

Summary
Nearly all information about patients with chronic lymphocytic leukaemia (CLL) who develop diffuse large B-cell lymphoma (Richter syndrome [RS]) is derived from retrospective case series or patients treated on clinical trials. We used the Mayo Clinic CLL Database to identify patients with newly diagnosed CLL (1/2000–7/2011). Individuals who developed biopsy-proven RS during follow-up were identified. After median follow-up of 4 years, 37/1641 (2.3%) CLL patients developed RS. The rate of RS was approximately 0.5%/year. Risk of RS was associated with advanced Rai stage at diagnosis (p<0.001), high-risk FISH (p<0.0001), unmutated IGHV (p=0.003), and expression of ZAP-70 (p=0.02) and CD38 (p=0.001). The rate of RS doubled in patients treated for CLL (1%/year). Stereotyped B-cell receptors (odds-ratio=4.2; p=0.01) but not VH4–39 was associated with increased risk of RS. Treatment with combination of purine analogues and alkylating agents increased the risk of RS 3-fold (odds-ratio= 3.26, p=0.0003). Median survival after RS diagnosis was 2.1 years. The RS prognosis score stratified patients into three risk groups with median survivals of 0.5 years, 2.1 years and not reached. Both underlying characteristics of the CLL clone and subsequent CLL therapy influence the risk of RS. Survival after RS remains poor and new therapies are needed.
Keywords: transformation, aggressive lymphoma, stem cell transplantation, purine analogues, RS survival score
Introduction
Although chronic lymphocytic leukaemia (CLL) is classified as a low grade B-cell malignancy, approximately 2–8% of patients will experience transformation into a more aggressive B-cell lymphoma, most commonly diffuse large B-cell lymphoma (DLBCL) (Armitage, et al 1978, Foucar and Rydell 1980, Robertson, et al 1993). This was originally described by Maurice Richter in 1928 with the occurrence of rapidly fatal generalized lymphadenopathy and hepatosplenomegaly in association with CLL (Richter 1928). Forty years later, the term “Richter syndrome” (RS) was introduced to describe DLBCL occurring in patients with CLL (Lortholary, et al 1964).
Patients with RS exhibit an aggressive clinical phenotype because of the combined effect of chemoresistance and rapid disease kinetics (Rossi, et al 2011). The median survival of patients with a diagnosis of RS is only about 1–2 years (Fan, et al 2012, Mauro, et al 1999, Robak, et al 2004, Tsimberidou, et al 2006). Although case series have reported on the efficacy of stem cell transplantation (SCT) in patients who develop RS, the proportion of RS patients who make it SCT has not been reported in an unselected RS cohort. In a recent large retrospective analysis describing the outcomes of 59 RS patients after SCT, 34 (58%) received an autologous SCT and 25 (42%) received an allogeneic SCT (Cwynarski, et al 2012). However, this analysis does not report what proportion of CLL patients who later developed RS could not receive a SCT (either they were not SCT candidates or they did not survive to make it to SCT).
Nearly all the available information on RS in patients with CLL comes from case series and retrospective analysis of selected patients at the time of diagnosis of RS. There is limited information on the future risk of RS in an unselected cohort of newly diagnosed CLL patients. Additionally, since most of the reported case series of RS are from older series, the association of biological prognostic markers (i.e, immunoglobulin heavy chain gene somatic hypermutation [IGHV], genetic abnormalities detected by fluorescence in situ hybridization [FISH], and expression of CD38 and zeta-associated protein-70 [ZAP-70]) and the risk of RS is not very well defined. The relationship between the risk of development of RS and the underlying biology of the disease, the duration of the disease, and treatment of CLL is also not well understood. Understanding the possible role of more intense combination chemotherapy and chemoimmunotherapy for CLL in the development of RS is especially important(Catovsky, et al 2007, Eichhorst, et al 2006, Flinn, et al 2007, Hallek, et al 2010, Hillmen, et al 2007, Knauf, et al 2009, Tam, et al 2008, Wierda, et al 2011). To address these questions, we conducted a cohort study in newly diagnosed CLL patients.
Methods
Patients
The Mayo Clinic CLL Database includes all patients with a diagnosis of CLL evaluated within the Division of Haematology at Mayo Clinic, Rochester, MN since 1995, and who consented the use of their medical records for research purposes. All patients entered into the database met the NCI Working Group 1996 criteria (Cheson, et al 1996) for a diagnosis of CLL or a diagnosis of small lymphocytic lymphoma (SLL) according to the World Health Organization (WHO) criteria (Harris, et al 1999). Baseline demographics, clinical and prognostic characteristics are entered into the database at the time of the first visit and prospectively maintained. Clinical outcomes including date of first treatment, type of treatment administered and disease-related complications are also recorded.
With the approval of the Mayo Clinic Institutional Review Board and in accordance with federal regulations and the Declaration of Helsinki, we used this database to identify all patients diagnosed with CLL between January 2000 and July 2011. To reduce referral bias, only those patients who were seen at Mayo Clinic at the time of CLL diagnosis (≤12 months) were included in the present analysis. Only patients with biopsy-proven DLBCL that developed after CLL diagnosis were considered to have RS. All cases classified as RS had pathologic confirmation at Mayo Clinic. The Mayo Clinic Lymphoma Data Base was cross referenced to ensure that all cases of RS were included. In selected patients, in situ hybridization studies were performed on paraffin sections of the bone marrow biopsy or lymph node specimen using probes specific for EBV-encoded ribonucleic acid (RNA). All RS patients were scored using the survival score described by Tsimberidou and colleagues at the MD Anderson Cancer Centre (MDACC). Each of the following parameters were assigned one point: ECOG performance status >1, serum LDH >1.5 times the upper limit of normal, platelet count <100 × 109/L, tumour size >5cm and >1 prior therapies for CLL (Tsimberidou, et al 2006).
Statistical analysis
The date of RS was defined as the date of the biopsy showing DLBCL. The time to development of transformation was defined as date of CLL diagnosis to the date of diagnosis of RS. Overall survival from RS was defined as the time from diagnosis of RS to death. Chi-square, Fisher, or Kruskal-Wallis tests, where appropriate, were used for comparisons of the clinical characteristics and prior treatment exposure on the risk of RS. Survival analysis was performed by the Kaplan–Meier method. Cox proportional hazards models were used to evaluate associations between TFT or OS and patient prognostic factors. All statistical tests were two-sided. Statistical significance was defined as P-value <0.05. All statistical analyses were conducted using the SAS 9.2 software package (SAS Institute, Cary, NC).
Results
Patient Characteristics and Incidence of Richter Syndrome
Between January 2000 and July 2011, 1641 patients were seen at Mayo Clinic within 12 months of diagnosis of their CLL. After a median follow-up of 4 years (range, 0–12.3 years), 37 (2.3%) had biopsy-proven RS. The median time to RS among those who developed RS (n=37) was 1.8 years (range, 0–11.7 years). The incidence of RS as determined by Kaplan-Meier analysis at 5 and 10 years post CLL diagnosis were 2.1% and 4.8% respectively. The annual incidence rate of RS was approximately 0.5% per year for all CLL patients. Among those CLL patients who received therapy, the annual incidence rate of RS was approximately 1% per year. (Figure 1)
Figure 1.
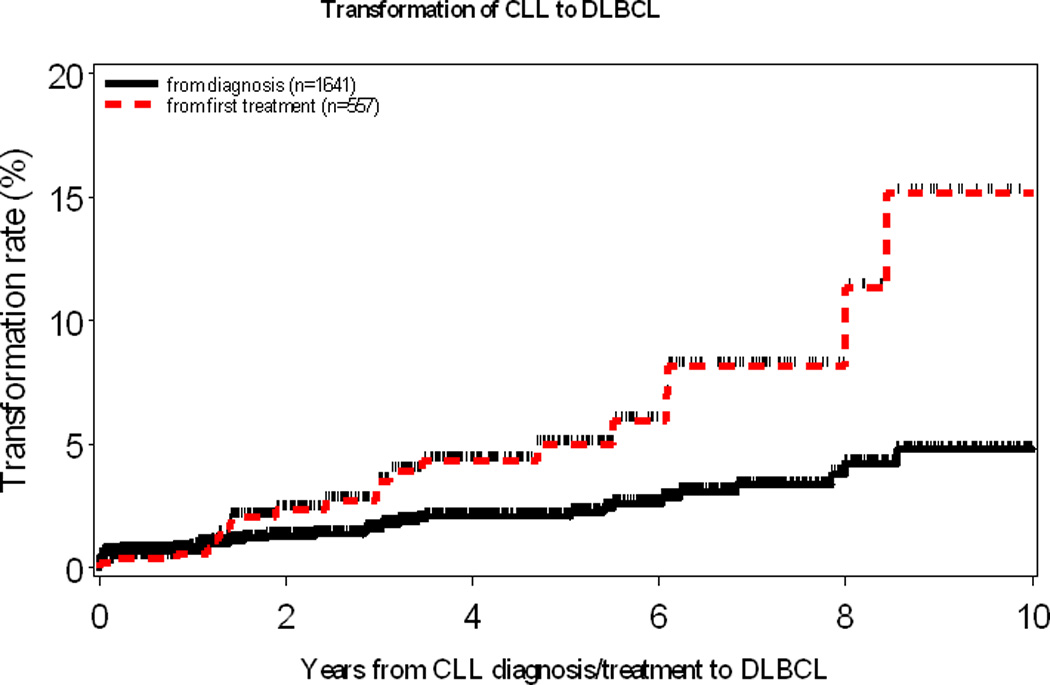
Rate of Richter syndrome in the entire cohort (black solid line). Rate of Richter syndrome in those who received CLL treatment (red dashed line, initiation of CLL treatment considered time zero)
The baseline characteristics of CLL patients who developed RS versus those who did not are shown in Table I. CLL patients who subsequently developed RS were more likely to have advanced Rai Stage at diagnosis (p<0.001). After adjusting for Rai stage, high serum beta-2 microglobulin, unmutated IGHV gene mutation status, unfavourable FISH results [del(11q22) or del(17p13)] and expression of CD38, ZAP-70 and CD49d, were independently associated with a higher risk of subsequent RS. Based on previous reports suggesting an association between IGHV4–39 family usage and higher risk of RS (Rossi, et al 2009b), we explored the rate of RS in the 29 patients with IGHV 4–39 in our cohort. IGHV4–39 gene usage was present in 0/14 (0%) RS patients compared to 29/797 (3.6%) non-RS patients (p=1.0). Stereotyped B-cell receptors have also been reported to be associated with higher risk of RS (Rossi, et al 2009b). Stereotyped receptors were found in 6/14 (43%) RS patients in our cohort as compared to 119/797 (15%) non-RS patients (OR=4.2, 95% Confidence Interval (CI) (1.5, 12.5), p=0.01), confirming this observation. This translated into a rate of RS of 5% (6/125) for patients with stereotyped receptors as compared to 1% (8/686) for those without. When evaluating the time to RS, the risk was greater in the cases with stereotyped receptors compared to those without (HR=4.8, 95% CI (1.6, 13.8), p=0.0004).
Table I.
Comparison of baseline characteristics at the time of CLL diagnosis between patients who later develop Richter syndrome versus those who do not
| Characteristic | RS n (%) |
No RS n (%) |
p-value | |
|---|---|---|---|---|
| Number of Patients | 37 | 1604 | ||
| Median Age, years (range) | 61 (21–85) | 64 (25–97) | 0.19 | |
| Sex (male) | 29 (78) | 1047 (65) | 0.09 | |
| Median white cell count (×109/L) (range) | 14.8 (2.6–415.5) | 13.9 (0.5–732.1) | 0.85 | |
| Median absolute lymphocyte count (×109/L) (range) | 9.1 (0.7–386.4) | 8.4 (0.0–673.5) | 0.79 | |
| Median absolute B-cell count (×109/L) (range) | 14.3 (1.4–378.3) | 7.0 (0.0–666.7) | 0.02 | |
| Median haemoglobin (gm/dL) (range) | 13.5 (6.8–16.0) | 13.9 (4.9–17.9) | 0.11 | |
| Median platelets (×109/L) (range) | 190 (71–493) | 199 (4–1229) | 0.43 | |
| Median beta-2 microglobulin (mg/L) (range) | 3.1 (1.2–13.1) | 2.3 (0.9–21.7) | 0.02 | |
| Rai Stage | Low (0) | 10 (27) | 822 (52) | <0.001 |
| Intermediate (I, II) | 19 (51) | 683 (43) | ||
| High (III, IV) | 8 (22) | 89 (6) | ||
| Missing | 0 | 10 | ||
| ECOG Performance Status | 0 | 27 (73) | 1394 (87) | 0.02 |
| 1–2 | 10 (27) | 192 (11) | ||
| 3–4 | 0 (0) | 14 (1) | ||
| IGHV Mutation Status | Mutated | 5 (24) | 531 (56) | 0.003 |
| Unmutated | 16 (76) | 419 (44) | ||
| Missing | 16 | 654 | ||
| IGHV 4–39 usage | Yes | 0 | 29 | 1.0 |
| No | 14 | 797 | ||
| Missing | 23 | 778 | ||
| Stereotyped B-cell receptor | Yes | 6 | 119 | 0.01 |
| No | 8 | 678 | ||
| Missing | 23 | 807 | ||
| ZAP-70 | Negative | 8 (42) | 664 (65) | 0.02 |
| Positive | 11 (58) | 333 (35) | ||
| Missing | 18 | 607 | ||
| CD49d | Negative | 3 (23) | 512 (69) | 0.0004 |
| Positive | 10 (77) | 227 (31) | ||
| Missing | 24 | 865 | ||
| CD38 | Negative | 12 (43) | 957 (71) | 0.001 |
| Positive | 16 (57) | 387 (29) | ||
| Missing | 9 | 260 | ||
| FISH category | Nil | 6 (21) | 282 (25) | <0.001 |
| 13q- | 3 (11) | 455 (41) | ||
| Trisomy 12 | 6 (21) | 214 (19) | ||
| 11q- | 6 (21) | 95 (8) | ||
| 17p- | 7 (25) | 48 (4) | ||
| Other | 0 (0) | 13 (1) | ||
| Missing | 9 | 497 | ||
Abbreviations Used: ECOG: Eastern Oncology Co-operative Group; IGHV immunoglobulin heavy chain variable gene; FISH: fluorescence in-situ hybridization
Treatment of CLL and Risk of Richter Syndrome
After median follow-up of 4 years, 578 (35%) patients received treatment for CLL. RS occurred prior to CLL treatment in 17 (46%) patients and after treatment of CLL in 20 (54%) patients. Except for a higher proportion of ZAP-70 expression (73% vs. 27%, p=0.04) in RS patients who received prior CLL therapy compared to RS patients who did not receive CLL therapy, there were no significant differences between these two groups. Table II shows the association between treatment of CLL with purine analogues, alkylating agents and monoclonal antibodies and subsequent development of RS (based on univariable analysis). Exposure to a combination of purine analogues and alkylating agents (odds ratio [OR]= 3.26, 95% C.I. =1.67–6.37, p-value=0.0003) was associated with an increased risk of RS. In contrast, exposure to single-agent purine nucleoside analogue, single-agent alkylating agent and monoclonal antibody therapy alone were not associated with a higher risk of RS.
Table II.
Association between prior treatment for CLL and subsequent development of Richter syndrome (based on univariable analysis)
| Drug Class | RS n |
No RS n |
Odds Ratio (95% CI) |
p-value | |
|---|---|---|---|---|---|
| Purine nucleoside analogue but not alkylating agent | Yes | 0 | 38 | 0.72 (0.04–12.08) | 0.42 |
| No | 20 | 1132 | |||
| Alkylating agent but not purine analogue agent | Yes | 1 | 119 | 0.49 (0.06–3.67) | 0.48 |
| No | 20 | 1162 | |||
| Combination of purine nucleoside analogue and alkylating agent | Yes | 16 | 285 | 3.26 (1.67–6.37) | 0.0003 |
| No | 20 | 1162 | |||
| Alemtuzumab ± rituximab* | Yes | 3 | 88 | 2.15 (0.62–7.49) | 0.21 |
| No | 17 | 1074 | |||
: without exposure to purine analogue or alkylating agent
Characteristics of Richter Syndrome Patients
The characteristics of patients with RS (n=37) at the time of transformation included the following: the median age was 65 years (range, 29–88 years), median WBC was 8.9 × 109/L (range, 0.6–118), median platelet count was 188 × 109/L (range, 11–569), and serum lactate dehydrogenase (LDH) was >1.5 times the upper limit of normal in 20/35 (54%) patients with available data. Although EBV in-situ hybridization studies were performed on 11 samples, EBV was detected in only 1 (9%) patient. The majority of patients (26/37, 70%) received R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) as initial therapy for RS. Other treatments included CHOP (n=2); ESHAP (etoposide, cytarabine, cisplatin and methylprednisolone) (n=1); R-HyperCVAD (rituximab, cyclophosphamide, vincristine, doxorubicin and dexamethasone) (n=1); R-DHAP (rituximab, cytarabine, cisplatin and dexamethasone) (n=1); R-CEPP (rituximab, cyclophosphamide, etoposide, procarbazine and prednisone) (n=1); RICE (rituximab, ifosfamide, carboplatin and etoposide) (n=1); ProMACE-CytaBOM (cyclophosphamide, doxorubicin, etoposide, bleomycin, vincristine, methotrexate and prednisone) (n=1) and unknown (n=1). Two patients (5%) died before the start of definitive therapy. Only 5 (14%) patients were ultimately able to undergo SCT: 3 (8%) autologous SCT, 1 (3%) allogeneic SCT and 1 (3%) autologous SCT followed by an allogeneic SCT. The most common reasons for patients not to proceed to SCT were as follows: failure to achieve response to treatment (n=10, 31%), co-morbid health conditions (n=10, 31%) and age >70 years (n=4, 13%).
Survival of Richter Syndrome Patients
The median OS after development of RS was 2.1 years (Figure 2A). As of last follow-up, 17 (46%) patients with RS had died. Causes of death were: progressive DLBCL (n=10), treatment-related complication (infection, n=4) and other/unknown (n=3). Patients who were treated for their CLL prior to development of RS had a worse OS compared to those patients who did not receive therapy (1 year vs. 3.3 years, p= 0.03, Figure 2B). The OS of all patients with RS using the prognostic score described by Tsimberdiou and colleagues is shown in Figure 3. Median survival in the low, intermediate, and high-risk groups were not reached, 2.1, and 0.5 years, respectively. The unadjusted hazard ratios (HR) and their corresponding confidence intervals (CI) for these three groups of patients are as follows: high-risk vs low-risk HR=13.7 (95% CI: 3.3–56.9), p=0.0003; and intermediate-risk vs low-risk HR=1.9 (95% CI: 0.6–6.4), p=0.30. No difference in survival was observed when the intermediate risk patients were sub-classified as low-intermediate or intermediate-high by the RS score.
Figure 2.
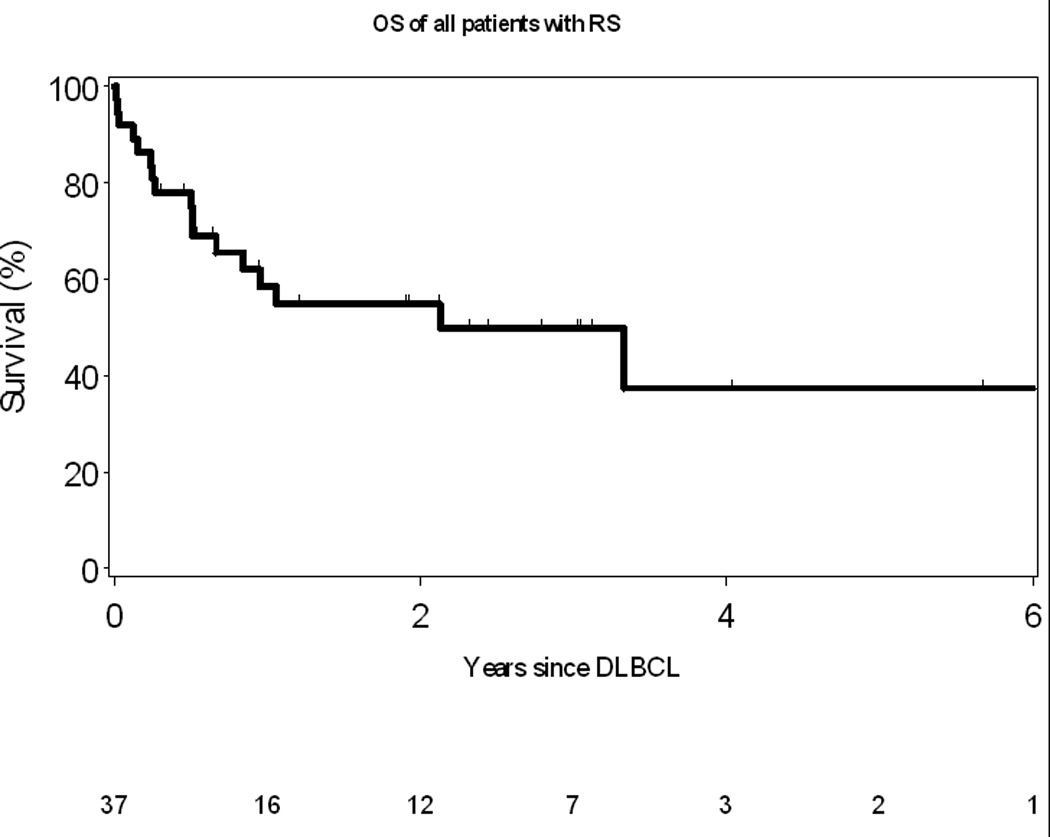
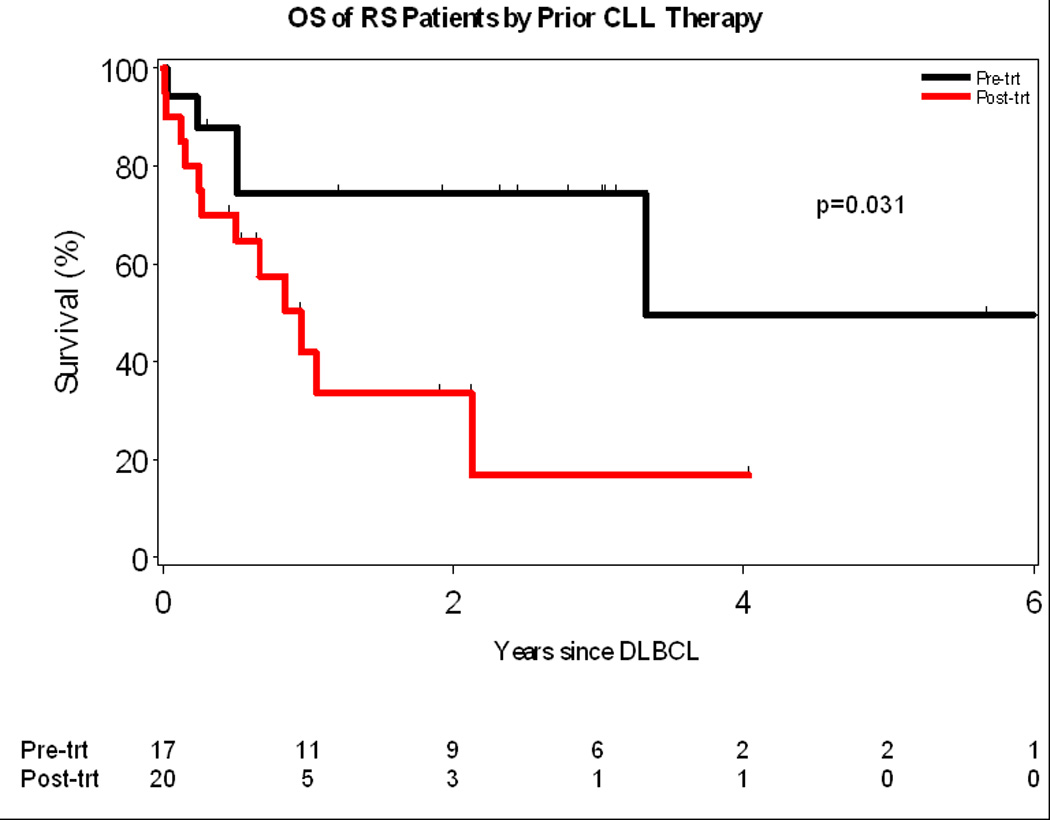
A: Overall Survival of all patients with Richter Syndrome
B: Overall Survival of RS patients who received prior therapy for CLL compared to those who did not
Figure 3.
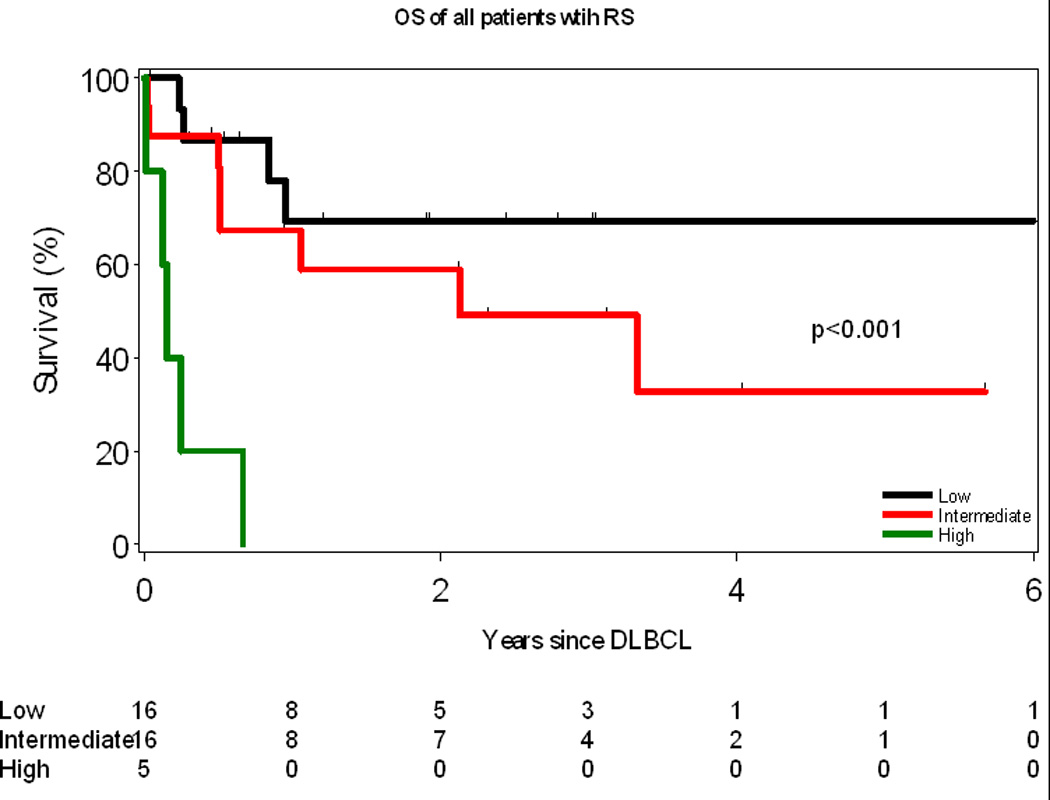
Overall Survival of all Richter syndrome patients according to the RS score
*: RS score as described by Tsimberidou et al. Each of the following characteristics at the time of diagnosis of RS gets one point: ECOG performance status of 2–4, LDH >1.5× normal, PLT <100, Tumour size >5 cm and >1 prior therapy for CLL. Patients are then assigned to one of three risk groups based on their calculated RS score: 0 or 1, low risk; 2 or 3, intermediate risk; 4 or 5, high risk.
Discussion
We report the first cohort study evaluating the frequency and characteristics of RS in a large sample of CLL patients seen at the time of diagnosis and followed prospectively. The rate of biopsy-proven RS in this cohort of >1600 newly diagnosed CLL patients was approximately 0.5% per year (incidence at 5-years: 2.1%, and at 10-years: 4.8%). The rate of RS was doubled in those patients who received treatment for their CLL (1% per year, incidence at 5-years: 5.0%, and at 10-years: 15.2%). Risk factors for RS at the time of CLL diagnosis were advanced Rai stage (III/IV), positive expression of ZAP-70, CD38 and CD49d, unmutated IGHV, and either del(11q22.3) or del(17p13) on FISH. Although treatment of CLL increased the risk of RS, nearly half the patients developed RS prior to receiving any therapy for CLL. In patients who received treatment for CLL, therapy that included a combination of purine nucleoside analogues and alkylating agents was associated with a higher rate of RS. Additionally, survival of RS patients who received therapy for their CLL was significantly shorter than those who were untreated.
The median OS of all RS patients in our study was 2.1 years, but the range of 0.1 – 12.0 years suggests considerable variability. To determine if the prognosis of newly diagnosed RS could be better predicted, we tested the validity of the MDACC scoring method (Tsimberidou, et al 2006). To our knowledge, this is the second independent validation of this model (Rossi, et al 2011) which could stratify patients into risk groups with widely different median survival using simple and widely available clinical and laboratory characteristics (high-risk patients: <1 year; low-risk patients: not reached; the difference between intermediate-risk and low-risk was not statistically significant, possibly due to limited sample size).
Data from case series of CLL patients referred to a transplant centre suggest that 40–50% of patients with RS are ultimately able to proceed to SCT.(Tsimberidou, et al 2006) Such analyses are limited by referral bias since treating physicians refer those patients they believe are transplant candidates and have responded to initial therapy for RS. Although the vast majority (35 out of 37 [95%]) patients in our cohort who developed RS were treated aggressively with multi-agent chemotherapy, only 5 patients (14%) were able to proceed to SCT. To the best of our knowledge, the data reported represent the first analysis of the proportion of RS patients derived from a CLL cohort assembled at diagnosis who are actually able to be treated with SCT. This information underscores the critical need to develop better debulking therapies to make SCT accessible to more patients. Additionally, it provides further impetus to develop novel treatment strategies that could improve survival for RS patients who are not candidates for SCT due to age, performance status, or co-morbid conditions.
How do these findings compare to other studies? In a retrospective analysis of approximately 4000 patients referred to MDACC at various time-points in the course of their CLL between 1975 and 2005, RS was diagnosed in 5.1% patients by clinical criteria, of whom 3.7% had biopsy-proven RS (Tsimberidou, et al 2006). The median time to development of RS was not reported. Mauro et al reported an RS incidence of 1.8% in a cohort of 1011 CLL patients referred to their institution over a period of 10 years but did not specify how many of these had biopsy-proven RS (Mauro, et al 1999). In a retrospective Italian case series of 185 CLL patients, the actuarial incidence of RS at 10 years was 16.2% (Rossi, et al 2008). An aggressive tissue excisional biopsy policy was thought to contribute to the increased incidence observed in this later series. Our study enhances the understanding of the incidence of RS by using a prospective cohort study design to follow patients seen at the time of CLL diagnosis. In addition, all cases of RS in our cohort were confirmed by biopsy. The RS incidence of 2.1% at 5 years is likely a more accurate estimate of RS in patients with newly diagnosed CLL and is in keeping with other large studies.
Over the last decade, traditional CLL prognostic factors such as Rai Stage, lymphocyte doubling time and lymph node size have been supplanted with the use of novel biological parameters including IGHV mutation status, expression of ZAP-70 and CD38, and identification of genetic abnormalities by FISH. In a multivariable analysis analysing the impact of clinical and biological risk factors for the development of RS in 185 CLL patients, Rossi et al identified lymph node size ≥3 cm and adverse FISH category (absence of del13q) as independent adverse prognostic markers at the time of CLL diagnosis (Rossi, et al 2008). Our study is one of the first to report on the risk of RS in a cohort of newly diagnosed CLL patients where comprehensive prognostic markers are available. Unmutated IGHV, high-risk FISH category [del(11q22) or del(17p13)], and positive expression of ZAP-70, CD49d and CD38 were all associated with an increased risk of RS. These observations provide initial insight into the biological characteristics of CLL cells that may be associated with an increased risk of RS and merit additional studies.
The effect of CLL therapy on the future risk of RS remains a fundamental and unanswered question. The Cancer and Leukaemia Group B (CALGB) 9011 study compared fludarabine, chlorambucil or a combination of the two in previously untreated CLL and reported RS in 7% patients (Solh, et al). After a median follow-up of almost 10 years, the CALGB 9712 study which examined the administration of rituximab either concurrently or sequentially with fludarabine therapy reported RS in 4% patients (Woyach, et al 2011). The rate of RS in the CLL4 trial (Catovsky, et al) (fludarabine versus fludarabine and cyclophosphamide) and CLL8 trial (Fischer, et al 2012) (fludarabine and cyclophosphamide versus fludarabine, cyclophosphamide and rituximab) was 1.6% and 4.1%, respectively, although follow-up for these trials is significantly shorter than the CALGB studies. These studies are unable to determine whether the risk is primarily driven by the treatment itself or the biological characteristics of CLL that predict a greater likelihood of requiring treatment in the first place. Our study is unique in that we prospectively followed a large cohort of newly diagnosed patients and studied the impact of both biological characteristics at diagnosis and treatment exposure during the course of follow-up on the risk of RS. Exposure to the combination of purine nucleoside analogues and alkylators was associated with an increased risk of RS whereas risk did not appear to be increased in patients exposed to only one of these drug classes.
Nearly half of the patients in this series developed RS before requiring initial therapy for CLL. This indicates the underlying risk of developing DLBCL in CLL patients independent of treatment. Although a small previous report of 20 patients suggested that up to 35% of patients with IGHV4–39 gene usage ultimately experience RS,(Rossi, et al 2009b) we were unable to confirm this observation. In fact, none of the 29 patients with IGHV4–39 gene usage in our series experienced RS. Rossi et al also reported that 21/174 (14%) CLL patients with stereotyped B-cell receptor developed RS compared to 18/579 (4%) patients with non-stereotyped B-cell receptor.(Rossi, et al 2009b) Our study confirms these findings, although only 6/125 (5%) CLL patients with stereotyped B-cell receptor developed RS in our series compared to 8/686 (1%) patients who did not have a stereotyped B-cell receptor. Other biological characteristics that may relate to risk of RS include shortened telomeres (Rossi, et al 2009a), CD38 gene polymorphism (Aydin, et al 2008), and NOTCH1 mutation (Rossi, et al 2012), in addition to the parameters identified by us in this study. We suspect that a combination of host characteristics, biology of the CLL B-cell clones, their interaction with the tumor microenvironment and the treatments that are administered during the course of the disease will influence risk of RS. Continued efforts to develop models that accurately identify CLL patients at greatest risk for RS may allow tailoring of treatment strategies to reduce risk (such as avoiding particular drugs that may increase risk in susceptible patients).
There are several limitations to our study. Since the new prognostic parameters were not available during the entire study period, we were not able to study their impact on the rate of RS in all patients. A number of treatment advances also occurred during the study period – including the introduction of chemoimmunotherapy and the introduction of newer monoclonal antibody therapies. Since only 37 cases of RS were identified, we had limited ability to perform comprehensive modelling of several risk factors for RS. A number of recent studies have also shown that the clonal relationship of the DLBCL to the underlying CLL has critical prognostic implications in RS. Patients with clonally unrelated DLBCL have survival similar to those with de novo DLBCL while cases where the DLBCL and CLL are clonally related have significantly worse outcomes (Rossi, et al 2011). Unfortunately, such information is not assessed outside of a research setting and is therefore not available in our cohort.
In summary, our study shows that the rate of biopsy-proven RS in a cohort of consecutive patients with newly diagnosed CLL was 0.5% per year (incidence at 5-years post CLL diagnosis: 2.1%). The median time to development of RS after CLL diagnosis was 1.8 years and median OS after development of RS was 2.1 years. Exposure to the combination of purine nucleoside analogues and alkylating agents was associated with substantial increased risk of RS. Only 1 in 7 CLL patients with RS was able to receive SCT, and newer treatment approaches are needed. A combination of simple laboratory and clinical characteristics can predict survival after development of RS.
ACKNOWLEDGEMENTS
Dr Shanafelt is a scholar of the Leukaemia and Lymphoma Society.
The conduct of this research was supported in part by the Predolin Foundation.
Tait D. Shanafelt: Has received research funding from Genentech, Glaxo-Smith-Kline, Cephalon, Hospira, Celgene, Polyphenon E International.
Clive S. Zent: Has received research funding from GlaxoSmithKline, Novartis, Genentech, Genzyme and Biothera.
Footnotes
Presented at the 54th Annual American Society of Haematology Meeting, Atlanta, GA, December 2012.
AUTHORSHIP
Sameer Parikh and Tait Shanafelt were the principal investigators, collected and analysed data and wrote the study and manuscript. Kari Rabe and Susan Slager collected and analysed data and performed statistical analyses. Timothy Call, Clive Zent, Thomas Habermann, Wei Ding, Jose Leis, and Neil Kay helped conceive the study, and participated in manuscript preparation. Susan Schwager collected and analysed data. Curtis Hanson and William Macon performed pathological review on specimens. All authors participated in data interpretation and critical review of the final paper.
DISCLOSURES
The following authors have financial disclosures:
All other authors: None.
REFERENCES
- Armitage JO, Dick FR, Corder MP. Diffuse histiocytic lymphoma complicating chronic lymphocytic leukemia. Cancer. 1978;41:422–427. doi: 10.1002/1097-0142(197802)41:2<422::aid-cncr2820410207>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Aydin S, Rossi D, Bergui L, D'Arena G, Ferrero E, Bonello L, Omede P, Novero D, Morabito F, Carbone A, Gaidano G, Malavasi F, Deaglio S. CD38 gene polymorphism and chronic lymphocytic leukemia: a role in transformation to Richter syndrome? Blood. 2008;111:5646–5653. doi: 10.1182/blood-2008-01-129726. [DOI] [PubMed] [Google Scholar]
- Catovsky D, Richards S, Matutes E, Oscier D, Dyer MJ, Bezares RF, Pettitt AR, Hamblin T, Milligan DW, Child JA, Hamilton MS, Dearden CE, Smith AG, Bosanquet AG, Davis Z, Brito-Babapulle V, Else M, Wade R, Hillmen P. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet. 2007;370:230–239. doi: 10.1016/S0140-6736(07)61125-8. [DOI] [PubMed] [Google Scholar]
- Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O'Brien S, Rai KR. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87:4990–4997. [PubMed] [Google Scholar]
- Cwynarski K, van Biezen A, de Wreede L, Stilgenbauer S, Bunjes D, Metzner B, Koza V, Mohty M, Remes K, Russell N, Nagler A, Scholten M, de Witte T, Sureda A, Dreger P. Autologous and Allogeneic Stem-Cell Transplantation for Transformed Chronic Lymphocytic Leukemia (Richter's Syndrome): A Retrospective Analysis From the Chronic Lymphocytic Leukemia Subcommittee of the Chronic Leukemia Working Party and Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Journal of Clinical Oncology. 2012;30:2211–2217. doi: 10.1200/JCO.2011.37.4108. [DOI] [PubMed] [Google Scholar]
- Eichhorst BF, Busch R, Hopfinger G, Pasold R, Hensel M, Steinbrecher C, Siehl S, Jager U, Bergmann M, Stilgenbauer S, Schweighofer C, Wendtner CM, Dohner H, Brittinger G, Emmerich B, Hallek M. Fludarabine plus cyclophosphamide versus fludarabine alone in first-line therapy of younger patients with chronic lymphocytic leukemia. Blood. 2006;107:885–891. doi: 10.1182/blood-2005-06-2395. [DOI] [PubMed] [Google Scholar]
- Fan L, Wang L, Zhang R, Fang C, Zhu DX, Wang YH, Zou ZJ, Li JY, Xu W. Richter transformation in 16 of 149 Chinese patients with chronic lymphocytic leukemia. Leukemia & lymphoma. 2012;53:1749–1756. doi: 10.3109/10428194.2012.664845. [DOI] [PubMed] [Google Scholar]
- Fischer K, Bahlo J, Fink A-M, Busch R, Bottcher S, Mayer J, Dreger P, Maurer C, Engelke A, Kneba M, Dohner H, Eichhorst BF, Wendtner C-M, Stilgenbauer S, Hallek M. Extended Follow up of the CLL8 Protocol, a Randomized Phase-III Trial of the German CLL Study Group (GCLLSG) Comparing Fludarabine and Cyclophosphamide (FC) to FC Plus Rituximab (FCR) for Previously Untreated Patients with Chronic Lymphocytic Leukemia (CLL): Results On Survival, Progression-Free Survival, Delayed Neutropenias and Secondary Malignancies Confirm Superiority of the FCR Regimen. ASH Annual Meeting Abstracts. 2012;120:435. [Google Scholar]
- Flinn IW, Neuberg DS, Grever MR, Dewald GW, Bennett JM, Paietta EM, Hussein MA, Appelbaum FR, Larson RA, Moore DF, Jr, Tallman MS. Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:793–798. doi: 10.1200/JCO.2006.08.0762. [DOI] [PubMed] [Google Scholar]
- Foucar K, Rydell RE. Richter's syndrome in chronic lymphocytic leukemia. Cancer. 1980;46:118–134. doi: 10.1002/1097-0142(19800701)46:1<118::aid-cncr2820460120>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von Grunhagen U, Bergmann M, Catalano J, Zinzani PL, Caligaris-Cappio F, Seymour JF, Berrebi A, Jager U, Cazin B, Trneny M, Westermann A, Wendtner CM, Eichhorst BF, Staib P, Buhler A, Winkler D, Zenz T, Bottcher S, Ritgen M, Mendila M, Kneba M, Dohner H, Stilgenbauer S. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999;17:3835–3849. doi: 10.1200/JCO.1999.17.12.3835. [DOI] [PubMed] [Google Scholar]
- Hillmen P, Skotnicki AB, Robak T, Jaksic B, Dmoszynska A, Wu J, Sirard C, Mayer J. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:5616–5623. doi: 10.1200/JCO.2007.12.9098. [DOI] [PubMed] [Google Scholar]
- Knauf WU, Lissichkov T, Aldaoud A, Liberati A, Loscertales J, Herbrecht R, Juliusson G, Postner G, Gercheva L, Goranov S, Becker M, Fricke HJ, Huguet F, Del Giudice I, Klein P, Tremmel L, Merkle K, Montillo M. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:4378–4384. doi: 10.1200/JCO.2008.20.8389. [DOI] [PubMed] [Google Scholar]
- Lortholary P, Boiron M, Ripault P, Levy JP, Manus A, Bernard J. Chronic Lymphoid Leukemia Secondarily Associated with a Malignant Reticulopathy: Richter's Syndrome. Nouvelle revue francaise d'hematologie. 1964;4:621–644. [PubMed] [Google Scholar]
- Mauro FR, Foa R, Giannarelli D, Cordone I, Crescenzi S, Pescarmona E, Sala R, Cerretti R, Mandelli F. Clinical Characteristics and Outcome of Young Chronic Lymphocytic Leukemia Patients: A Single Institution Study of 204 Cases. Blood. 1999;94:448–454. [PubMed] [Google Scholar]
- Richter MN. Generalized Reticular Cell Sarcoma of Lymph Nodes Associated with Lymphatic Leukemia. The American journal of pathology. 1928;4:285–292. 287. [PMC free article] [PubMed] [Google Scholar]
- Robak T, Blonski JZ, Gora-Tybor J, Kasznicki M, Konopka L, Ceglarek B, Komarnicki M, Lewandowski K, Hellmann A, Moskwa A, Dmoszynska A, Sokolowska B, Dwilewicz-Trojaczek A, Tomaszewska A, Sulek K, Calbecka M. Second malignancies and Richter's syndrome in patients with chronic lymphocytic leukaemia treated with cladribine. European journal of cancer. 2004;40:383–389. doi: 10.1016/j.ejca.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Robertson LE, Pugh W, O'Brien S, Kantarjian H, Hirsch-Ginsberg C, Cork A, McLaughlin P, Cabanillas F, Keating MJ. Richter's syndrome: a report on 39 patients. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1993;11:1985–1989. doi: 10.1200/JCO.1993.11.10.1985. [DOI] [PubMed] [Google Scholar]
- Rossi D, Cerri M, Capello D, Deambrogi C, Rossi FM, Zucchetto A, De Paoli L, Cresta S, Rasi S, Spina V, Franceschetti S, Lunghi M, Vendramin C, Bomben R, Ramponi A, Monga G, Conconi A, Magnani C, Gattei V, Gaidano G. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. British journal of haematology. 2008;142:202–215. doi: 10.1111/j.1365-2141.2008.07166.x. [DOI] [PubMed] [Google Scholar]
- Rossi D, Lobetti Bodoni C, Genuardi E, Monitillo L, Drandi D, Cerri M, Deambrogi C, Ricca I, Rocci A, Ferrero S, Bernocco E, Capello D, De Paoli L, Bergui L, Boi M, Omede P, Massaia M, Tarella C, Passera R, Boccadoro M, Gaidano G, Ladetto M. Telomere length is an independent predictor of survival, treatment requirement and Richter's syndrome transformation in chronic lymphocytic leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 2009a;23:1062–1072. doi: 10.1038/leu.2008.399. [DOI] [PubMed] [Google Scholar]
- Rossi D, Rasi S, Spina V, Fangazio M, Monti S, Greco M, Ciardullo C, Fama R, Cresta S, Bruscaggin A, Laurenti L, Martini M, Musto P, Forconi F, Marasca R, Larocca LM, Foa R, Gaidano G. Different impact of NOTCH1 and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. British journal of haematology. 2012;158:426–429. doi: 10.1111/j.1365-2141.2012.09155.x. [DOI] [PubMed] [Google Scholar]
- Rossi D, Spina V, Cerri M, Rasi S, Deambrogi C, De Paoli L, Laurenti L, Maffei R, Forconi F, Bertoni F, Zucca E, Agostinelli C, Cabras A, Lucioni M, Martini M, Magni M, Deaglio S, Ladetto M, Nomdedeu JF, Besson C, Ramponi A, Canzonieri V, Paulli M, Marasca R, Larocca LM, Carbone A, Pileri SA, Gattei V, Gaidano G. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009b;15:4415–4422. doi: 10.1158/1078-0432.CCR-08-3266. [DOI] [PubMed] [Google Scholar]
- Rossi D, Spina V, Deambrogi C, Rasi S, Laurenti L, Stamatopoulos K, Arcaini L, Lucioni M, Rocque GB, Xu-Monette ZY, Visco C, Chang J, Chigrinova E, Forconi F, Marasca R, Besson C, Papadaki T, Paulli M, Larocca LM, Pileri SA, Gattei V, Bertoni F, Foa R, Young KH, Gaidano G. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117:3391–3401. doi: 10.1182/blood-2010-09-302174. [DOI] [PubMed] [Google Scholar]
- Solh M, Rai KR, Peterson BL, Kolitz JE, Appelbaum FR, Tallman MS, Belch A, Larson RA, Morrison VA. The impact of initial fludarabine therapy on transformation to Richter syndrome or prolymphocytic leukemia in patients with chronic lymphocytic leukemia: analysis of an intergroup trial (CALGB 9011) Leukemia & lymphoma. 2013;54(2):252–254. doi: 10.3109/10428194.2012.710327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam CS, O'Brien S, Wierda W, Kantarjian H, Wen S, Do KA, Thomas DA, Cortes J, Lerner S, Keating MJ. Lon-g-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112:975–980. doi: 10.1182/blood-2008-02-140582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimberidou AM, O'Brien S, Khouri I, Giles FJ, Kantarjian HM, Champlin R, Wen S, Do KA, Smith SC, Lerner S, Freireich EJ, Keating MJ. Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:2343–2351. doi: 10.1200/JCO.2005.05.0187. [DOI] [PubMed] [Google Scholar]
- Wierda WG, Kipps TJ, Durig J, Griskevicius L, Stilgenbauer S, Mayer J, Smolej L, Hess G, Griniute R, Hernandez-Ilizaliturri FJ, Padmanabhan S, Gorczyca M, Chang CN, Chan G, Gupta I, Nielsen TG, Russell CA. Chemoimmunotherapy with O-FC in previously untreated patients with chronic lymphocytic leukemia. Blood. 2011;117:6450–6458. doi: 10.1182/blood-2010-12-323980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach JA, Ruppert AS, Heerema NA, Peterson BL, Gribben JG, Morrison VA, Rai KR, Larson RA, Byrd JC. Chemoimmunotherapy With Fludarabine and Rituximab Produces Extended Overall Survival and Progression-Free Survival in Chronic Lymphocytic Leukemia: Long-Term Follow-Up of CALGB Study 9712. Journal of Clinical Oncology. 2011;29:1349–1355. doi: 10.1200/JCO.2010.31.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]