

Abstract
Objective
Facioscapulohumeral muscular dystrophy (FSHD) is associated with D4Z4 repeat contraction on human chromosome 4q35. This genetic lesion does not result in complete loss or mutation of any gene. Consequently, the pathogenic mechanisms underlying FSHD have been difficult to discern. In leading FSHD pathogenesis models, D4Z4 contractions are proposed to cause epigenetic changes, which ultimately increase expression of genes with myopathic potential. Although no gene has been conclusively linked to FSHD development, recent evidence supports a role for the D4Z4-encoded DUX4 gene in FSHD. In this study, our objective was to test the in vivo myopathic potential of DUX4.
Methods
We delivered DUX4 to zebrafish and mouse muscle by transposon-mediated transgenesis and adeno-associated viral vectors, respectively.
Results
Overexpression of DUX4, which encodes a transcription factor, caused abnormalities associated with muscular dystrophy in zebrafish and mice. This toxicity required DNA binding, because a DUX4 DNA binding domain mutant produced no abnormalities. Importantly, we found the myopathic effects of DUX4 were p53 dependent, as p53 inhibition mitigated DUX4 toxicity in vitro, and muscles from p53 null mice were resistant to DUX4-induced damage.
Interpretation
Our work demonstrates the myopathic potential of DUX4 in animal muscle. Considering previous studies showed DUX4 was elevated in FSHD patient muscles, our data support the hypothesis that DUX4 overexpression contributes to FSHD development. Moreover, we provide a p53-dependent mechanism for DUX4 toxicity that is consistent with previous studies showing p53 pathway activation in FSHD muscles. Our work justifies further investigation of DUX4 and the p53 pathway in FSHD pathogenesis.
Facioscapulohumeral muscular dystrophy (FSHD) is a complex autosomal dominant disorder characterized by progressive and asymmetric weakness of facial, shoulder, and limb muscles.1 Symptoms typically arise in adulthood with most patients showing clinical features before age 30 years. About 5% develop symptoms as infants or juveniles and are generally more severely affected.2,3 Clinical presentation can vary from mild (some limited muscle weakness) to severe (wheelchair dependence). Historically, FSHD was classified as the third most common muscular dystrophy, affecting 1 in 20,000 individuals worldwide.1 However, recent data indicate that FSHD is the most prevalent muscular dystrophy in Europe, suggesting that its worldwide incidence may be underestimated.2
Typical FSHD cases (FSHD1A; heretofore referred to as FSHD) are linked to heterozygous chromosomal deletions that decrease the copy number of 3.3 kilobase (kb) D4Z4 repeats on human chromosome 4q35.4,5 Simplistically, normal individuals have 11 to 100 tandem repeated D4Z4 copies on both 4q35 alleles, whereas patients with FSHD have 1 normal and 1 contracted allele containing 1 to 10 repeats.4 In addition, FSHD-associated D4Z4 contractions must occur on specific disease-permissive chromosome 4q35 backgrounds.6–9 Importantly, no genes are completely lost or structurally mutated as a result of FSHD-associated deletions. Thus, although the disease was formally classified in 1954,1 and the primary genetic defect identified in 1992,5 the pathogenic mechanisms underlying FSHD remain unresolved.
In leading FSHD pathogenesis models, D4Z4 contractions are proposed to cause epigenetic changes that permit expression of genes with myopathic potential.10 As a result, aberrant overexpression of otherwise silent or near-silent genes may ultimately cause muscular dystrophy. This model is consistent with data showing that normal 4q35 D4Z4 repeats have heterochromatin characteristics, whereas FSHD-linked D4Z4 repeats contain marks more indicative of actively transcribed euchromatin.5,11–16 These transcription-permissive epigenetic changes, coupled with the observation that complete monosomic D4Z4 deletions (ie, zero repeats) do not cause FSHD,17 support the hypothesis that D4Z4 repeats harbor potentially myopathic open reading frames (ORFs), which are abnormally expressed in FSHD muscles. This notion was initially considered in 1994, when a D4Z4-localized ORF, called DUX4, was first identified.12,14 However, the locus had some characteristics of an unexpressed pseudogene, and DUX4 was therefore summarily dismissed as an FSHD candidate. For many years thereafter, the search for FSHD-related genes was mainly focused outside the D4Z4 repeats, and although some intriguing candidates emerged from these studies, no single gene has been conclusively linked to FSHD development.18–30 This slow progress led to the re-emergence of DUX4 as an FSHD candidate in 2007, and several recent findings support its potential role in FSHD pathogenesis.20,24,25,29,31,32
First, D4Z4 repeats are not pseudogenes. The DUX4 locus produces 1.7kb and 2.0kb full-length mRNAs with identical coding regions, and D4Z4 repeats also harbor smaller sense and antisense transcripts, including some resembling microRNAs.24,25,29 Importantly, overexpressed DUX4 transcripts and a ~50kDa full-length DUX4 protein were found in biopsies and cell lines from FSHD patients.19,20,24,25,29,33 These data are consistent with the transcriptional de-repression model of FSHD pathogenesis. In addition, unlike pseudogenes, D4Z4 repeats and DUX4 likely have functional importance, because tandem arrayed D4Z4 repeats are conserved in at least 11 different placental mammalian species (nonplacental animals lack D4Z4 repeats), with the greatest sequence conservation occurring within the DUX4 ORF.19 Second, overexpressed DUX4 is toxic to tissue culture cells and embryonic progenitors of developing lower organisms in vivo.25,29,31,32 This toxicity occurs at least partly through a proapoptotic mechanism, indicated by caspase-3 activation in DUX4 transfected cells, and presence of terminal deoxynucleotide transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL)-positive nuclei in developmentally arrested Xenopus embryos injected with DUX4 mRNA at the 2-cell stage.25,31,32 These findings are consistent with studies showing that some proapoptotic proteins, including caspase-3, are present in FSHD patient muscles.26,34 In addition to stimulating apoptosis, DUX4 may negatively regulate myogenesis. Human DUX4 inhibited differentiation of mouse C2C12 myoblasts in vitro, potentially by interfering with PAX3 and/or PAX7, and caused developmental arrest and reduced staining of some muscle markers when delivered to progenitor cells of zebrafish or Xenopus embryos.25,29,31,32 Finally, aberrant DUX4 function was directly associated with potentially important molecular changes seen in FSHD patient muscles. Specifically, full-length human DUX4 encodes a ~50kDa double homeodomain transcription factor, and its only known target, PITX1, was elevated in DUX4-overexpressing FSHD patient muscles.20,24,35 These data support the hypothesis that DUX4 catalyzes numerous downstream molecular changes, which are incompatible with maintaining normal muscle integrity.
In summary, recent studies implicated DUX4 as a leading FSHD candidate gene that is overexpressed in FSHD tissue, and generally toxic to tissue culture cells and embryonic progenitors of nonmammalian organisms, possibly through activation of downstream genes involved in apoptosis. However, the in vivo myopathic potential of DUX4 in adult placental mammalian muscle, which most closely resembles the human FSHD condition, has not been tested. Here, we demonstrate the in vivo myopathic potential of DUX4, using zebrafish and mice. We present evidence that DUX4 overexpression causes histological and functional features consistent with muscular dystrophy. Importantly, we show that DUX4-mediated toxicity requires DUX4 DNA binding and activation of p53-dependent apoptosis. Our comprehensive in vivo data are consistent with the hypothesis that DUX4 over-expression contributes to FSHD.
Subjects and Methods
DUX4 Epitope Tagging and Adeno-Associated Virus Production
Details are described in Supplementary Methods.
Western Blot
Western blots were performed using standard procedures, detailed in Supplementary Methods.
Cell Death Assay
Caspase 3/7 activity was measured using the Apo-ONE Homogeneous caspase-3/7 Assay (Promega, Madison, WI). HEK293 cells (60,000 cells/well) were transfected as described in the text and plated simultaneously on 96-well plates. Where indicated, Bax channel blocker, caspase-1 inhibitor VI, or the p53-inhibitor pifithrin-α, (all from Calbiochem, San Diego, CA) were immediately added to media at 5μM, 160μM, and 100μM concentrations, respectively, and media were changed 4 hours later. Caspase 3/7 activity was measured 48 hours post-transfection using a fluorescent plate reader (Spectra max M2, Molecular Devices, Sunnyvale, CA). Individual assays were performed in triplicate (n = 6 and n = 3 for noninhibitor and inhibitor studies, respectively), and data were reported as mean caspase activity relative to the pCINeo control.
Zebrafish Transgenesis and Histology
Tol2 fish expression plasmids and transposase RNA were injected into 1-cell stage zebrafish embryos as previously described.36 Clonal lines were not generated, and all phenotypes were quantified from individually injected animals. Body morphology phenotypes were assessed by microscopy, and embryos were fixed in 4% paraformaldehyde/phosphate-buffered saline overnight at 4°C and paraffin-embedded. Five-micrometer sections were deparaffinized and rehydrated before hematoxylin and eosin (H&E) staining. For body morphology counts, n = 53 hrGFP- and 49 DUX4-injected embryos. For tissue sectioning, n = 5 representative animals per group.
Adeno-Associated Virus Injections
Six- to 8-week-old C57BL/6 females and 8- to 12-month-old Trp53−/− males and females received 8 × 108 or 3 × 1010 DRP units of adeno-associated virus (AAV) 6 bilaterally via direct 30 microliter intramuscular (IM) injection into the tibialis anterior (TA). In vivo transduction was determined in AAV.CMV.hrGFP injected using a fluorescent dissecting microscope (MZ16FA, Leica, Wetzlar, Germany) at ×4.63 magnification.
Grip Strength Testing
Muscle grip strength was assessed weekly as indicated (Columbus Instruments, Columbus, OH). Three separate trials were recorded per limb group, and force measurements were averaged (n = 5 animals per group). Data are reported as mean hindlimb:forelimb ratios ± standard error of the mean (SEM).
Histological Analysis
TA muscles were dissected from IM injected mice at indicated times postinjection for histological analysis (n = 6 muscles per group each time point at 8 × 108 DNase resistant particles (DRP) units). Ten-micrometer cryosections were generated and H&E stained as previously described.37 Fiber diameter and central nuclei quantifications (±SEM) were determined from muscles injected 2 and 4 weeks prior (n = 5 muscles per group; 5 representative ×20 photomicrographs per section), using AxioVision 4.7 software (Zeiss, Thornwood, NY). Fiber size distribution histograms represent percentage of total fibers analyzed.
Immunohistochemistry
Immunofluorescence staining of cryosections was performed using standard protocols, detailed in Supplementary Methods. DUX4 or caspase-3 were detected by indirect immunofluorescence using rabbit polyclonal anti-V5 (1:2,500; Chemicon, Temecula, CA) or caspase-3 (1:1,000; Abnova, Taipei, Taiwan) primary antibodies and Alexa-594 or -488 coupled goat antirabbit immunoglobulin G (IgG) secondary antibodies (1:1,000; Molecular Probes, Eugene, OR). Alternatively, V5 antibody (Chemicon) or caspase-3 antibodies (Cell Signaling Technology, Beverly, MA) were directly labeled using the Zenon Rabbit IgG Labeling Kit (Molecular Probes) following manufacturer’s instructions. Apoptotic nuclei were detected by TUNEL assay (In Situ Cell Death Detection Kit, Fluorescein; Roche, Indianapolis, IN) following manufacturer’s instructions. All slides were mounted in Vectashield (Vector Laboratories, Burlingame, CA) plus DAPI.
Real-Time Polymerase Chain Reaction Array
RT2Profiler Mouse Apoptosis PCR Arrays (SABiosciences, Frederick, MD) were performed and analyzed following manufacturer’s instructions. Details are provided in Supplementary Methods.
Statistical Analysis
All statistical analyses were performed in GraphPad Prism 5 (GraphPad Software, La Jolla, CA) using indicated statistical tests.
Results
Epitope-Tagged DUX4 Causes Apoptosis In Vitro
We hypothesized that DUX4 overexpression is an underlying pathogenic insult in FSHD. In this study, our ultimate goal was to examine the in vivo effects of DUX4 overexpression in adult mouse muscle. As an initial step, we developed DUX4 expression vectors, and validated their protein expression and cytotoxic potential in vitro. To simplify DUX4 protein detection, we first added a C-terminal V5-epitope tag to normal human DUX4 cDNA, and then confirmed that the V5 tag did not impact DUX4 expression and proapoptotic function in tissue culture (Supplementary Fig 1). All references to DUX4 in experiments hereafter refer to DUX4 with a C-terminal V5 tag.
DUX4 Is Detrimental to Developing Zebrafish Muscle
We next evaluated the in vivo effects of DUX4 overexpression in animal muscle. We initially screened for DUX4 myotoxicity in zebrafish, because they are rapidly produced and were previously used to investigate the pathogenesis of other human muscular dystrophies.38
We used the Tol2 transposon system and the MHCK7 promoter to generate transgenic zebrafish with striated muscle-specific hrGFP or DUX4 expression (Fig 1).36 The MHCK7 promoter turned on 3 days postinjection, and consistent with previous reports in mice, was active in zebrafish skeletal muscle and heart.39 By 4 days, 47% of MHCK7.DUX4-injected embryos had gross body malformations, and 81% had strikingly abnormal muscle histology, whereas most MHCK7.hrGFP-injected fish were normal (81% histology; 93% body shape). The abnormalities in MHCK7.DUX4-injected fish were consistent with those reported in other zebrafish models of muscular dystrophy.38 In addition, misexpression of DUX4 in the heart caused cardiac hypertrophy in some fish, whereas hrGFP did not. Although the expression patterns of DUX4 are unknown, cardiac defects are not normally seen in patients with FSHD, and the heart abnormalities in fish were likely artifacts of MHCK7-directed cardiac DUX4 expression. Nevertheless, these data demonstrated that striated muscles in general are susceptible to DUX4-induced myotoxicity. Importantly, in contrast to previous studies showing that ubiquitous DUX4 expression in developing lower eukaryotes caused embryonic arrest, our data demonstrate that embryos expressing DUX4 specifically in muscle are viable.
FIGURE 1.
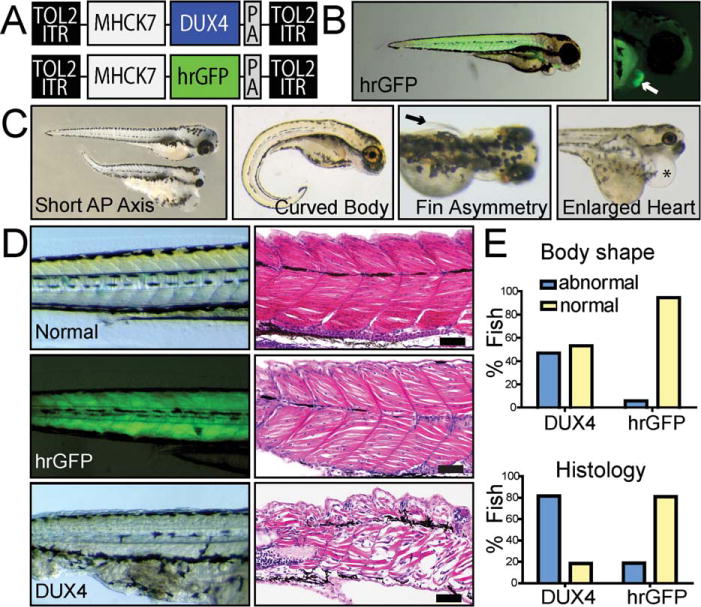
DUX4 overexpression is detrimental to developing zebrafish muscle. (A) Tol2 zebrafish expression constructs contained striated muscle-specific MHCK7 promoter-driven DUX4 or hrGFP. ITR = inverted terminal repeat from Tol2 transposon; PA = SV40 polyA signal. (B) hrGFP epifluorescence showed MHCK7 activity in zebrafish muscle, which turned on 3 days postinjection. This lag in MHCK7 promoter expression allowed culling of abnormal embryos arising from nonspecific plasmid toxicity within the first 2 days postinjection. (C) MHCK7.DUX4 caused body malformation defects including short anterior-posterior (AP) axes, curved bodies, asymmetrically undeveloped pectoral fins (arrow indicates fin), or combinations of these morphologies. Some fish also showed cardiac hypertrophy (asterisk indicates heart) due to MHCK7-mediated DUX4 expression in the myocardium. (D) Hematoxylin and eosin staining of zebrafish body muscle shows MHCK7.DUX4 zebrafish had undefined somite boundaries, absent sarcomeric banding, and myofiber disorganization/degeneration. In contrast, MHCK7.hrGFP had no significant impact on gross body formation or somite/myofiber organization compared to normal zebrafish embryos. The only abnormal phenotypes seen in hrGFP fish were short AP axes, whereas undeveloped pectoral fins and abnormal body shapes were never present. Scale bars = 50mm. (E) Quantification of abnormal muscle phenotypes in zebrafish pictured in C–D. All DUX4-injected fish with abnormal body morphology also showed histological defects.
DUX4 Toxicity Requires DNA Binding In Vitro
We hypothesized that DUX4-mediated apoptosis in vitro (see Supplementary Fig 1) and myotoxicity in developing vertebrate muscle (see Fig 1) were directly related to its ability, as a transcription factor, to bind promoter DNA and stimulate transcription of downstream genes. However, because in vivo overexpression of otherwise inert proteins can sometimes be toxic to striated muscle,40 we also considered the possibility that DUX4-induced abnormalities were artifacts of nonspecific overexpression unrelated to its transcriptional activity. To rule out the latter, we generated a mutant DUX4 construct containing alanine substitution mutations in the first DUX4 DNA binding domain, to produce a structurally intact but functionally deficient protein (DUX4.-HOX1; Fig 2A). Transfection of a CMV.DUX4.HOX1 expression plasmid into HEK293 cells produced a DUX4.HOX1 protein migrating at the same apparent molecular weight (~50kDa) as normal DUX4, but with consistently higher expression levels (see Fig 2B). Importantly, unlike DUX4, the DUX4.HOX1 mutant did not cause apoptosis in vitro and showed reduced binding to a DUX4-binding site in the PITX1 gene (see Fig 2C, D). These results suggested that DUX4 DNA binding was required to elicit proapoptotic effects in vitro, and that DUX4-mediated cytotoxicity required a specific transcriptional function of DUX4. We used the DUX4.HOX1 mutant as a control in subsequent in vivo experiments.
FIGURE 2.
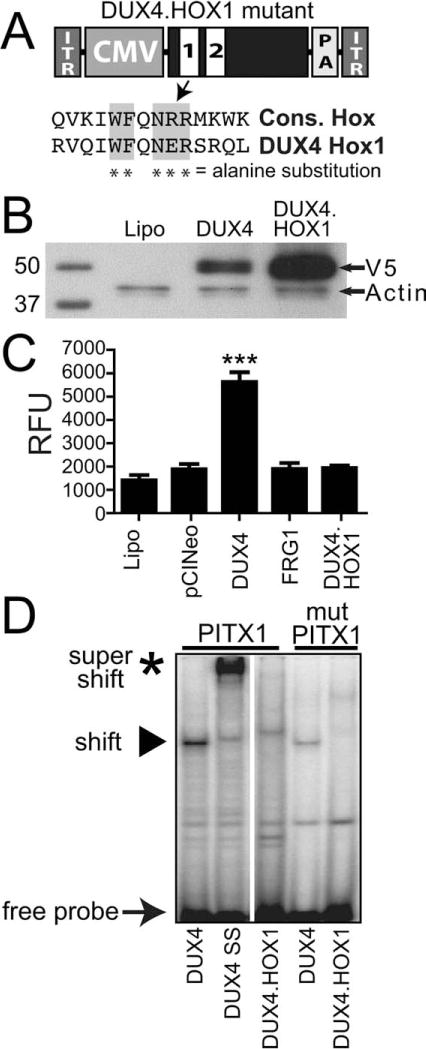
DUX4 toxicity requires DNA binding. (A) Structure of DUX4 adeno-associated virus (AAV) expression construct. White boxes indicate homeodomains (labeled 1 and 2). ITR = AAV inverted terminal repeats; CMV = cytomegalovirus promoter; PA = SV40 polyA signal. Alignment with a consensus homeodomain (Cons. Hox) identified 5 important residues required for DNA binding. *Indicates residues mutated to alanines in DUX4.HOX1 DNA binding mutant. (B) Western blot using extracts from transfected HEK293 cells showed DUX4.HOX1 protein was expressed at expected molecular weight (~50kDa) and consistently produced at higher levels than normal DUX4 in vitro. Lipo = HEK293 cells transfected with Lipofectamine-2000™ (Invitrogen, Carlsbad, CA) but no DNA. (C) Unlike DUX4, the DUX4.HOX1 mutant did not cause apoptosis in vitro, as indicated by lack of caspase-3/7 activation following transfection into HEK293 cells. ***Indicates significant differences from Lipofectamine controls, p < 0.0001 (analysis of variance; n = 3 independent experiments performed in triplicate). RFU = relative fluorescent units from caspase-3/7assay. (D) Electrophoretic mobility shift assay. Lanes 1 and 2 (DUX4 and DUX4 SS, respectively) show a shifted and super-shifted oligonucleotide (oligo) corresponding to the DUX4 binding site in the PITX1 promoter.20 Lane 3, DUX4.HOX1 has lower affinity for PITX1 promoter oligo. Mutation of the PITX1 binding site (mutPITX1) further reduces binding by DUX4, as previously reported, whereas no binding occurs between DUX4.HOX1 and the mutPITX1 site. Arrow indicates free PITX1 promoter probe; arrowhead indicates DUX4-bound PITX1 promoter sequence; asterisk indicates DUX4-PITX1 promoter complex supershifted with V5 antibody. SS = supershift.
DUX4 Causes Muscle Degeneration in Adult Mouse Muscle
Because FSHD is typically an adult-onset disorder of skeletal muscle, we next investigated the myotoxic potential of DUX4 overexpression in adult, postmitotic mouse muscle. To do this, we bilaterally injected 3 × 1010 DRP AAV6.DUX4, AAV6.DUX4.HOX1, or AAV6.hrGFP vectors into the TA muscles of 6- to 8-week old C57BL/6 mice. One week postinjection, we confirmed widespread TA muscle transduction by hrGFP epifluorescence and assessed histopathological changes in muscles injected with each group of vectors (Fig 3). We found that DUX4-injected muscles developed strikingly massive lesions containing degenerating myofibers and infiltrating mononuclear cells, indicating severe damage. In contrast, muscles injected with hrGFP or DUX4.HOX1 controls showed no evidence of damage at identical vector doses. Moreover, DUX4-injected animals were weaker than controls at 1 and 2 weeks postinjection, but recovered strength by 3 weeks, which was likely due to loss of non-integrating AAV vectors in degenerating myofibers and subsequent normal muscle regeneration. Although these initial experiments demonstrated the myopathic potential of DUX4, the massive lesions we observed at the high 3 × 1010 DRP dose were inconsistent with more subtle and focal damage typically associated with FSHD. We hypothesized that our high-dose vector was accelerating the development of pathology, which was reported in other vector-based models of disease.41,42 We therefore delivered lower DUX4 vector doses (8 × 108 DRP), which caused milder and less widespread muscle degeneration consistent with what is observed in FSHD patients. In contrast, DUX4.HOX1- or hrGFP-transduced muscles showed no histological abnormalities even by 4 weeks postinjection, our latest time point. We found that DUX4-induced degeneration was accompanied by significant muscle regeneration, which is a feature typical of muscular dystrophy. Specifically, 2 and 4 weeks postinjection, myofibers injected with low-dose vectors had smaller mean diameters, broad size variability, and dramatically increased numbers of centrally located nuclei compared to saline-, DUX4.HOX1-, or hrGFP-injected controls, which remained normal. We confirmed DUX4 and DUX4.HOX1 transgene expression by real-time PCR and immunofluorescence staining of serial muscle cryosections using rabbit polyclonal primary V5 antibodies (Supplementary Figs 2 and 3; Fig 4). DUX4 and DUX4.HOX1 mRNAs and protein were expressed at similar levels (see Supplementary Fig 2), and as expected for a transcription factor,35 DUX4 was primarily localized to myonuclei (see Supplementary Fig 3), but we also found cytoplasmic DUX4 staining in degenerating myofibers (see Supplementary Fig 3). This cytoplasmic staining was specific, because we did not see complete overlap of stained myofibers using a second rabbit polyclonal antibody. In contrast, the DUX4.HOX1 protein was restricted to myonuclei, and we never found myofibers expressing cytoplasm-localized DUX4.HOX1.
FIGURE 3.
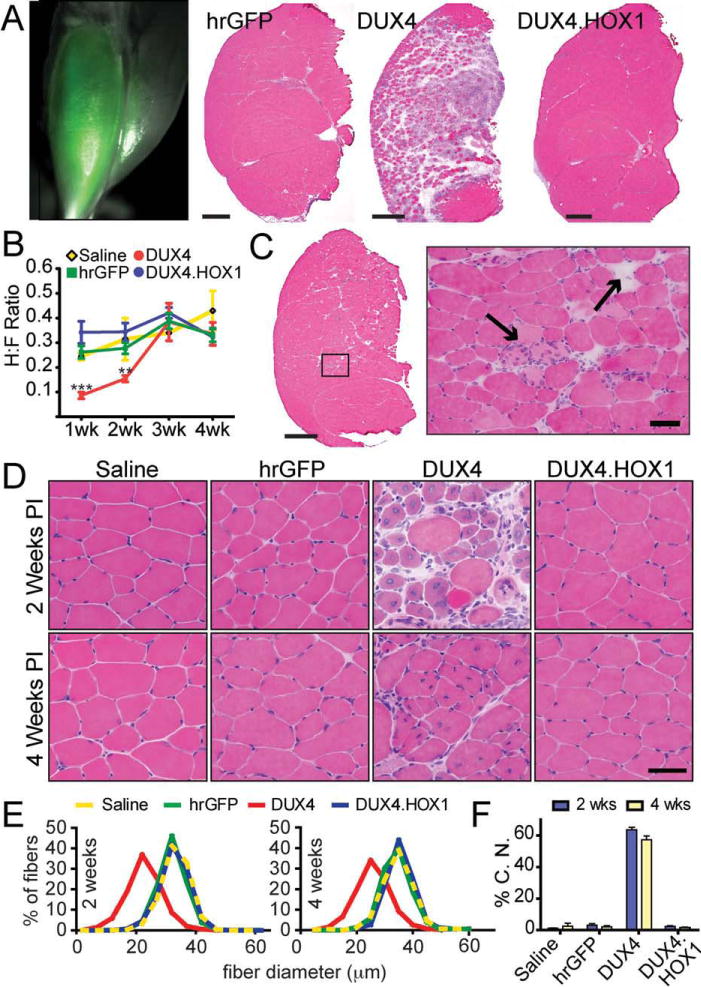
DUX4 is toxic to adult mouse muscle in vivo. (A) hrGFP epifluorescence shows AAV6 transduction of adult mouse tibialis anterior (TA) 1 week postinjection. Hematoxylin and eosin (H&E) staining shows DUX4 caused massive myofiber degeneration and mononuclear cell infiltration that was not present in DUX4.HOX1 or hrGFP controls at high vector dose (3 × 1010 DRP). Scale bars = 500mm. (B) DUX4, but not DUX4.HOX1 or controls, significantly reduced TA muscle grip strength 1 and 2 weeks postinjection. n = 5 mice per group. ***p < 0.001; **p < 0.01; 2-way analysis of variance with Bonferroni post hoc test. H:F ratio indicates hindlimb (transduced) to forelimb (untransduced) grip strength ratios. (C) At lower doses (8 × 108 DRP), DUX4 caused myofiber degeneration (by 1 week, shown here) that recapitulated the focal dystrophic lesions seen in facioscapulohumeral muscular dystrophy patients. Arrows point to degenerating myofibers, indicated by loss of acidophilic staining in H&E stains. Scale bars: left panel = 500mm; right panel = 50mm. (D) H&E staining revealed abundant centrally located nuclei and myofiber size variability only in DUX4-injected muscles. PI = indicates postinjection. Scale bar = 50mm. (E) Distribution of fiber diameter as a percentage of total fibers counted during sampling. DUX4 transduced muscles had more small-bore fibers compared to all controls, which is characteristic of regenerating dystrophic muscle. (F) DUX4-injected muscles had significantly higher percentages of centrally located nuclei (%C.N.) at both time points, which is another feature of dystrophic muscle, p < 0.001 (chi-square). All injections for panels C, D, and E delivered 8 × 108 DRP of AAV6 vectors.
FIGURE 4.
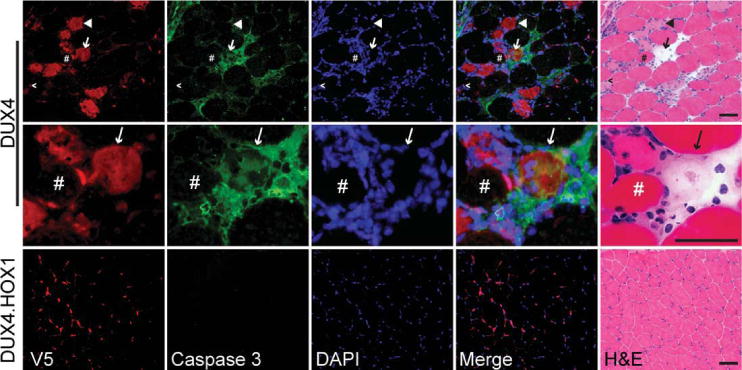
DUX4-transduced myofibers are caspase-3 positive. Top panels show DUX4+/caspase-3+ degenerating myofibers indicated by arrow, and shown in higher power in middle panels. Rabbit V5 antibody stain shows DUX4 was present in the nucleus but also had cytoplasmic localization in degenerating myofibers. In contrast, DUX4.HOX1 protein was exclusively nuclear. Some degenerating myofibers were caspase-3 negative but expressed DUX4 in the nucleus (caret) or cytoplasm (arrowhead). In contrast, several normal myofibers were DUX4+/caspase-3 negative (pound sign). Bottom panels, caspase-3 staining was absent in histologically normal muscle expressing DUX4.HOX1. The rabbit polyclonal caspase-3 primary antibody used here (Abnova; PAB0242) detects total caspase-3. Antibodies specifically recognizing cleaved caspase-3 showed similar staining patterns, as demonstrated in Supplementary Figure 3. DAPI (4′,6-diamidino-2-phenylindole) stains nuclear DNA. Scale bars = 50μm. H&E = hematoxylin and eosin.
DUX4 Causes Apoptosis through a p53-Dependent Mechanism
The presence of Bax and caspase-3 proteins in affected myofibers from FSHD patients supports that apoptosis may at least partly contribute to FSHD-associated muscle wasting.26,34 Because DUX4 induces apoptosis in vitro (see Supplementary Fig 1; Fig 2),25,31 we next investigated the possibility that DUX4 caused cell death through an apoptotic mechanism in adult placental mammal muscle in vivo, thereby potentially linking DUX4-mediated cell death mechanisms to known FSHD-associated pathology. As a general screen for apoptosis, we detected DNA fragmentation by TUNEL-staining muscle cryosections from AAV6.DUX4- or AAV6.DUX4.HOX1-injected mice. One week postinjection, only DUX4-expressing muscles contained TUNEL-positive nuclei (see Supplementary Fig 3), which is consistent with TUNEL-positive nuclei found in DUX4-injected early Xenopus embryos.32 We found several TUNEL-positive nuclei that were also DUX4-positive. Because intramuscularly delivered AAV6 preferentially transduces muscle cells but not inflammatory mononuclear cells,43,44 and we delivered our vectors to wild-type muscles lacking inflammatory infiltrates at the time of injection, we concluded that any DUX4+/TUNEL+ nuclei were present within myofibers. Nevertheless, it is possible that some TUNEL-positive nuclei were present within infiltrating immune cells, which normally undergo apoptotic cell death. Moreover, TUNEL stains may also indicate necrotic cell death. For these reasons, we more closely examined the status of apoptotic pathways in DUX4- and DUX4.HOX1-transduced muscle, first using real-time PCR arrays of 85 different genes involved in apoptosis (Supplementary Table). We found that 36 genes (42%) were significantly increased (>1.5-fold, p < 0.05) in DUX4-injected muscles, compared to DUX4. HOX1 controls, and 12 genes (33%) were members of the p53 pathway (Table). Because DUX4-induced degeneration was associated with mononuclear cell infiltration, and inflammatory cells eventually undergo apoptosis, it is possible that these proapoptotic genes were changed primarily in the immune infiltrates. To determine if apoptosis was occurring in DUX4-expressing myofibers, we stained serial cryosections from DUX4- or DUX4.HOX1-transduced muscles with antibodies to caspase-3, because it was the most highly upregulated p53 pathway gene in DUX4-expressing muscle, and was previously associated with human FSHD (see Table).26,34 Muscles expressing DUX4.HOX1 were histologically normal and lacked caspase-3 staining, whereas DUX4-transduced muscles contained numerous caspase-3 positive, degenerating myofibers (see Fig 4). Moreover, DUX4 was expressed in essentially every degenerating myofiber, and similar to our observations using TUNEL staining (see Supplementary Fig 3), we identified differentially stained cells that were likely in different apoptotic stages. Specifically, several myofibers expressing abundant cytoplasmic DUX4 and caspase-3 protein were nearly devoid of acidophilic eosin staining. Cells with this pattern were likely near the terminal stages of apoptosis, because caspase-3 activation is a late event during the apoptotic cascade and cytoplasmic localization of the normally nuclear-sequestered DUX4 protein suggests apoptotic nuclear breakdown. We also found degenerating myofibers that were caspase-3 negative but otherwise expressed DUX4 in the nucleus and/or cytoplasm. These cells may be in early apoptotic stages upstream of caspase-3 activation. Finally, we identified several histologically normal myofibers containing DUX4-positive myonuclei and no caspase-3 expression. The lack of complete overlap of DUX4 and caspase-3, using 2 different rabbit polyclonal antibodies, supports the specificity of our antibody stains. Together, our real-time PCR and immunostaining data suggested that DUX4-induced cell death occurs through a p53 pathway-dependent mechanism.
TABLE.
Significantly Changed p53 Pathway Genes in DUX4-Injected Muscles
| Gene Symbol | Fold Change | p |
|---|---|---|
| Casp3 | 7.87 | 0.013 |
| Birc5 | 6.98 | 0.0068 |
| Bax | 4.86 | 0.0082 |
| Casp1 | 4.85 | 0.034 |
| Apaf1 | 4.44 | 0.016 |
| Trp63 | 3.93 | 0.0035 |
| Bid | 3.78 | 0.0022 |
| Casp9 | 3.76 | 0.0097 |
| Bak1 | 3.40 | 0.030 |
| Trp53 | 3.06 | 0.029 |
| Bad | 2.68 | 0.036 |
| Casp7 | 2.57 | 0.0065 |
To test this hypothesis, we first determined whether p53 pathway inhibition could prevent or blunt DUX4-induced caspase-3/7 activation in vitro. We chose to chemically inhibit p53, caspase-1, and Bax because these genes are key components of signaling cascades that ultimately lead to caspase-3–associated apoptosis,45–49 and all were activated by DUX4 overexpression in mouse muscle (see Table). For this experiment, we separately pretreated HEK293 cells with chemical inhibitors to the aforementioned genes, transfected cells with DUX4 or DUX4.HOX1 expression plasmids, and measured caspase-3 activity 48 hours later. Consistent with our previous findings, DUX4 alone caused significant caspase-3/7 activation, whereas DUX4.HOX1 did not. Importantly, we found that p53, caspase-1, or Bax inhibition prevented or significantly reduced caspase-3/7 activation by DUX4 in vitro (Fig 5). To confirm these results in vivo, we injected low-dose (8 × 108 DRP) AAV6.DUX4 or AAV6.DUX4.HOX1 into TA muscles of C57BL/6 or p53 knockout mice (Trp 53 −/−; B6.129S2-Trp53tm1Tyj/J, Jackson Laboratories, Bar Harbor, ME),50 confirmed expression by real-time PCR and immunostaining (see Supplementary Fig 2), and examined muscle histopathology 2 weeks postinjection. As expected, DUX4 was toxic to wild-type muscle, indicated by widespread presence of regenerating myofibers containing centrally located nuclei (see Fig 3), whereas muscles from p53 knockout mice appeared normal and showed no indications of the massive muscle degeneration and regeneration typified by DUX4-transduced wild-type muscles. This is most obviously observed quantitatively by the comparatively small percentage of myofibers containing centrally located nuclei in DUX4-injected Trp53 −/− mice (4%) relative to wild-type muscles (see Fig 4; 63%) 2 weeks postinjection. Moreover, the Trp53 −/− central nuclei values do not differ significantly from control-injected wild-type muscles, and may arise from physical damage caused by the injection needle. These findings strongly support that DUX4 toxicity is p53 pathway dependent.
FIGURE 5.
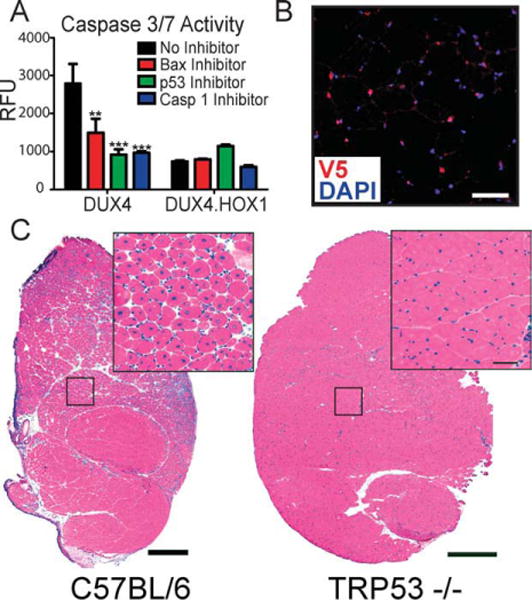
DUX4 causes apoptosis through a p53-dependent mechanism. (A) DUX4-induced apoptosis is significantly reduced by Bax, p53, or caspase-1 inhibition, in vitro. **p < 0.01; ***p < 0.001 (analysis of variance). RFU = relative fluorescent units from caspase-3/7 assay. (B) V5 immunofluorescence and DAPI staining showed DUX4 expression in Trp53 −/− mouse myonuclei 2 weeks after injection. Scale bar = 50mm. (C) Trp53 −/− muscles are resistant to DUX4-induced degeneration, indicated by normal muscle histology in DUX4-transduced muscles, 2 weeks postinjection. In contrast, low-dose AAV6.DUX4 (8 × 108 DRP) caused massive myofiber degeneration and subsequent regeneration 2 weeks postinjection. Scale bar = 500mm.
Discussion
According to the most widely accepted pathogenesis model, FSHD is caused by genetic and epigenetic abnormalities that create a favorable environment for overexpression of genes with myopathic potential. Since 1992, several important publications described FSHD-associated genetic and epigenetic changes that are consistent with this model,4,5,11,12,15,23,51,52 but the downstream transcriptional abnormalities contributing to FSHD pathogenesis are unclear. This uncertainty has not arisen from a lack of investigation; indeed, numerous groups identified potential FSHD gene candidates based on 4q35 localization or expression changes in gene profiling experiments.12,18,20,21,23,24,27–30,53–57 However, a legitimate FSHD candidate gene should minimally satisfy 3 main criteria, and to date no single gene has met each requirement. Specifically, at a minimum, an FSHD candidate gene should: (1) show consistent overexpression in muscles from FSHD patients; (2) have the capability to damage muscles when overexpressed in vivo, and (3) be activated specifically in preferentially affected FSHD muscles (eg, facial, shoulder-girdle, limb muscles) and/or nonmuscle areas of pathology (retina, inner ear).
DUX4 emerged as an intriguing FSHD candidate because of its position within the D4Z4 repeats, and, importantly, several recent studies showed it was overexpressed in affected muscles and cell lines from FSHD patients.20,25,29,33 Thus, these initial reports demonstrated that DUX4 satisfies the first criteria for an FSHD candidate gene, although additional DUX4 expression studies in FSHD patient biopsies are required to make this assertion more definitive. The main focus of our study was to determine whether DUX4 satisfied the second requirement of an FSHD candidate, by possessing sufficient in vivo myopathic potential to be worthy of further study in FSHD pathogenesis. We reasoned that the best approach to assess this was by overexpressing DUX4 in adult mouse muscles, for 3 reasons. First, based on the transcriptional de-repression model of FSHD pathogenesis, it is necessary to study FSHD candidate genes by overexpressing them. Second, FSHD is typically an adult-onset muscular dystrophy with comparatively little or no nonmuscle pathology, which supports the hypothesis that FSHD genes are expressed solely in areas that show pathology (ie, primarily muscles but perhaps also retinal vasculature and inner ear). Third, D4Z4 repeats, which are inextricably linked to FSHD development, are only present in placental mammals, including mice.19 Thus, directing DUX4 expression in adult mouse muscle is the most feasibly relevant model system for testing its myopathic potential in humans. Importantly, using AAV6 vectors to deliver DUX4 to muscle, we demonstrated that DUX4 has the potential to damage adult mouse muscle in vivo. Our findings strongly support the hypothesis that DUX4 plays a role in FSHD pathogenesis. We also uncovered a novel mechanism of DUX4 toxicity involving the p53 pathway (discussed in greater detail below).
Although our study represents the first direct demonstration of the myopathic potential of DUX4 in an adult mammalian muscle model, we are not the first to show the harmful effects of DUX4 overexpression in general. Several recent studies demonstrated that unregulated or ubiquitously overexpressed DUX4 was toxic to plurior multipotent progenitor cells of early Xenopus or zebrafish embryos (which lack D4Z4 repeats), or in cultured mammalian cells.20,25,29,31,32 The general toxicity of DUX4 raises an important issue about its specificity, because one could argue that if DUX4 is indeed involved in FSHD pathogenesis, its toxic effects should be restricted solely to mammalian muscle. Thus, general toxicity could suggest that DUX4 is not involved in FSHD but is instead an artifact caused possibly by nonspecific protein overload and/or interference with the normal function of other similar homeodomain transcription factors. Indeed, precedence exists for both possibilities; overexpression of otherwise inert GFP protein was nonspecifically toxic to striated muscle,40 and at sublethal doses, DUX4 inhibited C2C12 cell differentiation by competing with PAX3/PAX7.31 Our DUX4.HOX1 mutant ruled out the former, as high levels of this protein caused no abnormalities in vitro or in vivo, but not the latter, because its reduced DNA binding ability would likely preclude its interference with other similar transcription factors. Regardless, the general toxicity of DUX4 does not rule out its involvement in FSHD pathogenesis. Considering our novel finding that DUX4 activates p53-dependent cell death (see Table and Fig 5), its toxicity to nonmuscle cells and embryos of lower organisms is not surprising, because the p53 pathway is conserved in zebrafish, Xenopus, and most nontumor mammalian cell lines. Interestingly, the only cell line with known resistance to high levels of DUX4 protein expression is derived from a human rhabdomyosarcoma tumor typically associated with loss of p53 tumor suppressor function, which is consistent with our finding that DUX4 does not damage muscles from p53 null mice.25,58 Thus, the ability of DUX4 to activate conserved cell death pathways argues for its contribution to FSHD if it is preferentially expressed in areas of FSHD pathology (the third criteria for an FSHD candidate gene; as yet undetermined for DUX4). Our zebrafish data demonstrate this point. Ubiquitous DUX4 overexpression was incompatible with normal zebrafish and Xenopus embryonic development in previous studies,29,32 but we showed that muscle-directed DUX4 expression produced viable zebrafish with varying degrees of dystrophic abnormalities, including asymmetrical defects, which are a hallmark of FSHD (see Fig 1).1 Finally, it is not unprecedented for overexpressed disease genes, particularly those definitively linked to a specific genetic disorder, to cause more aggressive and widespread phenotypes in animal models than what are typically seen in humans. For example, the most widely used mouse model for Huntington disease (HD; R6/2 model), which overexpresses a mutant human huntingtin gene, recapitulates the striatal neuron dysfunction and motor abnormalities that are hallmarks of the human disease.59 Nevertheless, striatal pathology in R6/2 mice is significantly more aggressive than in humans, and they also display widespread phenotypes that are not present in typical HD patients, including reduced fertility, very early death, epilepsy, diabetes, cardiac dysfunction, and neuromuscular junction defects.59 More recently developed models that more faithfully genocopy human HD do not display such widespread abnormalities.59 This case study analogy illustrates that even a definitive human disease gene like HD can cause unexpected widespread toxicity in animal models. Our data strongly demonstrate the in vivo myopathic potential of DUX4; considering our discussion above, our study further supports the hypothesis that DUX4 overexpression is an underlying pathogenic event in FSHD. In future studies, it will be important to address the third criteria of an FSHD candidate gene by testing the myopathic potential of DUX4 using natural human D4Z4-derived promoter and polyA elements.
We also reported the novel finding that the proapoptotic effects of DUX4 were p53 dependent (see Table, Fig 5). Our data therefore support a mechanism for DUX4 promyopathic activity, which could also explain its general toxicity to most cells in which it is overexpressed. Although previous discoveries that p53 pathway components (Bax and caspase-3) are activated in FSHD muscles are consistent with our DUX4/p53 findings, it will be important in future studies to better define p53 pathway involvement in FSHD. In addition to searching for p53 pathway activation in FSHD muscles and cell lines, additional mechanistic data are needed to determine whether DUX4 directly activates the p53 promoter or does so indirectly through activation of intermediary gene products. One potential mechanism for the latter may involve DUX4 activation of the PITX1 gene, which activates p53 and is increased with DUX4 in FSHD patient biopsies.20,60
Although DUX4 is clearly a transcription factor, its normal biological roles are unknown. Here, we reported that DUX4 activates the p53 pathway and caspase-3, which play important roles in skeletal muscle differentiation and regeneration.61,62 It is therefore possible that DUX4 normally functions to regulate skeletal muscle development, but in FSHD, its over- or misexpression negatively impacts muscle development and regeneration through chronic p53 pathway activation and/or interference with other homeodomain transcription factors (like PAX3/PAX7). If DUX4 is expressed in muscle progenitors, as suggested,31 chronic DUX4-induced damage could reduce the pool of proliferating satellite cells, causing regeneration defects that could, over time, manifest as the progressive weakness that typifies FSHD. Thus, understanding where and when DUX4 is normally expressed in vivo will help further define its potential mechanistic role in FSHD pathogenesis.
Finally, our data do not rule out the involvement of other FSHD-associated potentially myopathic genes,22,28 independent of DUX4. It is possible that DUX4 overexpression is one of multiple pathogenic insults that conspire to produce FSHD pathologies.22 Nevertheless, our data show that DUX4 is highly toxic to mammalian muscle, at least partly through activation of p53-dependent cell death. Because it is a transcription factor, even small perturbations in its expression could dramatically alter p53 signaling or expression of other genes required to maintain normal muscle integrity. Thus, additional characterization of DUX4-controlled pathways, including p53, may help define mechanisms contributing to muscular dystrophy and ultimately provide targets for therapeutic intervention of FSHD.
Acknowledgments
This work was financially supported by the Facioscapulohumeral Society Landsman Charitable Trust Research Fellowship Grant (FSHD-LCT-002, to S.Q.H.) and Conners and Jacobs Families Research Fellowship Grant (FSHS-JJFR-001, to S.Q.H.), and a National Institutes of Health (NIH) KL2 Clinical and Translational Scholar Award (KL2 RR025754, to S.Q.H.). Additional support was provided by startup funds to S.Q.H. and a Graduate Student Fellowship to L.M.W. from the Research Institute at Nationwide Children’s Hospital Research Foundation.
We thank members of the Harper laboratory for assistance and support, Dr. S. Hauschka for providing the MHCK7 promoter, Dr. P. Martin for Trp53−/− mice, Dr. Yi-Wen Chen for DUX4 antibodies, Mr. D. P. Perez for advice and support, and Dr. K. R. Clark and TRINCH Viral Vector Core Facility members for assistance with AAV production.
Footnotes
Additional supporting information can be found in the online version of this article.
Potential Conflicts of Interest
J.Y.: grants/grants pending, NIH. S.Q.H.: grants/grants pending, NIH.
References
- 1.Flanigan KM. Myology. 3. New York, NY: McGraw-Hill Medical Publishing Division; 2004. Facioscapulohumeral muscular dystrophy and scapuloperoneal disorders; pp. 1123–1133. [Google Scholar]
- 2.Prevalence of rare diseases: bibliographic data. Orphanet. 2008 Available at: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf Accessed May 2010.
- 3.Klinge L, Eagle M, Haggerty ID, et al. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2006;16:553–558. doi: 10.1016/j.nmd.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandem repeated unit. Hum Mol Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 5.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 6.Bakker E, Wijmenga C, Vossen RH, et al. The FSHD-linked locus D4F104S1 (p13E–11) on 4q35 has a homologue on 10qter. Muscle Nerve. 1995;2:S39–S44. [PubMed] [Google Scholar]
- 7.Lemmers RJ, van der Maarel SM, van Deutekom JC, et al. Inter-and intrachromosomal sub-telomeric rearrangements on 4q35: implications for facioscapulohumeral muscular dystrophy (FSHD) aetiology and diagnosis. Hum Mol Genet. 1998;7:1207–1214. doi: 10.1093/hmg/7.8.1207. [DOI] [PubMed] [Google Scholar]
- 8.Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–894. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas NS, Wiseman K, Spurlock G, et al. A large patient study confirming that facioscapulohumeral muscular dystrophy (FSHD) disease expression is almost exclusively associated with an FSHD locus located on a 4qA-defined 4qter subtelomere. J Med Genet. 2007;44:215–218. doi: 10.1136/jmg.2006.042804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Greef JC, Frants RR, van der Maarel SM. Epigenetic mechanisms of facioscapulohumeral muscular dystrophy. Mutat Res. 2008;647:94–102. doi: 10.1016/j.mrfmmm.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Greef JC, Lemmers RJ, van Engelen BG, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–1459. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 12.Hewitt JE, Lyle R, Clark LN, et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum Mol Genet. 1994;3:1287–1295. doi: 10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 13.Jiang G, Yang F, van Overveld PG, et al. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum Mol Genet. 2003;12:2909–2921. doi: 10.1093/hmg/ddg323. [DOI] [PubMed] [Google Scholar]
- 14.Lyle R, Wright TJ, Clark LN, Hewitt JE. The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics. 1995;28:389–397. doi: 10.1006/geno.1995.1166. [DOI] [PubMed] [Google Scholar]
- 15.van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 16.Zeng W, de Greef JC, Chen YY, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD) PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tupler R, Berardinelli A, Barbierato L, et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet. 1996;33:366–370. doi: 10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celegato B, Capitanio D, Pescatori M, et al. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics. 2006;6:5303–5321. doi: 10.1002/pmic.200600056. [DOI] [PubMed] [Google Scholar]
- 19.Clapp J, Mitchell LM, Bolland DJ, et al. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:264–279. doi: 10.1086/519311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci U S A. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenberg I, Eran A, Nishino I, et al. Distinctive patterns of micro-RNA expression in primary muscular disorders. Proc Natl Acad Sci U S A. 2007;104:17016–17021. doi: 10.1073/pnas.0708115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabellini D, D’Antona G, Moggio M, et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 23.Gabellini D, Green MR, Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 24.Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 25.Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Laoudj-Chenivesse D, Carnac G, Bisbal C, et al. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J Mol Med. 2005;83:216–224. doi: 10.1007/s00109-004-0583-7. [DOI] [PubMed] [Google Scholar]
- 27.Osborne RJ, Welle S, Venance SL, et al. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. 2007;68:569–577. doi: 10.1212/01.wnl.0000251269.31442.d9. [DOI] [PubMed] [Google Scholar]
- 28.Reed PW, Corse AM, Porter NC, et al. Abnormal expression of mu-crystallin in facioscapulohumeral muscular dystrophy. Exp Neurol. 2007;205:583–586. doi: 10.1016/j.expneurol.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Snider L, Asawachaicharn A, Tyler AE, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet. 2009;18:2414–2430. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winokur ST, Chen YW, Masny PS, et al. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet. 2003;12:2895–2907. doi: 10.1093/hmg/ddg327. [DOI] [PubMed] [Google Scholar]
- 31.Bosnakovski D, Xu Z, Gang EJ, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wuebbles RD, Long SW, Hanel ML, Jones PL. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- 33.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandri M, El Meslemani AH, Sandri C, et al. Caspase 3 expression correlates with skeletal muscle apoptosis in Duchenne and facioscapulo human muscular dystrophy. A potential target for pharmacological treatment? J Neuropathol Exp Neurol. 2001;60:302–312. doi: 10.1093/jnen/60.3.302. [DOI] [PubMed] [Google Scholar]
- 35.Ostlund C, Garcia-Carrasquillo RM, Belayew A, Worman HJ. Intracellular trafficking and dynamics of double homeodomain proteins. Biochemistry. 2005;44:2378–2384. doi: 10.1021/bi047992w. [DOI] [PubMed] [Google Scholar]
- 36.Kawakami K, Takeda H, Kawakami N, et al. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell. 2004;7:133–144. doi: 10.1016/j.devcel.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Harper SQ, Hauser MA, DelloRusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 38.Thornhill P, Bassett D, Lochmuller H, et al. Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP) Brain. 2008;131:1551–1561. doi: 10.1093/brain/awn078. [DOI] [PubMed] [Google Scholar]
- 39.Salva MZ, Himeda CL, Tai PW, et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 40.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescent protein can cause dilated cardiomyopathy. Nat Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- 41.Franich NR, Fitzsimons HL, Fong DM, et al. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington’s disease. Mol Ther. 2008;16:947–956. doi: 10.1038/mt.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang F, Rendahl KG, Manning WC, et al. AAV-mediated expression of vascular endothelial growth factor induces choroidal neovascularization in rat. Invest Ophthalmol Vis Sci. 2003;44:781–790. doi: 10.1167/iovs.02-0281. [DOI] [PubMed] [Google Scholar]
- 43.Blankinship MJ, Gregorevic P, Allen JM, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Zhu T, Qiao C, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 45.Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 46.Degli Esposti M, Dive C. Mitochondrial membrane permeabilisation by Bax/Bak. Biochem Biophys Res Commun. 2003;304:455–461. doi: 10.1016/s0006-291x(03)00617-x. [DOI] [PubMed] [Google Scholar]
- 47.Gupta S, Radha V, Furukawa Y, Swarup G. Direct transcriptional activation of human caspase-1 by tumor suppressor p53. J Biol Chem. 2001;276:10585–10588. doi: 10.1074/jbc.C100025200. [DOI] [PubMed] [Google Scholar]
- 48.Sadasivam S, Gupta S, Radha V, et al. Caspase-1 activator Ipaf is a p53-inducible gene involved in apoptosis. Oncogene. 2005;24:627–636. doi: 10.1038/sj.onc.1208201. [DOI] [PubMed] [Google Scholar]
- 49.Thalappilly S, Sadasivam S, Radha V, Swarup G. Involvement of caspase 1 and its activator Ipaf upstream of mitochondrial events in apoptosis. FEBS J. 2006;273:2766–2778. doi: 10.1111/j.1742-4658.2006.05293.x. [DOI] [PubMed] [Google Scholar]
- 50.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 51.de Greef JC, Wohlgemuth M, Chan OA, et al. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology. 2007;69:1018–1026. doi: 10.1212/01.wnl.0000271391.44352.fe. [DOI] [PubMed] [Google Scholar]
- 52.Flanigan KM, Coffeen CM, Sexton L, et al. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord. 2001;11:525–529. doi: 10.1016/s0960-8966(01)00201-2. [DOI] [PubMed] [Google Scholar]
- 53.Arashiro P, Eisenberg I, Kho AT, et al. Transcriptional regulation differs in affected facioscapulohumeral muscular dystrophy patients compared to asymptomatic related carriers. Proc Natl Acad Sci U S A. 2009;106:6220–6225. doi: 10.1073/pnas.0901573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Masny PS, Chan OY, de Greef JC, et al. Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur J Hum Genet. 2010;18:448–456. doi: 10.1038/ejhg.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rijkers T, Deidda G, van Koningsbruggen S, et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J Med Genet. 2004;41:826–836. doi: 10.1136/jmg.2004.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Deutekom JC, Lemmers RJ, Grewal PK, et al. Identification of the first gene (FRG1) from the FSHD region on human chromosome 4q35. Hum Mol Genet. 1996;5:581–590. doi: 10.1093/hmg/5.5.581. [DOI] [PubMed] [Google Scholar]
- 57.Winokur ST, Barrett K, Martin JH, et al. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul Disord. 2003;13:322–333. doi: 10.1016/s0960-8966(02)00284-5. [DOI] [PubMed] [Google Scholar]
- 58.Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- 59.Heng MY, Detloff PJ, Albin RL. Rodent genetic models of Huntington disease. Neurobiol Dis. 2008;32:1–9. doi: 10.1016/j.nbd.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Liu DX, Lobie PE. Transcriptional activation of p53 by Pitx1. Cell Death Differ. 2007;14:1893–1907. doi: 10.1038/sj.cdd.4402209. [DOI] [PubMed] [Google Scholar]
- 61.Fernando P, Kelly JF, Balazsi K, et al. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2002;99:11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen BD, Rampalli S, Burns LE, et al. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci U S A. 2010;107:4230–4235. doi: 10.1073/pnas.0913089107. [DOI] [PMC free article] [PubMed] [Google Scholar]