

Abstract
Objectives:
To evaluate the efficacy and safety of 18 months of tafamidis treatment in patients with early-stage V30M transthyretin familial amyloid polyneuropathy (TTR-FAP).
Methods:
In this randomized, double-blind trial, patients received tafamidis 20 mg QD or placebo. Coprimary endpoints were the Neuropathy Impairment Score–Lower Limbs (NIS-LL) responder analysis (<2-point worsening) and treatment-group difference in the mean change from baseline in Norfolk Quality of Life–Diabetic Neuropathy total score (TQOL) in the intent-to-treat (ITT) population (n = 125). These endpoints were also evaluated in the efficacy-evaluable (EE; n = 87) population. Secondary endpoints, including changes in neurologic function, nutritional status, and TTR stabilization, were analyzed in the ITT population.
Results:
There was a higher-than-anticipated liver transplantation dropout rate. No differences were observed between the tafamidis and placebo groups for the coprimary endpoints, NIS-LL responder analysis (45.3% vs 29.5% responders; p = 0.068) and change in TQOL (2.0 vs 7.2; p = 0.116) in the ITT population. In the EE population, significantly more tafamidis patients than placebo patients were NIS-LL responders (60.0% vs 38.1%; p = 0.041), and tafamidis patients had better-preserved TQOL (0.1 vs 8.9; p = 0.045). Significant differences in most secondary endpoints favored tafamidis. TTR was stabilized in 98% of tafamidis and 0% of placebo patients (p < 0.0001). Adverse events were similar between groups.
Conclusions:
Although the coprimary endpoints were not met in the ITT population, tafamidis was associated with no trend toward more NIS-LL responders and a significant reduction in worsening of most neurologic variables, supporting the hypothesis that preventing TTR dissociation can delay peripheral neurologic impairment.
Classification of evidence:
This study provides Class II evidence that 20 mg tafamidis QD was associated with no difference in clinical progression in patients with TTR-FAP, as measured by the NIS-LL and the Norfolk QOL-DN score. Secondary outcomes demonstrated a significant delay in peripheral neurologic impairment with tafamidis, which was well tolerated over 18 months.
Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a rare inherited amyloidosis that presents as a progressive sensorimotor and autonomic polyneuropathy.1,2 Axonal degeneration begins in small myelinated and unmyelinated fibers, resulting in sensory symptoms,3,4 progressing to larger myelinated fibers, causing muscle weakness and motor impairment.4 Gastrointestinal disturbances are a common autonomic manifestation, with malabsorption and cachexia developing in late-stage disease.1,4 Death occurs within a decade of symptom onset.3,4
TTR is a homotetrameric plasma protein comprising 127–amino acid monomers produced primarily by the liver. TTR has 2 thyroxine-binding sites and orthogonal retinol-binding protein/vitamin A complex sites.5,6 Mutations in TTR destabilize the tetramer, facilitating dissociation, the initial, rate-limiting step in amyloidogenesis (figure 1).7 This enables monomers to misfold and misassemble into amyloid.7 More than 100 TTR mutations have been linked to TTR-FAP,8 the most common of which is Val30Met (V30M).1 Evidence suggests that TTR amyloidogenesis leads to neurodegeneration and TTR-FAP.9,10
Figure 1. The TTR amyloidogenesis cascade is blocked by tafamidis-mediated kinetic stabilization of tetrameric TTR.
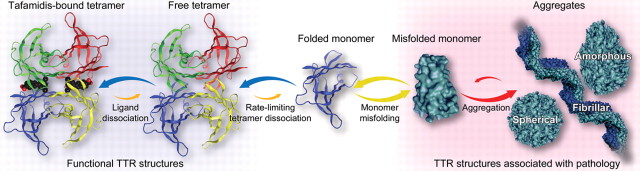
Tafamidis, depicted as the black space-filling structure with a red carboxyl group, binds to tetrameric TTR (far left), slowing TTR tetramer dissociation, which is the rate-limiting step for TTR amyloid fibril formation. Thus, the TTR-tafamidis complex is locked in a functional, nonamyloidogenic state, rendering the neurodegenerative amyloidogenesis cascade inaccessible. TTR = transthyretin.
The current standard of care for patients with TTR-FAP is liver transplantation, which replaces the source of mutant TTR with a genetically normal organ.11 However, the high perioperative mortality12 and morbidity associated with chronic immunosuppression13 highlight the need for safe, effective alternatives.
Interallelic trans-suppressor mutations inhibit amyloid formation via kinetic stabilization of tetrameric TTR and prevent TTR-FAP.9,14 Tafamidis, a small molecule that occupies the thyroxine-binding sites with negative cooperativity, kinetically stabilizes the tetramer.15 Thus, it was hypothesized that tafamidis would halt or slow neurodegeneration in TTR-FAP.
The primary objectives of this study were to evaluate the effect of 18 months of tafamidis (20 mg QD) on disease progression and assess its safety in patients with the V30M TTR mutation. A secondary objective was to determine the pharmacodynamic stabilization effect of tafamidis on human V30M TTR.
METHODS
Patients.
Men and women with TTR-FAP were enrolled at 8 sites in 7 countries (Argentina, Brazil, France, Germany, Portugal, Spain, Sweden). Key inclusion criteria were age 18 to 75 years, documented V30M TTR mutation, biopsy-confirmed amyloid deposits, and peripheral or autonomic neuropathy with a Karnofsky performance status ≥50. Key exclusion criteria were the presence of primary amyloidosis, other causes of sensorimotor neuropathy, absence of a recordable sensory threshold for vibration perception in both feet, liver function test abnormalities, prior liver transplantation, renal insufficiency (creatinine clearance <30 mL/min), NY Heart Association classification ≥3, any comorbidity anticipated to limit survival to <18 months, and chronic use of non–protocol-approved nonsteroidal anti-inflammatory drugs.
Study protocol.
Patients were randomized by a central computerized telerandomization system, in a 1:1 ratio, to self-administer once-daily tafamidis 20 mg [2-(3, 5-dichloro-phenyl)-benzoxazole-6-carboxylic acid] as a 1:1 meglumine [d-glucitol, 1-deoxy-1-(methylammonium)] salt or matching placebo. Dose and interval were determined using a pharmacokinetic/pharmacodynamic model to achieve serum tafamidis:TTR ratios of 1:1 to 2:1. The active drug was provided in soft-gelatin capsules containing a suspension of tafamidis and excipients. The packaging, appearance, and constitution of the placebo capsules were identical to those of the active-drug capsules except for the absence of tafamidis.
Study medication was initiated the day after the baseline visit. Patients returned to the clinical sites during the double-blind treatment period at weeks 2, 4, 8, and 12 and at months 6, 9, 12, and 18.
Outcome measures.
As with many rare diseases, there were no validated outcome measures for TTR-FAP. Therefore, measures of disease progression with demonstrated sensitivity and specificity in another axonal degenerative neuropathy—diabetic polyneuropathy (DPN)—were used.16,17 Detailed descriptions of the outcome measures are provided in appendix e-1 on the Neurology® Web site at www.neurology.org.
The Neuropathy Impairment Score–Lower Limbs (NIS-LL)16 quantifies the motor, sensory, and reflex functions in the lower limbs, which are most affected in early-stage TTR-FAP. To reduce variability, the NIS-LL for each patient was assessed by the same neurologist throughout the study. For each study visit, the NIS-LL was assessed twice within a 7-day period with an interval of at least 24 hours between tests. The 2 assessments were averaged to provide the visit score.
The Norfolk Quality of Life–Diabetic Neuropathy Questionnaire (Norfolk QOL-DN)17 is a 35-item, patient-reported questionnaire which provides a total quality of life (TQOL) score ranging from −2 (best possible quality of life [QOL]) to 138 (worst possible QOL).
Summated scores, which are obtained by summing multiple objective measures of nerve fiber impairment, have been used to detect disease progression in other neuropathies.18,19 The summated 7 nerve tests normal deviates (Σ7 NTs nds), which measures primarily large-fiber function, is scored from −26 (extreme normal function) to 26 (extreme abnormal function), and the summated 3 nerve tests small-fiber normal deviates (Σ3 NTSF nds), which measures small-fiber function, is scored from −11.2 (extreme normal function) to 11.2 (extreme abnormal function). For statistical analyses, individual test data were expressed as normal deviates based on healthy subject cohort data from the Mayo Clinic, Rochester, MN.
Modified body mass index (mBMI), a measure of wasting and autonomic gastrointestinal function, was calculated as the product of the BMI and serum albumin concentration (g/L).
TTR tetramer stabilization was assessed using a validated immunoturbidimetric assay performed on patients' plasma samples.15,20
Statistical analysis.
Primary endpoints.
The coprimary efficacy endpoints at month 18 were NIS-LL response to treatment (“responders” were patients with an increase from baseline in NIS-LL of <2 points21,22) and the least-squares mean (LS Mean) change from baseline in the Norfolk QOL-DN total (TQOL) scores. The primary efficacy analyses were performed on the intent-to-treat (ITT) population (all randomized patients who received at least 1 dose of study medication and who had ≥1 postbaseline assessment for both coprimary endpoints or who discontinued due to liver transplantation). For patients with postbaseline assessments, the last-observation-carried-forward method was used to impute missing data at month 18. Patients who discontinued due to liver transplantation were categorized as NIS-LL nonresponders. A χ2 test for proportions assessed treatment comparability for the NIS-LL responder outcome. An analysis of covariance (ANCOVA) with baseline as covariate assessed the treatment group difference in LS Mean change from baseline TQOL scores. The assumptions of the ANCOVA were assessed and met. Analyses of the coprimary endpoints were performed in an efficacy-evaluable (EE) population consisting of ITT patients who completed the study per protocol. This EE population was prespecified as it was anticipated that the majority of patients enrolled would be on the liver transplant list and that many would undergo liver transplantation during the study if a donor organ became available.
Absolute risk reduction (ARR) in the ITT population was calculated as the treatment group difference in the percentage of NIS-LL responders, and the number needed to treat (NNT) was calculated as the reciprocal of the ARR.
Secondary endpoints.
Multiple secondary endpoints were used to assess the efficacy of tafamidis, including change from baseline at months 6, 12, and 18 in NIS-LL, TQOL, Σ7 NTs nds, Σ3 NTSF nds, and mBMI. Analyses of the secondary endpoints were conducted in the ITT population using a repeated-measures analysis of variance model that included fixed effects for treatment, month, their interaction, and patient as a random effect. Only observed values were used. Within each treatment group, a 1-sample t test was used to determine whether the change from baseline in TQOL was significantly different from 0.
Post hoc models were used to investigate whether muscle weakness (measured by the NIS-LL) progressed in a distal-to-proximal fashion.
Sample size, based on the coprimary endpoints, assumed 2-sided tests, α = 0.05, 90% power, and a discontinuation rate of <10%. Response rates of 20% for placebo and 50% for tafamidis (a 30% difference) were anticipated, and a χ2 test was assumed for the NIS-LL analysis. A true difference of 0.6 SD was assumed between the groups in TQOL, where SD was the square root of the mean squared error of the ANCOVA model for change from baseline scores. Each group required 58 patients.
Standard protocol approvals, registrations, and patient consent.
This study (ClinicalTrials.gov: NCT00409175) was approved by the institutional review boards at each site. All patients provided written informed consent.
RESULTS
Patient disposition.
A total of 162 patients were screened and 128 were randomized to tafamidis (n = 65) or placebo (n = 63). Eighty-eight (69%) were on liver transplant waiting lists at enrollment (see figure e-1). Thirteen patients in each group (21%) discontinued treatment to undergo liver transplantation; 19 (73%) discontinued prior to the 12-month assessment.
Baseline demographics and clinical characteristics.
In general, patients had early-stage neurologic disease, greater involvement of small than large nerve fibers, relatively well-preserved nutritional status, and some impairment in QOL. Baseline characteristics of the tafamidis and placebo groups were similar (table e-1).
Coprimary endpoints.
In the ITT population at month 18, there was a trend toward more NIS-LL responders in the tafamidis group than in the placebo group (45.3% vs 29.5%; p = 0.068; figure 2A). The ARR was 15.8% (95% confidence interval [CI] −0.9% to 32.5), resulting in an NNT of 6.3 patients. Treatment group differences (−5.2-point difference; p = 0.116; 95% CI −11.8 to 1.3) in the LS Mean change from baseline in TQOL score at month 18 in the ITT population were not significant (figure 2B).
Figure 2. Coprimary endpoints in the ITT population and secondary analysis in the EE population.
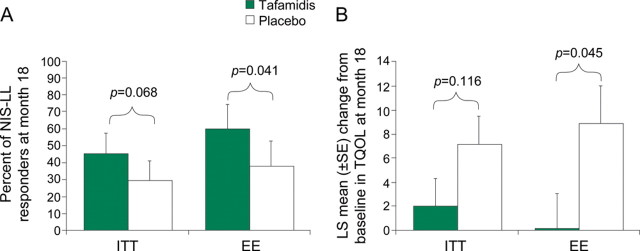
(A) Percentage of patients in each treatment group classified as NIS-LL responders at month 18 based on an increase of <2 points in NIS-LL overall score in both the ITT (primary analysis) and EE (prespecified secondary analysis) populations. EE = efficacy-evaluable; ITT = intent-to-treat; NIS-LL = Neuropathy Impairment Score–Lower Limbs. (B) LS Mean (±SE) change from baseline at month 18 in the TQOL score from the Norfolk QOL-DN in the same populations. LS Mean = least-squares mean; QOL-DN = Quality of Life–Diabetic Neuropathy Questionnaire; TQOL = Norfolk Quality of Life–Diabetic Neuropathy total score.
In the prespecified analyses of coprimary endpoints in the EE population, significantly more patients in the tafamidis group than in the placebo group were NIS-LL responders (60.0% vs 38.1%; p = 0.041; figure 2A). The LS Mean change from baseline in TQOL for tafamidis-treated patients was 0.1 point compared with 8.9 points for patients receiving placebo, a significant difference of 8.8 points (p = 0.045) (figure 2B). The within-treatment comparison (ITT population) demonstrated statistically significant worsening in QOL in patients who received placebo (LS Mean change from baseline, 7.2 ± 2.4; p = 0.002), but no change in tafamidis-treated patients (2.0 ± 2.3; p = 0.384).
Secondary endpoints.
In the repeated-measures analyses of secondary endpoints in the ITT population, tafamidis-treated patients demonstrated 52% less neurologic deterioration at month 18 than patients who received placebo, with a difference of 3 NIS-LL points (2.81 vs 5.83; p = 0.027) (figure 3A). This difference was due primarily to significantly more muscle weakness in the placebo group (p = 0.013). Patients who received placebo had significantly greater muscle weakness than tafamidis-treated patients at distal sites such as the hallux (p = 0.009) and ankle (p = 0.016), but not at more proximal joints, such as the knee (p = 0.054) and hip (p = 0.835). Nerve function was preserved in tafamidis-treated patients, but worsened in patients who received placebo. The latter group experienced 5 times greater mean deterioration in small-fiber function (Σ3 NTSF nds; p = 0.005) (figure 3B), with no trend toward more deterioration in large-fiber function (Σ7 NTs nds; p = 0.066) (figure 3C). Nutritional status at 18 months significantly improved in tafamidis-treated patients (mBMI increase from baseline [LS Mean ± SE] +39.3 ± 11.5) compared with a worsening mBMI in patients who received placebo (−33.8 ± 11.8; p < 0.0001) (figure 3D). Figure 3E depicts no trend toward preserved TQOL in the tafamidis group (p = 0.209). TTR stabilization at 18 months was demonstrated in 98% of tafamidis-treated patients and none of the patients who received placebo (p < 0.0001).
Figure 3. Secondary endpoints in the ITT population.

This figure shows the LS Mean (±SE) changes from baseline at months 6, 12, and 18 for the NIS-LL (A), small- (Σ3 NTSF nds) and large- (Σ7 NTs nds) nerve fiber function (B and C, respectively), mBMI (D), and TQOL (E). Analyses were performed using observed cases. ITT = intent-to-treat; LS Mean = least-squares mean; mBMI = modified body mass index; NIS-LL = Neuropathy Impairment Score–Lower Limbs; TQOL = Norfolk Quality of Life–Diabetic Neuropathy total score; Σ7 NTs nds = summated 7 nerve tests normal deviates; Σ3 NTSF nds = summated 3 nerve tests small-fiber normal deviates.
Adverse events.
The overall incidence of nonserious adverse events (AEs) was similar in both groups (table e-2), with AEs leading to drug discontinuation in 4 tafamidis-treated patients (6.2%) and 3 receiving placebo (4.8%). The incidence of serious AEs was similar in the tafamidis group (9.2%) and placebo group (7.9%). The only serious AE reported by >1 patient was urinary tract infection, which was reported by 2 tafamidis-treated patients. Complications following liver transplantation led to the deaths of 2 patients in the tafamidis group (cardiac tamponade postpacemaker insertion; unknown cause) and 3 patients in the placebo group (sepsis; hepatic failure; unknown cause). There were no clinically relevant effects on laboratory measures, including thyroid function.
DISCUSSION
This randomized, placebo-controlled, double-blind trial in patients with TTR-FAP assessed the ability of tafamidis to stabilize the TTR tetramer and evaluated its effect on clinical progression over 18 months. For the coprimary endpoints, the differences between treatments failed to achieve the prespecified statistical significance. Nevertheless, based on analyses in the EE population and secondary endpoints that demonstrated a significant reduction in neurologic deterioration, preservation of nerve fiber function, improved nutritional status, maintenance of QOL, and TTR stabilization, we contend that tafamidis had a beneficial effect on disease progression in patients with V30M TTR-FAP.
Designing the trial of a novel investigational agent in a rare and progressive disease for which no approved pharmacotherapeutic agent exists is inherently difficult.23 Without previous studies or extensive literature on the natural disease history to guide trial design, the choice of outcome measures, study duration, and statistical analyses (including power calculations and sample size determination) presents a substantial challenge. Moreover, for a therapy designed to influence disease progression, it is also necessary to evaluate the consistency of the treatment effect across multiple endpoints, each of which measures different aspects of the disease.
The NIS-LL is validated in DPN and measures motor, sensory, and reflex function in the limbs most affected in early-stage TTR-FAP.22 Like TTR-FAP, DPN involves small- and large-fiber neuropathy resulting in peripheral and autonomic symptoms. There are emerging data that suggest the NIS-LL is useful in assessing disease severity and can differentiate between disease stages in TTR-FAP.24,25 The NIS-LL responder analysis was chosen as a coprimary endpoint based on its use in a DPN registration trial21 and an expert consensus report that deemed a 2-point change to be the smallest change that is recognizable by a physician.22 More patients receiving tafamidis than placebo were NIS-LL responders in the ITT and EE populations, with the difference achieving statistical significance in the latter. The treatment effect in the EE population (22%) was nearer to the anticipated 30% than that observed in the ITT population (15%). Although an evaluable population analysis may overestimate treatment effects, the observed effect size is supported by the change from baseline in NIS-LL at 18 months in the ITT population. The 3-point difference between the treatment groups represented approximately 50% less neurologic deterioration in the tafamidis-treated patients, which was attributable to differences in muscle strength. This suggests that tafamidis treatment long-term may slow progression to ambulatory difficulties.
In addition to the effect of tafamidis on NIS-LL, the 55% and 84% preservation of large– and small–nerve fiber function, respectively, suggests that tafamidis may directly slow neurodegeneration. A 50% decrease in neurophysiologic deterioration is clinically meaningful26 and should result in better long-term outcomes in treated patients. Finally, one likely consequence of the preserved neurologic function by tafamidis was the trend to maintained QOL, as compared with the decline in QOL seen in the patients administered placebo over the 18 months. In contrast, QOL assessed in post-transplant patients is inconsistent; in some studies patients report satisfaction with the procedure while other studies report lower health-related QOL, particularly when compared with patients transplanted due to underlying liver disease.27–30
The mBMI predicts survival after liver transplantation and correlates highly with pretransplant neurologic function and duration of gastrointestinal symptoms.31 The patients in the present study had relatively normal baseline mBMI, consistent with early-stage disease. Tafamidis-treated patients experienced improvements in mBMI, while the patients who received placebo showed worsening. Continued worsening of mBMI over time is indicative of cachexia, a positive prognostic factor for mortality in TTR-FAP.32
Eighteen months of tafamidis treatment was well tolerated, with AE profile similar to placebo. The safety profile of tafamidis contrasts with that of liver transplantation, which has a reported 10% perioperative mortality.12
While this trial demonstrates the promise of tafamidis as a treatment for individuals with TTR-FAP, we acknowledge its limitations, most notably the inability to achieve statistical significance in the coprimary endpoints. The lower-than-expected treatment effect size for NIS-LL responder analysis in the ITT population was likely due to the higher-than-anticipated discontinuation rate due to liver transplantation (21% observed vs 10% estimated). Current clinical practice is to perform liver transplantation as early in the course of TTR-FAP as possible, and the timing in the current study suggests that patients chose transplantation when a donor organ became available and not as salvage therapy. For this reason, and due to the a priori designation of these patients as nonresponders in the ITT population, it is likely that the study was underpowered to demonstrate a statistical difference. The results in patients completing the 18-month treatment per protocol (EE population) provide an accurate measure of the treatment effects of tafamidis over that period of time.
Another potential limitation concerns the duration of the trial. While sufficient to observe effects on neurologic outcomes, a period of 18 months did not allow the assessment of longer-term outcomes, such as the occurrence of AEs arising from long-term TTR stabilization or the impact on survival, ambulation, and non-neurologic manifestations of disease, including cardiomyopathy. As such, patients were followed in a 12-month open-label extension study in which all received tafamidis 20 mg QD. In addition, the longer-term outcomes of tafamidis-treated patients will be followed in the Transthyretin Amyloidosis Outcomes Survey (THAOS), an observational registry established to improve understanding of the disease ( www.thaos.net).
Eighteen months of treatment with tafamidis 20 mg QD was well-tolerated by patients, and although the coprimary endpoints were not met, the totality of the results demonstrate the potential of tafamidis to slow neurologic deterioration and maintain nutritional status compared with placebo. These findings support the hypothesis that preventing TTR tetramer dissociation by tafamidis-mediated kinetic stabilization results in slowing of the neurodegenerative process and preservation of neurophysiologic function, which ultimately translates to maintenance of QOL. Future studies will address the effect of tafamidis on longer-term outcomes of TTR-FAP patients, including survival, ambulation, and cardiomyopathy.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Drs. Aaron and Etta Vinik from Eastern Virginia Medical School, Strelitz Diabetes Center, Norfolk, VA, for the use of the Norfolk Quality of Life-Diabetic Neuropathy Questionnaire in the tafamidis clinical trials program. Peter J. Dyck, MD, P. James B. Dyck, MD, Wolfgang Singer, MD, and Michelle L. Mauermann, MD, from the Mayo Clinic, Rochester, MN, assisted with the evaluation and interpretation of key secondary endpoints. Editorial support was provided by Stephen Towers, PhD, and Ali Shandiz, PhD, at Scientific Strategy Partners and was funded by Pfizer Inc.
GLOSSARY
- AE
adverse event
- ANCOVA
analysis of covariance
- ARR
absolute risk reduction
- CI
confidence interval
- DPN
diabetic polyneuropathy
- EE
efficacy-evaluable
- ITT
intent-to-treat
- LS Mean
least-squares mean
- mBMI
modified body mass index
- NIS-LL
Neuropathy Impairment Score–Lower Limbs
- NNT
number needed to treat
- QOL
quality of life
- QOL-DN
Quality of Life–Diabetic Neuropathy Questionnaire
- TQOL
total quality of life
- TTR-FAP
transthyretin familial amyloid polyneuropathy
Footnotes
Editorial, page 730
Supplemental data at www.neurology.org
Coinvestigators are listed on the Neurology® Web site at www.neurology.org.
AUTHOR CONTRIBUTIONS
Dr. Coelho: drafting/revising the manuscript for content, including medical writing for content, study concept or design, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Maia: drafting/revising the manuscript for content, including medical writing for content, and analysis or interpretation of data and acquisition of data. Dr. Martins da Silva: drafting/revising the manuscript for content, including medical writing for content, analysis or interpretation of data, and acquisition of data. Dr. Waddington Cruz: drafting/revising the manuscript for content, including medical writing for content, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Planté-Bordeneuve: drafting/revising the manuscript for content, including medical writing for content, study concept or design, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Lozeron: revising the manuscript for content, including medical writing for content, and acquisition of data. Dr. Suhr: drafting/revising the manuscript for content, including medical writing for content, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Campistol: drafting/revising the manuscript for content, including medical writing for content, study concept or design, and analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Conceição: drafting/revising the manuscript for content, including medical writing for content, study concept or design, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Schmidt: drafting/revising the manuscript for content, including medical writing for content, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Dr. Trigo: drafting/revising the manuscript for content, including medical writing for content, acquisition of data, and study supervision or coordination. Dr. Kelly: drafting/revising the manuscript for content, including medical writing for content, and study concept or design. Dr. Labaudinière: drafting/revising the manuscript for content, including medical writing for content, study concept or design, analysis or interpretation of data, and contribution of vital reagents/tools/patents. Dr. Chan: analysis or interpretation of data and statistical analysis and drafting/revising the manuscript for content. Mr. Packman: drafting/revising the manuscript for content, including medical writing for content, study concept or design, and analysis or interpretation of data. Dr. Wilson: drafting/revising the manuscript for content, including medical writing for content, and analysis or interpretation of data. Dr. Grogan: drafting/revising the manuscript for content, including medical writing for content, study concept or design, and analysis or interpretation of data.
STUDY FUNDING
Support provided by FoldRx which was acquired by Pfizer Inc in October 2010, NIH grant DK 46335, and FDA Orphan Drug grant FD-R-00(03414-01).
DISCLOSURE
T. Coelho's institution has received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010; T. Coelho has served on the scientific advisory board of Pfizer Inc and received funding from Pfizer Inc for scientific meeting expenses (travel, accommodations, and registration). She currently serves on the THAOS (natural history disease registry) scientific advisory board. L. Maia has received research support from a Portuguese government foundation, Fundação para a Ciência e a Tecnologia (FCT grant no. SFRH/BD/66216/2009). A. Martins da Silva has received support from Bayer-Schering Pharma AG, Biogen Idec Inc., Merck Serono S.A., and sanofi-aventis for MS clinical research done in Centro Hospitalar do Porto/Hospital de Santo António–Oporto. M. Waddington Cruz received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, as a clinical investigator; has served on the scientific advisory board of Pfizer Inc; received funding from Pfizer Inc for scientific meeting expenses (travel, accommodations, and registration); and received research support from the National Institutes of Health. She currently serves on the THAOS (natural history disease registry) scientific advisory board. V. Planté-Bordeneuve received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, as a clinical investigator, and serves on the THAOS (natural history disease registry) scientific advisory board, but did not receive compensation for this involvement. P. Lozeron has received honoraria from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, and travel support from Bayer Schering Pharma, Biogen, CSL Behring, Serono, and Teva. O. Suhr served as an advisor for Alnylam Pharmaceuticals, Isis Pharmaceuticals, and Pfizer Inc as well as having served as an advisor and receiving support as a clinical investigator from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010. He currently serves on the THAOS (natural history disease registry) scientific advisory board. J. Campistol received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, as a clinical investigator and has also received fellowships and grants from Astellas, Novartis, Roche, and Wyeth (Pfizer). I.M. Conceição received honoraria from serving on the scientific advisory board of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, and served as primary investigator, and received research support from, FoldRx Pharmaceuticals/Pfizer Inc. She currently serves on the THAOS (natural history disease registry) scientific advisory board. H.H.-J. Schmidt received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, as a clinical investigator. P. Trigo received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, as a clinical investigator. J. Kelly is a founder, shareholder (and option holder), and paid consultant for FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010. R. Labaudinière was an employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010. J. Chan was an employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, during the conduct of this trial and preparation of the manuscript. J. Packman was an employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in October 2010, during the conduct of this trial and preparation of the manuscript. A. Wilson was an employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in 2010, during the conduct of this trial and preparation of the manuscript. D. Grogan was an employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc in 2010, during the conduct of this trial and preparation of the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hund E, Linke RP, Willig F, Grau A. Transthyretin-associated neuropathic amyloidosis: pathogenesis and treatment. Neurology 2001;56:431–435. [DOI] [PubMed] [Google Scholar]
- 2.Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol 2003;17:909–927. [DOI] [PubMed] [Google Scholar]
- 3.Planté-Bordeneuve V, Lalu T, Misrahi M, et al. Genotypic-phenotypic variations in a series of 65 patients with familial amyloid polyneuropathy. Neurology 1998;51:708–714. [DOI] [PubMed] [Google Scholar]
- 4.Coutinho P, Martins da Silva A, Lopes Lima J, Resende Barbosa A. Forty years of experience with type I amyloid neuropathy: review of 483 cases. In: Glenner GG, Pinho e Costa P, Falcao de Freitas A. eds. Amyloid and Amyloidosis. Amsterdam: Excerpta Medica; 1980:88–98. [Google Scholar]
- 5.Blake CC, Geisow MJ, Swan ID, Rerat C, Rerat B. Structure of human plasma prealbumin at 2.5 A resolution: a preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J Mol Biol 1974;88:1–12. [DOI] [PubMed] [Google Scholar]
- 6.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science 1995;268:1039–1041. [DOI] [PubMed] [Google Scholar]
- 7.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci USA 2002;99(suppl 4):16427–16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 2003;10:160–184. [DOI] [PubMed] [Google Scholar]
- 9.Almeida MR, Alves IL, Terazaki H, Ando Y, Saraiva MJ. Comparative studies of two transthyretin variants with protective effects on familial amyloidotic polyneuropathy: TTR R104H and TTR T119M. Biochem Biophys Res Commun 2000;270:1024–1028. [DOI] [PubMed] [Google Scholar]
- 10.Koike H, Misu K, Sugiura M, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology 2004;63:129–138. [DOI] [PubMed] [Google Scholar]
- 11.Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 1991;40:242–246. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto S, Wixner J, Obayashi K, et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients' survival. Liver Transpl 2009;15:1229–1235. [DOI] [PubMed] [Google Scholar]
- 13.Winkler M, Brinkmann C, Jost U, Oldhafer K, Ringe B, Pichlmayr R. Long-term side effects of cyclosporine-based immunosuppression in patients after liver transplantation. Transplant Proc 1994;26:2679–2682. [PubMed] [Google Scholar]
- 14.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003;299:713–716. [DOI] [PubMed] [Google Scholar]
- 15.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA 2012;109:9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dyck PJ, Davies JL, Litchy WJ, O'Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study cohort. Neurology 1997;49:229–239. [DOI] [PubMed] [Google Scholar]
- 17.Vinik EJ, Hayes RP, Oglesby A, et al. The development and validation of the Norfolk QOL-DN, a new measure of patients' perception of the effects of diabetes and diabetic neuropathy. Diabetes Technol Ther 2005;7:497–508. [DOI] [PubMed] [Google Scholar]
- 18.Dyck PJ, Litchy WJ, Daube JR, Harper CM, Davies J, O'Brien PC. Individual attributes versus composite scores of nerve conduction abnormality: sensitivity, reproducibility, and concordance with impairment. Muscle Nerve 2003;27:202–210. [DOI] [PubMed] [Google Scholar]
- 19.Dyck PJ, O'Brien PC, Litchy WJ, Harper CM, Klein CJ. Monotonicity of nerve tests in diabetes: subclinical nerve dysfunction precedes diagnosis of polyneuropathy. Diabetes Care 2005;28:2192–2200. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Packman J, Labaudinière R, Bulawa C. Novel immunoturbidimetric method to monitor transthyretin (TTR) stability in plasma. Amyloid 2006;13:67.16911960 [Google Scholar]
- 21.Apfel SC, Schwartz S, Adornato BT, et al. Efficacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy: a randomized controlled trial. rhNGF Clinical Investigator Group. JAMA 2000;284:2215–2221. [DOI] [PubMed] [Google Scholar]
- 22.Diabetic polyneuropathy in controlled clinical trials: Consensus Report of the Peripheral Nerve Society. Ann Neurol 1995;38:478–482. [DOI] [PubMed] [Google Scholar]
- 23.Tambuyzer E. Rare diseases, orphan drugs and their regulation: questions and misconceptions. Nat Rev Drug Discov 2010;9:921–929. [DOI] [PubMed] [Google Scholar]
- 24.Kim DH, Zeldenrust SR, Low PA, Dyck PJ. Quantitative sensation and autonomic test abnormalities in transthyretin amyloidosis polyneuropathy. Muscle Nerve 2009;40:363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coelho T, Merkies I, Vinik A, et al. Relationship between objective measures of neuropathy and quality of life in stages of severity of transthyretin familial amyloid polyneuropathy. Amyloid 2010;17(suppl 1):138. [Google Scholar]
- 26.Arezzo JC. The use of electrophysiology for the assessment of diabetic neuropathy. Neurosci Res Commun 1997;21:13–23. [Google Scholar]
- 27.Drent G, Graveland CW, Hazenberg BP, Haagsma EB. Quality of life in patients with familial amyloidotic polyneuropathy long-term after liver transplantation. Amyloid 2009;16:133–141. [DOI] [PubMed] [Google Scholar]
- 28.Jonsén E, Suhr O, Athlin E, Wikström L. Quality of life after liver transplantation in patients with familial amyloidotic polyneuropathy. Amyloid 1996;3:124–129. [Google Scholar]
- 29.Jonsen E, Suhr OB, Tashima K, Athlin E. Early liver transplantation is essential for familial amyloidotic polyneuropathy patients' quality of life. Amyloid 2001;8:52–57. [DOI] [PubMed] [Google Scholar]
- 30.Telles-Correia D, Cortez-Pinto H, Barbosa A, Mega I, Monteiro E. Quality of life following liver transplantation: a comparative study between familial amyloid neuropathy and liver disease patients. BMC Gastroenterol 2009;9:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suhr OB, Holmgren G, Steen L, et al. Liver transplantation in familial amyloidotic polyneuropathy: follow-up of the first 20 Swedish patients. Transplantation 1995;60:933–938. [PubMed] [Google Scholar]
- 32.Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med 1994;235:479–485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.