

Abstract
Microbial components such as lipopolysaccharide (LPS) bind to Toll-like receptors (TLR) and activate innate and inflammatory responses. Responses to LPS and other microbial components are limited by the activation of negative feedback mechanisms that reduce responsiveness to subsequent LPS exposure, often termed LPS tolerance. Our laboratory has previously shown that calcineurin, a phosphatase known for its activation of T cells via NFAT, negatively regulates the TLR pathway in macrophages; consequently, calcineurin inhibitors (FK506 and Cyclosporin A) mimic TLR ligands in activating the TLR pathway, NF-κB, and associated innate and inflammatory responses. This study investigated the physiological consequences of calcineurin inactivation for LPS-induced inflammatory responses in vitro and in vivo using two models: calcineurin inhibition by FK506 (tacrolimus) and myeloid cell-specific calcineurin deletion. Activation of dendritic cells and macrophages with FK506 in vitro was shown to induce a state of reduced responsiveness to LPS (i.e., a form of LPS tolerance). Similarly, macrophages from FK506-treated mice or from mice in which the calcineurin B1 (CnB1) subunit was conditionally knocked out in myeloid cells were found to have diminished LPS-induced inflammatory responses. In addition, mice with CnB1-deficient myeloid cells and mice undergoing FK506 treatment showed improved survival and recovery when challenged with high doses of systemic LPS compared to controls. These results demonstrate that inactivation of calcineurin in macrophages and other myeloid cells by inhibition or deletion can induce a form of LPS tolerance and protect the host from LPS toxicity in vivo.
Keywords: monocytes/macrophages, dendritic cells, cell activation, endotoxin shock, LPS tolerance, calcineurin, FK506
Introduction
The mammalian innate immune system uses a variety of strategies to recognize and defend against invading pathogens. The recognition of “microbial non-self” is thought to be a universal strategy of innate immunity as it is found in all studied multicellular organisms. Mammalian Toll-like receptors (TLR), homologous to the Toll-1 anti-fungal receptor in Drosophila, are expressed by many cell types, including dendritic cells and macrophages, which express an abundant array of TLRs and play a key role in innate immunity by binding pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS) 1, 2. LPS, also called endotoxin, is a component of the cell membrane of Gram-negative bacteria and is one of the most potent activators of innate immune responses. LPS binds to TLR4, leading to activation of downstream pathways that can activate NF-κB and other transcription factors such as IRF3 3, 4. NF-κB is a major and essential activator of inflammatory processes, inducing expression and secretion of cytokines (TNF- α, IL-12, IL-1α, IL-1β, IL-6), adhesion molecules (ELAM-1, VCAM-1, ICAM-1), chemokines (IL-8, MCP-1, RANTES), enzymes (matrix metalloproteinases, cyclooxygenase-2 (COX-2), inducible nitric oxide synthase), and other mediators 5.
Innate immune responses are highly regulated. For example, to minimize harmful consequences of excessive response to bacterial infection, such as septic shock, LPS-induced production of inflammatory mediators is limited by the activation of negative feedback mechanisms that lead to unresponsiveness or “tolerance” to subsequent LPS exposure (often referred to as endotoxin tolerance or LPS tolerance). When animals and humans are exposed to even small doses of LPS, inhibitory mechanisms are initiated that cause them to become less susceptible to the lethal effects of subsequent exposure to LPS 6, 7. One study showed that rats that had been pretreated with endotoxin survived a challenge dose of endotoxin that was 100% lethal for naïve rats 8. This diminished susceptibility was shown to be associated with an absent or decreased cellular response to LPS. Priming of macrophages or dendritic cells with LPS can render them unresponsive or “tolerant” for up to several days following initial exposure 9–11, leaving them unable to respond to subsequent exposure to LPS and other TLR ligands, sometimes termed `homotolerance' and `heterotolerance', respectively 8, 12, 13. Additionally, macrophages from patients who have survived exposure to endotoxin during infection have decreased or absent response to endotoxin exposure in vitro 14, 15, which has also been shown for macrophages from healthy volunteers injected with LPS 16.
Calcineurin, also called protein phosphatase 2B (PP2B), was originally discovered as a major calmodulin-binding protein in the brain over twenty years ago 17. Calcineurin was subsequently determined to be the only known Ca2+ and calmodulin-activated serine/threonine phosphatase, and it is highly conserved from yeast to man 18. Calcineurin is composed of a catalytic subunit, CnA (69-–62 kD), and a regulatory subunit, CnB (18–19 kD). CnA exists as three isoforms in vertebrates, of which CnAα and CnAβ are ubiquitous expressed, while CnAγ is only expressed in testis and brain. There are two CnB isoforms: CnB1 is ubiquitously expressed, while CnB2 is only found in testis 19–21.
The discovery of the effectiveness of calcineurin inhibitors cyclosporin A (CsA) and tacrolimus (FK506) as immunosuppressants has revolutionized transplantation 22–26. The identification of calcineurin as the target of these drugs 27 led to the discovery that calcium-flux activated calcineurin is responsible for the dephosphorylation and activation of NFAT, required for the activation of T-cell responses 28–30. More recently the activation of NFAT by calcineurin has also been shown to be involved in B-cell responses 31. Currently, patients undergoing transplantation often receive calcineurin inhibitors as part of their immunosuppression regimen to prevent rejection. Calcineurin inhibitors are also used as treatment for inflammatory skin diseases 32 and autoimmune disease 32–34.
In addition to the central role of calcineurin in activating adaptive immune responses, our lab has demonstrated that calcineurin is involved in regulating the Toll-like receptor pathway in some cell types, including monocytes, macrophages, astrocytes, and fibroblasts 35, 36. However, in these cells calcineurin plays a negative regulatory role; calcineurin inhibitors have activating, pro-inflammatory effects, in contrast to the inhibitory effects seen in T cells. Calcineurin inhibitors do not block LPS-induced cytokine expression in macrophages; instead, in the absence of LPS or other stimuli they induce expression of a number of pro-inflammatory macrophage effector genes. We have shown that treatment of peritoneal macrophages with CsA or FK506 activates NF-κB, mRNA expression of pro-inflammatory genes TNF-α, IL-1α, IL-1β, IL-12, and iNOS and secretion of TNF-α and IL-12 when given with IFN-γ 35. Also, calcineurin inhibitors and siRNA knockdown of calcineurin activated the intermediates of the IL-1R/TLR-mediated NF-κB activation pathway (both MyD88-dependent and MyD88-independent) in macrophages. Conversely, expression of a constitutively-active form of calcineurin was shown to block macrophage activation by TLR ligands. The results of knockout and immunoprecipitation experiments suggest that TLR-proximal signaling components (some of the TLR and the adapter proteins MyD88 and TRIF) are the key direct or indirect targets of calcineurin 36.
The pro-inflammatory effects of calcineurin inhibitors predict several possible physiological consequences of calcineurin inhibition in vivo. Our previously-published in vitro findings that calcineurin inhibitors activate the TLR pathway in macrophages and other innate immune cells suggest that inhibition of calcineurin in these cells in vivo could lead to an ongoing systemic inflammatory response or to sensitization to LPS and/or pathogens. Alternatively, calcineurin inhibitors might induce a form of LPS tolerance in macrophages and dendritic cells. Studies reported here show the latter to be the case. Exposure to calcineurin inhibitors in vitro or in vivo induces tolerance to LPS in macrophages and denditritic cells, and in vivo studies show that prolonged inactivation of calcineurin in myeloid cells by inhibition or deletion leads to protection against LPS-induced toxicity. These findings suggest that treatment with calcineurin inhibitors may suppress innate and inflammatory responses as well as adaptive immune responses.
Materials and Methods
Animals
CD11b-cre+ mice were generously provided by Jean Vacher 37. The CD11b-cre line used in the crosses contains ~4 copies of the cre transgene integrated on the Y chromosome under control of the CD11b promoter. Cnb1f/Δ mice were generously provided by Gerald Crabtree 38. Cnb1f/Δ mice carry on one chromosome the Cnb1f allele, in which the Cnb1 gene is flanked by loxP sites, and on the other a deletion of the Cnb1 gene, i.e. a null Cnb1 allele (Cnb1Δ). CD11b-cre+; Cnb1f/Δ mice were generated by breeding Cnb1f/Δ mice with CD11b-cre+ mice. Because only males carry the transgene, all experiments involving CD11b-cre+; Cnb1f/Δ mice included only male mice. Cnb1 screening of tail DNA was done with the following primers, CNB15′ΔB, 5′-CAATGCAGTCCGCTGTAGTTC-3′; LOXP3′AMP/SEQ, 5′- GACAGCTATACAGAGAAACCCTG-3′; LOXP3′AMP, 5′-AGCCTCCACATACACAGATAC-3′, and where necessary, Cre recombinase transgenic screening using the following primers, forward, 5'AATGCTTCTGTCCGTTTGC 3' and reverse, 5'-CGGCAACACCATTTTTTCTG-3'. The Cnb1+ product, 189 bp, Cnb1f, 286 bp, Cnb1Δ, 168 bp, and cre, 617 bp. All CD11b-cre+; Cnb1f/Δ mice used in experiments were screened to confirm that peritoneal macrophages, which are 80–90% positive for CD11b surface expression, were negative for CnB1 protein expression using western blot or PCR. For use in FK506 injection experiments, C57BL/6J female mice were obtained from Jackson Laboratories. All mice used were age matched, and were generally 6–8 weeks old. All animal studies were approved by the Administrative Panel for Laboratory Animal Care.
Reagents
FK506 lyophilized powder (LC Labs) was reconstituted in either 100% ethanol for in vitro treatments or 80% ethanol, 20% cremaphor solution for in vivo treatments and stored at −20°C. For injection the FK506 solution was diluted into a vehicle (75% propanediol, 25% water) to 0.2–1.0 mg/ml. The vehicle control solution for in vivo experiments consisted of ethanol and cremaphor mixture diluted into 75% propanediol, 25% water. All FK506 and vehicle solutions were tested for LPS contamination using the Limulus Amebocyte Lysate (LAL) test (Cambrex Bio Science Walkersville). LPS lyophilized powder (List Biological Labs) was dissolved and diluted in phosphate buffered saline (PBS). As lethality varied among lots of LPS, when a lethal dose of LPS was desired for challenge, each lot of LPS was tested for LD50 before each experiment by injecting naïve mice with varying doses of LPS (5–10 mice per dose group) and assessing survival. The lowest dose that yielded 80–100% death was used for the LPS challenge dose in the subsequent experiment.
LPS challenge
All mice were observed every hour for the first 24–48 hours following the lethal LPS challenge. Mice were observed for characteristics of LPS toxicity (ocular exudates, diarrhea, increased respiratory rate) as well as moribund status (inability to sustain body temperature, ataxia or tremors, diaphragmatic breathing). In order to assess recovery, mice were given a score from 0–3 based on ambulation as an indication of overall health status: 0 = unable to walk, 1= take steps to walk, 2 = able to walk quickly but not run, and 3 = able to run/normal ambulation.
FK506 treatment protocol and experimental groups
C57BL/6J mice received a subcutaneous injection of FK506 (5 mg/kg) or vehicle control solution of the same volume every 24 hours for 3 days (3 injections total). The third injection was given simultaneously with an intraperitoneal (i.p.) injection of LPS challenge lethal dose (10–20 mg/kg) or a high sub-lethal dose (5–10 mg/kg). Another group of naïve mice were given a sub-lethal i.p. injection of LPS (1.0 mg/kg) on the second injection day, 24 hours before the LPS challenge injection, as the positive control. All mice were observed as indicated above.
Isolation of peritoneal macrophages and bone marrow-derived dendritic cells
Peritoneal macrophages were isolated as previously described 35. Bone marrow was obtained from femurs taken from C57BL/6J mice and flushed with medium. Dendritic cells were differentiated using a protocol adapted from Lutz et al. 39 with the following modifications: Day 0: bone marrow cells were plated at 2 ×106 cells/plate in 9 ml of standard medium with 20 ng/ml of GM-CSF. Day 3: 1 ml of media with 100 ng GM-CSF was added to the plate to a final concentration of 10 ng/ml of GM-CSF. Day 6: 5 ml were removed from each plate and centrifuged to pellet floating cells. Floating cells were resuspended in medium with 20 ng/ml GM-CSF and added back to the plate to a final concentration of 10 ng/ml GM-CSF. Day 8: repeat steps for day 6. Day 10: floating cells (dendritic cells) were gently removed from each plate and plated for future experiments. Each mouse (2 femurs) yielded ~ 80–90 ×106 floating cells which were 60–80% CD11c positive by FACS (data not shown).
In vitro assay for LPS tolerance induction
RAW264.7 mouse macrophage cell line, BMDC, and peritoneal macrophages were plated at 1×106 cells/well in standard medium and treated with either solvent, 0.1 μg/ml LPS or FK506 at 1 or 10 μg/ml and incubated at 37°C for 12–24 hours. Supernatants were removed and cells were washed 3X with PBS, treated with medium alone or 2 μg/ml LPS for tolerance induction and incubated for another 12–24 hours. Supernatants were assayed for IL-12 and TNF-α using sandwich ELISA.
Antibodies
Capture and biotinylated detection antibodies against mouse cytokines used in ELISA were purchased from BD-Pharmingen except anti-IL-12p40 C15.6 coating antibody, which was purified in our lab from hybridoma culture. HRP-linked antibodies used on western blots were purchased from Amersham Biosciences, and Anti-CnB1 antibody from Sigma.
ELISA
To quantify the levels of cytokines present in cell supernatants, plates were coated with 1–2 μg/ml of primary antibody. Cytokines of interest were then detected with 0.5 μg/ml of biotinylated antibody, followed by addition of Streptavidin-HRP conjugate. Colorimetric reactions were generated using ABTS (BD-Pharmingen) at optical densities of 405 nm.
Cytometric bead array
In some experiments the levels of TNF-α in serum samples and supernatants were measured using a cytometric bead array (CBA) from BD-Pharmingen according to the manufacturers instructions. Flow cytometry was performed on a Coulter Epics XL-MCL machine.
Cytokine RTPCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen). 1 μg of total RNA was reverse transcribed using Murine Leukemia Virus (MLV) reverse transcriptase (GIBCO). Resulting cDNA was subsequently amplified using the following specific primers: (TNF-α forward, 5'-TTCTATGGCCCAGACCCTCA-3', reverse, 5'-TCCCAGGTATATGGGCTCATA-3'; IL-1β forward, 5'-GGCTGCTTCCAAACCTTTGA-3', reverse, 5'-GAAGACACGGATTCCATGGT-3'; IL-12p40 forward, 5'-TGGCCAATGTGGGAGCTGGA-3', reverse, 5'-TTGGTGCTTCACACTTCAGGA-3'; GAPDH forward, 5'-TCGTGGAGTCTACTGGCGT-3', reverse, 5'-GCCTGCTTCACCACCTTCT-3'). PCR conditions were optimized and varied for each primer pair.
Nuclear extracts and EMSA
Nuclear extracts were prepared as previously described 35. Radiolabeling of NF-κB oligonucleotides with [γ-32P]ATP, EMSA, and supershift assays were performed, according to the manufacturer's protocols (Promega).
Immunoblotting
Whole cell lysates were prepared after various incubation conditions. Cells were washed in ice cold PBS and suspended in ice cold RIPA lysis buffer (50 mM TRIS-HCl; 1 mM EDTA; 1 mM EGTA; 150 mM NaCl; 1 % Nonidet P-40; 0.5 mM phenymethylsulfonyl fluoride; 2 μg/ml aprotinin; 2 μg/ml leupeptin; 2 μg/ml pepstatin; 1 mM dithiothreitol; 1 mM sodium fluoride; 10 mM sodium orthvonidate). Samples were agitated for 30 min, and then centrifuged for 8 min at 8,000 rpm at 4°C. Equal amounts of protein were separated by SDS-PAGE on 10–12 % acrylamide gels. The proteins were then transferred to a PVDF membrane (Amersham). The membrane was placed in blocking buffer (PBS, 0.5 % Tween 20, 5 % milk powder) and subsequently incubated with primary antibody and HRP-conjugated secondary antibody, and detected with ECL chemiluminescence kit (Amersham).
Results
FK506 induces production of inflammatory cytokines and activates NF-κB in dendritic cells
Previously published findings from this laboratory showed that the calcineurin inhibitors FK506 and CsA activate NF-κB in macrophages leading to the expression and secretion of pro-inflammatory mediators 35. This finding has now been extended to mouse bone marrow-derived dendritic cells (BMDC) treated with FK506. Assays for secreted protein (Figure 1A) and RNA (Figure 1B) demonstrated the induction of the inflammatory mediators IL-12, TNF-α, IL-1, and COX-2 in BMDC treated with FK506. The activation of pro-inflammatory mediators seen in these results is supported by the finding that FK506 activates NF-κB in BMDC (Figure 1C) as previously shown in macrophages 35, 36.
Figure 1. FK506 activates bone marrow-derived dendritic cells.

Dendritic cells (BMDCs) were differentiated from bone marrow of C57BL/6 mice. BMDCs were stimulated in vitro with solvent, lipopolysaccharide (LPS), or FK506 (FK) at the indicated concentrations (μg/ml). A. After 16 hour stimulation BMDC supernatants were assayed for IL-12p40/p70 using ELISA. Data represent the mean ± SD of three independent experiments. *, p < 0.02 by Student's t-test when compared to the untreated control. B. After a 2 hr stimulation cDNA was made from RNA extracted from BMDCs. RTPCR was performed for IL-12p40, TNF-α, COX-2, IL-1β, and GAPDH (loading control). Data is represented of three independent experiments. C. After 2 hr stimulation, nuclear extracts were prepared from BMDCs and used for NF-κB EMSA. *NS, non-specific band. Similar results were obtained in several independent experiments.
FK506 induces a form of LPS tolerance in macrophages and dendritic cells in vitro
In addition to the initial induction of pro-inflammatory gene expression, the activation of TLR4 by LPS induces multiple negative feedback mechanisms that act on TLR-activated signaling pathways, which render cells tolerant to subsequent exposure to LPS 40, 41. As FK506 and LPS both induce inflammatory mediators via TLR-mediated signaling pathways 35, 36, whether FK506 is also able to induce a similar LPS tolerant state was examined. LPS tolerance in macrophages has been thought to be most closely associated with decreased TNF production 7, 8, whereas in dendritic cells, tolerance is better associated with decreased IL-12 production 13; therefore, macrophage TNF responses and dendritic cell IL-12 responses were assessed. Both peritoneal macrophages (Figure 2A) and the macrophage cell line RAW264.7 (Figure 2B) showed decreased production of TNF-α in response to LPS after being pre-treated 20 hours earlier with either FK506 or LPS. Similar effects of pre-treatment with FK506 or LPS were observed for BMDC IL-12 production (Figure 2C). These impaired responses constitute a form of LPS tolerance. Cell counts obtained when supernatants were harvested confirmed that diminished responses were not due to differences in cell viability (data not shown).
Figure 2. FK506 induces a form of LPS tolerance in macrophages and dendritic cells in vitro.
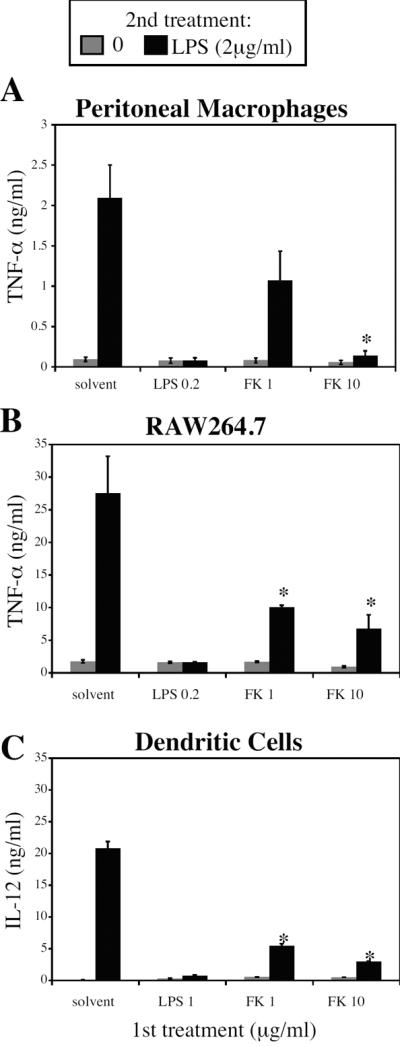
A. Peritoneal macrophages, B. RAW264.7 macrophage cell line, and C. bone marrow-derived dendritic cells were treated for 20 hours with an initial dose (1st treatment) of LPS or FK506 at the indicated concentrations. Cells were then washed and treated with either medium alone (gray bars) or 2 μg/ml of LPS (black bars). Supernatants were collected and analyzed for TNF-α or IL-12 p40/p70 production by ELISA. Results shown represent the mean ± SD of three independent experiments. *, p < 0.05 by Student's t-test when compared to untreated control.
Macrophages from mice treated with FK506 exhibit tolerance to LPS in vitro
Macrophages and peripheral blood monocytes from septic patients have been shown to have a decreased response to LPS when stimulated in vitro 7; therefore, we investigated whether macrophages from FK506-treated mice also exhibit a similar decreased response, reflecting a form of in vivo LPS tolerance. As expected, resident peritoneal macrophages from mice that had received a prior injection of LPS produced significantly less IL-12 and TNF-α in response to an in vitro treatment with LPS or FK506 compared to macrophages from mice receiving only PBS (Figure 3A). Significantly, macrophages from mice given FK506 injections over three days also produced lower amounts of IL-12 and TNF-α in response to an in vitro treatment with LPS or FK506 compared to those from mice receiving only the vehicle control (Figure 3A). In addition, NF-κB activation was reduced in macrophages from FK506-treated mice compared to cells from the vehicle-treated mice (Figure 3B).
Figure 3. Macrophages from FK506-treated mice exhibit reduced responsiveness to LPS in vitro.

C57BL/6 mice were injected with FK506 treatment regimen (see Materials and Methods), LPS priming dose (1 mg/kg), the FK506 vehicle control (Vehicle), or PBS control. A. Peritoneal macrophages were harvested from injected mice and stimulated in vitro with solvent, 2 μg/ml LPS, or 10 μg/ml FK506. IL-12 p40/p70 and TNF-α levels in cell supernatants were determined by ELISA. Results represent the mean ± SEM of three independent experiments. *, p < 0.03 by Student's t-test when compared to appropriate control. B. Peritoneal macrophages were harvested from injected mice and stimulated in vitro with solvent or 2 μg/ml LPS, and nuclear extracts were tested for NF-κB levels by EMSA. *NS, non-specific band.
FK506 protects against LPS-induced toxicity in vivo
The phenomenon of LPS tolerance has also been observed in vivo; it contributes to various processes in human and animal subjects that can be either protective (i.e., reducing septic shock) or detrimental (i.e., reducing protective immunity to pathogens) to the host. To determine whether the induction of tolerance to LPS by FK506 could be protective against LPS-induced toxicity in an animal model, mice were injected with either an FK506 regimen or one low dose of LPS (1 mg/kg) and subsequently challenged with either a lethal (15–20 mg/kg) or sub-lethal (7.5–10 mg/kg) dose of LPS. The mice were then observed for signs of LPS toxicity in a blind study for up to 48 hours post LPS challenge. Mice showing signs of morbidity were euthanized. Mice pre-treated with FK506 or LPS had a significantly better survival rate when challenged with the lethal dose of LPS than did those that received vehicle only (Figure 4A). In addition, mice pre-treated with FK506 or LPS exhibited improved recovery and reduced illness, as determined by a qualitative score based on health status, from a sublethal dose of LPS compared to mice given vehicle only (Figure 4B).
Figure 4. FK506 protects against LPS-induced toxicity in vivo.

C57BL/6 mice were injected with FK506 treatment regimen (see Materials and Methods), LPS priming dose (1 mg/kg), or the FK506 vehicle control (Vehicle). A. After treatment, mice were challenged with a lethal dose (20 mg/kg) of LPS and observed in a blind study. Each experimental group consisted of 10 animals. Results shown are representative of three independent experiments. p values shown on the graph were calculated using the Kaplan-Meier log rank test. B. After treatment, mice were challenged with a sublethal dose (7.5 mg/kg) of LPS and observed in a blind study. Mice were given a score of 0, 1, 2, or 3, as an assessment of general health. A score of 2 or better was considered good health and/or substantial recovery. Each experimental group consisted of 5 animals. Results represent mean ±SEM of three independent experiments. *, p < 0.05 by Student's t-test when compared to solvent control.
Macrophages deficient in CnB1 exhibit a form of LPS tolerance in vitro
Mice that are genotypically CD11b-cre+;Cnb1f/Δ, generated to have myeloid cells deficient in the CnB1 subunit, are fertile and show no gross abnormalities. Approximately 30% of all CD11b-cre+;Cnb1f/Δ adult mice possessed CnB1-deficient peritoneal macrophages, as determined by PCR (Figure 5A) or western blot (Figure 5B); therefore, mice were screened for macrophage CnB1 expression prior to their assignment to experimental groups. All mice designated as CnB1-deficient in the data shown here were CD11b-cre+;Cnb1f/Δ mice confirmed to have CnB1-deficient peritoneal macrophages by western blot or PCR. CnB1-deficient peritoneal macrophages from CD11b-cre+;Cnb1f/Δ mice showed decreased activation by LPS in vitro. CnB1-deficient macrophages showed decreased TNF-α secretion (Figure 5C) and IL-12 secretion (Figure 5D) compared to wild-type when stimulated with LPS. Also, as would be predicted, CnB1-deficient macrophages produced little or no cytokines in response to calcineurin inhibition by FK506 treatment unlike wild-type macrophages (Figure 5C and 5D). In contrast, cells from the CD11b-cre+;Cnb1f/Δ mice with CnB1-positive macrophages behaved similar to wild-type cells (data not shown). There was no significant difference in the cell counts from CnB1-deficient macrophages compared to wild-type (data not shown).
Figure 5. Macrophages deficient in Cnb1 exhibit reduced responsiveness to LPS in vitro.

A. Cnb1 genotyping of DNA isolated from tail sample or peritoneal macrophages (pMϕ) by agarose gel electrophoresis of PCR products. B. Whole cell extracts from pMϕ were analyzed for CnB1 protein levels by western blot. Each lane represents an individual mouse. * = mice identified as having CnB1-deficient macrophages. C and D. Peritoneal macrophages were isolated from individual mice: wild-type (WT), n=2, or CD11b-Cre+; Cnb1f/Δ (CnB1-deficient) mice (confirmed to have CnB1-negative macrophages by western blot of whole cell extracts), n=2, or, and treated with medium alone (0), LPS at the indicated concentrations (μg/ml), or 10 μg/ml FK506 for 16 hours. TNF-α levels were measured by cytometric bead array. IL-12 p40/p70 levels were measured using ELISA. Data are representative of three independent experiments. *, p < 0.03; **, p < 0.002 by student's t-test.
Mice with CnB1-deficient macrophages are less susceptible to LPS-induced toxicity in vivo
The data demonstrating tolerance to LPS in CnB1-deficient macrophages presented in Figure 5 along with reported findings showing that LPS tolerance in vivo is mediated by macrophages 42 suggests that the ability of injected FK506 to protect mice from LPS-induced toxicity and mortality (Figure 4) was due to the induction of a form of LPS tolerance secondary to calcineurin inactivation in the macrophages of these mice. This led us to investigate whether the deletion of Cnb1 (which would reduce calcineurin activity) in macrophages of mice would also yield protection from LPS in vivo. To test this hypothesis, both wild-type and CD11b-cre+;Cnb1f/Δ mice with CnB1-deficient macrophages (designated CnB1-deficient mice) were challenged with a lethal dose (20 mg/kg) of LPS and observed in a blind study for 24 hours post LPS challenge. Mice showing signs of morbidity were euthanized. The CnB1-deficient mice showed a better survival rate when challenged with lethal doses of LPS compared to wild-type control mice (Figure 6A). In addition, sera from CnB1-deficient mice challenged with LPS showed significantly lower levels of TNF-α compared to sera from wild-type mice (Figure 6B).
Figure 6. Mice with CnB1-deficient macrophages are less susceptible to LPS-induced toxicity.
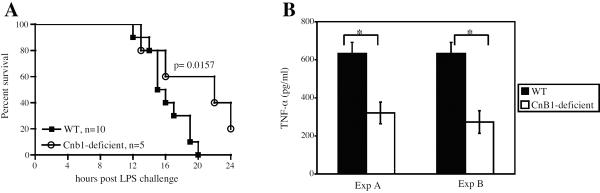
A. Wild-type (WT) and CD11b-Cre+; Cnb1f/Δ mice with CnB1-deficient macrophages (CnB1-deficient) were challenged with a lethal dose (20 mg/kg) of LPS and observed in a blind study. Data shown are representative of three independent experiments. p values shown were calculated using the Kaplan-Meier log rank test. B. After LPS challenge, sera were taken from moribund mice (WT and CnB1-deficient) just before euthanasia. TNF was assayed using cytometric bead array. Data represent the mean±SEM. *, p < 0.02 by Student's t-test. The serum TNF concentrations seen in these experiments are comparable to other published results 10.
Discussion
Although the role of calcineurin in T-cell activation and adaptive immunity is well established, little has been reported about its role in innate immune responses. Our lab previously reported data showing that calcineurin inhibitors activate NF-κB and innate and inflammatory responses in mouse peritoneal macrophages, the WEHI-3 mouse myelomonocytic cell line, primary mouse astrocytes, and the L929 fibroblast cell line 35. More recently we have extended these findings to mouse macrophage cell line RAW 264.7, human monocytic cell lines, U937 and THP-1, 36, and bone marrow derived dendritic cells (this study). Results described in this report show that inactivation of calcineurin with the calcineurin inhibitor, FK506, or by cell-specific deletion of CnB1, induces a state of reduced responsiveness to LPS, reminiscent of LPS tolerance in macrophages and dendritic cells in vitro. In addition, FK506-treated mice and mice with CnB1-deficient macrophages exhibit an LPS-tolerant phenotype and are partially protected from LPS-induced toxicity.
In the induction of LPS tolerance, initial activation of the TLR pathway leads to gene activation followed by activation of negative feedback pathways and subsequent unresponsiveness to LPS. In our in vitro studies, macrophages and dendritic cells became less responsive to LPS following pretreatment with FK506. Also, macrophages from FK506-treated mice showed diminished NF-κB activation in response to LPS, and decreased production of TNF and IL-12 in response to LPS as well as FK506. Similarly, the CnB1-deficient macrophages showed decreased production of TNF in response to LPS. Collectively these results indicate that reduced calcineurin activity results in LPS tolerant state in macrophages. These findings are consistent with earlier reports that calcineurin inhibitor treatment leads to decreased inflammatory responses. Dendritic cells differentiated in the presence of CsA showed decreased expression of costimulatory molecules, associated with a reduction in nuclear translocation of NF-κB 43, and also reduced IL-12 production following activation 44.
In addition to the in vitro findings, our in vivo studies showed that treatment with FK506 improves both survival and recovery in mice challenged with LPS. As expected, mice pre-treated with LPS, which is known to induce LPS tolerance strongly, exhibited the most protection. Also, mice with CnB1-deficient macrophages had improved survival and decreased serum TNF after challenge with a lethal dose of LPS. Although we had previously shown that reduced calcineurin activity mediated by calcineurin inhibitors or siRNA leads to an initial increase in NF-κB activation and associated induction of pro-inflammatory cytokines35, 36, the persistent inactivation of calcineurin by inhibition or deletion, as in the models presented in this report, also results in the induction of tolerance to subsequent exposure to LPS, similar to the effects of LPS itself.
Many mechanisms contributing to LPS tolerance have been described 7, 40, 41 and are likely candidates for FK506-induced tolerance to LPS, as FK506 activates the same signaling pathways downstream of TLR 36. Interestingly, the macrophages from LPS-treated mice showed decreased cytokine production in response to in vitro exposure to FK506 as well as to LPS; thus FK506 and LPS induce tolerance to each other. These observations suggest that the mechanism(s) of tolerance seen here reflect negative feedback pathways activated via the TLR signaling pathways, which are triggered by both LPS and calcineurin inhibitors. As we reported recently 36, calcineurin inhibitors mimic TLR ligands in that they act at TLR-proximal steps to activate both MyD88-dependent and MyD88-independent signaling pathways, leading to activation of NF-κB and other transcription factors. The finding that deletion of calcineurin in macrophages also induces tolerance to both LPS and FK506 supports the conclusion that the induction of tolerance by FK506 is a result of its inhibition of calcineurin, and subsequent activation of the TLR signaling pathways.
The incidence of sepsis has recently been estimated to be 751,000 cases per year with a mortality of approximately 30% in the United States 45. Sepsis is also a leading cause of death in infants and children with 42,000 cases per year in the United States 46. A recent study reported that in early sepsis, during what is called systemic inflammatory response syndrome (SIRS), endotoxin was detected in the blood of 52.1% of patients 47. Patients with sepsis have been found to have high levels of circulating inflammatory mediators such as TNF and IL-6 7. Those who survive the initial SIRS develop a compensatory immune deactivation with profound reduction in immune response, sometimes referred to as immunoparalysis 48. Although there are murine models that better emulate the environment of sepsis compared to LPS-induced shock 49–51, it is still believed that endotoxin tolerance is a good model for the numerous alterations in cellular responses that occur in the immunoparalysis of sepsis and SIRS 52. It has been recently reported that induction of endotoxin tolerance can enhance survival in sepsis 53.
Our findings show that inactivation of calcineurin by inhibition or deletion in murine macrophages and dendritic cells in vitro or macrophages in vivo induces decreased responsiveness to LPS. Inactivation of calcineurin in vivo also provides partial protection against LPS-induced morbidity and mortality in mice. These results suggest that the modulation of calcineurin activity affects survival during endotoxin exposure and possibly sepsis. Further studies in specific sepsis models might reveal a use for FK506 and other calcineurin inhibitors in decreasing mortality from sepsis.
Currently, patients undergoing transplantation often receive calcineurin inhibitors as part of their immunosuppression regimen to prevent rejection, and calcineurin inhibitors are also used to treat inflammatory disorders, such autoimmune disease 33, 34 and inflammatory skin disease 32. The efficacy of CsA and FK506 in these situations has been assumed to reflect their potent inhibition of T-cell activation, which also has been the explanation for a common detrimental side effect of calcineurin inhibitors: the increased risk of opportunistic infections 54. It has also been shown that SIRS-associated LPS tolerance also can cause increased susceptibility to infection 55, 56. This study has demonstrated the induction of a form of LPS tolerance in macrophages and dendritic cells by the calcineurin inhibitor, FK506, in in vitro and in vivo models. In a similar way, treatment with calcineurin inhibitors, as occurs in many patients today, may induce alterations in innate immune responses in addition to suppression of adaptive immune responses. Lipopolysaccharide tolerance in its classical form (induced by prior in vivo exposure to LPS) can increase resistance to infection, 57, 58 likely due to increases in some types of innate immune mechanisms such as pathogen binding and antigen presentation.59 However, LPS tolerance has also been shown to reduce expression of many pro-inflammatory mediators,59 thus reducing the benefit of these responses in the host. Calcineurin inactivation, as shown in this report, creates a form of LPS tolerance characterized by reduced production of pro-inflammatory mediators IL-12 and TNF, which may contribute to increased susceptibility to infections in patients treated with calcineurin inhibitors. These findings support the need to discover additional immunosuppressants, especially inhibitors that specifically target T cells, to avoid alterations in protective innate immune responses.
Acknowledgements
The authors thank G. Crabtree, J. Neilson, M. Winslow, and E. Gallo for providing reagents, mice, and helpful discussions and J. Vacher for providing mice. This study was supported by funds from Stanford University. C.J. was supported by NIH Medical Scientist Training Program grant GM07365.
References
- 1.Medzhitov RM. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–146. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 2.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med. 2006;84:712–725. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 4.Roach JC, Smith KD, Strobe KL, Nissen SM, Haudenschild CD, Zhou D, Vasicek TJ, Held GA, Stolovitzky GA, Hood LE, Aderem A. Transcription factor expression in lipopolysaccharide-activated peripheral-blood-derived mononuclear cells. Proc Natl Acad Sci U S A. 2007;104:16245–16250. doi: 10.1073/pnas.0707757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 6.Proctor RA, Will JA, Burhop KE, Raetz CRH. Protection of mice against lethal endotoxemia by a lipid A precursor. Infect Immun. 1986;52:905–907. doi: 10.1128/iai.52.3.905-907.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West MA, Heagy W. Endotoxin tolerance: A review. Crit Care Med. 2002;30(1 Supp):S64–S73. [PubMed] [Google Scholar]
- 8.Sanchez-Cantu L, Rode HN, Christou NV. Endotoxin tolerance is associated with reduced secretion of tumor necrosis factor. Arch Surg. 1989;124:1432–1436. doi: 10.1001/archsurg.1989.01410120082016. [DOI] [PubMed] [Google Scholar]
- 9.Lepe-Zuniga JL, Klostergaard J. Tolerance to endotoxin in vitro: Independent regulation of interleukin-1, tumor necrosis factor and interferon alpha production during in vitro differentiation of human monocytes. Lymphokine Res. 1990;9:309–319. [PubMed] [Google Scholar]
- 10.Mathison JC, Virca GD, Wolfson E, Tobias PS, Glaser K, Ulevitch RJ. Adaptation to bacterial lipopolysaccharide controls lipopolysaccharide-induced tumor necrosis factor production in rabbit macrophages. J Clin Invest. 1990;85:1108–1118. doi: 10.1172/JCI114542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: Dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 12.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by toll-like receptor TLR2 amd TLR4 agonists in murine macrophages: Effects of TLR “homotoerance” and “heterotolerance” on NF-kappaB signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 13.Karp CL, Wysocka M, Ma X, Marovich M, Factor RE, Nutman T, Armant M, Wahl L, Cuomo P, Trinchieri G. Potent suppression of IL-12 production from monocytes and dendritic cells during endotoxin tolerance. Eur J Immunol. 1998;28(10):3128–3136. doi: 10.1002/(SICI)1521-4141(199810)28:10<3128::AID-IMMU3128>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 14.Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: Is there a clinical relevance? J Endotoxin Res. 2003;9:101–107. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 16.Granowitz EV, Porat R, Mier JW, Orencole SF, Kaplanski G, Lynch EA, Ye K, Vannier E, Wolff SM, Dinarello CA. Intravenous endotoxin suppresses the cytokine response of peripheral blood mononuclear cells of healthy humans. J Immunol. 1993;151:1637–1645. [PubMed] [Google Scholar]
- 17.Klee CB, Crouch TH, Krinks MH. Calcineurin: A calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci U S A. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibasaki F, Hallin U, Uchino H. Calcineurin as a multifunctional regulator. J Biochem. 2002;131:1–15. doi: 10.1093/oxfordjournals.jbchem.a003063. [DOI] [PubMed] [Google Scholar]
- 19.Rusnak F, Mertz P. Calcineurin: Form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 20.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 21.Crabtree GR. Generic signals and specific outcomes: Signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–614. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- 22.Borel JF. Comparative study of in vitro and in vivo drug effects on cell mediated toxicity. Immunology. 1976;31:631–641. [PMC free article] [PubMed] [Google Scholar]
- 23.Kino T, Hatanaka H, Hashimoto M, Nishiyama M, Goto T, Okuhara M, Kohsaka M, Aoki H, Imanaka H. FK-506, a novel immunosuppressant isolated from a streptomyces. I. fermentation, isolation, and physico-chemical and biological characteristics. J Antibiot (Tokyo) 1987;40:1249–1255. doi: 10.7164/antibiotics.40.1249. [DOI] [PubMed] [Google Scholar]
- 24.Kino T, Hatanaka H, Miyata S, Inamura N, Nishiyama M, Yajima T, Goto T, Okuhara M, Kohsaka M, Aoki H. FK-506, a novel immunosuppressant isolated from a streptomyces. II. immunosuppressive effect of FK-506 in vitro. J Antibiot (Tokyo) 1987;40:1256–1265. doi: 10.7164/antibiotics.40.1256. [DOI] [PubMed] [Google Scholar]
- 25.Goto T, Kino T, Hatanaka H, Nishiyama M, Okuhara M, Kohsaka M, Aoki H, Imanaka H. Discovery of FK-506, a novel immunosuppressant isolated from streptomyces tsukubaensis. Transplant Proc. 1987;19:4–8. [PubMed] [Google Scholar]
- 26.Tanaka H, Kuroda A, Marusawa H, Hashimoto M, Hatanaka H, Kino T, Goto T, Okuhara M. Physicochemical properties of FK-506, a novel immunosuppressant isolated from streptomyces tsukubaensis. Transplant Proc. 1987;19:11–16. [PubMed] [Google Scholar]
- 27.Friedman J, Weissman I. Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: One in the presence and one in the absence of CsA. Cell. 1991;66:799–806. doi: 10.1016/0092-8674(91)90123-g. [DOI] [PubMed] [Google Scholar]
- 28.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Albers MW, Wandless TJ, Luan S, Alberg DG, Belshaw PJ, Cohen P, MacKintosh C, Klee CB, Schreiber SL. Inhibition of T cell signaling by immunophilin-ligand complexes correlates with loss of calcineurin phosphatase activity. Biochemistry. 1992;31:3896–3901. doi: 10.1021/bi00131a002. [DOI] [PubMed] [Google Scholar]
- 30.O'Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O'Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 31.Winslow MM, Gallo EM, Neilson JR, Crabtree GR. The calcineurin phosphatase complex modulates immunogenic B cell responses. Immunity. 2006;24:141–152. doi: 10.1016/j.immuni.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Woo DK, James WD. Topical tacrolimus: A review of its uses in dermatology. Dermatitis. 2005;16:6–21. [PubMed] [Google Scholar]
- 33.Tse KC, Lam MF, Tang SC, Tang CS, Chan TM. A pilot study on tacrolimus treatment in membranous or quiescent lupus nephritis with proteinuria resistant to angiotensin inhibition or blockade. Lupus. 2007;16:46–51. doi: 10.1177/0961203306073167. [DOI] [PubMed] [Google Scholar]
- 34.Kitahara K, Kawai S. Cyclosporine and tacrolimus for the treatment of rheumatoid arthritis. Curr Opin Rheumatol. 2007;19:238–245. doi: 10.1097/BOR.0b013e328099af80. [DOI] [PubMed] [Google Scholar]
- 35.Conboy IM, Manoli D, Mhaiskar V, Jones PP. Calcineurin and vacuolar-type H+ - ATPase modulate macrophage effector functions. PNAS. 1999;96:6324–6329. doi: 10.1073/pnas.96.11.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang YJ, Kusler B, Otsuka M, Hughes M, Suzuki N, Suzuki S, Yeh WC, Akira S, Han J, Jones PP. Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol. 2007;179:4598–4607. doi: 10.4049/jimmunol.179.7.4598. [DOI] [PubMed] [Google Scholar]
- 37.Ferron M, Vacher J. Targeted expression of cre recombinase in macrophages and osteoclasts in transgenic mice. Genesis. 2005;41:138–145. doi: 10.1002/gene.20108. [DOI] [PubMed] [Google Scholar]
- 38.Nielson JR, Winslow MM, Crabtree AG. Essential role of calcineurin B1 in thymocyte positive but not negative selection. Immunity. 2004;20:255–266. doi: 10.1016/s1074-7613(04)00052-4. [DOI] [PubMed] [Google Scholar]
- 39.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 40.Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res. 2004;10:71–84. doi: 10.1179/096805104225003997. [DOI] [PubMed] [Google Scholar]
- 41.Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: Molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12:133–150. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- 42.Freudenberg MA, Galanos C. Induction of tolerance to lipopolysaccharide (LPS)-D-galactosamine lethality by pretreatment with LPS is mediated by macrophages. Infect Immun. 1988;56:1352–1357. doi: 10.1128/iai.56.5.1352-1357.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JI, Ganster RW, Geller DA, Burckart GJ, Thomson AW, Lu L. Cyclosporine A inhibits the expression of costimulatory molecules on in vitro-generated dendritic cells: Association with reduced nuclear translocation of nuclear factor kappa B. Transplantation. 1999;68:1255–1263. doi: 10.1097/00007890-199911150-00007. [DOI] [PubMed] [Google Scholar]
- 44.Sauma D, Fierro A, Mora JR, Lennon-Dumenil AM, Bono MR, Rosemblatt M, Morales J. Cyclosporine preconditions dendritic cells during differentiation and reduces IL-2 and IL-12 production following activation: A potential tolerogenic effect. Transplant Proc. 2003;35:2515–2517. doi: 10.1016/j.transproceed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the united states: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Watson RS, Carcillo JA. Scope and epidemiology of pediatric sepsis. Pediatr Crit Care Med. 2005;6:S3–5. doi: 10.1097/01.PCC.0000161289.22464.C3. [DOI] [PubMed] [Google Scholar]
- 47.Venet C, Zeni F, Viallon A, Ross A, Pain P, Gery P, Page D, Vermesch R, Bertrand M, Rancon F, Bertrand JC. Endotoxaemia in patients with sever sepsis or septic shock. Intensive Care Med. 2000;26:538–544. doi: 10.1007/s001340051201. [DOI] [PubMed] [Google Scholar]
- 48.Volk HD, Reinke P, Docke WD. Clinical aspects: From systemic inflammation to 'immunoparalysis'. Chem Immunol. 2000;74:162–177. doi: 10.1159/000058753. [DOI] [PubMed] [Google Scholar]
- 49.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 50.Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24(Suppl 1):7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- 51.Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007;81:137–143. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]
- 52.Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: Endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, Zingarelli B. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock. 2008 doi: 10.1097/shk.0b013e318162c190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh N. Infectious complications in organ transplant recipients with the use of calcineurin-inhibitor agent-based immunosuppressive regimens. Curr Opin Infect Dis. 2005;18:342–345. doi: 10.1097/01.qco.0000172698.52408.be. [DOI] [PubMed] [Google Scholar]
- 55.Mason CM, Dobard E, Summer WR, Nelson S. Intraportal lipopolysaccharide suppresses pulmonary antibacterial defense mechanisms. J Infec Dis. 1997;176:1293–1302. doi: 10.1086/514125. [DOI] [PubMed] [Google Scholar]
- 56.Severn A, Xu D, Doyle J, Leal LM, O'Donnell CA, Brett SJ, Moss DW, Liew FY. Pre-exposure of murine macrophages to lipopolysaccharide inhibits the induction of nitric oxide synthase and reduces leishmanicidal activity. Eur J Immunol. 1993;23:1711–1714. doi: 10.1002/eji.1830230747. [DOI] [PubMed] [Google Scholar]
- 57.Rayhane N, Fitting C, Lortholary O, Dromer F, Cavaillon JM. Administration of endotoxin associated with lipopolysaccharide tolerance protects mice against fungal infection. Infect Immun. 2000;68:3748–3753. doi: 10.1128/iai.68.6.3748-3753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehner MD, Ittner J, Bundschuh DS, van Rooijen N, Wendel A, Hartung T. Improved innate immunity of endotoxin-tolerant mice increases resistance to salmonella enterica serovar typhimurium infection despite attenuated cytokine response. Infect Immun. 2001;69:463–471. doi: 10.1128/IAI.69.1.463-471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]