

Abstract
Introduction
The Herpesviridae are responsible for debilitating acute and chronic infections, and some members of this family are associated with human cancers. Conventional anti-herpesviral therapy targets the viral DNA polymerase and has been extremely successful; however, the emergence of drug-resistant virus strains, especially in neonates and immunocompromised patients, underscores the need for continued development of anti-herpes drugs. In this article, we explore an alternative target for antiviral therapy, the HSV helicase/primase complex.
Areas covered
This review addresses the current state of knowledge of HSV DNA replication and the important roles played by the herpesvirus helicase–primase complex. In the last 10 years several helicase/primase inhibitors (HPIs) have been described, and in this article, we discuss and contrast these new agents with established inhibitors.
Expert opinion
The outstanding safety profile of existing nucleoside analogues for a-herpesvirus infection make the development of new therapeutic agents a challenge. Currently used nucleoside analogues exhibit few side effects and have low occurrence of clinically relevant resistance. For HCMV, however, existing drugs have significant toxicity issues and the frequency of drug resistance is high, and no antiviral therapies are available for EBV and KSHV. The development of new anti-herpesvirus drugs is thus well worth pursuing especially for immunocompromised patients and those who develop drug-resistant infections. Although the HPIs are promising, limitations to their development into a successful drug strategy remain.
Keywords: antiviral compounds, DNA replication, helicase-primase, herpes simplex virus
1. Introduction
The human herpesviruses are responsible for lifelong debilitating and congenital infections, and some members of this family are associated with human cancers. The herpesvirus family comprises of three major classes: α, β and γ, that diverge in tissue tropism and many aspects of their interactions with their hosts. The human α-herpesviruses include human herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) and varicella-zoster virus (VZV). HSV infections are one of the most widespread infectious diseases in the world, affecting between 60 and 95% of the population [1]. HSV-1 and -2 are associated with a wide range of clinical manifestations including cold sores, genital lesions, kerititis, corneal blindness and very rarely, encephalitis. VZV causes chickenpox and shingles, but can also be associated with viral encephalitis and a painful condition known as postherpetic neuralgia. HSV-1 and -2 infections are also believed to be cofactors in increased HIV infection rates thus contributing to the HIV epidemic [2]. The human β-herpesviruses include cytomegalovirus (HCMV), HHV-6 and -7. HCMV is associated with mononucleosis and rare complications such as pneumonia, CNS disease and hepatitis in immunocompetent individuals; however, in immunocompromised hosts these complications are more prevalent. CMV frequently invades the eye, and CMV retinitis is a major problem. HCMV can cause significant morbidity and mortality especially in patients who receive a hematopoetic stem cell transplant or a solid organ transplant. In transplant patients, viral infection can lead to graft-versus-host disease as well as adverse effects on the host immune system. Furthermore, HCMV infection during pregnancy is an important cause of neonatal infection with significant morbidity in infants. HHV-6 is associated with fever, skin rashes, graft-versus-host disease, pneumonitis, encephalitis and meningitis. HHV-7 so far has not been linked to any disease; however, in immunocompromised individuals, it may act as an opportunistic agent. The γ-herpesviruses include Epstein–Barr virus (EBV) and Kaposi's sarcoma associated herpesvirus (KSHV). These viruses are associated with diseases including mononucleosis and also malignancies such as B-cell lymphomas, nasopharyngeal carcinoma and Kaposi's Sarcoma. Kaposi's Sarcoma is now recognized as one of the most common cancers in many sub-Saharan African countries [3]. Thus, the human herpesviruses are associated with significant disease, underscoring the need for safe and effective antiviral strategies. Despite differences between the three classes of human herpesviruses, the mechanisms by which they replicate their DNA during productive (‘lytic’) infection are largely conserved. Viral enzymes involved in DNA replication have provided a rich store of useful targets for antiviral therapy.
2. Currently used anti-herpesvirus drugs
The survival of all organisms including viruses depends on their ability to produce an exact copy of their genetic material. Currently, the primary target of clinically used anti-herpes drugs is the viral polymerase (Pol). For the α-herpesviruses, these include the nucleoside prodrugs acyclovir, penciclovir and their orally bioavailable pre-prodrugs valaciclovir and famciclovir, along with the pyrophosphate analogue foscarnet [4]. The nucleoside prodrug ganciclovir is the primary treatment for CMV infection in immunocompromised individuals [5]. For all of the nucleoside prodrugs the active form is the nucleoside triphosphate. These Pol inhibitors have proven extremely effective against HSV-1 and -2; however, they are not as effective against VZV as they are against HSV-1 and -2, necessitating higher and more frequent dosing [6,7]. Although clinically significant resistance to these agents is relatively rare in immunocompetent individuals, drug resistance is much more likely in immunocompromised patients [8-10]. Thus, drug-resistant clinical isolates are likely to be more problematic in transplant patients undergoing immunosuppressive therapy, especially when infection occurs in the body at a site that is not easily accessed by the immune system such as during herpes keratitis and encephalitis or in neonates who lack a mature immune system.
Foscarnet (phosphonoformic acid) inhibits herpes DNA replication by binding in the pyrophosphate binding site of the herpes Pol and blocking polymerization of additional dNTPs. The nucleoside analogues take advantage of the promiscuity of the herpes Pol such that the Pol incorporates the analogues and chain termination ensues either immediately (acyclovir) or shortly thereafter (penciclovir, ganciclovir). Acyclovir uses a particularly clever mechanism to prevent its removal after incorporation – once the Pol has incorporated acyclovir, the enzyme binds the next correct dNTP to form an E-DNA–dNTP complex where the DNA is locked in the Pol active site but the bound dNTP cannot be incorporated due to the lack of a 3′-OH on the acyclovir [11].
Nucleoside analogues (acyclovir, penciclovir and their orally bioavailable pre-prodrugs valaciclovir and famciclovir) are particularly effective against HSV-1 and -2 for two key reasons: i) the drugs have a truly remarkable safety record and ii) although drug resistance has been reported, especially in immune-compromised individuals [12-14], the number of cases is not high [8]. The extraordinary safety of the nucleoside analogues results from several features. The nucleosides themselves are nontoxic; they are only poorly converted to the triphosphate in cells that are not infected by a herpesvirus because cells do not efficiently convert these nucleosides into the nucleoside monophosphate. Furthermore, even if converted to the triphosphate these analogues are relatively poor inhibitors of cellular DNA Pols [15]. The unique ability of these nucleoside analogues to avoid clinically relevant resistance issues results from resistant viruses being relatively non-pathogenic [8]. Additionally, in those cases where resistance does occur, foscarnet often proves effective. These nucleoside analogues have thus proven extraordinarily effective against HSV-1 and -2 infection. A bicyclic nucleoside analogue (FV-100) has been described with highly specific antiviral activity against VZV [16]. Its mechanism of action is not clear but it appears to rely on the VZV thymidine kinase. No toxicity was found in Phase I human trials [17], and a Phase II clinical trial comparing FV-100 to valacyclovir in the treatment of shingles is underway [18].
Another nucleoside analogue, ganciclovir is licensed to treat HCMV; however, in contrast to the outstanding safety profile of the nucleoside analogues for treating -herpesviruses, ganciclovir has the opposite profile [19]. It is cytotoxic due to severe effects of even small amounts of ganciclovir triphosphate on DNA replication. Remarkably, once incorporated into on cellular DNA the ganciclovir can poison cellular DNA replication in S-phases following treatment [19]. Consequently, up to 10% of the patients taking ganciclovir for CMV infection must stop treatment. Furthermore, resistance to ganciclovir develops very frequently [20], presumably aided by the fact that CMV-infections generally occur in immunocompromised individuals. The human γ-herpesviruses (EBV and KSHV) are also responsible for complications in immunocompromised patients. EBV and KSHV are associated with serious complications in immunosuppressed patients causing EBV-associated post-transplant lymphoproliferative disorders and KSHV-associated Kaposi sarcoma, primary effusion lymphoma and some cases of Castleman disease. Current treatment for these disorders is to discontinue or reduce immunosuppressive therapy [21], although valganciclovir was recently shown to decrease KSHV replication [22]. At this time, however, no effective anti-herpesvirus therapies are available for the γ-herpesviruses EBV and KSHV or the β-herpesviruses HHV-6 and -7.
In this article, we will explore an alternative target for anti-herpesviral therapy, the viral helicase/primase (H/P) complex. This enzyme complex, like the Pol, is common to all members of the herpesvirus family suggesting that it may be possible to develop broad-spectrum, anti-herpes therapies. As will be described in greater detail below, several compounds have been described that inhibit the helicase/primase (HPIs) from the α-herpesviruses. To date, however, none of the new HPIs have been shown to be effective against the helicase–primase complexes from β- or γ-herpesviruses.
3. Overview of lytic HSV-1 infection
The earliest steps in HSV-1 infection involve attachment to cell surface heparan sulfate (HS) followed by fusion of the lipid envelope with the plasma membrane of the host cells [23]. Following entry, viral capsids translocate along microtubules [24] to the nuclear pores where they dock and eject their genomes into the nucleus [25]. A combination of viral and cellular transcription factors are recruited to viral genomes resulting in a highly regulated cascade of gene expression consisting of three well-defined kinetic classes of genes: immediate early, early and late. Viral gene expression, DNA replication and encapsidation occur within globular domains in the nucleus termed replication compartments [26-28]. The products of DNA replication are longer-than-unit length concatemers consisting of tandem repeats of the viral genome [29]. Concatemer production is an essential step for the generation of progeny virus as the packaging machinery must recognize longer-than-unit-length concatemers during the encapsidation step. Although it was previously thought that linear virion DNA circularized in infected cells and that concatemers were generated by rolling circle replication [30], this model has never been directly confirmed. Several lines of evidence suggest that HSV DNA replication may require DNA recombination [31,32]. The larger-than-unit-length viral genomes are cleaved into unit-length fragments and packaged into preformed procapsids [33,34]. The HCMV terminase, the enzyme responsible for cleavage, has been exploited as a target leading to clinical trials of the inhibitor Letermovir [35,36]. DNA-containing capsids exit the nucleus by budding through the nuclear membrane to the cytoplasm where they acquire their final envelope and mature glycoproteins by a series of envelopment and de-envelopment steps [37].
4. Overview of HSV DNA replication
The genome of HSV is large and structurally complex containing three origins of DNA replication; oriL is located in the UL sequence [38], and two copies of oriS are located in the two inverted repeats of the c sequence [39]. OriL and OriS contain an A-/T-rich region surrounded by recognition sites for the origin-binding protein, UL9 [38]. The HSV-1 genome encodes seven essential trans-acting replication proteins and several additional non-essential replication proteins [40,41]. These essential genes encode three core replication modalities including a single strand DNA binding protein (known as ICP8 or UL29), a two subunit DNA Pol (UL30 and UL42), and a three subunit H/P complex (UL5, UL8 and UL52). In addition to the six core replication proteins that are shared among all human and animal herpesviruses, HSV and the other α-herpesviruses also encode an origin binding protein (UL9) believed to function during initiation of viral DNA synthesis.
4.1 HSV DNA replication occurs in two phases
The first step involves an UL9-dependent step presumed to occur at one of the three replication origins. The analysis of temperature-sensitive (ts) mutants has revealed that UL9 is indispensable early in HSV-1 infection but does not appear to be required late in infection, once DNA synthesis has initiated [42,43]. Thus, the second phase appears to be independent of UL9 and has been proposed to occur by rolling circle replication or by a recombination-dependent mode of DNA replication analogous to the replication program of the bacteriophages T4 and lambda.
4.2 DNA replication and recombination may be linked
Recombination is a frequent event between two co-infecting HSV genomes not only in the laboratory [44-46], but also in animal infection models and in human populations [47-49]. Recombination may thus contribute to genetic diversity in the herpesviruses. Several lines of evidence support the notion that DNA replication and recombination are linked [50-52]. Viral DNA replication results in the accumulation of head-to-tail concatemers that are highly branched, with X- and Y-junctions [53-56]. Furthermore, pulsed-field gel electrophoresis (PFGE) suggests that HSV DNA replication products adopt a complex, perhaps branched, structure which cannot enter the gel, even after digestion with a restriction enzyme which recognizes a single site within the HSV genome [57-59]. Genomic inversions are detectable as soon as newly replicated DNA is detected, also supporting linkage between recombination and replication [58-60]. Alternatively, the large amount of recombination may reflect a high requirement for recombinatorial DNA repair during herpes replication (e.g., double-strand DNA break repair). It is also possible that this recombinatorial DNA repair simultaneously serves to repair DNA damage as well as initiate the synthesis of new DNA strands.
4.3 Relationship between HSV DNA replication and repair
The structure of the viral genomes that enter the nucleus at the outset of infection might be expected to induce a cellular DNA damage response: not only does the incoming linear genome contain double strand ends which resemble double strand breaks, but packaged viral genomes also contain nicks and gaps [32]. HSV has evolved a complex relationship with cellular pathways involved in DNA repair, activating some pathways and inactivating others [31,61-65].
4.4 Initiation of HSV DNA replication
Viral DNA synthesis initiates at one of the three viral origins of replication with UL9 and ICP8 acting in conjunction to distort the AT rich origin spacer region. Distortion at the origins is thought to promote the loading of the HSV-1 H/P complex that unwinds duplex DNA at the replication fork and generates short RNA primers to initiate both leading and lagging strand DNA synthesis. The HSV-1 H/P is a heterotrimer consisting of the UL5, UL8 and UL52 gene products that are essential for viral DNA replication in cell culture [66-70]. Active primase is required for the recruitment of HSV DNA Pol to viral foci in infected cells [71]. This may indicate that Pol can be recruited only to a replication fork that already contains a RNA primer-template. Alternatively, however, Pol may recognize a conformational change in the viral scaffold induced by an active primase subunit capable of binding DNA and/or other protein–protein interactions. The HSV Pol is tethered at the replication fork by protein–protein interactions, not protein–nucleic acid interactions [72].
4.5 Leading and lagging strand DNA synthesis
Once the HSV Pol and its accessory protein are recruited to the replication fork, it is thought that leading- and lagging-strand DNA synthesis occur in a coordinated fashion analogous to other cellular and viral systems [73]. Okazaki fragments during herpes replication have not been demonstrated in vivo. However, model replication systems using only purified herpes proteins and a minicircle template resulted in the production of Okazaki fragments around 400 nucleotides long [74].
5. HSV H/P
The three-component HSV-1 H/P complex (UL5, UL52 and UL8) exhibits 5′ – 3′ helicase, primase and ssDNA-dependent NTPase activities. The UL5 subunit (99 kDal) contains seven motifs that are conserved in helicase super family I (Figure 1) [68]. All seven helicase motifs are essential for viral growth and DNA synthesis in infected cells and for leading strand synthesis in an in vitro assay [75]. Consistent with its putative role as the elongation helicase, UL5 is needed continuously during DNA replication [69,70]. The UL52 subunit (114 kDal) contains two conserved regions shared with other known primases: a signature DID motif required for catalytic activity [76,77] and a putative zinc-binding domain associated with DNA binding in prokaryotic and eukaryotic primases (Figure 1) [71,72,78]. While a subcomplex containing UL5 and UL52 exhibits NTPase, helicase and primase activities [79-81], neither subunit by itself exhibit either helicase or primase activity (see below). The UL8 subunit (80 kDal) has no known catalytic functions but can facilitate nuclear uptake of the complex and modulates the enzymatic functions of the UL5 and UL52 subcomplex [81-85]. UL8 increases the rate of primer synthesis by the UL5/UL52 subcomplex by increasing the rate of primer initiation [86]. Interactions of UL8 with other components of the HSV replication machinery including UL9, ICP8 and the UL30 DNA Pol suggest that it may play a role integrating activities at the fork [87-90].
Figure 1. Domain structure of the three subunits of the HSV helicase/primase (UL5, UL8 and UL52).

The conserved motifs of UL5 are shown as seven black boxes shared within superfamily 1 members. Two conserved UL52 domains are shown: the LVFDID catalytic domain (608 – 630) and putative zinc binding domain at the C-terminus (residues 998 – 1028). UL8 is a 751 aa protein with no known functional motifs.
5.1 Replication fork dynamics
In most well-studied DNA replication systems, replicative helicases play central roles not only in the assembly of the replisome but also in the regulation of replication fork dynamics such as processivity and coordination of leading-and lagging-strand synthesis [91]. Interactions between the replicative helicase and its partner proteins are essential for all aspects of replisome dynamics, and potential interaction partners have been identified by their ability to stimulate helicase activity in vitro. One major class of helicase interaction partners is the primases. During DNA synthesis, leading- and lagging-strand Pols must be coordinated, and the primase must be regulated to synthesize primers on the lagging strand and even on the leading strand both during initiation of replication and in the event of the stalling of the leading strand Pol [90-92]. Primase activity is often stimulated by associating with its cognate helicase, and the processivity of many helicases is stimulated by the presence of primase [93]. In some replication systems such as E. coli and T4, the primase is an independent protein; whereas, in the case of T7, the primase and helicase are encoded on a single polypeptide. In the case of the herpesviruses, the catalytic cores for the helicase and primase reside on different subunits of the same complex (UL5 and UL52); however, UL5 and UL52 exhibit a remarkably complex interdependence. For instance, UL5 and UL52 must be co-translated to generate a functional complex: if UL52 is expressed without UL5, it is insoluble, and if UL5 is expressed without UL52, it is inactive [79]. Although one can generate a soluble UL5/UL8 complex, it still lacks any enzymatic activity [86]. In addition, mutations in the putative zinc finger at the C-terminus of UL52 influence not only primase but also ATPase, helicase and DNA-binding activities of the UL5/UL52 subcomplex [72,75,78,94,95]. It is possible that UL5 and UL52 may share a DNA-binding site created by interaction between the two subunits. Alternatively, UL52 binding to DNA through the zinc finger may be necessary for loading UL5 onto DNA. In a recent series of kinetic and photo-crosslinking studies in which the UL5, UL52 and UL8 subunits were expressed in various pair-wise combinations it was shown UL52 likely contains the entire primase active site but also contributes to helicase activity. Furthermore, it appears that UL5 contacts DNA and contributes amino acids needed for the initiation of primer synthesis [86], thereby confirming a complex interdependence between the UL5 and UL52 subunits. This complex inter-relationship between the two subunits and H/P activity suggests that it should be possible to inhibit these activities with ligands that bind to either UL5 or UL52.
5.2 Stoichiometry of H/P at the fork
Most replicative helicases form hexameric rings that encircle one or both strands of the substrate DNA during unwinding. These include both viral initiator proteins such as papillomavirus E1, SV40 large T-antigen and T7gp4 as well as the E. coli DnaB and eukaryotic mcm proteins [88]. Interestingly, the HSV H/P is unlikely to form a hexamer as UL5 is a member of the SF1 helicase superfamily generally thought to exist as monomers in solution. Consistent with this, the HSV H/P behaves as a single heterotrimer by gel filtration [80]. However, in the presence of forked DNA and AMP–PNP or ATP, higher-order complexes can form [96]. Furthermore, two or more H/P heterotrimers are needed in order to observe efficient primase activity [96]. In these experiments, primase exhibited a cooperative dependence on protein concentration while helicase activity did not. DNA-dependent multimerization is reminiscent of the behavior of other SF1 family helicases that unwind dsDNA only if two or more helicase polypeptides are present [97-99]. In the case of the SF1 UvrD helicase, unwinding in vitro appears to require at least a dimeric UvrD complex in which one subunit binds the ssDNA/dsDNA junction, while the second subunit binds the ssDNA tail [98]. In other SF1 helicases, direct protein–protein interactions between the helicase protomers are unnecessary to achieve enhanced unwinding. Although helicase activity per se did not appear to exhibit cooperative behavior, it remains possible that the presence of two or more H/P complexes enhances the processivity of the H/P complex. Interestingly, this also provides a simple mechanism for generating a two-Pol replisome at the replication fork (Figure 2A and B).
Figure 2. HSV replication fork and model for model for coordinated leading- and lagging-strand DNA synthesis.
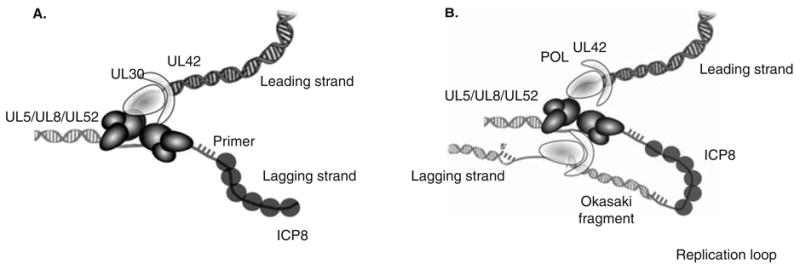
A. An HSV-1 replication fork would be expected to contain the helicase–primase complex (UL5/UL52/UL8) at the fork: UL5 would be expected to unwind duplex DNA ahead of the fork, and UL52 would be expected to lay down RNA primers that the two-subunit DNA polymerase (UL30/UL42) extends. The HSV-1 Pol also carries out leading-strand synthesis. ICP8 (UL29, SSB) presumably binds to ssDNA generated during HSV DNA synthesis. B. Coupled leading- and lagging-strand synthesis was recently reconstituted in vitro and requires HSV polymerase, helicase–primase and ICP8 [74]. In this model, coupled leading-and lagging-strand DNA synthesis requires two heterotrimers of helicase/primase and two molecules of the DNA polymerase. The leading- and lagging-strand polymerase molecules are proposed to communicate with each other through interactions between UL30 and the helicase–primase. A replication loop is formed in the lagging-strand to align it with the leading-strand. The lagging-strand DNA polymerase initiates Okazaki fragment synthesis using RNA primers (hatched bars).
5.3 H/P interactions with DNA Pol
The interaction of the replicative helicase with the DNA Pol is one of the most critical among the several protein–protein interactions within the replisome. Coordination of leading-and lagging-strand DNA synthesis requires communication between the replicative helicases, the leading- and lagging-strand Pols and the primase. For example, during lagging-strand synthesis, primase and Pol must be coupled to effectively produce an Okazaki fragment. Conversely, during leading-strand synthesis, coupled activity between helicase and Pol activity are expected to be critical. In some cases such as T7, the replicative helicase interacts directly with the DNA Pol [100] while in E. coli, the interaction is indirect as the helicase and the Pol interaction is mediated by the tau subunit of the clamp loader [102]. Interactions and communication between helicases, Pols and primases act to regulate the rate and processivity of DNA synthesis [92]. Several lines of evidence indicate functional interactions between the HSV H/P and the HSV Pol. The recruitment of the HSV Pol (UL30/UL42) complex to prereplicative sites requires an active primase [71]. This implies that conformational changes that occur in an active H/P complex and/or the RNA primer itself are required for HSV Pol to be recruited to prereplicative sites. The UL8 subunit of H/P interacts directly with the catalytic subunit of HSV Pol (UL30), thus providing a mechanism for direct binding of the two complexes [102].
6. HSV helicase–primase as a target for antiviral therapy
6.1 Spectrum of activities of HPIs
In the last 10 years, the HSV H/P has emerged as an excellent target for antiviral therapy as several classes of inhibitors of the UL5/UL8/UL52 helicase–primase complex have been reported. The new inhibitors are chemically diverse and include thiazole, thiazoleamide, thiazoleurea and thiazolylphenyl derivatives (Figure 3). High throughput screening assays against helicase activity resulted in the identification of T157602, a 2-aminothiazole compound [103]. T157602 also inhibits primase activity with an IC50 similar to that for helicase inhibition, consistent with the interdependence of the two activities. A thiazolylphenyl-containing compound, BILS 179 BS, was also discovered in a high throughput screen against the helicase and inhibits helicase, primase and ATPase activities [104]. BILS 179 BS reduced recurrent skin lesions in mice and was effective against HSV-2 genital disease in a mouse model [104] and was more effective than acyclovir in these studies. Structural analogues of BILS 179 BS (BILS 45 BS and BILS 22 BS) were also effective against acyclovirresistant virus [105,106]. While they showed efficacy in animal studies, the clinical development of these compounds has been discontinued. Another class of HPIs are the thiazoleamide compounds discovered using a cell-based virus replication assay [107]. One example of this class of compounds, BAY 57-1293, showed almost 200-fold greater potency against HSV than acyclovir in vitro with an IC50 of 0.02 μM; furthermore, it showed superior in vivo activities in guinea pig and mouse models when compared with valacyclovir [107-109]. BAY 57-1293, now known as AIC316 or Pritelivir, is undergoing a second clinical efficacy trial in which it will be compared with Valacyclovir in reducing genital HSV shedding [110]. Pritelivir is effective against HSV-1 and -2 but not other human herpesviruses. A newer compound ASP2151, an oxadiazolephenyl derivative, was designed based on the 2-aminothiazole compound (T157602) described by Spector et al. [103]. ASP2151 (Amenamevir, Figure 3) inhibits the single-stranded DNA-dependent ATPase, helicase and primase activities associated with the HSV-1 helicase–primase complex and exhibits antiviral activity against HSV-1, HSV-2 and VZV [111]. In mice oral ASP2151 was more effective than oral valacyclovir at reducing intradermal HSV-1 titers and for treating herpes simplex keratitis [112,113]. A Phase II study showed that ASP2151 was a safe and effective treatment for genital HSV infection [114,115]; however, another recent Phase I clinical trial was discontinued because of adverse events [116]. Thus, the future of this drug as a potential herpes treatment remains unclear.
Figure 3. Structures of HPIs.

T157602 (1,3-thiazol-2-amine); BAY 57-1293 (AIC316 or Pritelivir) N-methyl-N-(4-methyl-5-sulfamoyl-1, 3-thiazol-2-yl)-2-[4-(pyridin-2-yl)phenyl]acetamide); ASP2151 (Amenamevir) - N-(2,6-dimethylphenyl)-N-(2-{[4-(1,2,4-oxadiazol-3-yl)phenyl]amino}-2-oxoethyl)etrahydro-2-H-thiopyran-4-carboxamide 1,1-dioxide; BILS 22 BS N-(2-{[4-(2-amino-1,3-thiazol-4-yl)phenyl]amino}-2-oxoethyl)-N-benzylbenzamide.
6.2 Resistant mutations have been mapped to both UL5 and UL52 subunits
The specificity of these new HPIs is indicated by the ability to select drug-resistant mutants. The relative ease with which the virus becomes resistant to all HPIs suggests that they may preexist in clinical isolates [106], as is the case with acyclovir resistance. In fact, mutations resistant to BAY-57-1293 have now been confirmed using PCR analysis in clinical isolates that had never been exposed to the drug [117]. Mutations in both the UL5 and UL52 subunits have been reported which are resistant to all of the new H/P inhibitors; however, by a large margin, most of these mutations have been mapped to UL5. The first reported resistant mutations were found in the UL5 gene of HSV-1 and included residues Gly352, Met355 and Lys356 Figure 4A [104,107,108]. Since then a number of other resistant mutations in UL5 have been reported against BAY 57-1293, BILS 179 BS and ASP2151, and interestingly they all appear to map to the same region of the UL5 protein which is just downstream of motif IV of the helicase subunit (Figure 4A, [104,107,108]). These results suggest that these inhibitors likely act through specific interactions with the UL5 protein and employ similar mechanisms. Some resistant mutants show wild type levels of pathogenicity in mice compared to the parent strain, while others appear to be attenuated [103,106,118]. Three isolates resistant to BILS 22BS, a potent analog of BILS 45 BS, all exhibited pathogenicity similar to the wild type HSV-1 KOS from which they were derived [106], and T157602-resistant mutants did not significantly impair their growth [103]. T157602-resistance mutants contain a single amino acid substitution and show resistance to both helicase and primase inhibition, consistent with the UL5/UL52 complex containing a single binding site for the drug. On the other hand several mutations resistant to BAY57-1293 including Asn342Arg, Gly352Arg and Met355Thr showed attenuated in vitro growth, and several exhibited decreased in vivo pathogenicity compared with parent strains [118].
Figure 4.

A. UL5 amino acid substitutions in HSV-1 mutants resistant to HPIs. Several HPI-resistant mutations have been mapped to the UL5 gene. The amino acid substitutions are shown for mutants resistant to T157602, BAY 57-1293, BILS 22BS or ASP2151 [104,107,108,119,122]. B. UL52 Amino acid substitutions in HSV mutants resistant to HPIs. Arg367His was found in a ASP2151-resistant mutant [122] and A899T was found in a BAY 57-1293-resistant mutant [121]
6.2.1 UL5 mutations resistant to HPIs map near motif IV
As described above, UL5 contains seven predicted helicase motifs that have been shown to be essential in cell culture for viral growth [69] and in vitro for helicase and ATPase activity (Figure 1) [75]. Single point mutations in these motifs have been expressed in insect cells infected with recombinant baculovirus, and UL5/UL52 complexes analyzed for helicase and ATPase activity. Mutants in motifs I and II are severely compromised for ATPase and helicase activities [75]. Interestingly, mutants in motifs III, IV, V and VI exhibit only a 3 – 6-fold decrease in ssDNA stimulated ATPase activity but have no helicase activity. That these mutants can hydrolyze ATP but exhibit no helicase activity indicate they uncouple the favorable G of ATP hydrolysis from the unfavorable G of DNA unwinding. Although the three-dimensional structure of UL5 is not known, structures have been solved for several other SF1 family helicases. The conserved motifs in one prototype SF1 helicase, Rep, lie along the interface between two RecA like domains [119]. These results may indicate that mutations in motifs III, IV, V and VI are unable to carry out conformational changes required for DNA unwinding [75]. Interestingly, most of the HPI mutations in UL5 are located just downstream of motif IV (Figure 4A), suggesting that this region may be part of a conformationally active region of the protein. Biswas et al. have suggested that mutations such as Asn342Lys result in the steric or allosteric modification of the HPI binding pocket conferring resistance to the inhibitor [118]. The observation that this mutant exhibits slower growth and reduced virulence may also suggest that the mutation interferes with helicase–primase activity [118]; however, this has not been demonstrated directly.
6.2.2 UL52 mutations resistant to HPIs
In contrast to the many drug resistance mutations associated with the UL5 gene, only a few mutations have been found in UL52 [107]. A single mutation in UL52 (Ala899Thr, HSV-1) confers resistance to BAY 57-1293 (Figure 4B); however, this mutant remains sensitive to BILS 22 BS [121]. This again suggests that the BILS compounds may have different binding sites and/or mechanisms than BAY 57-1293. One ASP2151 resistant mutant has been isolated and found to contain an Arg367His substitution [122]. This ASP2151-resistant mutant showed attenuated growth in tissue culture and reduced pathogenicity in vivo compared with the parent strain [121]. All of the resistance mutants were still susceptible to nucleoside analogue antiviral compounds. Interestingly, to date there have been no reports of UL52 mutants resistant to the thioazolphenyl derivatives such as the BILS-series suggesting that those agents do not directly interact with this subunit. Further experiments will be needed, however, to more precisely understand the mechanism of action of this interesting class of drugs.
6.3 Mechanism of action
Although the mechanism of action of these compounds against helicase and primase has not been determined, they all inhibit helicase, DNA-dependent ATPase and primase activities [104,109]. The fact that several of these compounds show a concomitant inhibition of helicase, primase and DNA-dependent ATPase activities [103,104] further demonstrates the complex interdependence of UL5 and UL52 subunits. The rapid isolation of UL5 mutants resistant to all known HPIs suggest that these compounds likely bind directly to the UL5 protein [104-106]. It was reported that T157602 does not function by blocking binding of the nucleotide cofactor or the DNA substrate [103]. Both T157602 and one of the BILS compounds (BILS 103 BS) were shown to stabilize the interaction between the HSV helicase–primase enzyme and DNA [104]. Pre-formed enzyme–DNA complexes were more resistant to NaCl-induced disruption leading to the suggestion that these compounds may increase the affinity of enzyme to its substrate and that this prevents the enzyme from translocating along DNA [103,104]. Since both primase and helicase activity require movement along the DNA, this would provide a simple mechanism for simultaneously inhibiting both activities even though the catalytic cores for each activity reside on different subunits. On the other hand, UL52 mutants resistant to HPIs are relatively rare and have only been observed following growth of virus in the presence of BAY57-1293 and ASP2151 [120,121] and not with the BILS compounds. BAY57-1293 and ASP2151 may thus act by a different mechanism from the BILS compounds and/or their binding site may also include the primase subunit, UL52 [121,122]. Furthermore, mutants resistant to these two compounds map to different parts of the UL52 protein (Figure 4B), suggesting that they bind to different portions of the protein. Additional work is clearly needed to elucidate the precise inhibitory mechanisms of these compounds.
7. Conclusion
Of the many viral gene products encoded by herpesviruses, the most successful target for antiviral therapy has been the DNA Pol. Although acyclovir and related nucleoside analogues are widely used to combat HSV, VZV and HCMV, there are many reasons to pursue the development of additional strategies including the emergence of drug resistant mutants and the lack of effective treatment options for HCMV, EBV and other β- and γ-herpesviruses. In addition to providing alternate methods for controlling drug resistant variants of HSV, compounds that inhibit viral DNA replication provide novel tools for elucidating the mechanism of DNA replication [62,72,124,125].
8. Expert opinion
8.1 Challenges for the development of new therapies to combat herpesvirus infections
Although there is a clinical need, the outstanding safety profile of existing nucleoside analogues for α-herpesvirus infection will make it difficult to develop new anti-herpesvirus drugs. Nucleoside analogues exhibit few side effects, negligible occurrence of resistance in immunocompetent patients and are relatively inexpensive due to their lack of patent protection. These generate a substantial financial barrier to developing new drugs given the generally massive cost of drug development. On the other hand, for those cases where resistance to currently available drugs proves problematic, new drugs would clearly be advantageous. Additionally, new drugs might prove more effective than acyclovir, penciclovir and foscarnet for treating herpes encephalitis, a disease for which significant mortality and debilitating long-term effects remain major problems. In this last indication, however, these outcomes largely result from the non-specific initial symptoms and consequent delayed treatment.
For CMV infection, treatments other than ganciclovir are clearly needed. The primary limitation for developing new CMV treatments is the relatively small number of cases that occur, and these are primarily immunocompromised transplant patients or neonates. HIV-infected individuals had at one time provided a relatively large market for anti-CMV treatments; however, the remarkable success of new HIV therapies has largely eliminated opportunistic infections such as CMV. Better treatments for EBV and KSHV associated disorders are also needed; however, the markets for these indications may also be relatively small. Thus, although better treatments for HSV-associated encephalitis, VZV-infections and disorders associated with β- and γ-herpesvirus infection are clearly needed, the cost of developing new treatments, the relatively small markets and the success of existing drugs to combat herpesvirus infections has significantly dampened enthusiasm for herpesvirus programs at most pharmaceutical companies.
8.2 Limitations of current HPIs
In this article, we have outlined the current state of research for developing inhibitors of the helicase–primase complex. Although several promising compounds have been identified and two have undergone clinical trials, several questions remain about whether they will eventually have a major impact as a new treatment strategy. Although all herpesviruses encode a helicase–primase complex leading to hopes that HPIs would be broad spectrum and inhibit herpesviruses for which no good treatment options are available, the new HPIs have only been effective against the α-herpesviruses. Another potential limitation is that resistance may prove problematic, since resistant mutants pre-exist in clinical and laboratory isolates [117]; however, it should be noted that mutants resistant to nucleoside analogues also pre-exist in viral populations. Although BAY 57-1293 (AI316)-resistant isolates resulted in reduced viral growth in cell culture and in some cases reduced pathogenicity [118,125], other resistant isolates did not significantly impair viral growth [103,106]. If resistance mutants to particular drugs are associated with reduced viral fitness, this would diminish the likelihood of clinically important drug resistance, as is the case with many but not all acyclovir-resistant viruses. The mechanism of action of the various HPI inhibitors is unknown, necessitating detailed biochemical analysis; however, there seems to be little appetite for this type of analysis in the pharmaceutical industry. Lastly, the lack of structural information about the helicase–primase complex complicates efforts to use docking and rational drug design to improve existing compounds. In summary, developing HPIs into effective drugs to combat herpesvirus infections will require substantial work.
On the other hand, several points argue for continuing these studies. The initial Phase I and II trials of AIC316 (BAY 57-1293) are very promising [110,126], and it is possible that ASP2151 will undergo additional modifications resulting in safer and more effective compounds. New therapies will be useful in combating herpes isolates that have developed resistance to the DNA Pol inhibitors. Analogous to the multidrug approaches used to treating HIV, HPIs could be particular efficacious in combination with Pol inhibitors. Eventually HPI inhibitors may be discovered that have broad spectrum efficacy against all three families of herpesviruses. And lastly, inhibitors to H/P such as BAY-1293 have already been useful in the laboratory as a means to inhibit HSV DNA replication without inhibiting the Pol [62,72,123,124]. Thus, the eventual goals for development of effective HPI inhibitors should be to i) identify compounds that are more broad spectrum against all sub-families of herpesviruses, ii) determine more precisely the mechanism of action of the current HPIs and iii) better understand the consequence of the drug resistance mutants. It is promising that many of the HPI resistant mutants show reduced pathogenicity and inability to generate stable infection as is seen with Pol inhibitors.
8.3 Other potential targets for anti-herpesvirus therapy
In addition to HSV Pol and the H/P targets that inhibit viral DNA replication, other targets can be considered. Viral and cellular proteins required for viral gene expression, capsid assembly and encapsidation may provide exploitable targets such as immediate-early and early gene transactivators, encapsidation proteins such as the portal and the HCMV UL97 kinase. As mentioned above a compound that targets the HCMV terminase is undergoing clinical trials [35,36]. In addition, two other conserved viral proteins have recently been shown to play essential and novel roles in the HSV-1 life cycle. The HSV-1 alkaline nuclease, UL12, well conserved in all herpesviruses, has been shown to stimulate single strand annealing in HSV-infected cells [31] and is essential for virus production [128]. Another essential HSV protein, UL32, is required for genome encapsidation [27]. UL12 and UL32 are thus potential targets for future drug development. Furthermore, as we learn more about the roles of viral glycoproteins needed for attachment and penetration, it may be possible to develop drugs that can inhibit those important steps. Future drug discovery efforts are expected to combine bioinformatics, structural biology and high throughput screening. Another potentially promising strategy involves targeting essential cellular proteins which are required for viral processes such as gene expression, DNA replication and recombination, capsid assembly and packaging and viral egress [128]. One advantage of this strategy is that antiviral agents could be active against multiple viruses since it is becoming increasingly clear that the same cellular proteins are often required for replication of several different viruses. Another potential advantage is that it likely would be much more difficult for the virus to become resistant to drugs that target cellular proteins. One concern of course in developing agents that target cellular proteins would be possible cellular toxicity. Clearly herpesviruses offer a wide array of druggable targets although substantial hurdles remain in exploiting them.
Article highlights.
Overview of currently used anti-herpesvirus drugs.
Overview of HSV DNA replication.
Genetic and biochemical characterization of the HSV-1 H/P.
Spectrum of antiviral activity, resistance and mechanism of action of HPIs.
Expert opinion on future directions of the field.
This box summarizes key points contained in the article.
Acknowledgments
This work was supported by NIH grants AI-21747 (S.K.W.) and AI-059764 (R.D.K.).
Footnotes
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Chayavichitsilp P, Buckwalter JV, Krakowski AC, Friedlander SF. Herpes simplex. Ped Rev. 2009;30:119–29. doi: 10.1542/pir.30-4-119. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Raddad LJ, Magaret AS, Celum C, et al. Genital herpes has played a more important role than any other sexually transmitted infection in driving HIV prevalence in Africa. PLoS One. 2008;3:e2230. doi: 10.1371/journal.pone.0002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dedicoat M, Newton R. Review of the distribution of Kaposi's sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi's sarcoma. Br J Cancer. 2003;88:1–3. doi: 10.1038/sj.bjc.6600745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimberlin DW, Whitley RJ. Antiviral therapy of HSV-1 and -2. In: Arvin A, Campadelli-Fiume G, Mocarski E, et al., editors. Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press; Cambridge: 2007. [PubMed] [Google Scholar]
- 5.Crumpacker CS. Ganciclovir. N Engl J Med. 1996;335:721–9. doi: 10.1056/NEJM199609053351007. [DOI] [PubMed] [Google Scholar]
- 6.Arvin AM. Antiviral therapy for varicella and herpes zoster. Sem Ped Infect Dis. 2002;13:12–21. doi: 10.1053/spid.2002.29753. [DOI] [PubMed] [Google Scholar]
- 7.Wallace MR, Bowler WA, Murray NB, et al. Treatment of adult varicella with oral acyclovir. A randomized, placebo-controlled trial. Ann Intern Med. 1992;117:358–63. doi: 10.7326/0003-4819-117-5-358. [DOI] [PubMed] [Google Scholar]
- 8•.Bacon TH, Levin MJ, Leary JJ, et al. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev. 2003;16:114–28. doi: 10.1128/CMR.16.1.114-128.2003. Excellent review of why acyclovir and related copounds are so effective. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coen DM, Whitley RJ. Antiviral drugs and antiviral drug resistance. Curr Opinion Virol. 2011;1:545–7. doi: 10.1016/j.coviro.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Field HJ, Biswas S. Antiviral drug resistance and helicase-primase inhibitors of herpes simplex virus. Drug Resist Updat. 2011;14:45–51. doi: 10.1016/j.drup.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 11•.Reardon JE, Spector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J Biol Chem. 1989;264:7405–11. Demonstration of why the herpes polymerase cannot remove acyclovir from DNA. [PubMed] [Google Scholar]
- 12.Frobert E, Cortay JC, Ooka T, et al. Genotypic detection of acyclovir-resistant HSV-1: characterization of 67 ACV-sensitive and 14 ACV-resistant viruses. Antiviral Res. 2008;79:28–36. doi: 10.1016/j.antiviral.2008.01.153. [DOI] [PubMed] [Google Scholar]
- 13.Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55:459–72. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson C, Whitley R. Neonatal herpes simplex virus infections: where are we now? Adv Exp Med Biol. 2011;697:221–30. doi: 10.1007/978-1-4419-7185-2_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilsley DD, Lee SH, Miller WH, Kuchta RD. Acyclic guanosine analogs inhibit DNA polymerases alpha, delta, and epsilon with very different potencies and have unique mechanisms of action. Biochemistry. 1995;34:2504–10. doi: 10.1021/bi00008a014. [DOI] [PubMed] [Google Scholar]
- 16.McGuigan C, Pathirana RN, Migliore M, et al. Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus. J Antimicrob Chemother. 2007;60:1316–30. doi: 10.1093/jac/dkm376. [DOI] [PubMed] [Google Scholar]
- 17.Pentikis HS, Matson M, Atiee G, et al. Pharmacokinetics and safety of FV-100, a novel oral anti-herpes zoster nucleoside analogue, administered in single and multiple doses to healthy young adult and elderly adult volunteers. Antimicrob Agents Chemother. 2011;55:2847–54. doi: 10.1128/AAC.01446-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Squibb BM. A phase ii, multicenter, randomized, double-blind, parallel-group, comparative study of fv-100 vs. valacyclovir in patients with herpes zoster. Clinical Trial NCT00900783. 2010 Available from: http://clinicaltrials.gov/ct2/results?term=NCT00900783&Search=Search.
- 19.Rubsam LZ, Davidson BL, Shewach DS. Superior cytotoxicity with ganciclovir compared with acyclovir and 1-beta-D-arabinofuranosylthymine in herpes simplex virus-thymidine kinase-expressing cells: a novel paradigm for cell killing. Cancer Res. 1998;58:3873–82. [PubMed] [Google Scholar]
- 20.Cunha-Bang C, Kirkby N, Sonderholm M, et al. The time course of development and impact from viral resistance against ganciclovir in cytomegalovirus infection. Am J Transplant. 2013;13:458–66. doi: 10.1111/ajt.12042. [DOI] [PubMed] [Google Scholar]
- 21.Razonable RR, Zerr DM. Practice ASTIDCo. HHV-6, HHV-7 and HHV-8 in solid organ transplant recipients. Am J Transplant. 2009;9(Suppl 4):S97–100. doi: 10.1111/j.1600-6143.2009.02899_1.x. [DOI] [PubMed] [Google Scholar]
- 22.Casper C, Krantz EM, Corey L, et al. Valganciclovir for suppression of human herpesvirus-8 replication: a randomized, double-blind, placebo-controlled, crossover trial. J Infect Dis. 2008;198(1):23–30. doi: 10.1086/588820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6(5):401–10. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 24.Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol. 1997;136(5):1007–21. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ojala PM, Sodeik B, Ebersold MW, et al. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol. 2000;20(13):4922–31. doi: 10.1128/mcb.20.13.4922-4931.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Bruyn Kops A, Knipe DM. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell. 1988;55(5):857–68. doi: 10.1016/0092-8674(88)90141-9. [DOI] [PubMed] [Google Scholar]
- 27.Lamberti C, Weller SK. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J Virol. 1998;72(3):2463–73. doi: 10.1128/jvi.72.3.2463-2473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phelan A, Dunlop J, Patel AH, et al. Nuclear sites of herpes simplex virus type 1 DNA replication and transcription colocalize at early times postinfection and are largely distinct from RNA processing factors. J Virol. 1997;71(2):1124–32. doi: 10.1128/jvi.71.2.1124-1132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben-Porat T, Rixon FJ. Replication of herpesvirus DNA. IV: analysis of concatemers. Virology. 1979;94(1):61–70. doi: 10.1016/0042-6822(79)90438-0. [DOI] [PubMed] [Google Scholar]
- 30.Boehmer P. Herpes simplex virus DNA replication. Ann Rev Biochem. 1997 doi: 10.1146/annurev.biochem.66.1.347. [DOI] [PubMed] [Google Scholar]
- 31.Schumacher AJ, Mohni KN, Kan Y, et al. The HSV-1 Exonuclease, UL12, Stimulates Recombination by a Single Strand Annealing Mechanism. PLoS Pathog. 2012;8(8):e1002862. doi: 10.1371/journal.ppat.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkinson DE, Weller SK. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life. 2003;55(8):451–8. doi: 10.1080/15216540310001612237. [DOI] [PubMed] [Google Scholar]
- 33.Baines J, Weller SK. Cleavage and packaging of herpes simplex virus 1 DNA. In: Catalano C, editor. Viral genome packaging machines: genetics, structure and mechanism. Landes Bioscience; Austin, TX: 2005. [Google Scholar]
- 34.Conway JF, Homa F. Nucleocapsid structure, assembly and DNA packaging of herpes simplex viru. In: Weller SK, editor. Alphaheresviruses: molecular virology. Caister Academic Press; Norwich UK: 2011. [Google Scholar]
- 35.Goldner T, Hewlett G, Ettischer N, et al. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol. 2011;85(20):10884–93. doi: 10.1128/JVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lischka P, Hewlett G, Wunberg T, et al. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother. 2010;54(3):1290–7. doi: 10.1128/AAC.01596-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mettenleiter TC, Klupp BG, Granzow H. Herpesvirus assembly: a tale of two membranes. Curr Opin Microbiol. 2006;9(4):423–9. doi: 10.1016/j.mib.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Weller SK, Spadaro A, Schaffer JE, et al. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol Cell Biol. 1985;5(5):930–42. doi: 10.1128/mcb.5.5.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stow ND. Localization of an origin of DNA replication within the TRS/IRS repeated region of the herpes simplex virus type 1 genome. EMBO J. 1982;1(7):863–7. doi: 10.1002/j.1460-2075.1982.tb01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward S, Weller S. HSV-1 DNA Replication. Alphaherpesviruses: Mol Virol; 2011. [Google Scholar]
- 41.Weller SK, Coen DM. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol. 2012;4(9):a013011. doi: 10.1101/cshperspect.a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blumel J, Matz B. Thermosensitive UL9 gene function is required for early stages of herpes simplex virus type 1 DNA synthesis. J Gen Virol. 1995;76(Pt 12):3119–24. doi: 10.1099/0022-1317-76-12-3119. [DOI] [PubMed] [Google Scholar]
- 43.Schildgen O, Graper S, Blumel J, Matz B. Genome replication and progeny virion production of herpes simplex virus type 1 mutants with temperature-sensitive lesions in the origin-binding protein. J Virol. 2005;79(11):7273–8. doi: 10.1128/JVI.79.11.7273-7278.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown SM, Ritchie DA, Subak-Sharpe JH. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J Gen Virol. 1973;18(3):329–46. doi: 10.1099/0022-1317-18-3-329. [DOI] [PubMed] [Google Scholar]
- 45.Honess RW, Buchan A, Halliburton IW, Watson DH. Recombination and linkage between structural and regulatory genes of herpes simplex virus type 1: study of the functional organization of the genome. J Virol. 1980;34(3):716–42. doi: 10.1128/jvi.34.3.716-742.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaffer PA, Tevethia MJ, Benyesh-Melnick M. Recombination between temperature-sensitive mutants of herpes simplex virus type 1. Virology. 1974;58(1):219–28. doi: 10.1016/0042-6822(74)90156-1. [DOI] [PubMed] [Google Scholar]
- 47.Kintner RL, Allan RW, Brandt CR. Recombinants are isolated at high frequency following in vivo mixed ocular infection with two avirulent herpes simplex virus type 1 strains. Arch Virol. 1995;140(2):231–44. doi: 10.1007/BF01309859. [DOI] [PubMed] [Google Scholar]
- 48.Lingen M, Hengerer F, Falke D. Mixed vaginal infections of Balb/c mice with low virulent herpes simplex type 1 strains result in restoration of virulence properties: vaginitis/vulvitis and neuroinvasiveness. Med Microbiol Immunol (Berl) 1997;185(4):217–22. doi: 10.1007/s004300050033. [DOI] [PubMed] [Google Scholar]
- 49.Bowden R, Sakaoka H, Donnelly P, Ward R. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect Genet Evol. 2004;4(2):115–23. doi: 10.1016/j.meegid.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 50.Dutch RE, Bianchi V, Lehman IR. Herpes simplex virus type 1 DNA replication is specifically required for high-frequency homologous recombination between repeated sequences. J Virol. 1995;69(5):3084–9. doi: 10.1128/jvi.69.5.3084-3089.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dutch RE, Bruckner RC, Mocarski ES, Lehman IR. Herpes simplex virus type 1 recombination: role of DNA replication and viral a sequences. J Virol. 1992;66(1):277–85. doi: 10.1128/jvi.66.1.277-285.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber PC, Challberg MD, Nelson NJ, et al. Inversion events in the HSV-1 genome are directly mediated by the viral DNA replication machinery and lack sequence specificity. Cell. 1988;54(3):369–81. doi: 10.1016/0092-8674(88)90200-0. [DOI] [PubMed] [Google Scholar]
- 53.Jacob RJ, Roizman B. Anatomy of herpes simplex virus DNA VIII. Properties of the replicating DNA. J Virol. 1977;23(2):394–411. doi: 10.1128/jvi.23.2.394-411.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jean JH, Blankenship ML, Ben-Porat T. Replication of herpesvirus DNA. I. Electron microscopic analysis of replicative structures. Virology. 1977;79(2):281–91. doi: 10.1016/0042-6822(77)90355-5. [DOI] [PubMed] [Google Scholar]
- 55.Shlomai J, Friedmann A, Becker Y. Replication intermediates of herpes simplex virus DNA. Virology. 1976;69(2):647–59. doi: 10.1016/0042-6822(76)90493-1. [DOI] [PubMed] [Google Scholar]
- 56.Severini A, Scraba DG, Tyrrell DL. Branched structures in the intracellular DNA of herpes simplex virus type 1. J Virol. 1996;70(5):3169–75. doi: 10.1128/jvi.70.5.3169-3175.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinez R, Sarisky RT, Weber PC, Weller SK. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol. 1996;70(4):2075–85. doi: 10.1128/jvi.70.4.2075-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Severini A, Morgan AR, Tovell DR, Tyrrell DL. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology. 1994;200(2):428–35. doi: 10.1006/viro.1994.1206. [DOI] [PubMed] [Google Scholar]
- 59.Zhang X, Efstathiou S, Simmons A. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: implications for viral DNA amplification strategies. Virology. 1994;202(2):530–9. doi: 10.1006/viro.1994.1375. [DOI] [PubMed] [Google Scholar]
- 60.Lamberti C, Weller SK. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology. 1996;226(2):403–7. doi: 10.1006/viro.1996.0668. [DOI] [PubMed] [Google Scholar]
- 61.Mohni KN, Dee AR, Smith S, et al. Efficient herpes simplex virus 1 replication requires cellular ATR pathway proteins. J Virol. 2013;87(1):531–42. doi: 10.1128/JVI.02504-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mohni KN, Livingston CM, Cortez D, Weller SK. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J Virol. 2010;84(23):12152–64. doi: 10.1128/JVI.01643-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohni KN, Mastrocola AS, Bai P, et al. DNA Mismatch Repair Proteins Are Required For Efficient Herpes Simplex Virus Type I Replication. J Virol. 2011 doi: 10.1128/JVI.05487-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weitzman M, Weller SK. Interactions Between HSV-1 and the DNA Damage Response. Alphaherpesviruses: Mol Virol; 2011. [Google Scholar]
- 65.Weller SK. Herpes simplex virus reorganizes the cellular DNA repair and protein quality control machinery. PLoS Pathog. 2010;6(11):e1001105. doi: 10.1371/journal.ppat.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carmichael EP, Weller SK. Herpes simplex virus type 1 DNA synthesis requires the product of the UL8 gene: isolation and characterization of an ICP6::lacZ insertion mutation. J Virol. 1989;63(2):591–9. doi: 10.1128/jvi.63.2.591-599.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67•.Goldstein DJ, Weller SK. An ICP6:lacZ insertional mutagen is used to demonstrate that the UL52 gene of herpes simplex virus type 1 is required for virus growth and DNA synthesis. J Virol. 1988;62(8):2970–7. doi: 10.1128/jvi.62.8.2970-2977.1988. First isolation of an insertion mutant in UL52 subsequently shown to be the primase subunit of the helicase-primase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68•.Zhu L, Weller SK. UL5, a protein required for HSV DNA synthesis: genetic analysis, overexpression in Escherichia coli, and generation of polyclonal antibodies. Virology. 1988;166(2):366–78. doi: 10.1016/0042-6822(88)90507-7. First genentic analysis of UL5 gene subsuquently shown to be the helicase subunit of the helicase-primase. [DOI] [PubMed] [Google Scholar]
- 69••.Zhu LA, Weller SK. The six conserved helicase motifs of the UL5 gene product, a component of the herpes simplex virus type 1 helicase-primase, are essential for its function. J Virol. 1992;66(1):469–79. doi: 10.1128/jvi.66.1.469-479.1992. First genetic analysis of the helicase motifs of UL5 showing that they are all essential. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu LA, Weller SK. The UL5 gene of herpes simplex virus type 1: isolation of a lacZ insertion mutant and association of the UL5 gene product with other members of the helicase-primase complex. J Virol. 1992;66(1):458–68. doi: 10.1128/jvi.66.1.458-468.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carrington-Lawrence SD, Weller SK. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J Virol. 2003;77(7):4237–47. doi: 10.1128/JVI.77.7.4237-4247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72••.Chen Y, Livingston CM, Carrington-Lawrence SD, et al. A mutation in the human herpes simplex virus type 1 UL52 zinc finger motif results in defective primase activity but can recruit viral polymerase and support viral replication efficiently. J Virol. 2007;81(16):8742–51. doi: 10.1128/JVI.00174-07. First demonstration that the UL52 zinc finger is imortant for primase activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lehman IR, Boehmer PE. Replication of herpes simplex virus DNA. J Biol Chem. 1999;274(40):28059–62. doi: 10.1074/jbc.274.40.28059. [DOI] [PubMed] [Google Scholar]
- 74••.Stengel G, Kuchta RD. Coordinated leading and lagging strand DNA synthesis by using the herpes simplex virus 1 replication complex and minicircle DNA templates. J Virol. 2011;85(2):957–67. doi: 10.1128/JVI.01688-10. First demonstration of efficient coordinated leading and lagging strand DNA synthesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75••.Graves-Woodward KL, Gottlieb J, Challberg MD, Weller SK. Biochemical analyses of mutations in the HSV-1 helicase-primase that alter ATP hydrolysis, DNA unwinding, and coupling between hydrolysis and unwinding. J Biol Chem. 1997;272(7):4623–30. doi: 10.1074/jbc.272.7.4623. First demonstration that the helicase motifs of UL5 are essential for biochemical activities of the helicase-primase complex. [DOI] [PubMed] [Google Scholar]
- 76••.Dracheva S, Koonin EV, Crute JJ. Identification of the primase active site of the herpes simplex virus type 1 helicase-primase. J Biol Chem. 1995;270(23):14148–53. doi: 10.1074/jbc.270.23.14148. Along with Ref. [77], this paper identified the primase active site of UL52. [DOI] [PubMed] [Google Scholar]
- 77••.Klinedinst DK, Challberg MD. Helicase-primase complex of herpes simplex virus type 1: a mutation in the UL52 subunit abolishes primase activity. J Virol. 1994;68(6):3693–701. doi: 10.1128/jvi.68.6.3693-3701.1994. Along with Ref. [76], this paper identified the primase active site of UL52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78•.Chen Y, Carrington-Lawrence SD, Bai P, Weller SK. Mutations in the putative zinc-binding motif of UL52 demonstrate a complex interdependence between the UL5 and UL52 subunits of the human herpes simplex virus type 1 helicase/ primase complex. J Virol. 2005;79(14):9088–96. doi: 10.1128/JVI.79.14.9088-9096.2005. Demonstration of complex interdependence between UL5 and UL52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calder JM, Stow ND. Herpes simplex virus helicase-primase: the UL8 protein is not required for DNA-dependent ATPase and DNA helicase activities. Nucleic Acids Res. 1990;18(12):3573–8. doi: 10.1093/nar/18.12.3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80•.Dodson MS, Lehman IR. Association of DNA helicase and primase activities with a subassembly of the herpes simplex virus 1 helicase-primase composed of the UL5 and UL52 gene products. Proc Natl Acad Sci U S A. 1991;88(4):1105–9. doi: 10.1073/pnas.88.4.1105. Demonstration of complex interdependence between UL5 and UL52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sherman G, Gottlieb J, Challberg MD. The UL8 subunit of the herpes simplex virus helicase-primase complex is required for efficient primer utilization. J Virol. 1992;66(8):4884–92. doi: 10.1128/jvi.66.8.4884-4892.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82•.Barnard EC, Brown G, Stow ND. Deletion mutants of the herpes simplex virus type 1 UL8 protein: effect on DNA synthesis and ability to interact with and influence the intracellular localization of the UL5 and UL52 proteins. Virology. 1997;237(1):97–106. doi: 10.1006/viro.1997.8763. Demonstration that UL8 can facilitate the nuclear transport of UL5 and UL52 into the nucleus. [DOI] [PubMed] [Google Scholar]
- 83•.Tenney DJ, Hurlburt WW, Micheletti PA, et al. The UL8 component of the herpes simplex virus helicase-primase complex stimulates primer synthesis by a subassembly of the UL5 and UL52 components. J Biol Chem. 1994;269(7):5030–5. Important demonstration that UL8 can enhnce primer synthesis. [PubMed] [Google Scholar]
- 84.Falkenberg M, Bushnell D, Elias P, Lehman IR. The UL8 subunit of the heterotrimeric herpes simplex virus type 1 helicase-primase is required for the unwinding of single strand DNA-binding protein (ICP8)-coated DNA substrates. J Biol Chem. 1997;272(36):22766–70. doi: 10.1074/jbc.272.36.22766. [DOI] [PubMed] [Google Scholar]
- 85•.Hamatake R, Bifano M, Hurlburt WW, Tenney DJ. A functional interaction of ICP8, the herpes simplex virus single-stranded DNA-binding protein, and the helicase-primase complex that is dependent on the presence of the UL8 subunit. J General Virol. 1997;78(Pt 4):857–65. doi: 10.1099/0022-1317-78-4-857. Demonstration that UL8 can interact functionally with ICP8. [DOI] [PubMed] [Google Scholar]
- 86••.Cavanaugh NA, Ramirez-Aguilar KA, Urban M, Kuchta RD. Herpes simplex virus-1 helicase-primase: roles of each subunit in DNA binding and phosphodiester bond formation. Biochemistry. 2009;48(43):10199–207. doi: 10.1021/bi9010144. Important characterization of the roles of each subunit of the helicase-primase complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87•.Marsden HS, McLean GW, Barnard EC, et al. The catalytic subunit of the DNA polymerase of herpes simplex virus type 1 interacts specifically with the C terminus of the UL8 component of the viral helicase-primase complex. J Virol. 1997;71(9):6390–7. doi: 10.1128/jvi.71.9.6390-6397.1997. First demonstration of an interaction between UL8 and UL30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McLean GW, Abbotts AP, Parry ME, et al. The herpes simplex virus type 1 origin-binding protein interacts specifically with the viral UL8 protein. J Gen Virol. 1994;75(Pt 10):2699–706. doi: 10.1099/0022-1317-75-10-2699. [DOI] [PubMed] [Google Scholar]
- 89•.Muylaert I, Zhao Z, Andersson T, Elias P. Identification of conserved amino acids in the herpes simplex virus type 1 UL8 protein required for DNA synthesis and UL52 primase interaction in the virus replisome. J Biol Chem. 2012;287(40):33142–52. doi: 10.1074/jbc.M112.356782. Identificaiton of residues in UL8 required for interaction with UL52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kulczyk AW, Richardson CC. Molecular interactions in the priming complex of bacteriophage T7. Proc Natl Acad Sci USA. 2012;109(24):9408–13. doi: 10.1073/pnas.1207033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee SJ, Richardson CC. Choreography of bacteriophage T7 DNA replication. Curr Opin Chem Biol. 2011;15(5):580–6. doi: 10.1016/j.cbpa.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Patel SS, Pandey M, Nandakumar D. Dynamic coupling between the motors of DNA replication: hexameric helicase, DNA polymerase, and primase. Curr Opin Chem Biol. 2011;15(5):595–605. doi: 10.1016/j.cbpa.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frick DN, Richardson CC. DNA primases. Annu Rev Biochem. 2001;70:39–80. doi: 10.1146/annurev.biochem.70.1.39. [DOI] [PubMed] [Google Scholar]
- 94.Biswas N, Weller SK. A mutation in the C-terminal putative Zn2+ finger motif of UL52 severely affects the biochemical activities of the HSV-1 helicase-primase subcomplex. J Biol Chem. 1999;274(12):8068–76. doi: 10.1074/jbc.274.12.8068. [DOI] [PubMed] [Google Scholar]
- 95•.Biswas N, Weller SK. The UL5 and UL52 subunits of the herpes simplex virus type 1 helicase-primase subcomplex exhibit a complex interdependence for DNA binding. J Biol Chem. 2001;276(20):17610–19. doi: 10.1074/jbc.M010107200. Demonstration of the complex interdependence between UL5 and UL52. [DOI] [PubMed] [Google Scholar]
- 96••.Chen Y, Bai P, Mackay S, et al. Herpes simplex virus type 1 helicase-primase: DNA binding and consequent protein oligomerization and primase activation. J Virol. 2011;85(2):968–78. doi: 10.1128/JVI.01690-10. First demonstration that primase activity of the helicase-primase complex is dependent on concentration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang Y, Dou SX, Ren H, et al. Evidence for a functional dimeric form of the PcrA helicase in DNA unwinding. Nucleic Acids Res. 2008;36(6):1976–89. doi: 10.1093/nar/gkm1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maluf NK, Fischer CJ, Lohman TM. A Dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J Mol Biol. 2003;325(5):913–35. doi: 10.1016/s0022-2836(02)01277-9. [DOI] [PubMed] [Google Scholar]
- 99.Niedziela-Majka A, Chesnik MA, Tomko EJ, Lohman TM. Bacillus stearothermophilus PcrA monomer is a single-stranded DNA translocase but not a processive helicase in vitro. J Biol Chem. 2007;282(37):27076–85. doi: 10.1074/jbc.M704399200. [DOI] [PubMed] [Google Scholar]
- 100.Lee SJ, Marintcheva B, Hamdan SM, Richardson CC. The C-terminal residues of bacteriophage T7 gene 4 helicase-primase coordinate helicase and DNA polymerase activities. J Biol Chem. 2006;281(35):25841–9. doi: 10.1074/jbc.M604602200. [DOI] [PubMed] [Google Scholar]
- 101.Kim S, Dallmann HG, McHenry CS, Marians KJ. Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell. 1996;84(4):643–50. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 102•.Marsden HS, Cross AM, Francis GJ, et al. The herpes simplex virus type 1 UL8 protein influences the intracellular localization of the UL52 but not the ICP8 or POL replication proteins in virus-infected cells. J Gen Virol. 1996;77(Pt 9):2241–9. doi: 10.1099/0022-1317-77-9-2241. First demonstration that UL8 can facilitate the nuclear transport of UL5 and UL52 into the nucleus. [DOI] [PubMed] [Google Scholar]
- 103••.Spector FC, Liang L, Giordano H, et al. Inhibition of herpes simplex virus replication by a 2-amino thiazole via interactions with the helicase component of the UL5-UL8-UL52 complex. J Virol. 1998;72(9):6979–87. doi: 10.1128/jvi.72.9.6979-6987.1998. First small molecule inhibitor of helicase-primase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104••.Crute JJ, Grygon CA, Hargrave KD, et al. Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nat Med. 2002;8(4):386–91. doi: 10.1038/nm0402-386. Among the first description of helicase-primase inhibitors. [DOI] [PubMed] [Google Scholar]
- 105•.Duan J, Liuzzi M, Paris W, et al. Oral bioavailability and in vivo efficacy of the helicase-primase inhibitor BILS 45 BS against acyclovir-resistant herpes simplex virus type 1. Antimicrob Agents Chemother. 2003;47(6):1798–804. doi: 10.1128/AAC.47.6.1798-1804.2003. Early characerization of in vivo efficacy of new class of helicase-primse inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106••.Liuzzi M, Kibler P, Bousquet C, et al. Isolation and characterization of herpes simplex virus type 1 resistant to arninothiazolylphenyl-based inhibitors of the viral helicase-primase. Antiviral Res. 2004;64(3):161–70. doi: 10.1016/j.antiviral.2004.02.007. Early characterization of mutants resistant to a new class of helicase-primase inhibitors. [DOI] [PubMed] [Google Scholar]
- 107••.Kleymann G, Fischer R, Betz UA, et al. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat Med. 2002;8(4):392–8. doi: 10.1038/nm0402-392. Among the first description of helicase-primase inhibitors. [DOI] [PubMed] [Google Scholar]
- 108.Baumeister J, Fischer R, Eckenberg P, et al. Superior efficacy of helicase-primase inhibitor BAY 57-1293 for herpes infection and latency in the guinea pig model of human genital herpes disease. Antivir Chem Chemother. 2007;18(1):35–48. doi: 10.1177/095632020701800104. [DOI] [PubMed] [Google Scholar]
- 109.Biswas S, Jennens L, Field HJ. The helicase primase inhibitor, BAY 57-1293 shows potent therapeutic antiviral activity superior to famciclovir in BALB/c mice infected with herpes simplex virus type 1. Antiviral Res. 2007;75(1):30–5. doi: 10.1016/j.antiviral.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 110.AiCuris. A Double-blind, Double Dummy, Randomized Crossover Trial to Compare the Effect of AIC316 100 mg Once Daily Versus Valacyclovir 500 mg Once Daily on Genital HSV Shedding in HSV-2 Seropositive Adults. Clinical Trial NCT01658826. 2013 Available from: http://clinicaltrials.gov/ct2/show/NCT01658826.
- 111••.Chono K, Katsumata K, Kontani T, et al. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J Antimicrob Chemother. 2010;65(8):1733–41. doi: 10.1093/jac/dkq198. First report of ASP2151. [DOI] [PubMed] [Google Scholar]
- 112.Katsumata K, Chono K, Kato K, et al. Pharmacokinetics and Pharmacodynamics of ASP2151, a Helicase-Primase Inhibitor, in a Murine Model of Herpes Simplex Virus Infection. Antimicrob Agents Chemother. 2013;57(3):1339–46. doi: 10.1128/AAC.01803-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sasaki SI, Miyazaki D, Haruki T, et al. Efficacy of herpes virus helicase-primase inhibitor, ASP2151, for treating herpes simplex keratitis in mouse model. Br J Ophthalmol. 2013;97(4):498–503. doi: 10.1136/bjophthalmol-2012-302062. [DOI] [PubMed] [Google Scholar]
- 114.Astellas. A Phase II, Dose-Finding Study With ASP2151 in Subjects With Recurrent Episodes of Genital HErpes. Clinical trial NCT00486200. 2008 Available from: http://clinicaltrials.gov/show/NCT00486200.
- 115.Tyring S, Wald A, Zadeikis N, et al. ASP2151 for the treatment of genital herpes: a randomized, double-blind, placebo- and valacyclovir-controlled, dose-finding study. J Infect Dis. 2012;205(7):1100–10. doi: 10.1093/infdis/jis019. [DOI] [PubMed] [Google Scholar]
- 116.Astellas. A Phase 1, Randomized, Double-Blind, Multiple Dose, Multi-Center Study to Compare the Safety of ASP2151 to Valacylcovir and Placebo in Healthy Male and Female Subjects. Clinical Trial NCT00870441. 2010 Available from: http://clinicaltrials.cov/ct2/show/NCT00870441.
- 117••.Sukla S, Biswas S, Birkmann A, et al. Mismatch primer-based PCR reveals that helicase-primase inhibitor resistance mutations pre-exist in herpes simplex virus type 1 clinical isolates and are not induced during incubation with the inhibitor. J Antimicrob Chemother. 2010;65(7):1347–52. doi: 10.1093/jac/dkq026. Demonstration that mutants resistant to helicase-primase inhibitors pre-exist in clinical isolates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Biswas S, Miguel RN, Sukla S, Field HJ. A mutation in helicase motif IV of herpes simplex virus type 1 UL5 that results in reduced growth in vitro and lower virulence in a murine infection model is related to the predicted helicase structure. J Gen Virol. 2009;90(Pt 8):1937–42. doi: 10.1099/vir.0.011221-0. [DOI] [PubMed] [Google Scholar]
- 119.Korolev S, Hsieh J, Gauss GH, et al. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90(4):635–47. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 120.Biswas S, Kleymann G, Swift M, et al. A single drug-resistance mutation in HSV-1 UL52 primase points to a difference between two helicase-primase inhibitors in their mode of interaction with the antiviral target. J Antimicrob Chemother. 2008;61(5):1044–7. doi: 10.1093/jac/dkn057. [DOI] [PubMed] [Google Scholar]
- 121.Chono K, Katsumata K, Kontani T, et al. Characterization of virus strains resistant to the herpes virus helicase-primase inhibitor ASP2151 (Amenamevir) Biochem Pharmacol. 2012;84(4):459–67. doi: 10.1016/j.bcp.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 122.Kleymann G. Agents and strategies in development for improved management of herpes simplex virus infection and disease. Expert Opin Investig Drugs. 2005;14(2):135–61. doi: 10.1517/13543784.14.2.135. [DOI] [PubMed] [Google Scholar]
- 123.Livingston CM, DeLuca NA, Wilkinson DE, Weller SK. Oligomerization of ICP4 and rearrangement of heat shock proteins may be important for herpes simplex virus type 1 prereplicative site formation. J Virol. 2008;82(13):6324–36. doi: 10.1128/JVI.00455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wilkinson DE, Weller SK. Inhibition of the herpes simplex virus type 1 DNA polymerase induces hyperphosphorylation of replication protein A and its accumulation at S-phase-specific sites of DNA damage during infection. J Virol. 2005;79(11):7162–71. doi: 10.1128/JVI.79.11.7162-7171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Biswas S, Field HJ. Herpes simplex virus helicase-primase inhibitors: recent findings from the study of drug resistance mutations. Antivir Chem Chemother. 2008;19(1):1–6. doi: 10.1177/095632020801900101. [DOI] [PubMed] [Google Scholar]
- 126.AiCuris. A Double-blind Randomized Placebo Controlled Dose-finding Trial to Investigate Different Doses of a New Antiviral Drug in Subjects With Genital HSV Type 2 Infection. Clinical Trial NCT01047540. 2011 Available from: http://clinicaltrials.gov/ct2/results?term=NCT01047540&Search=Search.
- 127.Shao L, Rapp LM, Weller SK. Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology. 1993;196(1):146–62. doi: 10.1006/viro.1993.1463. [DOI] [PubMed] [Google Scholar]
- 128.Fauci AS, Challberg MD. Host-based antipoxvirus therapeutic strategies: turning the tables. J Clin Investigat. 2005;115(2):231–3. doi: 10.1172/JCI24270. [DOI] [PMC free article] [PubMed] [Google Scholar]