

Abstract
Development and repair of the skeletal system and other organs are highly dependent on precise regulation of the bone morphogenetic protein (BMP) pathway. The use of BMPs clinically to induce bone formation has been limited in part by the requirement of much higher doses of recombinant proteins in primates than were needed in cell culture or rodents. Therefore, increasing cellular responsiveness to BMPs has become our focus. We determined that an osteogenic LIM mineralization protein, LMP-1 interacts with Smurf1 (Smad ubiquitin regulatory factor 1) and prevents ubiquitination of Smads resulting in potentiation of BMP activity. In the region of LMP-1 responsible for bone formation, there is a motif that directly interacts with the Smurf1 WW2 domain and thus effectively competes for binding with Smad1 and Smad5, key signaling proteins in the BMP pathway. Here we show that the same region also contains a motif that interacts with Jun activation-domain-binding protein 1 (Jab1) which targets a common Smad, Smad4, shared by both the BMP and transforming growth factor-β (TGF-β) pathways, for proteasomal degradation. Jab1 was first identified as a coactivator of the transcription factor c-Jun. Jab1 binds to Smad4, Smad5, and Smad7, key intracellular signaling molecules of the TGF-β superfamily, and causes ubiquiti-nation and/or degradation of these Smads. We confirmed a direct interaction of Jab1 with LMP-1 using recombinantly expressed wild-type and mutant proteins in slot-blot-binding assays. We hypothesized that LMP-1 binding to Jab1 prevents the binding and subsequent degradation of these Smads causing increased accumulation of osteogenic Smads in cells. We identified a sequence motif in LMP-1 that was predicted to interact with Jab1 based on the MAME/MAST sequence analysis of several cellular signaling molecules that are known to interact with Jab-1. We further mutated the potential key interacting residues in LMP-1 and showed loss of binding to Jab1 in binding assays in vitro. The activities of various wild-type and mutant LMP-1 proteins were evaluated using a BMP-responsive luciferase reporter and alkaline phosphatase assay in mouse myoblastic cells that were differentiated toward the osteoblastic phenotype. Finally, to strengthen physiological relevance of LMP-1 and Jab1 interaction, we showed that overexpression of LMP-1 caused nuclear accumulation of Smad4 upon BMP treatment which is reflective of increased Smad signaling in cells.
Keywords: BMP-2, Smad, Smurf1, Jab1
Introduction
Members of the transforming growth factor-β (TGF-β) superfamily, BMPs and TGF-β, have important effects on osteoblast differentiation. Upon phosphorylation, the receptor-regulated Smad proteins (R-Smads) mediate TGF-b family signaling through binding to Smad4 which is a common Smad (Co-Smad) for both BMP and TGF-β pathways, translocating to the nucleus, and mediating transcription of various genes [1]. R-Smads and the Co-Smad are targeted for degradation by Smurf1 and Jab1, respectively (Fig. 1A). LIM mineralization protein-1 (LMP-1) is a novel intracellular LIM domain protein that has been shown by our group to enhance cellular responsiveness to BMP-2 by its association with Smurf1 [1]. In this study, we identified Jab1 as a second interacting partner of LMP-1. LMP-1 contains specific sequence motifs that interact with Smurf1 and Jab1 within its central osteogenic domain (Fig. 1B).
Fig. 1.

A Smurf1 and Jab1 regulate BMP pathway by targeting Smads for degradation. BMP-2-induces receptor-mediated phosphorylation of the receptor-regulated Smad proteins 1/5 (R-Smads). Activated Smads1/5 bind the common Smad (Co-Smad), Smad 4, translocate to nucleus, and initiate transcription of various osteogenic genes. R-Smads and Co-Smad are targeted for degradation by Smurf1 and Jab1, respectively. B LMP-1 protein sequence contains one PDZ domain and three LIM domains. There are specific sequence motifs that interact with Smurf1 and Jab1 within its unique central osteogenic domain
Jab1 is also involved in protein degradation pathways like Smurf1. Jab1 was originally identified as a c-Jun coactivator and subsequently discovered to be an integral component of the constitutive photomorphogenic-9 (COP9) signalosome complex involved in modulating signal transduction and protein stability in cells [2–4]. Jab1-induced Smad4 degradation results in reduced TGF-β and BMP-mediated gene transcription [5]. Jab1 plays an essential role in positively regulating cellular proliferation by functionally inactivating several key negative regulatory proteins and tumor suppressors through their subcellular localization, degradation, and deneddylation, including p53, Smad 4/7, and the cyclin-dependent kinase inhibitor p27Kip1 (p27) [6–8]. It is also capable of stabilizing certain proteins, including hypoxiainducible factor 1a (HIF-1α) and c-Jun, as well as acting as a transcriptional cofactor for c-myc, which is responsible for the transcriptional activation of genes involved in cell proliferation, angiogenesis, and invasion [2, 9, 10]. The human Jab1 protein consists of 334 amino acids and has a molecular mass of 37 kDa; there is only one known iso-form in humans [11]. Jab1 is evolutionarily conserved in humans, mice, fission yeast, and plants, which provides evidence that Jab1 is critical to cell survival and proliferation [12–14].
Here, we define the motif of LMP-1 that interacts with Jab1 using purified recombinant wild-type and mutant proteins both in biochemical-binding assays and cell-based assays in vitro. We show that LMP-1 blocks interaction of Jab1 with Smad4, causes increased nuclear accumulation of Smad4 upon BMP treatment; and, thus, enhances Smad-mediated BMP signaling.
Materials and methods
Bacterial strains and cloning of cDNAs in bacterial expression vectors
Escherichia coli XL1 blue and BL 21-codon plus (DE3)-RP (Stratagene) hosts were maintained on LB agar plates and grown at 37 °C in the presence of ampicillin at 100 mg/liter. All of the cloning methods were performed according to standard protocols. LMP-1, Smad1, and Smad5 cDNAs were cloned into TAT–HA vector. LMP-1 mutants were generated using the following primers: hLMP1-Smurf1-Mutant forward primer, 5′-ggcccggccctttggggcggcagcagcagctgacagcgccccgcaac-3′; and hLMP1-Smurf1-mutant reverse primer, 5′-gttgcggggcgctgtcagctgctgctgccgccccaa agggccgggcc-3′. Smurf1 cDNA was cloned into pTrcHis vector (Invitrogen). For generation of Smurf1DWW2 mutant, the following primers were used: hSMURF1WW2 forward primer, 5′-gtgtgaactgtgatgaacttaatcaccagtgccaactc-3′; and hSMURF1WW2 reverse primer, 5′-gagttggcactggt gattaagttcatcacagttcacac-3′. To mutate the JAB1-interacting sequence at amino acid position 151-154 (NTED) to AAAA in TAT/HA/LMP-1, TAT/HA/LMP-1 was digested with Aat II and Not I first to create an Aat II and Not I deletion; the two oligonucleotides designed for mutation were annealed, and an Alw NI and a Not I ends were formed at the ends of the double-stranded fragment; the Aat II–Alw NI fragment was recovered after digestion of LMP-1 cDNA, and these three fragments were ligated to form TAT/HA/LMP-1/Jab1-mutant. For the generation of Smurf1–Jab1-double mutant, the following smurf1 mutation primers were used with TAT/ HA/LMP-1/Jab1-mutant, Smurf1-mutant forward primer: 5′-cctttggggcggccgcggccgctgacagc-3′ and Smurf1-mutant reverse primer: 3′-ggaaaccccgccggcgccggcgactgtcg-5′. Muta-genesis was performed with a QuikChange site-directed mutagenesis kit (Stratagene).
Expression and purification of recombinant proteins
Expression and purification of recombinant proteins were performed as reported previously with some modifications [15]. Bacterial cultures were grown at 37 °C until the A600 reached 0.8. Isopropyl β-D-thiogalactopyranoside was added to 200 μM, and the culture was grown for another 8 h. The cells were harvested, and the pellets were suspended in ice-cold lysis buffer (20 mM phosphate buffer, pH 7.0, containing 50 mM Tris–HCl, pH 7.5, and 0.5 M NaCl). The uniform cell suspension was sonicated (Sonicator, model W-385, Heat Systems-Ultrasonics, Inc.) using 4 × 15 s bursts at minimum power output settings in ice with a 2-min interval between each burst. The lysate was centrifuged at 10,000×g at 4 °C, and the supernatant was applied to Sephacryl S-100/S-200 columns (HiPrep 16 × 60) using an AKTA fast protein liquid chromatography system with Unicorn 4.0 software (Amersham Biosciences) at a flow rate of 1 ml/min. Fractions (2–4 ml) were collected immediately after the void volume (35 ml). Aliquots from each fraction were assayed by slot blotting, SDS-PAGE, and western blotting. The fractions identified by western blots were pooled, dialyzed against 20 mM phosphate buffer, pH 7.5, containing NaCl (50 mM) and imidazole (20 mM), and applied to Ni2+ affinity resin (Probond, Invitrogen) previously equilibrated with 4 × 10 ml of buffer. Nonspecific proteins were washed off the column with 3 × 10 ml of 20 mM phosphate buffer, pH 6.0, containing NaCl (50 mM) and imidazole (20 mM). Affinity-bound proteins were eluted using three 10-ml washes with 20 mm phosphate buffer, pH 4.0, containing NaCl (50 mM). Fractions containing the desired protein (based on western blot) were pooled and then concentrated and desalted using centriprep devices (Amicon). The proteins were quantitated using Bio-Rad protein assay reagent. The yield of recombinant protein was routinely 0.5–1 mg of pure protein from every 2-l culture.
Biotinylation of protein ligands
Purified protein ligands were prepared at 10 mg/ml in 50 mM sodium borate buffer, pH 8.5, 0.5 M NaCl. Various amounts of sulfo-NHS-biotin (100 mM stock in dimethyl sulfoxide) were mixed with protein ligand to achieve a molar ratio of sulfo-NHS-biotin/protein ligand of 10.0 in a 100-μl reaction volume. After 2 h on ice with occasional shaking, the reaction was terminated with the addition of lysine to a final concentration of 20 mM. The unreacted free biotin was removed by gel filtration, and the concentrated labeled ligand was stored at −20 °C until use. Labeled LMP-1, its mutants and Jab1 were prepared by using a biotinylation kit from Pierce. The specific activity of biotin incorporation into proteins was normalized by quantitating biotin using the avidin-2-hydroxyazobenzene-4′-carboxylic acid assay as instructed by the manufacturer (Pierce).
Preparation of nuclear and cytoplasmic protein fractions
Human mesenchymal stem cell (hMSCs) pellets were suspended in buffer A (20 mM HEPES, pH 7.9, 10 mM KCl, 1 mM EGTA, 1 mM EDTA, 0.2 % Nonidet P-40, 10 % glycerol, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml protease inhibitor mix (Sigma)), incubated on ice for 10 min, and centrifuged. Supernatants (cytoplasmic fraction) were collected, and nuclear pellets were suspended in high salt buffer B (buffer A plus 600 mM KCl, 20 % glycerol), incubated on ice for 30 min, and centrifuged. Supernatants were collected as the nuclear fraction. The protein amounts were determined with Bio-Rad protein assay.
SDS-PAGE and western blotting
SDS-PAGE was performed using 10 % gels and transferred to nitrocellulose membranes. The membrane was blocked with milk protein, incubated with specific antibody, washed with Tris-buffered saline containing 0.1 % Tween 20 (TBST), incubated with anti-rabbit goat IgG-linked to horseradish peroxidase (PerkinElmer Life Sciences), and again washed with TBST. Chemiluminescent substrates were applied to the membrane, and the signal was detected by exposure to X-ray film. To demonstrate equal protein loading in each lane, a signal was developed for endogenous β-actin protein in all samples.
Biotin transfer assay for detection of LMP-1-interacting proteins
Sulfo-sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido)-hexanoamido]ethyl-1,3′-dithiopropionate (Pierce), a trifunctional cross-linking agent, was used to label LMP-1. The labeled protein was incubated as bait with nuclear proteins, and crosslinked to interacting proteins by UV (365 nm). Proteins that physically interact with LMP-1 retained the biotin group when suspended in SDS-PAGE reducing buffer. Biotin-containing target proteins were separated using neutravidin beads, detected by western blotting with neutravidin-HRP, and the signal was developed with chemiluminescent substrate. Corresponding protein bands were in-gel digested with trypsin. Tryptic peptides were recovered and concentrated, and their mass profile was analyzed by Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) at the Emory University Microchemical Facility. Confirmation of protein identification was carried out at ProtTech, Inc (Norristown, PA) by using the Nano-LC–MS/MS peptide sequencing technology. In brief, a solution sample was first reduced by adding 10 mM dithiothreitol (DTT) and alkylated by adding 20 mM iodoacetamide. Proteins were denatured by adding 8 M urea. After diluting sample to 2 M urea with 100 mM ammonium bicarbonate pH 8.5, proteins were digested by adding sequencing grade-modified trypsin (Promega, Madison, WI). The resulting peptides mixture was cleaned by PepClean spin column (Pierce, Rockford, IL), and analyzed by a Nano-LC–MS/MS system, in which a high-pressure liquid chromatography (HPLC) with a 75-μm-inner diameter reverse phase C18 column was online coupled with an ion trap mass spectrometer (Thermo, Palo Alto, CA). The mass spectrometric data acquired were used to search the most recent nonredundant protein database from GenBank (http://www.ncbi.nlm.nih.gov/) with ProtTech's proprietary software suite. The output from the database search was manually validated before reporting.
Slot-blot assay
Smurf1-LMP-binding assay
A 20 μl aliquot of purified Smurf1 (50 μg/ml) was blotted onto nitrocellulose in slot blot wells, and the wells were blocked with 0.5 % Tween 20 in TBST for 30 min. The biotinylated LMP-1 was mixed with varying concentrations of competing proteins and incubated in slot blot wells with Smurf1 for 90 min. The wells were washed, and the blots were blocked with TBST containing 0.5 % Tween 20. Control wells contained LMP-1 hapten (an antigenic peptide from the c-terminal end of the polypeptide chain) as a competitor peptide.
Jab1-Smad4-binding assay
A 20 μl aliquot of Jab1 (50 μg/ml) was blotted onto nitrocellulose in slot blot wells, and the wells were blocked with 0.5 % Tween 20 in TBST for 30 min. The biotinylated Smad4 was mixed with varying concentrations of competing LMP-1 wild-type or Jab1ΔMutant LMP-1 protein and incubated in slot blot wells with Jab1 for 90 min. The wells were washed, and the blots were blocked with TBST containing 0.5 % Tween 20. The blots were then incubated with horse radish peroxidase (HRP)-labeled avidin for 1 h. After washes the blots were incubated with ECL substrate solution, and the membranes were exposed to X-ray film for signal detection.
Protein A-based immunoprecipitation assay
Protein A-agarose beads were incubated with LMP-1 antibody or Jab1 antibody, washed three times, incubated with nuclear proteins, and washed again to remove unbound protein. The bound proteins were eluted by two washes in 0.1 M citric acid, pH 2.7. The eluates were neutralized with 1.0 M Tris base and concentrated by centricon tubes (Ambicon) prior to SDS-PAGE and western blotting.
Western blotting
The proteins were separated by SDS-PAGE and blotted onto a nitrocellulose membrane. The protein blots were blocked with 5 % milk protein and preincubated with purified LMP-1 or its mutants (10 μM) or TBST buffer. The blots were incubated with rabbit anti-LMP-1 or anti-Jab1 antibody at 1:500 or 1:5000 dilution, respectively. After washes, the blots were incubated with HRP-labeled anti-rabbit antibody. The washed blots were then incubated with ECL substrate solution, and the membranes were exposed to X-ray film for signal detection.
Cell culture reagents
Minimum essential medium (MEM), supplemented with 2 mM L-glutamine, 0.25 % trypsin, and penicillin/streptomycin (5,000 U/ml), was purchased from Gibco BRL (Gaithersburg, MD); BGJb bone culture medium, glucocorticoid, triamcinolone acetonide, β-glycerophosphate, and ascorbic acid were purchased from Sigma Chemical Co. (St. Louis, MO); collagenase was purchased from Worthington Enzymes (Freehold, NJ); heat-inactivated fetal bovine serum (FBS) was purchased from Hyclone Laboratories, Inc. (Logan, UT). Tissue culture flasks and plates were purchased from Corning, Inc. (Corning, NY). Timed pregnant Sprague–Dawley rats were purchased from Charles River Laboratories, Inc. (Raleigh, NC), and athymic rats (rnu/rnu) were purchased from Harlan (Indianapolis, IN).
Isolating fully mature and functional osteoblasts is challenging for bone tissue engineering and regenerative medicine. Human mesenchymal stem cells (hMSCs) or myoblastic C2C12 cells that can be triggered toward osteoblastic phenotype are often preferred alternatives and are thus chosen for our studies. Human MSCs at passage 2 (catalog #PT-2501, Cambrex Bio Sciences, Walkersville, MD) were grown at 37 °C in 5 % CO2 in MSC basal medium supplemented with Singlequots (Cambrex Bio Sciences), split at confluence, and plated at 30,000 cells/ well in 6-well dishes at passage 4. The next day treatments were applied in the presence of 50 μM ascorbic acid and 5 mM β-glycerol phosphate (Sigma-Aldrich). The medium was changed every 3–4 days with reapplication of treatments where appropriate. The cells were transduced for 30 min with adenoviral constructs in 0.3 ml of serum-free medium. For detection of Smad4 in western blots, hMSCs at passage 4 were seeded at 30,000 cells/well in a 6-well plate. The next day, the cells were infected with Ad35LMP-1 (1–10 pfu/cell) and incubated with or without BMP-2 (100 ng/ml) for 8 h.
Mouse C2C12 cells and Dulbecco's modified Eagle's medium (DMEM) were purchased from ATCC (Manassas, VA). The C2C12 cells at passages 5–10 were subcultured in T-75 cm2 flasks in DMEM supplemented with 10 % FBS at 37 °C in 5 % CO2 with humidification. When the flasks reached 80 % confluence, the cells were trypsinized and seeded in triplicate at 200,000 cells/well in a 6-well plate for quantitative real-time RT-PCR and alkaline phosphatase (ALP) assays or at 50,000 cells/well in a 12-well plate for the dual-luciferase reporter assay.
siRNA treatment of cells
Mouse C2C12 cells were transfected with Lipofectamine RNAiMAX Reagent (Invitrogen) and either irrelevant siRNA or Jab1 (5′-guauauggcugcauacaua[dT][dT]-3′) at 3 nM. Silencing of the gene and specificity was confirmed by determining mRNA levels and western blotting analysis using specific primary antibody and anti-rabbit secondary antibody (Santa Cruz).
RNA extraction
RNA was isolated from cells grown in 6-well plates using RNeasy mini kits (Qiagen). Briefly, the cells were disrupted in RNeasy lysis buffer (Qiagen) and passed over QiaShredder columns, and the eluate was brought to 35 % ethanol and passed over RNeasy columns. The RNA was eluted from the membrane with water. All the RNA samples were DNasetreated either using the Qiagen RNase-free DNase during the RNeasy procedure or after final harvest of the RNA using the Ambion DNA-free kit. After completion of the digestion, 5 μl of DNase inactivation buffer was added, and the samples were centrifuged for 1 min. The RNA containing supernatant was removed and stored at −70 °C. Each sample consisted of RNA isolated from two wells of a 6-well plate.
Real time reverse transcription-polymerase chain reaction
Two μg of total RNA was reverse transcribed in a 100-μl total volume containing 50 mM KCl, 10 mM Tris, pH 8.3, 5.5 mM MgCl2, 0.5 mM each dNTPs, 0.125 μM random hexamer, 40 units RNase inhibitor, and 125 units MultiScribe (Applied Biosystems). In control samples, the RNase inhibitor and MultiScribe were omitted. The samples were incubated for 10 min at 25 °C, 30 min at 48 °C, and then 5 min at 95 °C to inactivate the enzyme. Real time PCR was then performed on 5 μl of the resulting cDNA in a total volume of 25 μl containing 12.5 μl of 2×SYBR Green PCR Master Mix (Applied Biosystems), and 0.8 μM each primer. The forward primer for alkaline phosphatase was 5′-CGGCCCTGAGTCTGACAAAG-3′, and the reverse primer was 5′-CTCGTCACAAGCAGGG TCAA-3.′ PCR conditions were: 2 min at 50 °C, 10 min at 95 °C, and 45 cycles of 95 °C for 15 s followed by 1 min at 62 °C. PCR was also performed on a 1:800 dilution of the cDNA with 18 S primers for normalization of the samples. Relative RNA levels were calculated using the ΔΔCt method (Applied Biosystems).
Alkaline phosphatase (ALP) assay
The C2C12 cells were plated at 200,000 cells/well in 6-well plates and grown overnight in DMEM containing 10 % FBS. On day 2, the culture medium was replaced with DMEM containing 2 % FBS, and the cells were treated with various concentrations of TAT-proteins for 24 h. On day 3, the medium was replaced with fresh DMEM containing 2 % FBS, and the cells were treated with 50 ng/ml of BMP-2 for 72 h. The cells were washed with phosphate-buffered saline (PBS) and lysed by addition of lysis buffer (10 mM Tris–HCl pH 8.0, 1 mM MgCl2, and 0.5 % Triton X-100). The cell lysates were centrifuged for 5 min at 13,000×g. The supernatant was removed and assayed for ALP activity and protein amount. The ALP activity was measured in triplicate using an ALP assay kit (Sigma-Aldrich, St. Louis, MO) in microtiter plates. The protein amount was determined with Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA) using bovine serum albumin (BSA) as a standard. The ALP activity (nmoles of p-nitrophenol per ml) was normalized to the protein amount (nmoles of p-nitrophenol per μg).
Dual luciferase reporter assay
The BMP-specific Smad1-driven 9×GCCG (a consensus-binding sequence for Smad1) reporter plasmid was kindly provided by Dr. Miyazono (The Institute of Japanese Foundation for Cancer Research, Tokyo). The C2C12 cells were trypsinized and seeded in triplicate wells at 50,000 cells/well in 12-well plates on day 1. On day 2, the cells were cotransfected with the 9×GCCG-luciferase-reporter construct and the renilla-luciferase control vector using SuperFect (Qiagen, Valencia, CA) for 24 h. A total of 1 μg of plasmids was used for cotransfection in each well, and the concentration of renilla-luciferase vector was 1/15 of the 9×GCCG-reporter plasmid. On day 3, medium was replaced with DMEM containing 2 % FBS, and the cells were treated with various concentrations of recombinant LMP-1 proteins. On day 4, the cells were treated with BMP-2. On day 5, the luciferase activities were measured in 20 μl of cell-lysate using the dual-luciferase assay system (Promega, Madison, WI) with a luminometer (Lumi-Count; Packard Bioscience, Meriden, CT) following the manufacturer's instructions. The luciferase activity was expressed as relative units of luciferase (RUL; a ratio of firefly luciferase to renilla luciferase activity).
The luciferase assay system is designed to allow analysis of mammalian cells containing plasmid-coded genes for firefly and renilla luciferases, grown in culture plates. The activities of firefly (Photinus pyralis) and renilla (Renilla reniformis, also known as sea pansy) luciferases are measured sequentially. The firefly luciferase reporter is measured first by adding luciferase assay reagent II to generate a “glow-type” luminescent signal. After quantifying the firefly luminescence, this reaction is quenched, and the renilla luciferase reaction is initiated by simultaneously adding Stop & Glo Reagent to the same tube. The Stop & Glo reagent also produces a “glow-type” signal from the renilla luciferase, which decays slowly over the course of the measurement. In the assay system, both reporters yield linear assays with subattomole sensitivities and no endogenous activity of either reporter in the experimental host cells. The ratio of activity of luciferases normalizes the transfection efficiency.
Statistics and calculations
Results are presented as the mean of three determinations (n) with error bars representing the standard error of the mean (SEM). Experimental results that are visually represented are from consistent experiments where one representative experimental result is shown. Statistical significance (P < 0.05) was calculated using a one-way analysis of variance (ANOVA) with Bonferroni Post Hoc test (equal variances assumed) or Dunnett's T3 Post Hoc test (equal variances not assumed) using Statistical Products for Social Sciences Version 16.0 (SPSS 16.0) for Windows (SPSS, Chicago, IL) to compare various treatments in multigroup analysis. Statistical probability of P < 0.05 was considered significant.
Results
Validation of a BMP-2 reporter assay for screening activity of the recombinant TAT–LMP-1 protein
We demonstrated previously that TAT-tagged LMP-1 protein and its mutants enter the cells with comparable efficacy using fluorescently labeled proteins (15). In order to have a rapid assay to determine the effect of LMP-1 on the BMP-2 pathway, we developed a BMP-2 promoter reporter assay in which the promoter contains nine copies of the Smad1-binding sequence (9×GCCG). As shown in Fig. 2A, BMP alone induced the luciferase reporter activity 2–16-fold over no BMP control at a dose range of 1–25 ng/ml in a dose dependent manner. Similarly, under these conditions, the TAT–LMP-1 protein potentiated the BMP-induced response (about 2-fold) dose dependently over BMP-alone control (Fig. 2B).
Fig. 2.

A Validation of a BMP-2 reporter assay for screening activity of TAT-LMP-1. The dose-dependent induction of luciferase activity by BMP-2 in C2C12 cells transfected with BMP-specific and Smad1-driven 9×GCCG reporter plasmid is shown. The reporter responded dose dependently to BMP-2. B Potentiation of BMP-induced response by LMP-1. A dose response to BMP-2 by the reporter construct, and clear potentiation of BMP-2 responsiveness by purified recombinant TAT-LMP-1 protein is observed. Luciferase activities were determined in triplicate
LMP-1/Smurf1 interaction does not account for total LMP-1 activity
LMP-1 interacts with Smurf1 and enhances BMP-2 efficacy. To understand whether this LMP-1 effect was entirely dependent on its interaction with Smurf1, we prepared a mutant of wild-type TAT–LMP-1 (wild-type) fusion protein that lacks the Smurf1-binding motif (LMP-1ΔSmurf1) and assessed relative luciferase activity of the mutant in a previously validated BMP-specific Smad1-dependent reporter assay (Fig. 3). To our surprise, the mutant protein retained the ability to partially (about 50 %) enhance BMP-2 activation (5 ng/ml) of the reporter construct, despite loss of binding to Smurf1 in slot blot assays. This suggested that LMP-1 interaction with additional proteins was likely required for its full activity. Thus, we directed our efforts toward identifying other novel binding partners of LMP-1.
Fig. 3.

LMP-1/Smurf1 interaction does not account for total LMP-1 activity. The mutant protein of LMP-1 that fails to bind Smurf1 retained the ability to partially enhance BMP-2 activation (5 ng/ml) of the reporter construct in C2C12 cells, despite loss of binding to Smurf1 in slot blot assays
Identificatio of Jab1 as LMP-1-binding protein
Recombinant LMP-1 was labeled with sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido)-hexanoamido]ethyl-1, 3-dithiopropionate-biotin transfer reagent and incubated with lysates of hMSC cells. Biotin transfer to interacting proteins was accomplished as described under “Materials and methods.” Biotinylated proteins were enriched using neutravidin beads, separated by SDS-PAGE, and detected on western blots using HRP-labeled neutravidin and ECL. Bands were excised for tryptic digestion and MALDI–TOF, and Nano-LC–MS/MS analyses were performed. Table 1 shows petides that were sequenced in two separate tryptic digests. A representative scan of Nano-LC–MS/MS is shown in Fig. 4A. The identity of Jab1 was confirmed in western blots using Jab1-specific antibodies on immunoprecipitates obtained by antibiotin antibody. Western blots show the presence of both Smurf1 and Jab1 in immunoprecitates using horse radish peroxidase-labeled neutravidin (lane 1), Smurf1 with Smurf1 antibody (lane 2), and Jab1 with Jab1 antibody (lane 3), respectively (Fig. 4B).
Table 1.
Identification of Jabl in tryptic digests of two separate samples
| Tryptic digest #1 |
Tryptic digest #2 |
||
|---|---|---|---|
| Mass | Peptide sequence | Mass | Peptide sequence |
| 827.55 | ISALALLK | ||
| 1103.51 | IEDFGVHCK | 1103.51 | IEDFGVHCK |
| 1129.57 | LEQSEAQLGR | ||
| 1361.64 | GSFMLGLETHDR | ||
| 1404.71 | SGGNLEVMGLMLGK | ||
| 1409.69 | QYYALEVSYFK | 1409.69 | QYYALEVSYFK |
| 1539.84 | QQQEILAAKPWTK | 1539.84 | QQQEILAAKPWTK |
| 1639.9 | TTIEAIHGLM SQVIK | 1639.9 | TTIEAIHGLM SQVIK |
| 2089.98 | VNAQAAAYEYMAAYIENAK | 2089.98 | VNAQAAAYEYMAAYIENAK |
| 2132.04 | GYKPPDEGPSEYQTIPLNK | ||
| 2282.05 | TWELANNMQEAQSIDEIYK | ||
| 2410.14 | VDGETMIIMDSFALPVEGTETR | ||
The table shows results from LC-MS/MS (tandem MS) based on independent peptide sequencing. Therefore, all peptides listed here are actual peptides present in a sample instead of “potential candidates,” which are often seen in a MALDI-TOF-based peptide mapping. Mass: the calculated molecular weight of each peptide based on its amino acid sequence. Peptide sequence column shows the sequence of each peptide identified
Fig. 4.

A LMP-1 specifically interacts with Jab1. LMP-1 is associated with a 37-kDa protein. Recombinant LMP-1 was labeled with sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido)-hexanoamido]ethyl-1,3-dithiopropionate-biotin transfer reagent and incubated with nuclear lysates of hMSC cells. Biotin transfer to interacting proteins was accomplished as described under “Materials and methods.” Biotinylated proteins were enriched using neutravidin beads, separated by SDS-PAGE, and detected on western blots using HRP-labeled neutravidin and ECL. Bands were excised for tryptic digestion and NanoLC-MS/MS analysis. B The identity of Jab1 was confirmed in western blots using Smurf-1 and Jab1-specific antibodies on immunoprecipitates obtained by antibiotin antibody. Western blots show the presence of both Smurf1 and Jab1 with neutravidin-horse radish peroxidase (lane 1), Smurf1 with Smurf1 antibody (lane 2) and Jab1 with Jab1 antibody (lane 3). The 85 and 37 kDa bands were identified as Smurf1 and Jab1, respectively, as shown by western blotting
LMP-1 directly binds to Jab1
To determine whether LMP-1 directly binds Jab1, we performed binding assays with purified recombinant proteins. Cytoplasmic proteins from human mesenchymal stem cells (hMSCs) were separated by SDS-PAGE and blots were probed with biotin-labeled LMP-1 (Fig. 5 lane 1). The bound biotin-LMP-1 was detected using neutravadin-HRP. Lane 1 shows that LMP-1 is capable of binding directly to two proteins (85 and 37 kDa). The identity of these two bands was confirmed by staining with antibody specific to Smurf1 (lane 2) and Jab1 (lane 3), respectively. These blots provide evidence that LMP-1 contains a Jab1-interacting motif, in addition to the Smurf1-interacting motif. A natural variant of LMP which lacks the central region responsible for Jab1 interaction was also in immunoprecipitations as control. As expected, this variant did not pull down Jab1 protein when western blotting was performed using Jab1 antibody. LMP-1 failed to bind Jab1 under denatured conditions suggesting that a tertiary conformation of LMP-1 is required for Jab1 binding (data not shown).
Fig. 5.

LMP-1 and Jab1 coimmunoprecipitate from nuclear extracts of C2C12 cells with either LMP-1 antibody (LMP-1-ab) or Jab1 antibody (Jab1-ab), and protein A beads. Nuclear proteins were immunoprecipitated (IP) using LMP-1 antibody or Jab1 antibody. The immunoprecipitated proteins were concentrated and analyzed on western blots (WB) with LMP-1 or Jab1 antibody. The bands were at 50 or 37 kDa as expected for LMP1 or Jab1 protein, respectively
LMP-1 and Jab1 coexist as a cellular complex
To determine if LMP-1 and Jab1 coexist as binding partners in cell, we performed immunoprecipitations using either LMP-1 or Jab1 antibodies in lysates of mouse myoblastic cells. The immunoprecipitates of nuclear lysates of C2C12 cells obtained with Jab1 antibody contained LMP-1 and also the immunoprecipitates obtained with LMP-1 antibody contained Jab1 protein as shown by western blotting (Fig. 5). These data demonstrate that an association between Jab1 and LMP-1 occurs in cells under physiological conditions.
Mutation of the Smurf1-interaction motif or the Jab1-interaction motif in LMP-1 results in loss of binding to the respective target proteins
To determine the region of LMP-1 that interacts with Jab1, we performed LMP-1 protein sequence analyses using a motif discovery tool (MEME/MAST). Jab1-binding regions were detected in the known Jab1-binding partners p53, Smad4, rLHR, p27(kip1), cullin, and c-jun and a consensus Jab1-interacting sequence derived. We then determined that the consensus Jab1-interacting sequence was present at amino acid position 161 in LMP-1 (Table 2) and confirmed this by construction of a mutant LMP-1, in which the residues NTED were mutated to AAAA within the putative Jab1-interacting region. Binding of the wild-type and mutant proteins to Jab1 was tested in slot blot assays.
Table 2.
Consensus Jab1-interacting residues are identified from natural target proteins of Jab1 factor. Using MEME (motif discovery tool) the corresponding Jab1-binding motif is identified in LMP-1 sequence. Note: The primary amino acid sequence exhibits limited homology although the predicted tertiary structure exhibits higher homology in this region. Amino acids within a structurally conserved region of each Jab1-binding protein are shown
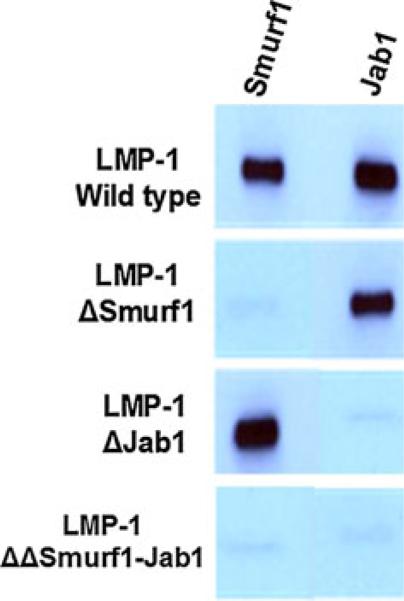
|
In slot blot-binding assays performed with purified recombinant proteins, the biotinylated TAT–LMP-1 (wild-type) bound to both Smurf1 and Jab1 that were immobilized onto nitrocellulose blots. Mutation of the Smurf1-interacting motif in LMP-1 (LMP-1ΔSmurf1) resulted in loss of binding to Smurf1 without affecting its binding affinity for Jab1. Similarly, mutation of the Jab1-interacting motif (LMP-1ΔJab1) resulted in loss of binding of LMP-1 to Jab1 without affecting its interaction with Smurf1 (Fig. 6). As expected, the double mutant (ΔSmurf1ΔJab1) lacking the required motifs for Smurf1 and Jab1 completely failed to bind these target proteins. In this experiment the specific activity of the biotin labeling was normalized by estimating the number of biotins per protein molecule by means of 4′-hydroxyazobenzene-2′-carboxylic acid (HABA) assay kit (Pierce). This confirms that the LMP-1 mutants do not bind Smurf1/Jab1, as expected, and validates the use of mutants to understand the importance of interaction of LMP-1 with Jab1 and Smurf1 in osteoblast differentiation.
Fig. 6.
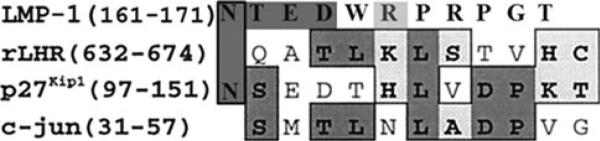
Mutation of the Smurf1-interaction motif or the Jab1-interaction motif in LMP-1 results in loss of binding to the respective target proteins. Mutation of the Smurf1-interacting motif in LMP-1 (LMP-1ΔSmurf1) resulted in loss of binding to Smurf1 without affecting its binding affinity for Jab1. Similarly, mutation of the Jab1-interacting motif (LMP-1ΔJab1) resulted in loss of binding of LMP-1 to Jab1 without affecting its interaction with Smurf1. A double mutant (ΔΔ Smurf1–Jab1) lacking the required motifs for Smurf1 and Jab1 fails to bind these target proteins
LMP-1 interactions with both Jab1 and Smurf1 are required for full LMP-1 activity
We assessed the activity of purified recombinant LMP-1 and its mutant proteins in the BMP-reporter assay. The Jab- noninteracting LMP mutant (LMP1ΔJab1), like the Smurf-noninteracting LMP mutant (LMP-1ΔSmurf1), showed significant loss of BMP-2-potentiating activity as measured by relative luciferase activity (Fig. 7A). As expected, nearly half of BMP-potentiating activity of LMP-1 was lost in each Smurf1 or Jab1 mutant. The double mutant (ΔSmurf1ΔJab1) lacking the required motifs for Smurf1 and Jab1 exhibits complete loss of activity. This indicated that both Smurf1 and Jab1 interactions are necessary for LMP-1 to fully potentiate BMP-2 activity.
Fig. 7.

A LMP-1 interactions with both Jab1 and Smurf1 are required for full LMP-1 activity as observed in relative activities of recombinant LMP-1 proteins in potentiating BMP-induced reporter activity. The Jab-noninteracting LMP mutant (LMP1ΔJab1), like the Smurf-noninteracting LMP mutant (LMP-1ΔSmurf1), showed significant loss of BMP-2-potentiating activity as measured by relative luciferase activity. A double mutant (ΔΔ Smurf1–Jab1) lacking the required motifs for Smurf1 and Jab1 shows complete loss of BMP- potentiating activity. B Alkaline phosphatase assay confirms both Smurf1 and Jab1 interactions are required for full LMP-1 activity. While the ΔSmurf1 or ΔJab1 mutants of LMP-1 exhibited partial enhancement of BMP-induced ALP activity, the double mutant (ΔΔ Smurf1Jab1) exhibited little or no potentiating activity on BMP. This indicated that both Smurf1 and Jab1 interactions are necessary for LMP-1 to exhibit maximal BMP-2 potentiating activity. The data points were determined in triplicate
Alkaline phosphatase assay confirms both Smurf1 and Jab1 interactions are required for full LMP-1 activity and validates the BMP-reporter assay
To confirm the physiologic relevance of the BMP-reporter assay, we measured alkaline phosphatase activity in response to BMP-2 (50 ng/mL) in C2C12 cells (Fig. 7B). Cells were transduced with various forms of TAT–LMP-1 for 24 h before treatment with BMP-2 for 3 days. Treatment with full length TAT–LMP-1 (wild-type) increased BMP-2 induction of alkaline phosphatase activity nearly 4-fold while the TAT–LMP-1 mutants lacking either the Smurf1 or Jab1-interacting motifs showed only partial enhancement. As expected, the double mutant (ΔSmurf1ΔJab1) lacking the required motifs for Smurf1 and Jab1 completely fails to exhibit the potentiating activity on BMP-induced ALP activity. These findings with a more physiologically relevant enzyme marker, closely mimicked the BMP-reporter assay results observed above.
Jab1 knockdown by siRNA causes elevation of alkaline phosphatase mRNA
We have previously shown that knocking down LMP-1 expression by antisense oligonucleotide potently inhibited osteoblast differentiation as measured by osteocalcin secretion and mineralized bone nodule formation in primary rat osteoblast cultures [16]. To establish a functional relationship between Jab1 levels and osteogenic potential in C2C12 cells, we determined the relative levels of alkaline phosphatase mRNA in response to Jab1 knockdown by siRNA in C2C12 cells. The C2C12 cells were transfected with control or Jab1 siRNA for 6 h followed by a treatment with or without BMP-2 at a final concentration of 100 ng/ml. RNA was isolated 24 and 48 h after BMP-2 treatment for RT-PCR as described in “Materials and methods.” As shown in Fig. 8, Panels A and B, we observed a reduced level of Jab1 protein and an elevated level of BMP-induced alkaline phosphatase mRNA, respectively, in C2C12 cells treated with Jab1 siRNA. This finding establishes the functional importance of Jab1 in induction of osteoblastogenesis.
Fig. 8.

Knockdown of Jab1 by siRNA elevates Jab1 mRNA levels in C2C12 cells. The C2C12 cells were transfected with control or Jab1 siRNA. Then cells were treated with or without BMP2 at 100 ng/ml. RNA was isolated 24 h after BMP2 treatment for RT-PCR as described in “Materials and methods.” Panel A Reduction of Jab1 protein expression in western blot using Jab1-specific antibody. Panel B Increase in BMP-induced ALP mRNA level upon Jab1 siRNA treatment
LMP-1 blocks binding of Jab1 to Smad4
To confirm that LMP-1 binding to Jab1 interferes with Jab1 and Smad4 interaction, we performed in vitro binding assays in slot blots using recombinantly expressed and purified Jab1, Smad4 and wild-type/mutant LMP-1 proteins. In the absence of competing LMP-1, we observed maximal binding of Jab1 and Smad4. This signal was dose dependently reduced in the presence of wild-type LMP-1 protein at concentrations of protein 10 μM or higher as shown in Fig. 9.
Fig. 9.
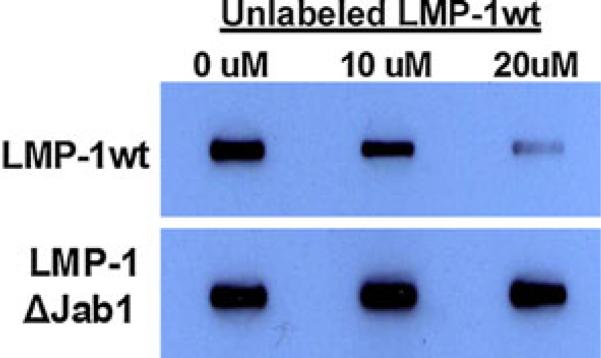
LMP-1 blocks binding of Jab1 to Smad4. Jab1 was immobilized onto nitrocellulose in slot blot wells. After blocking, the biotinylated Smad4 was mixed with varying concentrations (0–20 μM) of competing LMP-1 wild-type or Jab1Δ mutant LMP-1 protein and incubated in slot blot wells with Jab1. The signal for Jab1–Smad4 binding was developed as described in “Materials and methods.” wild-type LMP-1 inhibited Smad4 binding to Jab1 dose dependently
Overexpression of LMP elevates nuclear Smad4 levels
The most relevant physiologic question is whether blockage of Smad4 binding to Jab1 causes nuclear accumulation of Smad4, in hMSCs, which are the initiating cells in adult osteogenesis. Nuclear accumulation of Smad4 is associated with increased Smad signaling. We overexpressed LMP-1 by infecting MSC cells with adeno-virus carrying the LMP-1 gene. We then performed SDS-PAGE separation of nuclear proteins, and the blots were probed with Smad4 specific antibody. The 66-kDa band represents nuclear Smad4 which can be seen to increase at 8 h after LMP-1 treatment in response to BMP-2 treatment (100 ng/ml) (Fig. 10). Since Smad4 is required for both BMP and TGFβ effects on osteoblastogenesis, these findings suggest that LMP-1 enhancement of BMP-induced osteoblast formation depends, in part, on its interaction with Jab1 by competing with Smad4. The phosphorylated receptor Smads1, 5, or 8 oligomerize with Smad4, enter the nucleus, and induce osteogenic genes in the BMP pathway. An increase in nuclear Smad4 is an indicator of enhancement of this pathway.
Fig. 10.

Overexpression of LMP-1 causes nuclear accumulation of Smad4 protein in hMSCs. LMP-1 overexpression increases the efficacy of BMP-2 to induce nuclear translocation of Smad4 in hMSCs as measured in western blot. Human MSCs were treated with or without Ad5F35 LMP-1 (5 pfu/cell) and rhBMP-2 (100 ng/ml) for 8 h as described in “Materials and methods.” We observed a twofold increase in the amount of Smad4 detected in LMP-1 overexpressed cells compared with control cells. The phosphorylated receptor Smads1, 5 or 8 oligomerize with Smad4, enter the nucleus and induce osteogenic genes in the BMP pathway. An increase in nuclear Smad4 is an indicator of enhancement of this pathway
Discussion
The present study was undertaken to identify additional binding partners of LIM mineralization protein-1, an intracellular effector of BMP activity, which actively promotes BMP signaling in osteoblastic cells. This study demonstrates for the first time that LMP-1 physically interacts with Jab1 and is able to enhance BMP signaling. Previously, Jab1 was reported to physically interact with Smads 4, 5 and 7 [17–19] but not with Smads 1, 2, 3, and 6. Jab1 represents subunit 5 of the COP9 signalosome (CSN). Although the exact function of CSN is still unclear, the data are consistent with the notion that it has a substantial role as an interface between signal transduction and ubiquitin-mediated proteasomal degradation of proteins. The functional relevance of Jab1 and/or the COP9 complex to the skeleton is also unclear at present. Jab1-knockout mice die soon after implantation, most likely due to impaired general proliferative activity and increased apoptosis of cells [20]. In accordance with this, heterozygous animals show reduced skeletal growth. Our results suggest that Jab1 might have a role during skeletal development, at least in part by negatively modulating BMP signaling, which is important for skeletal growth. Results of our study provide evidence that there is direct interaction of Jab1 with LIM mineralization protein-1, an intracellular osteogenic protein which also interacts with Smads 1 and 5 and thereby modulates BMP signaling. Even if Jab1 is not as actively involved as Smurf1 in blocking of BMP signaling, its constant presence and BMP-blocking properties, together with its modulatory activity, make this molecule a unique target for therapeutic intervention for promoting BMP-induced osteogenic response in cells.
Using the optimized cell-based assay, we evaluated the activity of the recombinantly prepared proteins, TAT– LMP-1 and its mutants (LMP-1ΔSmurf1, LMP-1ΔJab1 and LMP-1ΔΔSmurf1Jab1 double mutant) that lack the binding motif(s) of Smurf1 or Jab1 or both. Both the wild-type and the mutant proteins contain an 11-amino acid HIV-TAT protein-derived membrane transduction domain to aid the recombinant proteins in cellular entry. The cell-based reporter assay confirmed that LMP-1 potentiates the BMP-induced stimulation of C2C12 cells toward the osteoblastic phenotype. The potentiating effect of LMP-1 was lost when specific motifs known to interact with Smurf1 or Jab1 were mutated. We validated the results obtained in the reporter assay by monitoring the expression of mRNA and activity of alkaline phosphatase which is widely accepted as an osteoblast differentiation marker gene.
Our results clearly show that both Smurf1 and Jab1 interactions are necessary for LMP-1 to be fully functional in its BMP-potentiating activity (Fig. 11). We show that LMP-1 accomplishes its BMP-potentiating activity by competing with Smad4 in binding to Jab1. We also show that overexpression of LMP-1 results in cellular accumulation of Smad4 which reflects increased Smad signaling upon BMP treatment. However, further studies need to be performed for further understanding how LMP-1 interaction specifically interferes with ubiquitination and subsequent degradation of target proteins that mediate BMP-induced responses in cell.
Fig. 11.
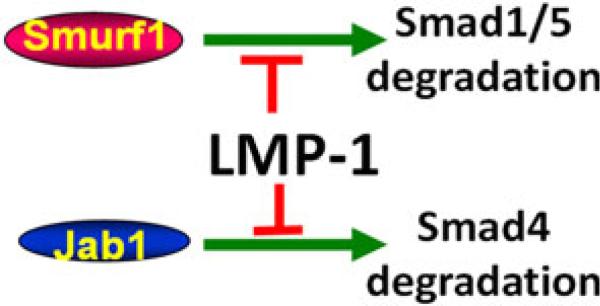
Smad1/5 and Smad4 are targeted by Smurf1 and Jab1 for degradation, respectively. LMP-1 interactions with Smurf1 and Jab1 are necessary to be fully functional in its BMP-potentiating activity by rescuing Smads1/5 and Smad4 from degradation. LMP-1 may also increase cellular responsiveness to TGF-β by rescuing Smad4 from degradation
Acknowledgments
All the biochemical studies in this study were performed at the Atlanta Veterans Affairs Medical Center and partly supported by the NIH Grant # R01 AR53093 (Boden) and a VA Merit award to Dr. Titus. The authors also thank Vandana Voleti for assistance in computational analyses. In the past and not related to this study, Dr. Boden had received compensation as a consultant for the Medtronic Sofamor Danek and for intellectual property. Emory University and some of the authors have/may receive royalties in the future related to LMP-1. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
Abbreviations
- BMP
Bone morphogenetic protein
- Jab1
Jun activation domain-binding protein 1
- RT-PCR
Reverse transcriptase polymerase chain reaction
- ALP
Alkaline phosphatase
- RUL
Relative units of luciferase
- FBS
Fetal bovine serum
- hMSCs
Human mesenchymal stem cells
- ECL
Enhanced chemiluminescence
- MOI
Multiplicity of infection
- Nano-LC-MS
Nano-liquid chromatography-mass spectrometry
Contributor Information
Sreedhara Sangadala, Atlanta VA Medical Center and Department of Orthopaedics, Emory University School of Medicine, Atlanta, GA 30329, USA; VAMC-Research Service, 1670 Clairmont Rd., Decatur, GA 30033, USA.
Katsuhito Yoshioka, Department of Orthopaedic Surgery, Kanazawa University School of Medicine, Kanazawa 920-8641, Japan.
Yoshio Enyo, Department of Orthopaedic Surgery, Kanazawa University School of Medicine, Kanazawa 920-8641, Japan.
Yunshan Liu, Atlanta VA Medical Center and Department of Orthopaedics, Emory University School of Medicine, Atlanta, GA 30329, USA.
Louisa Titus, Atlanta VA Medical Center and Department of Orthopaedics, Emory University School of Medicine, Atlanta, GA 30329, USA.
Scott D. Boden, Atlanta VA Medical Center and Department of Orthopaedics, Emory University School of Medicine, Atlanta, GA 30329, USA
References
- 1.Sangadala S, Boden SD, Viggeswarapu M, Liu Y, Titus L. LIM mineralization protein-1 potentiates bone morphogenetic protein responsiveness by a novel interaction with Smurf1 resulting in decreased ubiquitination of Smads. J Biol Chem. 2006;281:17212–17219. doi: 10.1074/jbc.M511013200. [DOI] [PubMed] [Google Scholar]
- 2.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996;383:453–457. doi: 10.1038/383453a0. [DOI] [PubMed] [Google Scholar]
- 3.Schwechheimer C, Deng XW. COP9 signalosome revisited: a novel mediator of protein degradation. Trends Cell Biol. 2001;11:420–426. doi: 10.1016/s0962-8924(01)02091-8. [DOI] [PubMed] [Google Scholar]
- 4.Chamovitz DA, Segal D. JAB1/CSN5 and the COP9 signalosome. A complex situation. EMBO Rep. 2001;2:96–101. doi: 10.1093/embo-reports/kve028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wan M, Cao X, Wu Y, Bai S, Wu L, Shi X, Wang N. Jab1 antagonizes TGF-beta signaling by inducing Smad4 degradation. EMBO Rep. 2002;3:171–176. doi: 10.1093/embo-reports/kvf024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh W, Lee EW, Sung YH, Yang MR, Ghim J, Lee HW, Song J. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J Biol Chem. 2006;281:17457–17465. doi: 10.1074/jbc.M601857200. [DOI] [PubMed] [Google Scholar]
- 7.Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, Dubiel W. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20:1630–1639. doi: 10.1093/emboj/20.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato J. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem. 2002;277:2302–2310. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- 9.Bae MK, Ahn MY, Jeong JW, Bae MH, Lee YM, Bae SK, Park JW, Kim KR, Kim KW. Jab1 interacts directly with HIF-1alpha and regulates its stability. J Biol Chem. 2002;277:9–12. doi: 10.1074/jbc.C100442200. [DOI] [PubMed] [Google Scholar]
- 10.Adler AS, Lin M, Horlings H, Nuyten DS, van de Vijver MJ, Chang HY. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421–430. doi: 10.1038/ng1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwok SF, Staub JM, Deng XW. Characterization of two subunits of Arabidopsis 19S proteasome regulatory complex and its possible interaction with the COP9 complex. J Mol Biol. 1999;285:85–95. doi: 10.1006/jmbi.1998.2315. [DOI] [PubMed] [Google Scholar]
- 12.Wei N, Tsuge T, Serino G, Dohmae N, Takio K, Matsui M, Deng XW. The COP9 complex is conserved between plants and mammals and is related to the 26S proteasome regulatory complex. Curr Biol. 1998;8:919–922. doi: 10.1016/s0960-9822(07)00372-7. [DOI] [PubMed] [Google Scholar]
- 13.Seeger M, Kraft R, Ferrell K, Bech-Otschir D, Dumdey R, Schade R, Gordon C, Naumann M, Dubiel W. A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J. 1998;12:469–478. [PubMed] [Google Scholar]
- 14.Wei N, Deng XW. Making sense of the COP9 signalsome: a regulatory protein complex conserved from Arabidopsis to human. Trends Genet. 1999;15:98–103. doi: 10.1016/s0168-9525(98)01670-9. [DOI] [PubMed] [Google Scholar]
- 15.Sangadala S, Okada M, Liu Y, Viggeswarapu M, Titus L, Boden SD. Engineering, cloning and functional characterization of recombinant LIM mineralization protein-1 containing an N-terminal HIV-derived membrane transduction domain. Protein Expr Purif. 2009;65:165–173. doi: 10.1016/j.pep.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Boden SD, Liu Y, Hair GA, Helms JA, Hu D, Racine M, Nanes MS, Titus L. LMP-1, a LIM-domain protein, mediates BMP-6 effects on bone formation. Endocrinology. 1998;139:5125–5134. doi: 10.1210/endo.139.12.6392. [DOI] [PubMed] [Google Scholar]
- 17.Wan M, Cao X, Wu Y, Bai S, Wu L, Shi X, et al. Jab1 antagonizes TGF-signaling by inducing Smad4 degradation. EMBO Rep. 2002;3:171–176. doi: 10.1093/embo-reports/kvf024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim BC, Lee HJ, Park SH, Lee SR, Karpova TS, McNally JG, et al. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor signaling by binding to Smad7 and promoting its degradation. Mol Cell Biol. 2004;24:2251–2262. doi: 10.1128/MCB.24.6.2251-2262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haag J, Aigner T. Jun activation domain–binding protein 1 binds Smad5 and inhibits bone morphogenetic protein signaling. Arthritis Rheum. 2006;54:3878–3884. doi: 10.1002/art.22261. [DOI] [PubMed] [Google Scholar]
- 20.Tomoda K, Yoneda-Kato N, Fukumoto A, Yamanaka S, Kato JY. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J Biol Chem. 2004;279:43013–43018. doi: 10.1074/jbc.M406559200. [DOI] [PubMed] [Google Scholar]