

Abstract
Purpose
The alkylating agent melphalan prolongs survival in multiple myeloma (MM) patients; however, it is associated with toxicities and development of drug-resistance. Here, we evaluated the efficacy of melphalan-flufenamide (Mel-flufen), a novel dipeptide prodrug of melphalan in MM.
Experimental Design
MM cell lines, primary patient cells, and the human MM xenograft animal model were utilized to study the antitumor activity of mel-flufen.
Results
Low doses of mel-flufen triggers a more rapid and higher intracellular concentrations of melphalan in MM cells than is achievable by free melphalan. Cytotoxicity analysis showed significantly lower IC50 of mel-flufen than melphalan in MM cells. Importantly, mel-flufen induces apoptosis even in melphalan-, and bortezomib-resistant MM cells. Mechanistic studies show that siRNA knockdown of aminopeptidase N, a key enzyme mediating intracellular conversion of mel-flufen to melphalan, attenuates anti-MM activity of mel-flufen. Furthermore, mel-flufen-induced apoptosis was associated with: 1) activation of caspases and PARP cleavage; 2) ROS generation; 3) mitochondrial dysfunction and release of cytochrome-c; and 4) induction of DNA damage. Moreover, mel-flufen inhibits MM cell migration and tumor-associated angiogenesis. Human MM xenograft studies showed a more potent inhibition of tumor growth in mice treated with mel-flufen than mice receiving equimolar doses of melphalan. Finally, combining mel-flufen with lenalidomide, bortezomib, or dexamethasone triggers synergistic anti-MM activity.
Conclusion
Our preclinical study supports clinical evaluation of mel-flufen to enhance therapeutic potential of melphalan, overcome drug-resistance, and improve MM patient outcome.
Keywords: Myeloma, Apoptosis, Novel therapeutics, alkylating agents
Introduction
Multiple myeloma (MM) remains incurable due to the development of a drug resistant phenotype after prolonged therapy (1, 2). For many years, combined melphalan (mustard-L-phenylalanine) and prednisone has been a mainstay of MM treatment in the non-transplant candidates. In transplant candidates, a treatment regimen comprising a high-dose melphalan (HDM) in conjunction with autologous stem cell transplantation (ASCT) has improved progression-free and overall survival in MM patients (3-5). More recent studies have combined melphalan and steroids with several novel agents such as bortezomib, thalidomide, or lenalidomide, as initial therapy of elderly newly diagnosed patients and have improved response extent and frequency, as well as prolonged progression free an overall survival (6-12). In a parallel fashion, integration of these novel therapies into the transplant paradigm as induction, consolidation, and maintenance has further improved outcome in this setting (13, 14). These studies exemplify the utility of melphalan in the current MM therapy, and provided impetus for the development of melphalan prodrug to increase tumor specificity, reduce toxicity and prevent drug-resistance.
Pharmacological screening of alkylating oligopeptides led to the identification of a novel melphalan-containing prodrug mel-flufen (L-melphalanyl-p-L-fluoro phenylalanine ethyl ester), a molecular entity with a more potent anti-tumor activity than parental drug melphalan despite identical alkylating capacity (Fig 1A) (15-17). Mel-flufen is rapidly incorporated into the tumor cells, followed by intracellular hydrolysis which in part is mediated by aminopeptidase N (ANPEP), an enzyme overexpressed in several tumor cell malignancies (18). Studies using solid tumor cell models showed that treatment with mel-flufen causes at least a 10-fold higher loading of melphalan which explain its higher tumor cell cytotoxicity (15-17, 19). To date, the mel-flufen activity against MM cells is undefined. In the present study, we examined the anti-tumor activity of mel-flufen in MM cells using both in vitro and in vivo model systems. Our studies show that mel-flufen is more potent than melphalan and can overcome resistance not only to melphalan, bur also to novel agents, providing the rationale for its clinical evaluation to improve patient outcome in MM.
Figure 1.

(A) Chemical structures of the melphalan-containing dipeptide mel-flufen and melphalan. (B) RPMI-8226 cells were treated with indicated concentrations of either mel-flufen or melphalan; samples were harvested at 0-5-15-30-60 and 120 min, followed by analysis for intracellular accumulation of melphalan using LC-MS. The peak area of mel-flufen and melphalan was analyzed at each time point after normalization of signal intensity in each samples using the internal fluorescein standard. Area under curve 0-120 min was calculated from the individual samples. The values are given relative to AUC for Mel-flufen 0.5 μM mean value. Data presented are means ± S.E.M (n = 3). (C) MM cell lines were treated with indicated doses of mel-flufen for 24h, and cytotoxicity was assessed using MTT assay (n = 3, mean ± SD; P < 0.005 for all cell lines). (D) MM.1R and RPMI-8226 cells were treated with mel-flufen (2 μM) for 24h and analyzed for apoptosis by AnnexinV/Propidium Iodide staining assay (n = 2, mean ± SD; P < 0.001). (E) MM.1R and RPMI-8226 MM cells were treated with mel-flufen {1μM and 3μM, respectively} for 24h; protein lysates were subjected to immunoblotting using indicated antibodies. (F) MM.1S cells were treated with mel-flufen in the presence or absence of BMSCs for 24h, and DNA synthesis was measured by 3H-TdR uptake (mean ± SD of triplicate cultures; P < 0.002 for all samples).
Material and Methods
Cell culture and reagents
MM cell lines including MM.1S (dexamethasone-sensitive), MM.1R (dexamethasone-resistant), RPMI-8226, LR-5 (melphalan-resistant derivative of RPMI-8226), KMS-12BM, and INA-6 (IL-6 dependent) were cultured with RPMI-1640 medium supplemented with 10% FBS, 2mM L-glutamine, 100 units/ml Penicillin and 100 ug/ml streptomycin. ANBL-6-bortezomib–sensitive (ANBL-6.WT) and -resistant (ANBL-6.BR) were kindly provided by Dr. Robert Orlowski (M.D. Anderson Cancer Center, Houston, TX). Tumor cells from MM patients were purified (greater than 95% purity) by CD138 positive selection using the Auto MACS magnetic cell sorter (Miltenyi Biotec Inc., Auburn, CA). Informed consent was obtained from all patients in accordance with the Helsinki protocol. Peripheral blood mononuclear cells (PBMCs) from healthy donors were maintained in culture medium, as above. Mel-flufen was obtained from Oncopeptides AB (Stockholm, Sweden). Melphalan was purchased from Sigma Chemical Company (St Louis, MO) and Apoteket AB, Sweden (Alkeran®, Apoteket AB, Sweden); bortezomib and lenalidomide were purchased from Selleck Chemicals LLC (Houston, TX); and Dex was obtained from Calbiochem (San Deigo, CA).
Measurement of intracellular concentrations of mel-flufen and melphalan
The intracellular concentration of melphalan in RPMI-8226 cells was assessed at various time points after treatment with freshly made solutions of mel-flufen or melphalan. For each treatment series RPMI-8226 cells were re-suspended at a concentration of 2.5 × 106 cells/ml in a total volume of 6 ml of complete pre-warmed RPMI media, and 1 ml sample was removed at 0, 5, 15, 30, 60 or 120 mins after addition of each drug. The 1 ml sample was added to 4 ml of pre cooled PBS and centrifuged for 5 min at 1,000 rpm; the resulting cell pellet was washed in 5 ml of pre cooled PBS and solubilized by adding 50 μl of ethanol/acetonitrile (1:1, v/v). The resulting precipitated cell debris were cleared by centrifugation at 10,000 rpm for 5 min and the supernatant was collected and frozen at -80 C until further analyses. The intracellular amount of mel-flufen or melphalan was measured in a aliquote of 25 μl of the sample which was mixed with 75 μl of an internal standard solution consisting of 1 μg/ml of fluorescein diluted in 1:1 (acetonitrile:ethanol), centrifuged for 4 minutes at 3700 rpm (Heraeus Biofuge 13). Supernatant was transferred to 200 μl HPLC vials and then analyzed for mel-flufen or melphalan content by LC-MS (SIM of 498 Da for mel-flufen, 305 Da for melphalan, and 333 Da for the fluorescein standard). The above analyses were performed at OncoTargeting AB, Uppsala, Sweden. The mean ± SEM of peak area of mel-flufen and melphalan at each time point was calculated. The area under the curve for 0-120 min of melphalan (AUC 0-120 min) was determined for each treatment and is shown relative to AUC 0-120 min of 0.5 μM of mel-flufen.
Cell viability, proliferation, and apoptosis assays
Cell viability was assessed by using colorimetric assay with 3-(4, 5-dimethylthiozol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Calbiochem), and cell proliferation analysis in co-culture experiments with patient-derived BMSCs was performed using thymidine incorporation, as described previously (20). Apoptosis was quantified using Annexin V-Fitc/Propidium iodide apoptosis detection kit, as per manufacturer’s instructions (BD Biosciences, San Jose, CA), followed by an analysis on FACS Calibur (BD Biosciences, San Jose, CA).
Aminopeptidase N (ANPEP) activity assay
Enzymatic activity of ANPEP was measured with the substrate L-alanine-4-nitro-anilide (Sigma–Aldrich) as previously described (21).
Immunoblotting
Western blot analysis was performed as previously described (22) using antibodies recognizing full length and cleaved forms of caspase-3, caspase-7, caspase-8, caspase-9, PARP (Cell signaling, Beverly, MA); as well as p53, γ-H2AX, ANPEP, and GAPDH (Abcam, Cambridge, MA).
Transient transfection assays
MM.1S cells were transiently transfected with genome control (scrambled) siRNA or siRNA against ANPEP (Smart pool siRNA, Dharmacon, Inc., Lafayette, CO) using Nucleofector Kit V, according to manufacturer’s instructions (Amaxa Biosystems, Cologne, Germany).
Human plasmacytoma xenograft model
All animal studies were approved by the DFCI Institutional Animal Care and Use Committee. The xenograft tumor model was performed as previously described (23, 24). CB-17 SCID mice were subcutaneously inoculated with 6.0 × 106 MM.1S cells in 100μl of serum free RPMI-1640 medium, and then randomized to treatment groups when tumors reached approximately ~100 mm3. Mice were treated intravenously with vehicle, mel-flufen, or melphalan with indicated concentrations and treatment duration times. Animals were euthanized when their tumors reached 2 cm3.
In vitro migration and capillary-like tube structure formation assays
Transwell Insert Assays (Chemicon, Billerica, MA) were utilized to measure migration, and in vitro angiogenesis was assessed by Matrigel capillary-like tube structure formation assay, as previously described (25). For endothelial tube formation assay, human vascular endothelial cells (HUVECs) were obtained from Clonetics (Walkersville, MD) and maintained in endothelial cell growth medium-2 (EGM2 MV SingleQuots, Clonetics) containing 5% FBS.
Statistical Analysis
Statistical significance of differences observed in drug-treated vs. control cultures was determined by using the Student’s t test. The minimal level of significance was P < 0.05. Tumor volume and survival in mice was measured using the GraphPad PRISM (GraphPad Software/version 5, SanDiego, CA). Isobologram analysis (26) was performed using “CalcuSyn” software program (Biosoft, Ferguson, MO and Cambridge, UK). Combination index (CI) values of < 1.0 indicate synergism and values > 1.0 antagonism.
Results and Discussion
Mel-flufen delivers high intracellular concentration of melphalan
RPMI-8226 MM cells were treated with either mel-flufen (0.5 μM, 1 μM, or 5 μM) or melphalan (10 μM or 100 μM) and the intracellular content of mel-flufen or melphalan was measured using HPLC-MS. Area under curve (AUC) 0-120 mins was calculated from melphalan Peak Area curves in each sample and relative AUC is shown in Figure 1B. Treatment of RPMI-8226 cells with mel-flufen led to high intracellular concentration of free melphalan as compared to cells exposed directly to melphalan (Fig 1B). A high concentration was reached as early as after 15 mins (data not shown). Importantly, treatment of RPMI-8226 cells with 5 μM of mel-flufen load the cells with much more melphalan than can be achieved using even 100 μM of melphalan (Fig 1B). These data suggest that mel-flufen allows for a more rapid and higher intracellular accumulation of melphalan in MM cells than is achievable by direct exposure to equimolar doses of melphalan. Our results using MM cells is consistent with similar observation in other cancer cell lines (19). Previous studies showed that lipophilicity and an early intracellular hydrolysis of mel-flufen by peptidases inside the cells to release melphalan contributes to achieving rapid and high intracellular concentrations of melphalan (19, 27, 28). Earlier findings also showed that in tumor cells a limited exposure time which simulate short half-life in vivo, proved more favorable for mel-flufen than for melphalan indicating a trapping mechanism through the enzymatic activation (29). Together, these results suggest that mel-flufen administration in MM patients is a more efficient therapeutic strategy for delivering higher concentrations of intracellular melphalan than by directly exposing cells to free melphalan.
Anti-MM activity of mel-flufen in vitro
Human MM cell lines (MM.1S, INA-6, RPMI-8226, MM.1R, Dox-40, ARP-1, ANBL-6) were treated with various concentrations of mel-flufen for 24h, followed by assessment of cell viability using MTT assays. A significant concentration-dependent decrease in viability of all cell lines was observed in response to mel-flufen treatment (Fig 1C). The cytotoxicity of mel-flufen was observed in MM cell lines sensitive and resistant to conventional and novel therapies, as well as representing distinct cytogenetic profiles. For example, we examined isogenic cell lines Dex-sensitive MM.1S and Dex-resistant MM.1R with t(14;16) translocation and c-maf overexpression; RPMI-8266 with TP53, K-Ras and EGFR mutations; and INA-6, an IL-6-dependent cell line with N-Ras activating mutation. The variable IC50 of mel-flufen observed against MM cell lines may be attributed to their distinct genetic background and/or drug resistance characteristics (30-33).
We next examined whether anti-MM activity of mel-flufen is due to induction of apoptosis. Treatment of MM.1R or RPMI-8226 cells with mel-flufen triggered accumulation of cells in early (Ann V+/PI-) and late stage (Ann V+/PI+) apoptosis (Fig 1D). Moreover, mel-flufen-induced apoptosis was associated with: 1) activation of caspase-3, caspase-7, caspase-8 and caspase-9, as well as PARP cleavage; 2) ROS generation; and 3) decrease in mitochondrial transmembrane potential (ΔΨm) accompanied by release of cytochrome-c (Fig 1E and Supplementary Fig 1A-1D). Studies using pan-caspase inhibitor (Z-VAD-FMK) showed attenuation of mel-flufen-induced cytotoxicity in three MM cell lines (Supplementary Fig 1E). These findings suggest that mel-flufen triggers both mitochondria-dependent and – independent apoptotic signaling pathways.
Interaction and adhesion of MM cells with bone marrow stromal cells (BMSCs) triggers cytokine secretion, which mediates paracrine growth of MM cells, as well as confers cell adhesion mediated drug resistance (CAM-DR) (34, 35). To determine whether mel-flufen can overcome these protective effects, MM.1S cells were cultured with or without BMSCs in the presence or absence of various concentrations of mel-flufen. A significant inhibition of BMSCs-induced proliferation of MM.1S cells was observed in response to mel-flufen treatment (Fig 1F). These data suggest that 1) mel-flufen not only directly targets MM cells, but also can overcome the cytoprotective effects of the MM-host BM microenvironment.
The mechanism(s) of melphalan-resistance include decreased melphalan uptake, reduced induction of DNA cross links, and/or increased repair of such DNA lessions which otherwise are converted to DNA single or double strand breaks. Since mel-flufen exhibit a higher potency and rapid kinetics of action in tumor cells as compared to melphalan, we examined whether these attributes impart mel-flufen the ability to overcome melphalan-resistance. We utilized previously characterized (36) melphalan-sensitive RPMI-8226 and its melphalan-resistant derivative of RPMI-8226 (LR5) MM cell lines. As seen in Figure 2A, mel-flufen induces cytotoxicity even in melphalan-resistant LR5 cells, whereas melphalan alone does not significantly affect the viability of LR5 cells at tested concentrations. Higher concentrations of melphalan (100 μM) showed similar resistance to melphalan in LR5 cells (data not shown). The mechanism(s) whereby mel-flufen overcomes melphalan-resistance remains to be examined; however, it is likely that mel-flufen induces a more potent and cumulative DNA damage and thereby can overcome the DNA crosslink repair capacity of MM cells. Besides reduced DNA damage, melphalan resistance is also linked to other factors, such as increased glutathione-S-transferase activity, cell-adhesion mediated drug resistance (CAM-DR), mitochondrial alterations, or impaired caspase activation (36-39); and many of these mechanism(s) may likely be circumvented by achieving a sustained and higher intracellular concentrations of melphalan with mel-flufen.
Figure 2.

(A) Melphalan-sensitive RPMI-8226 (WT) and melphalan-resistant LR-5 cells were treated with indicated concentrations of mel-flufen for 24h, and cytotoxicity was assessed using MTT assay (n = 3, mean ± SD; P < 0.005). (B) MM.1S and MM.1R cells were treated with mel-flufen or melphalan at the indicated concentrations for 24h, and cytotoxicity was measured by MTT assay (n = 3, mean ± SD; P < 0.005). (C) Bortezomib sensitive-ANBL6 {ANBL6.WT} and Bortezomib-resistant ANBL6 {ANBL6.BR} MM cells were treated with indicated concentrations of mel-flufen or melphalan, and cytotoxicity was measured using MTT assay (n = 3, means ± SD; P < 0.003). (D) Purified patient MM (CD138+) cells were treated with mel-flufen at indicated doses for 24h, and cell viability was measured using trypan blue assay (means ± SD of triplicate cultures; P < 0.001 for all patient samples).
We next examined the effects of mel-flufen in Dex- or proteasome inhibitor-resistant MM cells. We observed a significantly more potent anti-MM activity of mel-flufen than melphalan when tested against Dex-sensitive (MM.1S) and Dex-resistant (MM.1R) cells (Fig 2B). To determine whether mel-flufen can overcome bortezomib-resistance, we utilized bortezomib-sensitive (ANBL-6.WT) and bortezomib-resistant (ANBL-6.BR) MM cell lines (40). Interestingly, we found that while ANBL-6.WT cells are sensitive to melphalan, ANBL6.BR cells were relatively resistant to melphalan. Importantly, mel-flufen decreases the viability in ANBL-6.BR cells (Fig 2C), suggesting that it can overcome bortezomib resistance. Since the ANBL6.BR cells were derived after chronic exposure of ANBBL6.WT cells to low concentrations of bortezomib (acquired drug resistance), it is likely that, like LR5 (melphalan-resistant cells), ANBL6.BR cells have reduced DNA damage-associated signaling pathways. However, further studies in ANBL6.BR cells are needed to delineate the mechanism(s) whereby mel-flufen overcomes bortezomib-resistance, as well as mechanism(s) conferring cross-resistance to melphalan and bortezomib. Nevertheless, our data demonstrate that mel-flufen, but not melphalan, overcomes bortezomib-resistance.
We next determined whether mel-flufen similarly affects purified patient MM cells. Tumor cells from five MM patients, including those relapsing after multiple prior therapies including bortezomib, lenalidomide, and Dex, were treated with mel-flufen. Patients were considered refractory to specific therapy when disease progressed on therapy or relapsed within two months of stopping therapy. A significant dose-dependent decrease in viability of all patient MM cells was noted after mel-flufen treatment (Fig 2D). These data show that mel-flufen induces cytotoxicity in tumor cells obtained from patient whose MM is resistant to bortezomib, Dex, or lenalidomide therapies. Importantly, mel-flufen at the IC50 for MM cells does not significantly affect the viability of normal PBMCs (data not shown), suggesting specific anti-MM activity and a favorable therapeutic index for mel-flufen.
Mechanism(s) mediating mel-flufen activity
Earlier studies established that 1) aminopeptidase N (ANPEP, also known as CD13) plays a key role in catalyzing the release of free melphalan from mel-flufen, and 2) hydrolysis of the peptide bond in mel-flufen by ANPEPs is a prerequisite for mel-flufen-induced cytotoxicity (19). Of note, the activity and/or the expression of ANPEP is elevated in many cancer types, and is associated with various characteristics of malignant phenotype including cell proliferation, cytokine secretion, tumor invasion and angiogenesis (18). These studies suggest ANPEP as a viable therapeutic target in cancer (18). As seen in Figure 3A and 3B, both ANPEP expression and activity are constitutively elevated in MM cells. Importantly, transfection of ANPEP siRNA, but not negative-control (scrambled) siRNA, significantly inhibited mel-flufen-induced apoptosis in MM.1S cells, whereas no marked difference in melphalan-induced cytotoxicity was evident (Fig 3C). The residual mel-flufen cytotoxic activity in ANPEP-siRNA-tranfected cells may be attributed to limitations of RNA-interference strategy, which usually results in incomplete gene loss. Another possibility is that mel-flufen or its intermediate metabolite in MM cells is substrate of other aminopeptidases, as reported in solid tumor cells (19). Nevertheless, our data provide the evidence that mel-flufen-triggered apoptosis in MM cells is facilitated, at least in part, via ANPEP.
Figure 3.
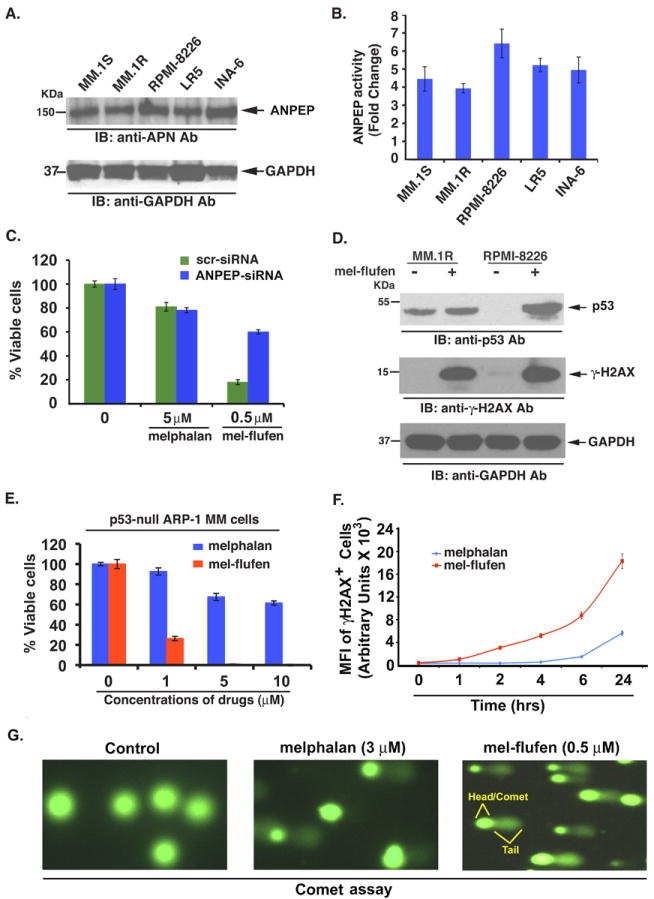
(A) Total protein lysates from MM.1S, MM.1R, RPMI-8226, LR-5 and INA6 cells were subjected to immunoblot analysis using anti-ANPEP, or anti GAPDH antibodies. (B) Bar graph shows the baseline ANPEP activity in various MM cell lines. (C) MM.1S cells were transfected with scr-siRNA or ANPEP-siRNA; 24h after transfection, cells were treated with indicated concentrations of melphalan or mel-flufen for 24h, followed by analysis of viability (n = 3, mean ± SD; P < 0.001 in scr- versus ANPEP siRNA-transfected cells in response to mel-flufen). Percent cell viability was normalized (as 100%) for scr- or ANPEP-siRNA controls, respectively. (D) MM.1R and RPMI-8226 MM cells were treated with mel-flufen (2 μM for MM.1R and 5 μM for RPMI-8226 cells) for 24h; protein lysates were prepared and subjected to immunoblotting using indicated antibodies. (E) p53-null ARP-1 MM cells were treated with indicated concentrations of either mel-flufen or melphalan for 24h, and cytotoxicity was assessed using MTT assay (n = 3, mean ± SD; P < 0.001). (F) MM.1S cells were treated with mel-flufen (1 μM) or melphalan (1 μM) for 1h, 2h, 6h, and 24h; cells were then washed and stained with Alexa-fluor 647 conjugated anti-H2AX antibody, followed by quantification of γ-H2AX-positive cells using flow cytometry (n = 3, mean ± SD; P < 0.005). (G) MM.1S cells were treated with melphalan (3 μM) or mel-flufen (0.5 μM) for 4h; harvested, and subjected to alkaline comet assay. For each slide, images were randomly captured by fluorescence microscopy and representative images from three independent experiments are shown.
Previous studies have shown that melphalan triggers DNA-damage response/repair signaling, associated with activation of p53 (41). We therefore next examined whether mel-flufen-induced apoptosis in MM cells correlates with induction of similar signaling cascades. Examination of mel-flufen-treated MM cells showed a robust increase in p53 (Fig 3D, upper panel) in RPMI-8226 cells, albeit to a lesser extent in MM.1R cells. Interestingly, our data (Fig 1C) show that mel-flufen trigger cytotoxicity even in p53-null ARP-1 MM cells, suggesting that functional p53 may not be obligatory for efficient induction of mel-flufen-induced apoptosis. Nevertheless, given the established role of p53 in melphalan response, we performed a side-by-side comparison of melphalan and mel-flufen activity using p53-null ARP-1 cells. As expected, we found that high concentrations of melphalan (5-10 μM) are required to achieve 30%-40% cell death; importantly, mel-flufen (0.5 μM) at the concentrations 10 fold lower than melphalan (5 μM) is able to triggers significant cytotoxicity in p53-null ARP-1 MM cells (Fig 3E). These data suggest that although mel-flufen increases p53 levels (Fig 3D), its cytotoxic activity in MM cells is not dependent on p53. Our findings have important clinical implications, since 10-15% of MM patients have p53 mutations/deletions at presentation which confer drug resistance, and majority of patients acquire this abnormality with disease progression; a therapeutic approach using mel-flufen would allow for potent anti-MM activity even in this patient population.
An early event in the response of mammalian cells to DNA double-strand breaks is the phosphorylation of histone H2AX (γ–H2AX) at the sites in proximity to DNA breaks (42). A robust induction of γ-H2AX was observed in mel-flufen-treated MM.1R and RPMI-8226 cells (Fig 3D, middle panel), suggesting that mel-flufen-induced DNA crosslinks were indeed converted to DNA double strand breaks. We next compared the potency of mel-flufen and melphalan in inducing DNA double strand breaks. MM.1S cells were treated with mel-flufen (1 μM) or melphalan (1 μM) for 1h, 2h, 6h, and 24h; cells were then washed and stained with Alexa-fluor 647 conjugated anti-H2AX (pS139) antibody, followed by quantification of γ-H2AX-positive cells using flow cytometry. Results show an early and potent induction of DNA double-strand breaks in mel-flufen- versus melphalan-treated MM.1S cells (Fig 3F).
In order to confirm the differential induction of DNA damage by mel-flufen and melphalan, we next performed alkaline comet assay. The comet assay is a single cell gel electrophoresis assay and utilizes the principle that damaged DNA, migrates, forming a “tail”, whereas undamaged DNA with intact supercoiled structure, does not migrate, forming the head of the comet. The intensity and length of the comet tail is proportional to extent of DNA damage. Results showed that even low concentrations of mel-flufen (0.5 μM) are able to trigger more potent and greater DNA damage than is observed in cells treated with higher concentrations (3 μM) of melphalan (Fig 3G, Supplementary Figure 2). Together, these results suggest that mel-flufen is more efficient inducer of DNA damage than melphalan.
Overall, our mechanistic studies shows that 1) mel-flufen-induced cytotoxicity is facilitated via ANPEP; 2) mel-flufen triggers DNA damage associated with induction of γ-H2AX, and p53; 3) while p53 is upregulated in response to mel-flufen treatment, the cytotoxic activity of mel-flufen is not dependent on p53, suggesting that there may be a p53-independent component to mel-flufen-induced cytotoxicity; and 4) mel-flufen-induced apoptosis is associated with activation of caspases and PARP cleavage. Of note, DNA damage response signaling is linked to activation of p53/caspases signaling axis (43), suggesting a potential crosstalk between these pathways during mel-flufen-induced apoptosis. It is possible that mel-flufen, like melphalan, triggers pleiotropic signaling pathways; however, due to the rapid intracellular accumulation characteristics of mel-flufen compared to melphalan, the kinetics of alterations in apoptotic response signaling may vary and this issue remains to be defined.
Effect of mel-flufen on migration of MM cells and angiogenesis
MM cell growth is associated with angiogenesis (44, 45). As noted above, ANPEP expression/activity is associated with maliganant phenotype, including angiogenesis (46); and importantly, mel-flufen is ANPEP-activated prodrug of melphalan. We examined the effect of mel-flufen on MM cell migration and angiogenesis using Transwell insert systems and in vitro tubule formation assays. Vascular endothelial growth factor (VEGF) is elevated in the MM BM microenvironment, and prior studies showed that VEGF triggers growth, migration, and angiogenesis in MM (44, 45, 47). We first examined whether mel-flufen affects VEGF-induced MM cell migration. VEGF alone markedly increases MM.1S cell migration; conversely, mel-flufen inhibits VEGF-dependent MM cell migration, evidenced by a decrease in the number of crystal violet-stained cells (Fig 4A and 4B). These cells were >95% viable prior to and after performing the migration assay, excluding the possibility that drug-induced inhibition of migration is due to cell death. Additional experiments show that melphalan also inhibits tumor-associated angiogenesis and MM cell migration, albeit at much higher concentrations than mel-flufen (data not shown). These findings are consistent with more potent and robust accumulation of intracellular melphalan upon mel-flufen treatment. These results suggest that mel-flufen may negatively regulate homing of MM cells to the BM.
Figure 4.

(A) Transwell insert assay: MM.1S cells were pretreated with mel-flufen (0.5 μM) for 12h; cells (>90% viable). Cells were then washed and incubated in serum-free medium for 2h (cell viability > 90%) and thereafter plated on a fibronectin-coated polycarbonate membrane in the upper chamber of Transwell inserts, and exposed for 4h to serum-containing medium in the lower chamber. Cells migrating to the bottom face of the membrane were fixed with 90% ethanol and stained with crystal violet (magnification: 10× /0.25 NA oil). A total of 3 randomly selected fields were examined for cells that had migrated from top to bottom chambers. Image is representative of 3 experiments with similar results. (B) Cells were plated as in ‘A’, and the effect of mel-flufen on migration was assessed in the presence or absence of rVEGF (1 μg). The bar graph represents quantification of migrated cells. (C) HUVECs were cultured in the presence or absence of mel-flufen (1 μM) for 16h, and then assessed for in vitro angiogenesis using Matrigel capillary-like tube structure formation assays (magnification: 4×/0.10 NA oil, media: EBM-2). Image is representative of 2 experiments with similar results. In vitro angiogenesis is evidenced by capillary tube branch formation (dark brown) (n=3, mean ± SD; P < 0.005). (D) HUVECs were cultured in the presence or absence of indicated concentrations mel-flufen for 16h, followed by assessment of in vitro angiogenesis as in “C”. The bar graph represents quantification of capillary-like tube structure formation by vehicle alone and mel-flufen-treated cells. Branch points in several random view fields/well were counted; values were averaged; and statistically significant differences were measured using student’s t-test (n = 2, mean ± SD; P = 0.03 for control versus meflufen-treated cells).
We next utilized in vitro capillary-like tube structure formation assays to examine the anti-angiogenic activity of mel-flufen. Angiogenesis was measured in vitro using Matrigel capillary-like tube structure formation assays in which human vascular endothelial cells (HUVECs) plated onto Matrigel differentiate and form capillary-like tube structures similar to in vivo neovascularization. This assay therefore provides evidence for anti-angiogenic effects of drugs. HUVECs were seeded in 96-well culture plates precoated with Matrigel; treated with vehicle (DMSO), mel-flufen for 16h; and then examined for tube formation using an inverted microscope. As seen in Fig 4C and 4D, tubule formation was markedly decreased in the mel-flufen-treated cells versus vehicle control. HUVEC cell viability was assessed using trypan blue exclusion assay, and <5% cell death was observed with mel-flufen treatment. Our results are consistent with earlier in vivo data showing that mel-flufen decreases the number of blood vessel formation in SH-SY5Y xenograft model (17). Taken together, our findings suggest that mel-flufen blocks MM cell migration and inhibit tumor-associated angiogenesis.
Anti-MM activity of mel-flufen in xenograft mouse model
Having shown that mel-flufen induces apoptosis in MM cells in vitro, we next examined the in vivo efficacy of mel-flufen using a human plasmacytoma MM.1S xenograft mouse model (23). Treatment of tumor-bearing mice with mel-flufen intravenously significantly inhibited MM tumor growth (P = 0.001) and prolonged survival (P < 0.001) of these mice (Fig 5A and 5B, respectively). Equimolar doses of melphalan also reduced tumor progression (Fig 5C), albeit to a lesser extent than mel-flufen. Moreover, mel-flufen-treated mice survived for a longer time than mice receiving equimolar doses of melphalan (P < 0.01; CI =95%) (Fig 5D). These in vivo data confirm our in vitro findings showing more potent anti-MM activity and tumor cell selectivity of mel-flufen versus melphalan.
Figure 5.

(A) Average and standard deviation of tumor volume (mm3) is shown from mice (n = 5/group) versus time (days) when tumour was measured. MM.1S cells (5 × 106 cells/mouse) were implanted in the rear flank of female mice (6 weeks of age). Tumor-bearing mice were randomized to treatment groups and treated intravenously with vehicle or mel-flufen (3 mg/kg) on a twice-weekly schedule for 2 weeks (mean tumor volume ± SD). A significant delay in tumor growth in mel-flufen-treated mice was noted compared to vehicle-treated control mice (P=0.008). Bars indicate mean ± SD. (B) Kaplan-Meier survival plot shows significant increase survival of mice receiving mel-flufen versus vehicle-alone-treated mice (P = 0.0002, Log-rank (Mantel-Cox) Test); median survival was 24 days in vehicle-treated mice versus 44 days in mel-flufen-treated mice (CI 95%). (C) MM.1S cells (5 × 106 cells/mouse) were implanted in the rear flank of female mice, as in ‘A’. Tumor-bearing mice were randomized to treatment groups and treated intravenously with vehicle (6 mice/group), mel-flufen (2 mg/kg) (6 mice/group) or equimolar dose of melphalan (2 mice/group) on a twice weekly schedule for 3 weeks (mean tumor volume ± SD). More potent anti-MM activity of mel-flufen was noted compared to either vehicle-, or melphalan-treatment. Bars indicate mean ± SD. (D) Kaplan-Meier survival plot shows significant (P < 0.05) increase in survival of mice receiving mel-flufen compare to vehicle-, or melphalan-treated mice.
Combined treatment with mel-flufen and lenalidomide, bortezomib, or dexamethasone induces synergistic anti-MM activity
We next examined whether mel-flufen can be combined with other drugs to enhance cytotoxicity and overcome melphalan-resistance. MM.1S cells were first treated with both mel-flufen and lenalidomide simultaneously across a range of concentrations for 24h, and then analyzed for viability using MTT assay. An analysis of synergistic anti-MM activity using the Chou and Talalay method (26) demonstrated that the combination of low concentrations of mel-flufen and lenalidomide triggered synergistic anti-MM activity, with a combination index (CI) < 1.0 (Fig 6A, Graph and Table). We next examined whether mel-flufen adds to the anti-MM activity of proteasome inhibitor bortezomib and conventional anti-MM agent Dex. As with lenalidomide, the combination of mel-flufen with bortezomib or Dex triggered synergistic anti-MM activity, evidenced by a significant decrease in viability of MM.1S cells (Fig 6B and 6C, Graphs and Tables). Importantly, a similar synergism was observed between mel-flufen and lenalidomide, bortezomib, or Dex in melphalan-resistant LR5 MM cells (Fig 6D-6F, Graphs and Tables). Although definitive evidence of decreased toxicity of combination therapy awaits results of clinical trials, the synergy observed in vitro may allow for use of lower doses and decreased toxicity.
Figure 6.
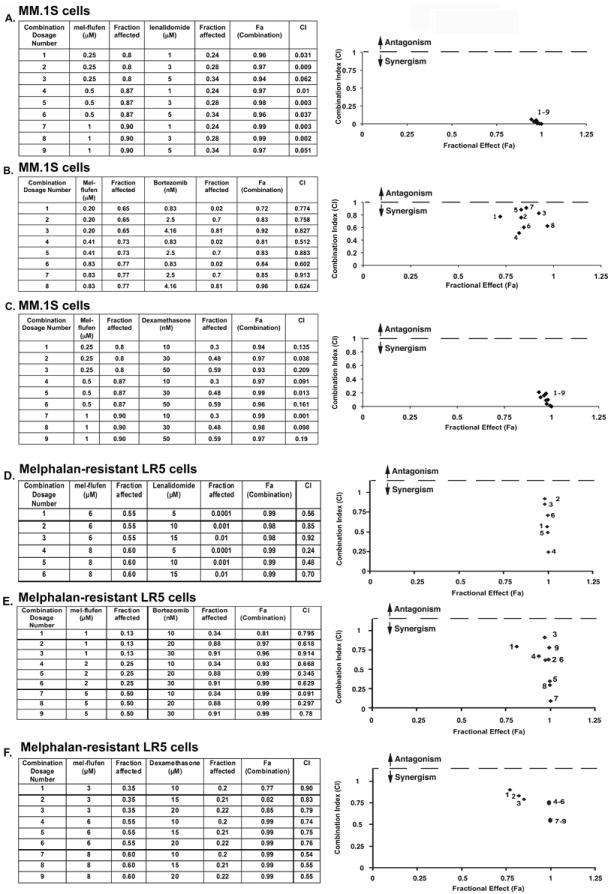
Combination of low doses of mel-flufen and lenalidomide, bortezomib, or dex trigger synergistic anti-MM activity. (A) MM.1S cells were treated for 24h with mel-flufen, lenalidomide, or mel-flufen plus lenalidomide; and then assessed for viability using MTT assays. Isobologram analysis shows the synergistic cytotoxic effect of mel-flufen and lenalidomide. The graph (right panel) is derived from the values given in the Table (left panel). The numbers 1-9 in graph represent combinations shown in the Table. Combination index (CI) < 1 indicates synergy. (B) MM.1S cells were treated for 24h with mel-flufen, bortezomib, or mel-flufen plus bortezomib; and then assessed for viability using MTT assays. Isobologram analysis shows the synergistic cytotoxic effect of mel-flufen and bortezomib. The graph (right panel) is derived from the values given in the Table (left panel). (C) MM.1S cells were treated for 24h with mel-flufen, Dex, or mel-flufen plus Dex; and then assessed for viability using MTT assays. Isobologram analysis shows the synergistic cytotoxic effect of mel-flufen and Dex. The graph (right panel) is derived from the values given in the Table (left panel). (D) Melphalan-resistant LR-5 cells were treated for 24h with mel-flufen, lenalidomide, or mel-flufen plus lenalidomide; and then assessed for viability using MTT assays. Isobologram analysis shows the synergistic cytotoxic effect of mel-flufen and lenalidomide. The graph (right panel) is derived from the values given in the Table (left panel). (E) Melphalan-resistant LR-5 cells were treated for 24h with mel-flufen, bortezomib, or mel-flufen plus bortezomib; and then assessed for viability using MTT assays. Isobologram analysis shows the synergistic cytotoxic effect of mel-flufen and bortezomib. The graph (right panel) is derived from the values given in the Table (left panel). (F) Melphalan-resistant LR-5 cells were treated for 24h with mel-flufen, Dex, or mel-flufen plus Dex; and then assessed for viability using MTT assays. Isobologram analysis shows the synergistic cytotoxic effect of mel-flufen and Dex. The graph (right panel) is derived from the values given in the Table (left panel).
Collectively, our preclinical studies therefore demonstrate potent in vitro and in vivo anti-MM activity of mel-flufen at doses that are well tolerated in human MM xenograft mouse models. These findings provide the framework for clinical trials of mel-flufen both as a single agent and together with lenalidomide, bortezomib, or Dex to increase response, overcome drug resistance, reduce side effects, and improve patient outcome in MM.
Supplementary Material
Translational Relevance.
The alkylating agent melphalan is actively utilized in MM therapy; however, dose-limiting toxicities and development of resistance limits its use. Recent studies have focused on developing a prodrug to enhance the therapeutic potential of melphalan. Melphalan-flufenamide (Mel-flufen) is an enzyme-activated prodrug of melphalan which allows for a more rapid and higher intracellular accumulation of melphalan in tumor cells than is achievable by direct exposure to equimolar doses of melphalan. Mel-flufen is undergoing evaluation in phase-I/IIa clinical trials in solid tumors. Here, we utilized both in vitro and in vivo MM xenograft models to show that mel-flufen is a more potent anti-MM agent than melphalan which can overcome conventional drug resistance. Moreover, the combination of mel-flufen with bortezomib, lenalidomide, or dexamethasone induces synergistic anti-MM activity. Our preclinical data therefore provide the framework for clinical evaluation of mel-flufen, either alone or in combination, to improve patient outcome in MM.
Acknowledgments
Grant Support: This investigation was supported by NIH grants P50100707, and CA078378 (DC, and KCA); Swedish Cancer Society, Swedish Research Council and Stockholm Cancer Society (KV, and RL). KCA is an American Cancer Society Clinical Research Professor.
Footnotes
Authors’ contributions: DC designed research, summarized and analyzed data, interpreted results, and wrote the manuscript; AR performed the experiments and interpreted data; KV, JS, and RL designed research, interpreted data, and reviewed the manuscript; CPP and PR provided clinical samples; and KCA analyzed data and wrote the manuscript.
Conflicts of Interest disclosure: KV, JS, and RL were employees and shareholders of Oncopeptde, AB. DC is consultant to Oncopeptide, AB. Other co-authors have no competing financial interests.
References
- 1.Anderson KC. Oncogenomics to target myeloma in the bone marrow microenvironment. Clin Cancer Res. 17:1225–33. doi: 10.1158/1078-0432.CCR-10-3366. [DOI] [PubMed] [Google Scholar]
- 2.Dimopoulos MA, San-Miguel JF, Anderson KC. Emerging therapies for the treatment of relapsed or refractory multiple myeloma. Eur J Haematol. 86:1–15. doi: 10.1111/j.1600-0609.2010.01542.x. [DOI] [PubMed] [Google Scholar]
- 3.Attal M, Harousseau JL, Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335:91–7. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 4.Barlogie B, Kyle RA, Anderson KC, Greipp PR, Lazarus HM, Hurd DD, et al. Standard chemotherapy compared with high-dose chemoradiotherapy for multiple myeloma: final results of phase III US Intergroup Trial S9321. J Clin Oncol. 2006;24:929–36. doi: 10.1200/JCO.2005.04.5807. [DOI] [PubMed] [Google Scholar]
- 5.Child JA, Morgan GJ, Davies FE, Owen RG, Bell SE, Hawkins K, et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med. 2003;348:1875–83. doi: 10.1056/NEJMoa022340. [DOI] [PubMed] [Google Scholar]
- 6.Falco P, Cavallo F, Larocca A, Rossi D, Guglielmelli T, Rocci A, et al. Lenalidomide-prednisone induction followed by lenalidomide-melphalan-prednisone consolidation and lenalidomide-prednisone maintenance in newly diagnosed elderly unfit myeloma patients. Leukemia. 2012 doi: 10.1038/leu.2012.271. [DOI] [PubMed] [Google Scholar]
- 7.Berenson JR, Yang HH, Sadler K, Jarutirasarn SG, Vescio RA, Mapes R, et al. Phase I/II trial assessing bortezomib and melphalan combination therapy for the treatment of patients with relapsed or refractory multiple myeloma. J Clin Oncol. 2006;24:937–44. doi: 10.1200/JCO.2005.03.2383. [DOI] [PubMed] [Google Scholar]
- 8.Palumbo A, Ambrosini MT, Benevolo G, Pregno P, Pescosta N, Callea V, et al. Bortezomib, melphalan, prednisone, and thalidomide for relapsed multiple myeloma. Blood. 2007;109:2767–72. doi: 10.1182/blood-2006-08-042275. [DOI] [PubMed] [Google Scholar]
- 9.Palumbo A, Waage A, Hulin C, Beksac M, Zweegman S, Gay F, et al. Safety of thalidomide in newly diagnosed elderly myeloma patients: an individual patient data meta-analysis of six randomized trials. Haematologica. 2012 doi: 10.3324/haematol.2012.067058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mateos MV, Richardson PG, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, et al. Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol. 2010;28:2259–66. doi: 10.1200/JCO.2009.26.0638. [DOI] [PubMed] [Google Scholar]
- 11.Mateos MV, Oriol A, Martinez-Lopez J, Gutierrez N, Teruel AI, de Paz R, et al. Bortezomib, melphalan, and prednisone versus bortezomib, thalidomide, and prednisone as induction therapy followed by maintenance treatment with bortezomib and thalidomide versus bortezomib and prednisone in elderly patients with untreated multiple myeloma: a randomised trial. Lancet Oncol. 2010;11:934–41. doi: 10.1016/S1470-2045(10)70187-X. [DOI] [PubMed] [Google Scholar]
- 12.Facon T, Mary JY, Hulin C, Benboubker L, Attal M, et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trial. Lancet. 2007;370:1209–18. doi: 10.1016/S0140-6736(07)61537-2. [DOI] [PubMed] [Google Scholar]
- 13.Rajkumar SV. Optimising bortezomib in newly diagnosed multiple myeloma. Lancet Oncol. 2010;11:909–10. doi: 10.1016/S1470-2045(10)70199-6. [DOI] [PubMed] [Google Scholar]
- 14.Ludwig H, Durie BG, McCarthy P, Palumbo A, San Miguel J, Barlogie B, et al. IMWG consensus on maintenance therapy in multiple myeloma. Blood. 2012;119:3003–15. doi: 10.1182/blood-2011-11-374249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gullbo J, Wallinder C, Tullberg M, Lovborg H, Ehrsson H, Lewensohn R, et al. Antitumor activity of the novel melphalan containing tripeptide J3 (L-prolyl-L-melphalanyl-p-L-fluorophenylalanine ethyl ester): comparison with its m-L-sarcolysin analogue P2. Mol Cancer Ther. 2003;2:1331–9. [PubMed] [Google Scholar]
- 16.Gullbo J, Lindhagen E, Bashir-Hassan S, Tullberg M, Ehrsson H, Lewensohn R, et al. Antitumor efficacy and acute toxicity of the novel dipeptide melphalanyl-p-L-fluorophenylalanine ethyl ester (J1) in vivo. Invest New Drugs. 2004;22:411–20. doi: 10.1023/B:DRUG.0000036683.10945.bb. [DOI] [PubMed] [Google Scholar]
- 17.Wickstrom M, Johnsen JI, Ponthan F, Segerstrom L, Sveinbjornsson B, Lindskog M, et al. The novel melphalan prodrug J1 inhibits neuroblastoma growth in vitro and in vivo. Mol Cancer Ther. 2007;6:2409–17. doi: 10.1158/1535-7163.MCT-07-0156. [DOI] [PubMed] [Google Scholar]
- 18.Wickstrom M, Larsson R, Nygren P, Gullbo J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2011;102:501–8. doi: 10.1111/j.1349-7006.2010.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wickstrom M, Viktorsson K, Lundholm L, Aesoy R, Nygren H, Sooman L, et al. The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan. Biochem Pharmacol. 2010;79:1281–90. doi: 10.1016/j.bcp.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 20.Hideshima T, Chauhan D, Shima Y, Raje N, Davies FE, Tai YT, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96:2943–50. [PubMed] [Google Scholar]
- 21.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, et al. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 1992;357:420–2. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8:407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 23.LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 24.Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–58. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2010;115:834–45. doi: 10.1182/blood-2009-03-213009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 27.Gullbo J, Dhar S, Luthman K, Ehrsson H, Lewensohn R, Nygren P, et al. Antitumor activity of the alkylating oligopeptides J1 (L-melphalanyl-p-L-fluorophenylalanine ethyl ester) and P2 (L-prolyl-m-L-sarcolysyl-p-L-fluorophenylalanine ethyl ester): comparison with melphalan. Anticancer Drugs. 2003;14:617–24. doi: 10.1097/00001813-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Gullbo J, Tullberg M, Vabeno J, Ehrsson H, Lewensohn R, Nygren P, et al. Structure-activity relationship for alkylating dipeptide nitrogen mustard derivatives. Oncol Res. 2003;14:113–32. doi: 10.3727/000000003771013071. [DOI] [PubMed] [Google Scholar]
- 29.Gullbo J, Wickstrom M, Tullberg M, Ehrsson H, Lewensohn R, Nygren P, et al. Activity of hydrolytic enzymes in tumour cells is a determinant for anti-tumour efficacy of the melphalan containing prodrug J1. J Drug Target. 2003;11:355–63. doi: 10.1080/10611860310001647140. [DOI] [PubMed] [Google Scholar]
- 30.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005;23:6333–8. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 31.Davies FE, Dring AM, Li C, Rawstron AC, Shammas MA, O’Connor SM, et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood. 2003;102:4504–11. doi: 10.1182/blood-2003-01-0016. [DOI] [PubMed] [Google Scholar]
- 32.Greenstein S, Krett NL, Kurosawa Y, Ma C, Chauhan D, Hideshima T, et al. Characterization of the MM.1 human multiple myeloma (MM) cell lines. A model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp Hematol. 2003;31:271–82. doi: 10.1016/s0301-472x(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 33.Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109:3489–95. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 34.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–67. [PMC free article] [PubMed] [Google Scholar]
- 35.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–12. [PubMed] [Google Scholar]
- 36.Hazlehurst LA, Enkemann SA, Beam CA, Argilagos RF, Painter J, Shain KH, et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res. 2003;63:7900–6. [PubMed] [Google Scholar]
- 37.Jones RB. Clinical pharmacology of melphalan and its implications for clinical resistance to anticancer agents. Cancer Treat Res. 2002;112:305–22. doi: 10.1007/978-1-4615-1173-1_15. [DOI] [PubMed] [Google Scholar]
- 38.McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2:483–90. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- 39.Spanswick VJ, Craddock C, Sekhar M, Mahendra P, Shankaranarayana P, Hughes RG, et al. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood. 2002;100:224–9. doi: 10.1182/blood.v100.1.224. [DOI] [PubMed] [Google Scholar]
- 40.Kuhn D, Bjorklund C, Magarotto V, Mathews J, Wang M, Baladandayuthapani V, et al. Bortezomib resistance is mediated by increased signaling through the insulin-like growth factor-1/Akt axis. ASH Annual Meeting Abstracts; 2009. p. 2739. [Google Scholar]
- 41.Lee CK, Wang S, Huang X, Ryder J, Liu B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 296:233–40. doi: 10.1016/j.canlet.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–70. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 44.Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, et al. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:3064–73. [PubMed] [Google Scholar]
- 45.Podar K, Tai YT, Lin BK, Narsimhan RP, Sattler M, Kijima T, et al. Vascular Endothelial Growth Factor-induced Migration of Multiple Myeloma Cells Is Associated with beta 1 Integrin- and Phosphatidylinositol 3-Kinase-dependent PKCalpha Activation. J Biol Chem. 2002;277:7875–81. doi: 10.1074/jbc.M109068200. [DOI] [PubMed] [Google Scholar]
- 46.Laitinen S, Wickstrom M, Fuchs R, Gerwins P, Larsson R, Gullbo J. Aminopeptidase N-activated prodrug melphalan-flufenamide inhibits angigenesis in vitro and in vivo. Mol Can Ther; Abstract AACR-NCI-EORTC international Conference: Molecular Tragets and Cancer Therapeutics; Nov, 2011. [Google Scholar]
- 47.Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Fonseca R, et al. Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003;17:2025–31. doi: 10.1038/sj.leu.2403084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.