [DOI] [PubMed] [Google Scholar]
- 126.Williamson P, Bevers EM, Smeets EF, Comfurius P, Schlegel RA, Zwaal RF. Continuous analysis of the mechanism of activated transbilayer lipid movement in platelets. Biochemistry. 1995;34:10448–10455. doi: 10.1021/bi00033a017. [DOI] [PubMed] [Google Scholar]
- 127.Bevers EM, Williamson PL. Phospholipid scramblase: an update. FEBS Lett. 2010;584:2724–2730. doi: 10.1016/j.febslet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 128.Basse F, Stout JG, Sims PJ, Wiedmer T. Isolation of an erythrocyte membrane protein that mediates Ca2+-dependent transbilayer movement of phospholipid. J Biol Chem. 1996;271:17205–17210. doi: 10.1074/jbc.271.29.17205. [DOI] [PubMed] [Google Scholar]
- 129.Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ. Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem. 1997;272:18240–18244. doi: 10.1074/jbc.272.29.18240. [DOI] [PubMed] [Google Scholar]
- 130.Stout JG, Basse F, Luhm RA, Weiss HJ, Wiedmer T, Sims PJ. Scott syndrome erythrocytes contain a membrane protein capable of mediating Ca2+-dependent transbilayer migration of membrane phospholipids. J Clin Invest. 1997;99:2232–2238. doi: 10.1172/JCI119397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Frasch SC, Henson PM, Nagaosa K, Fessler MB, Borregaard N, Bratton DL. Phospholipid flip-flop and phospholipid scramblase 1 (PLSCR1) co-localize to uropod rafts in formylated Met-Leu-Phe-stimulated neutrophils. J Biol Chem. 2004;279:17625–17633. doi: 10.1074/jbc.M313414200. [DOI] [PubMed] [Google Scholar]
- 132.Wiedmer T, Zhao J, Nanjundan M, Sims PJ. Palmitoylation of phospholipid scramblase 1 controls its distribution between nucleus and plasma membrane. Biochemistry. 2003;42:1227–1233. doi: 10.1021/bi026679w. [DOI] [PubMed] [Google Scholar]
- 133.Sun J, Nanjundan M, Pike LJ, Wiedmer T, Sims PJ. Plasma membrane phospholipid scramblase 1 is enriched in lipid rafts and interacts with the epidermal growth factor receptor. Biochemistry. 2002;41:6338–6345. doi: 10.1021/bi025610l. [DOI] [PubMed] [Google Scholar]
- 134.Ory S, Ceridono M, Momboisse F, Houy S, Chasserot-Golaz S, Heintz D, Calco V, Haeberle AM, Espinoza FA, Sims PJ, Bailly Y, Bader MF, Gasman S. Phospholipid scramblase-1-induced lipid reorganization regulates compensatory endocytosis in neuroendocrine cells. J Neurosci. 2013;33:3545–3556. doi: 10.1523/JNEUROSCI.3654-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu J, Dai Q, Chen J, Durrant D, Freeman A, Liu T, Grossman D, Lee RM. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res. 2003;1:892–902. [PubMed] [Google Scholar]
- 136.Francis VG, Majeed MA, Gummadi SN. Recovery of functionally active recombinant human phospholipid scramblase 1 from inclusion bodies using N-lauroyl sarcosine. J Ind Microbiol Biotechnol. 2012;39:1041–1048. doi: 10.1007/s10295-012-1105-1. [DOI] [PubMed] [Google Scholar]
- 137.Sahu SK, Aradhyam GK, Gummadi SN. Calcium binding studies of peptides of human phospholipid scramblases 1 to 4 suggest that scramblases are new class of calcium binding proteins in the cell. Biochim Biophys Acta. 2009;1790:1274–1281. doi: 10.1016/j.bbagen.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 138.Stout JG, Zhou Q, Wiedmer T, Sims PJ. Change in conformation of plasma membrane phospholipid scramblase induced by occupancy of its Ca2+ binding site. Biochemistry. 1998;37:14860–14866. doi: 10.1021/bi9812930. [DOI] [PubMed] [Google Scholar]
- 139.Sanchez-Magraner L, Posada IM, Andraka N, Contreras FX, Viguera AR, Guerin DM, Arrondo JL, Monaco HL, Goni FM. The C-terminal transmembrane domain of human phospholipid scramblase 1 is essential for the protein flip-flop activity and Ca(2)(+)-binding. J Membr Biol. 2014;247:155–165. doi: 10.1007/s00232-013-9619-7. [DOI] [PubMed] [Google Scholar]
- 140.Frasch SC, Henson PM, Kailey JM, Richter DA, Janes MS, Fadok VA, Bratton DL. Regulation of phospholipid scramblase activity during apoptosis and cell activation by protein kinase Cdelta. J Biol Chem. 2000;275:23065–23073. doi: 10.1074/jbc.M003116200. [DOI] [PubMed] [Google Scholar]
- 141.Merregaert J, Van Langen J, Hansen U, Ponsaerts P, El Ghalbzouri A, Steenackers E, Van Ostade X, Sercu S. Phospholipid scramblase 1 is secreted by a lipid raft-dependent pathway and interacts with the extracellular matrix protein 1 in the dermal epidermal junction zone of human skin. J Biol Chem. 2010;285:37823–37837. doi: 10.1074/jbc.M110.136408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhao J, Zhou Q, Wiedmer T, Sims PJ. Palmitoylation of phospholipid scramblase is required for normal function in promoting Ca2+-activated transbilayer movement of membrane phospholipids. Biochemistry. 1998;37:6361–6366. doi: 10.1021/bi980218m. [DOI] [PubMed] [Google Scholar]
- 143.Ben-Efraim I, Zhou Q, Wiedmer T, Gerace L, Sims PJ. Phospholipid scramblase 1 is imported into the nucleus by a receptor-mediated pathway and interacts with DNA. Biochemistry. 2004;43:3518–3526. doi: 10.1021/bi0356911. [DOI] [PubMed] [Google Scholar]
- 144.Chen MH, Ben-Efraim I, Mitrousis G, Walker-Kopp N, Sims PJ, Cingolani G. Phospholipid scramblase 1 contains a nonclassical nuclear localization signal with unique binding site in importin alpha. J Biol Chem. 2005;280:10599–10606. doi: 10.1074/jbc.M413194200. [DOI] [PubMed] [Google Scholar]
- 145.Rami A, Sims J, Botez G, Winckler J. Spatial resolution of phospholipid scramblase 1 (PLSCR1), caspase-3 activation and DNA-fragmentation in the human hippocampus after cerebral ischemia. Neurochem Int. 2003;43:79–87. doi: 10.1016/s0197-0186(02)00194-8. [DOI] [PubMed] [Google Scholar]
- 146.Inuzuka T, Inokawa A, Chen C, Kizu K, Narita H, Shibata H, Maki M. ALG-2-interacting Tubby-like protein superfamily member PLSCR3 is secreted by exosomal pathway and taken up by recipient cultured cells. Biosci Rep. 2013 doi: 10.1042/BSR20120123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fleming SD, Egan RP, Chai C, Girardi G, Holers VM, Salmon J, Monestier M, Tsokos GC. Anti-phospholipid antibodies restore mesenteric ischemia/reperfusion-induced injury in complement receptor 2/complement receptor 1-deficient mice. J Immunol. 2004;173:7055–7061. doi: 10.4049/jimmunol.173.11.7055. [DOI] [PubMed] [Google Scholar]